

Abstract
P-glycoprotein (Pgp) is an ATP-binding cassette (ABC) transporter that plays a major role in cardiovascular drug disposition by effluxing a chemically and structurally diverse range of cardiovascular therapeutics. Unfortunately, drug-drug interactions (DDIs) with the transporter have become a major roadblock to effective cardiovascular drug administration because they can cause adverse drug reactions (ADRs) or reduce the efficacy of drugs. Cardiovascular ion channel inhibitors are particularly susceptible to DDIs and ADRs with Pgp because they often have low therapeutic indexes and are commonly coadministered with other drugs that are also Pgp substrates. DDIs from cardiovascular ion channel inhibitors with the transporter occur because of inhibition or induction of the transporter and the transporter’s tissue and cellular localization. Inhibiting Pgp can increase absorption and reduce excretion of drugs, leading to elevated drug plasma concentrations and drug toxicity. In contrast, inducing Pgp can have the opposite effect by reducing the drug plasma concentration and its efficacy. A number of in vitro and in vivo studies have already demonstrated DDIs from several cardiovascular ion channel inhibitors with human Pgp and its animal analogs, including verapamil, digoxin and amiodarone. In this review, Pgp-mediated DDIs and their effects on pharmacokinetics for different categories of cardiovascular ion channel inhibitors are discussed. This information is essential for improving pharmacokinetic predictions of cardiovascular therapeutics, for safer cardiovascular drug administration and for mitigating ADRs emanating from Pgp.
Keywords: P-glycoprotein, drug-drug interactions, cardiovascular drugs, ion channel inhibitors, pharmacokinetics
INTRODUCTION
Cardiovascular drug prescriptions have significantly increased over the past decade with over 15% of patients on multidrug regimens (a.k.a. polypharmacy) (1). Of hospitalized patients on cardiovascular medications, ~4% exhibited serious adverse drug reactions (ADRs) (2). Cardiovascular ion channel inhibitors, which are used to treat cardiac arrhythmia and hypertension, represent a major contributor of cardiovascular drug ADRs (3, 4). These drugs target channels and enzymes that control the flow of ions in vascular smooth muscle cells and the cardiomyocytes (3). This serves to regulate cardiac inotropy, maintain the resting potential of cardiovascular cells and control blood pressure (3). Unfortunately, many of these drugs such as digoxin have a low therapeutic index, so any changes to their plasma concentration can potentially lead to ADRs (3). Many cardiovascular drug fatalities from ADRs are the result of drug-drug interactions (DDIs) with cardiovascular ion channel inhibitors (5–7). In one study the cardiovascular ion channel inhibitor digoxin was implicated in a majority of preventable DDIs (6). In another study, the cardiovascular ion channel inhibitor amiodarone, which is known to have several significant DDIs, had the second highest frequency of ADRs (7).
One of the main contributors to cardiovascular ion channel inhibitor DDIs and ADRs is the ATP-binding cassette (ABC) P-glycoprotein (Pgp) transporter (8). In general, Pgp is a promiscuous drug transporter that can bind multiple drugs simultaneously (9), which includes interactions with a chemically and structurally diverse range of cardiovascular drugs (10, 11). These interactions in combination with the narrow therapeutic indexes makes Pgp particularly susceptible to cardiovascular DDIs. The transporter is also prone to DDIs because of its function in absorption, elimination and distribution of drugs (11). This function leads to changes in cardiovascular ion inhibitor drug plasma concentrations and results in ADRs or reduced drug efficacy (9).
A general overview of cardiovascular drugs and Pgp was published (11), but no recent comprehensive review has been published that discusses Pgp-mediated DDIs from cardiovascular ion channel inhibitors and their clinical consequences. This review discusses a wider range of cardiovascular ion channel inhibitors and provides a more detailed analysis of the observed pharmacokinetics than (11). This review also describes the pharmacodynamics of cardiovascular ion channel inhibitors including their targets and mechanisms of action. Then we discuss Pgp-mediated transport of the cardiovascular ion channel inhibitor and DDIs observed with the drug. This is followed by a discussion and comments on the observed pharmacokinetics of coadministering the drug.
1. CARDIOVASCULAR DISEASES (CVD) AND CURRENT TREATMENTS
Cardiovascular disease (CVD) is a leading cause of death worldwide (12). CVD represents a class of diseases of the vascular system that can involve blood vessels such as coronary artery disease and stroke, or the heart, which includes congestive heart failure and hypertension (12). A number of treatments have been developed to treat CVD that target receptors, channels and enzymes of the cardiovascular system (13–19). Since hypertension represents a major risk factor for several diseases within CVD (20), several drug classes have been developed to lower blood pressure (e.g. 13). Some of the most effective treatments for hypertension have targeted the angiotensin-renin-aldosterone system, which is the signaling pathway for regulating blood pressure and fluid balance (13). There are also antihypertensive drugs that target the α and β adrenergic receptors that affect the action of catecholamines, norepinephrine and epinephrine (14–16). Loop diuretics represent a third class of antihypertensive drug that reduce blood pressure by decreasing fluid volume as a result of inhibiting the Na+-K+-2Cl− symporter in the kidneys (21). To treat strokes, cardiovascular drugs have been designed to reduce blood clotting by targeting proteins within the coagulation cascade or involved in platelet aggregation (17). High cholesterol, which represents a major risk factor for CVD, is most often treated with statin drugs (18). These drugs lower high cholesterol by targeting 3-hydroxy-3-methyl-glutaryl (HMG)-CoA reductase, which is the rate-limiting enzyme in cholesterol biosynthesis (18). The cardiovascular drugs that are the focus of this review are cardiovascular ion channel inhibitors. These drugs are used to treat a range of diseases within CVD, including hypertension, cardiac dysrhythmias and atrial fibrillation (19). These drugs affect vascular physiology by directly inhibiting the flow of ions through Ca2+, K+ or Na+ channels or indirectly by increasing intracellular Ca2+ concentration through inhibition of the Na+/K+ ATPase (19). In addition to the reviewed drugs, there are emerging therapies for CVD that target chemokines, high density lipoproteins and microRNA and there has been some progress towards the actual regeneration of cardiomyocytes (22).
2. CARDIOVASCULAR ION CHANNELS
Cardiovascular ion channels control the flow of ions and function to regulate the heart rhythm and blood pressure (23, 24). They are found in cardiomyocytes of the heart and vascular smooth muscle cells of the arteries and veins where heterogeneous expression of these channels promotes proper heart rhythms and blood pressure (23, 24). Mutations in genes that code for the ion channels or alterations in their expression level leads to inherited or acquired cardiac arrhythmia (23). Table 1 shows several types of cardiovascular ion channels, including Ca2+, Na+ and K+ channels. The table also shows the channel isoforms, their general functions and cardiovascular diseases associated with them.
Table 1.
Types and function of cardiovascular ion channels
| Ion Channel | Relevant Types | Physiological Function | Associated Cardiovascular Diseases | References |
|---|---|---|---|---|
| Ca2+ channels |
|
|
arrhythmias, Brugada syndrome, hypertension | (25, 26) |
| K+ channels |
|
|
atrial fibrillation, Brugada syndrome, hypertension, long and short QT syndrome | (26, 27) |
| Na+ channels |
|
|
atrial standstill, Brugada syndrome, cardiac conduction disorders, dilated cardiomyopathy, erthromelalgia, long QT syndrome, nonprogressive familial heart block | 26, 28, 29) |
| Na+/K+ ATPases |
|
|
atrial fibrillation, heart failure | (30) |
Calcium Channels
Cardiovascular Ca2+ channels are voltage-dependent channels that control the flux of Ca2+ into vascular smooth muscle cells and cardiomyocytes (25). Inward flux of Ca2+ ions by the channel triggers additional Ca2+ release from the sarcoplasmic reticulum (25). Calcium binds and induces a conformational change in the troponin-tropomyosin complex that facilitates interaction between the actin filament and myosin, and leads to a muscle contraction (25). There are two types of cardiovascular Ca2+ channels, long-lasting (L)-type and transient (T)-type Ca2+ channels, that activate the inward flux of Ca2+ ions through relatively high and low voltage potentials, respectively (25). The L-type Ca2+ channels function to excite and contract muscle cells, while T-type Ca2+ channels serve a cardiac pacemaking function and regulate arterial resistance (25). Defects in the ion channel have been associated with various cardiovascular disorders, including arrhythmias and hypertension (26)
Potassium Channels
Potassium ion transport by these proteins is accomplished by several mechanisms including Ca2+ activation, voltage and ATP (27). These channels are a major regulator of vascular smooth muscle cell voltage and resting potential (27). The channels also function to regulate the duration of the action potential in the cardiac muscle (27). Abnormal functioning of this channel has been associated with hypertension and Brugada syndrome, which leads to increased risk of cardiac death (26, 27).
Sodium Channels
Voltage-gated sodium channels (NaV) control the Na+ ion flux through Na+− induced conformational changes (28). In the heart, influx by these channels is responsible for the initial fast upstroke of the cardiac action potential (28). There is also recent evidence that these channels contribute to the contractile response of vascular smooth muscle cells (29). Defects in this channel are associated with a number of cardiomyopathies including long QT syndrome (26, 28).
Na+/K+ ATPase
This ATP-dependent enzyme effluxes Na+ ions out, while pumping K+ ions into cardiovascular cells (30). The enzymes help maintain the resting potential and cell volume through osmosis, and indirectly decrease intracellular Ca2+ concentration through the Na+/Ca2+ exchanger (30). Defects in the energy-dependent transporter are associated with heart failure and atrial fibrillation (30).
3. ION CHANNEL INHIBITORS AND THEIR MECHANISMS OF ACTION
Ion channel inhibitors are used to redistribute ions and restore the natural rhythm of the heart by inhibiting specific channels and transporters of the cardiovascular system (23). These inhibitors can be classified as calcium, sodium, or potassium channel inhibitors and cardiac glycosides, but may fit into multiple ion channel inhibitor categories and may have an array of electrophysiological actions (31). For example, the antiarrhythmic drug quinidine is a well-known sodium channel blocker, but it also exerts action against potassium channels and α-adrenergic receptors (31). For simplicity, ion channel inhibitors in this review will be discussed according to their main site of action. Table 2 summarizes the general categories of cardiovascular inhibitors, their mechanism of action, clinical uses and ADRs.
Table 2.
Characteristics of cardiovascular ion channel inhibitors
| Inhibitor Class | Examples | Mechanism | Clinical Uses | ADRs | References |
|---|---|---|---|---|---|
| Ca2+ channel inhibitors | amlodipine diltiazem mibefradil nicardipine nifedipine verapamil | Inhibit or interfere with Ca2+ influx by binding to specific sites on the channel | angina, arrhythmias, hypertension, coronary artery disease | bradycardia, hypotension | (19, 32, 33) |
| K+ channel inhibitors | amiodarone dronedarone | Interfere with K+ ion flow | cardiomyopathy, atrial fibrillation and flutter | arrhythmias, blue-gray hyperpigmentation, hepatotoxicity, pulmonary fibrosis | (19, 34, 35) |
| Na+ channel inhibitors | flecainide phenytoin quinidine | Stabilize the inactivated channel conformation through fast and slow mechanisms | atrial flutter, tachycardias | bradyarrhythmias, slow atrial flutter, proarrhythmia | (31) |
| Cardiac Glycosides | digitoxin digoxin ouabain | Inhibit Na+/K+ ATPase and indirectly increase intracellular [Ca2+] | atrial fibrillation and heart failure | atrioventricular block, bradycardia, gastrointestinal and neurological disorders, ventricular arrhythmias, | (36–38) |
Calcium Channel Inhibitors
These drugs are used to treat a range of cardiovascular disorders including hypertension and arrhythmias (32, 33). Calcium channel inhibitors (or blockers) disrupt the flow of L- and/or T-type Ca2+ channels (19, 32, 33). This disruption increases the intracellular Ca2+ concentration in vascular smooth muscle cells and cardiomyocytes, which leads to vasodilation and promotes a regular heart rhythm (32, 33). ADRs from toxic concentrations of calcium channel inhibitors result in bradycardia (abnormally slow heart rate) and hypotension that can lead to serious complications including death (32, 33).
Potassium Channel Inhibitors
Potassium channel inhibitors prolong repolarization and the action potential duration in cardiomyocytes by interfering with conduction through potassium channels (19, 34). In general, these inhibitors are used to treat cardiomyopathy and atrial fibrillation (19, 34). They have also been used to treat ventricular and tachycardia during cardiac arrest (34). These drugs exhibit a range of ADRs including worsening arrhythmias, blue-gray hyperpigmentation and sudden cardiac arrest (35).
Sodium Channel Inhibitors
Sodium channel blockers are used to suppress heart tachycardias and atrial flutter by decreasing the flow of Na+ ions through the sodium channel and reducing the action potential duration (31). Reduction of Na+ ion flow by these inhibitors is accomplished by stabilizing the inactivated state of the Na+ channel through fast and slow mechanisms (31). ADRs from these inhibitors include bradyarrhythmias (slowed heart rate) and slow atrial flutter (31).
Cardiac Glycosides
Cardiac glycosides are a special type of ion channel inhibitor and include digitalis glycosides such as digoxin, digitoxin and ouabain (36). Clinically, these drugs are used to treat atrial fibrillation and flutter, and some cases of heart failure (37, 38). These drugs target the Na+/K+ ATPase in the heart, which indirectly increases intracellular Ca2+ concentration by the effect of decreasing intracellular Na+ levels on the sodium-calcium exchanger (36). The increase in intracellular Ca2+ corresponds with an increase in inotropy or force of contraction of the heart (36). In addition to cardiovascular ADRs, gastrointestinal and neuropsychological disorders are commonly observed with this class of drug (37, 38).
4. CHARACTERISTICS OF PGP AND ITS EFFECTS ON ION CHANNEL INHIBITOR DISPOSITION
Pgp is a member of the ATP-binding cassette (ABC) transporter superfamily and can efflux a chemically and structurally diverse range of molecules (10, 11). Fig. 1A shows the X-ray crystal structure of mouse Pgp, which consists of two nucleotide-binding domains (NBDs), 12 transmembrane (TM) helices and a large 6000 Å3 drug binding cavity (8). Fig. 1B shows the generally accepted model for ATP-driven drug efflux by Pgp. The left side of the panel depicts drugs binding to Pgp from the cell membrane or the cytosol within the TM region of Pgp with the NBDs separated (8, 39). The binding of two ATP molecules shifts the NBDs together (right side of Fig. 1B) and releases the drug to the extracellular side of the membrane (8, 39). ATP is hydrolyzed into ADP and inorganic phosphate (Pi) resetting the NBDs back to their initial conformation for another round of drug binding and transport (left side of Fig. 1B) (8, 39).
Fig. 1.
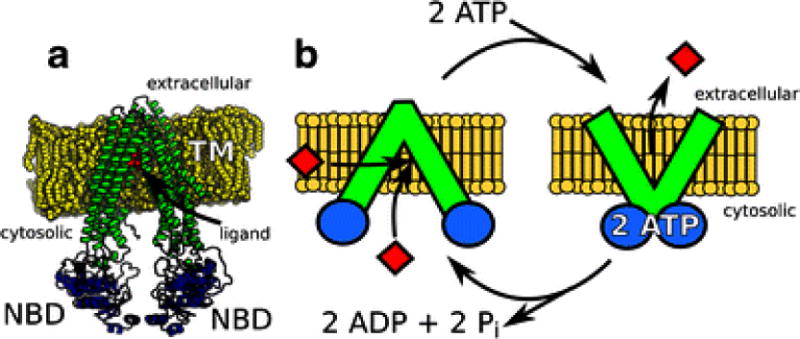
Structure and ATP-driven transport mechanism of Pgp. A) The X-ray crystal structure of mouse Pgp showing the transmembrane (TM, green), nucleotide-binding domains (NBDs, blue) and the position of QZ59-RRR, which is labeled “ligand” (red) in a phosphatidylcholine lipid bilayer (yellow) (8). B) Conformationally and ATP-driven model of efflux by Pgp. On the left of the panel, drug binding (red diamonds) occurs with the NBDs of Pgp separated. Binding of 2 ATP causes the NBDs to come together and leads to the release of drug (red diamonds) to the extracellular space. The extracellular and cytosolic sides of the cell membrane (yellow) are shown on the top and bottom of the membrane, respectively.
Pgp plays a key role in cardiovascular ion channel inhibitor DDIs by altering drug plasma concentrations and distribution, and by its cellular localization and expression levels. The transporter is found at relatively high concentrations on the lumenal side of enterocytes and reduces oral absorption and bioavailability by effluxing drugs back into the intestinal lumen (Fig. 2A) (40). The transporter is also found at relatively high concentrations on the lumenal side of kidney proximal tubule cells and the bile canalicular surface of hepatocytes decreasing drug plasma concentrations by excretion (Fig. 2B and 2C) (40). Pgp also affects drug distribution. Pgp is found on the blood side of epithelial cells located at the blood brain barrier (BBB) and reduces brain penetration of cardiovascular drugs (Fig. 2D) (40). Pgp is also present on the maternal side of placental trophoblasts preventing entry of cardiovascular drugs and protecting the unborn fetus (Fig. 2E) (41). Although there is Pgp present in the heart, it has little effect on drug disposition because of its relatively low concentration (42). Moreover, Pgp expression levels in all of these tissues can be influenced by genetic polymorphisms of Pgp, cardiomyopathy and different stages of pregnancy (43–45).
Fig. 2.
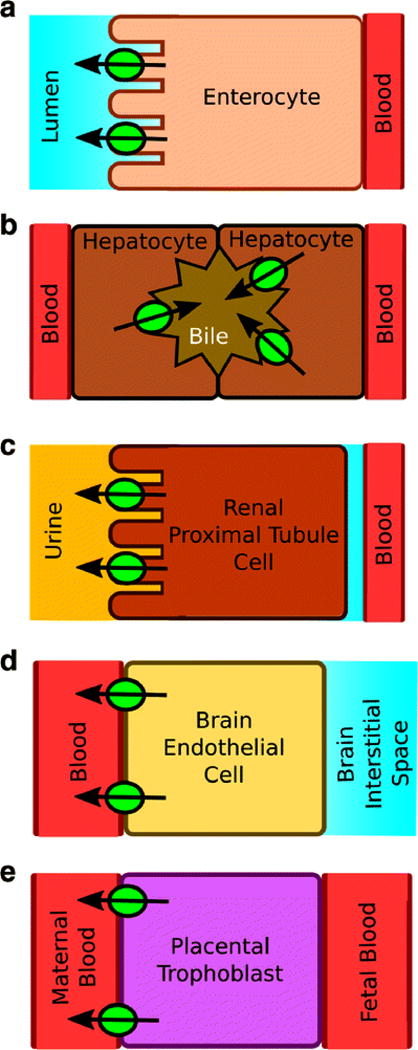
Pgp localization in A) an enterocyte, B) a hepatocyte, C) a kidney proximal tubule cell, D) a brain endothelial cell and E) a placental trophoblast. The green circles are Pgp and the arrows denote the direction of efflux.
Pgp-mediated DDIs result in significant changes in the drug pharmacokinetics by the inhibition or induction of the transporter. Inhibition of intestinal Pgp is saturable and will lead to an increase in oral absorption of Pgp substrates, while inhibition of excretory cells in the kidneys and the liver will reduce the clearance and increase the terminal elimination half-life (t1/2) of Pgp substrates (46). The combined inhibition will result in a net increase in the drug plasma concentration, and will lead to an increase in the peak drug plasma concentration (Cmax) and the individual’s exposure as reflected by the area under the curve (AUC) in the pharmacokinetics profile (46). This is particularly problematic for several cardiovascular ion channel inhibitors because of their relatively low therapeutic indexes and because elevated drug plasma concentration can lead to toxic drug plasma concentrations and serious ADRs (47). Inhibition of Pgp at the BBB or in the placental trophoblasts potentially increases penetration and toxic exposure to the brain and fetus, respectively (46). Pgp inhibition there can lead to changes in the drug’s distribution, which is reflected in the apparent volume of distribution (VD) (46). Induction of Pgp will have the opposite effect of Pgp inhibition by decreasing the drug plasma concentration and exposure, but might also significantly reduce a drugs’ efficacy. This will lead to a decrease in the Cmax and AUC in the pharmacokinetics profile (46). Pregnancy, age, sex and disease can also contribute to the pharmacokinetics and the clinically-observed DDIs (48).
5. IN VITRO ION CHANNEL INHIBITOR DDIs WITH PGP AND THE CORRESPONDING CLINICAL OBSERVATIONS
A number of in vitro studies have demonstrated that several cardiovascular ion channel inhibitors are substrates of and exhibit DDIs with Pgp. In some cases, the observed pharmacokinetics with the cardiovascular ion channel inhibitors seem to correlate with in vitro studies implying the involvement of Pgp. In other cases, the in vitro Pgp and the pharmacokinetics seem to contradict. In this section, in vitro DDI studies with Pgp and specific ion channel inhibitors are discussed and compared to the observed pharmacokinetics. The pharmacokinetic details associated with each DDI are summarized in Table 3.
Table 3.
Pgp-mediated DDIs of commonly prescribed cardiovascular ion channel inhibitors
| Drug | Sub.a | Inh.b | Ind. | Digoxin PK | Other | References |
|---|---|---|---|---|---|---|
| amiodarone | non | 6 μM | NA |
AUC: increases 66%c CLR: no change Cmax: increases 78%c VD: no change or decreases |
apixaband dabigatrand digitoxine daunorubicinf flecainidef rivaroxaband |
(55, 56, 58, 60–62) |
| amlodipine | non to good | NA | NA | Css: No change | simvastating | (65, 66, 68) |
| digoxin | good | - | Yes | - | quinidineh | (61, 69, 70, 72, 73, 78) |
| diltiazem | weak | 36 μM | NA |
AUC: increases 34%c Cmax: increases 31%c |
quinidineh | (61, 64, 82). |
| dronedarone | non | NA | NA |
AUC: increases 150% CLR: decreases 60% |
(59) | |
| flecainide | good | NA | NA | Cavg: increased ~20% | (57, 84) | |
| mibefradil | non | 7.5 μM | NA |
AUC: increases 31% Cmax: increases 41% |
atorvastating | (61, 87) |
| nicardipine | non | <1 μM | NA |
AUC: increases 6% Cmax: increases 6% |
(61, 64) | |
| nifedipine | non | <1 μM | NA |
AUC: increases 21%c Cmax: increases 5%c |
(61, 64) | |
| ouabain | X | No | Yes | NA | (71, 78, 80, 81) | |
| phenytoin | non to good | NA | NA |
AUC: decreases 23% CL: increases 27% CLR: no change VD: no change |
paclitaxeli | (90, 91, 93, 96) |
| quinidine | good | 21 μM | Yes |
AUC: increases 121%c CL: decreases 56% CLR: decreases 51% Cmax: increases 75% F: increases 16% t1/2: decreases 47% VD: decreases 38% |
edoxaband fentanylj flecainidef methadonej |
(57, 61, 70, 72, 98–102) |
| verapamil | non to good | 1–200 μM | NA |
AUC: increases 51% CL: decreases 34% CLH: decreases 62% CLR: decreases 21% Cmax: increases 44% t1/2: increases 31% VD: decreases 23% |
colchicinek dabigatrand prazosinl quinidineh vinblastinek |
(61, 64, 75, 86, 106, 112–114) |
Classification of a drug as a non-substrate (non), weak substrate (weak), good substrate (good) or non-ligand (X) to Pgp. A non-substrate had an efflux ratio = 1, a weak substrate had an efflux ratio >1 and <2, a good substrate had an efflux ratio >2 and a non-ligand was neither a substrate or inhibitor for Pgp.
In vitro inhibition concentration range of Pgp-mediated digoxin transport by the drug.
Average pharmacokinetic values.
Abbreviations:-, not applicable; AUC, area under the curve; Cavg, average drug plasma concentration; Css, steady-state drug plasma concentration; CL, total clearance CLH, extrarenal clearance; CLR, renal clearance; Cmax, peak drug plasma concentration; DEA, monodesethyl-amiodarone; Ind., inducer; Inh., inhibitor; NA, not available; Sub., Substrate; t1/2, terminal elimination half time; VD, apparent volume of distribution.
The more severe ADRs were myopathy (115),
digitalis-associated toxicities (116),
bleeding and thrombosis(119),
thrombocytopenia (120),
cardiac arrest (121),
orthostatic hypotension (126)l.
Amiodarone and Dronedarone
Amiodarone and dronedarone are potassium channel blockers used to treat cardiac dysrhythmias (49). Amiodarone is converted into the active metabolite monodesethyl-amiodarone (DEA) by cytochromes P450 in the liver (50). There is currently no evidence that amiodarone or dronedarone are actually transported by Pgp, but DEA was weakly transported by human Pgp in Caco-2 cells with an efflux ratio of 1.6 (51). These drugs are particularly prone to Pgp-mediated DDIs because of their unusually long elimination t1/2 (52, 53). While dronedarone has a t1/2 of ~24 hours (53), which is long by most standards, amiodarone and DEA have t1/2 of several days to over a month due to accumulation in adipose tissue (52, 54).
In vitro cell studies with porcine kidney epithelial cells overexpressing human Pgp have shown that both amiodarone and DEA inhibit transport of digoxin and the anticancer drug daunorubicin (55, 56). Amiodarone also inhibited transport of the sodium channel inhibitor flecainide in porcine kidney epithelial cells overexpressing human Pgp and in human intestinal epithelial LS180 cells (57).
These potassium channel inhibitors are also known to exhibit a number of DDIs in the clinic (e.g. 58, 59, 60). The pharmacokinetic consequences of amiodarone-digoxin DDIs have been the most thoroughly evaluated (e.g. 58, 61). Amiodarone causes ~70% increases in the Cmax and AUC of digoxin, while there were very little changes in VD of digoxin and surprisingly no significant decrease in the renal clearance (e.g. 58, 61). The authors explained the lack of renal clearance to an increase in intestinal absorption and a decrease in extrarenal clearance (58) implying the preferential inhibition of Pgp in the intestines and liver. Amiodarone also showed very strong DDIs with the related cardiac glycoside digitoxin leading to drug toxicity in several cases (62). Amiodarone was also found to increase the oral bioavailability of the anticoagulants, dabigatran, rivaroxaban and apixaban by ~10% through inhibition of intestinal Pgp (60, 63). In contrast, dronedarone showed even stronger DDIs with digoxin than amiodarone (59). The AUC of digoxin was almost 2-fold higher with dronedarone and there was a 60% decrease in renal clearance (59).
Amlodipine, Nicardipine and Nifedipine
The dihydropyridine drugs amlodipine, nicardipine and nifedipine are typically used in the treatment of hypertension and target the L-type Ca2+ channels (32). At pH 7.4, the drugs were Pgp ligands, but were not transported by Pgp (64, 65). At pH 6.5, amlodipine was efficiently transported by Pgp with an efflux ratio of ~10 (65), but it is unknown if nicardipine or nifedipine are also transported under these conditions. Digoxin transport by Pgp was inhibited by submicromolar concentrations of nifedipine and nicardipine (61). In the clinic, coadministration of nifedipine and digoxin lead to an increases in the Cmax and AUC in patients of 5% and 21%, respectively (61). DDIs from the coadministration of digoxin and nicardipine had a similar increase in Cmax, but the increase in the AUC was only ~6% (61). In contrast, despite its molecular similarity to nicardipine and amlodipine, amlodipine did not show significant clinical DDIs with digoxin (66). However, amlodipine did show clinical DDIs with simvastatin, which is a recognized Pgp substrate (67), with significant increases in the Cmax and AUC of simvastatin from 9.6 to 13.7 ng/ml and 34.3 to 43.9 ng • h/ml, respectively (68).
Digoxin
Digoxin is the most commonly prescribed cardiac glycoside and inhibits the Na+/K+ ATPase (47). The drug is primarily eliminated through the kidneys unmetabolized (38). Several in vitro studies with cells that express human Pgp have shown that digoxin is a good substrate for the transporter (e.g. 69, 70, 71). Because of digoxin’s low therapeutic index, the drug is administered at doses that it is unlikely to affect pharmacokinetic parameters of other drugs in the clinic (38). For example, digoxin only had minimal effects on the exposure of oral anticoagulant edoxaban (72). However, one study did find that the drug did increase the elimination t1/2 by ~20% and decreased renal clearance of quinidine at elevated doses, although digoxin’s affect on the AUC of quinidine was not statistically significant (73).
Therefore, most in vitro and in vivo studies have focused on inhibition of digoxin transport, which are known to occur with a number of Pgp ligands (61). In vivo, Pgp transport inhibition typically leads to significant increases in the AUC, Cmax, t1/2 and decreases in renal and extrarenal clearance of digoxin (61). Inhibition of intestinal Pgp often leads to increased oral absorption and bioavailability of the drug (61). Because Pgp is found at relatively high concentrations at the BBB (40), one might expect the VD of digoxin would increase significantly as well. In knockout mice lacking mouse Pgp, digoxin concentrations in the brain increased almost 30-fold versus wild type mice (74). Instead, the VD of digoxin often decreases in the presence of another drug (e.g. 75). One possibility is that Pgp inhibition at the BBB can be compensated by alternate efflux transporters including several isoforms of the multidrug resistance-associated protein (MRP) and the breast cancer resistance protein (BCRP) (76). This hypothesis is supported by that fact that digoxin is also a substrate for MRP2 (77). To complicate the digoxin pharmacokinetics further, digoxin can induce Pgp (78), which explains why Pgp-mediated DDIs between verapamil and digoxin were reduced after long-term coadministration (79).
Digitalis-related molecules
Digitalis-related molecules are functionally and structurally similar to digoxin (36), and include digitoxin, bufalin and strophanthidin. These molecules are generally good substrates of human Pgp like digoxin (80, 81). Digitoxin and bufalin both inhibited digoxin transport in Caco-2 cells containing human Pgp (71). Digitoxin also inhibited Pgp-mediated secretion of quinidine in the rat small intestine (82).
Diltiazem
Diltiazem is a benzothiazepine drug that targets L-type Ca2+ channels and is used in the treatment of hypertension and certain types of arrhythmia (32). The drug is known to be a relatively weak substrate of the transporter with an efflux ratio of 1.64 (64). Studies showed that diltiazem inhibited both quinidine and digoxin transport (61, 82). In the clinic, coadministration of diltiazem and digoxin to patients moderately increased the Cmax and AUC by about 30% (61).
Flecainide
Flecainide is an antiarrhythmic agent that specifically inhibits the NaV1.5 Na+ channel, which affect the fast depolarization phase of the cardiac action potential (31, 83). In vitro studies have shown that it is transported by Pgp with an efflux ratio of ~2 (57) and a clinical study showed that it increased blood plasma concentrations of digoxin by ~20% (84).
Mibefradil
Mibefradil is a non-specific inhibitor of both L- and T-type voltage-gated Ca2+ channels (85). The drug was weakly transported by Pgp in porcine kidney epithelial cells overexpressing the human and mouse Pgp (86). The drug appears to inhibit digoxin transport in human Pgp containing Caco-2 cells with low micromolar potency (61). In the clinic, the drug increases the Cmax and AUC of digoxin by 41 and 31%, respectively (61). The drug also shows strong Pgp-mediated DDIs with the cholesterol-lowering drug atorvastatin with a 4-fold increase in the AUC from 134 to 594 ng • h/ml when coadministered with mibefradil (87, 88).
Ouabain
Ouabain functions like other digitalis drugs (36) and is similar in structure to it, but only has a single sugar functional group rather than three. No Pgp-mediated transport of ouabain has been observed with mammalian cells expressing human Pgp (80, 81). Ouabain also did not inhibit digoxin in human epithelial Caco-2 cells (71). However, Pgp-mediated DDIs with this drug may occur indirectly through induction of the transporter (78).
Phenytoin
Phenytoin a sodium channel inhibitor that is used to treat abnormal heart rhythms and used as an alternative to digoxin (89). In vitro studies with human Pgp overexpressing cell lines and the drug have shown that it ranges from being a non-substrate to a substrate (c.f. 90, 91). In vivo, the drug has been shown to be a Pgp substrate with rats using Pgp-specific inhibitors (92). The drug inhibits transport of the anti-cancer drug paclitaxel in bovine retinal endothelial cells (93). However, in the clinic, phenytoin-paclitaxel interactions do not appear to result from Pgp-mediated DDIs, but from cytochrome P450-mediated DDIs due to cytochrome P450 induction (94, 95). Coadministration of this drug with digoxin in patients decreased the AUC and increased in the total clearance of digoxin around 20–30%, but no significant effects on the volume distribution or the renal clearance of digoxin was observed (96). This pharmacokinetic outcome is also consistent with cytochrome P450-mediated DDIs.
Quinidine
The sodium channel inhibitor quinidine is a stereoisomer of the anti-malarial drug quinine (31). In vitro and in vivo studies have demonstrated that this drug is a good substrate for Pgp (97). Quinidine is known to inhibit transport of several Pgp substrates (e.g. 57, 69). Several in vitro studies with cells expressing human Pgp have demonstrated that quinidine inhibits digoxin transport (61). In the clinic, coadministration of digoxin with quinidine decreased the terminal elimination t1/2, total and renal clearance, and the VD of digoxin in patients, while increasing its absorption and bioavailability (61, 98, 99). In mammalian cells expressing Pgp, quinidine inhibited transport of the sodium channel inhibitor flecainide (57). In the clinic, quinidine reduced the renal clearance of flecainide from 10.6 to 8.1 ml/min/kg (100). Quinidine also increased the AUC and Cmax of the oral anticoagulant edoxaban from 1577 to 2575 ng • h/ml and 223 to 390 ng/ml, respectively (72), and increased the oral bioavailability of methadone and fentanyl (101, 102). In addition to Pgp inhibition, quinidine also exhibits DDIs through Pgp induction (70).
6. CURRENT STRATEGIES FOR OVERCOMING PGP-MEDIATED DDIs IN THE CLINIC
In the clinic, finding alternative drug combinations to avoid DDIs all together is the most preferable strategy. For example, one can administer mibefradil and pravastatin, which does not elicit Pgp-mediated DDIs, instead of mibefradil and atorvastatin (87, 103). In many cases, this approach may not always be feasible, so methods have been developed to minimize ADR from DDIs.
The first step to minimize Pgp-mediated DDIs is to identify drugs that are known to exhibit DDIs (48, 104). For drugs that are coadministered, low therapeutic index drugs can be administered at subtherapeutic doses and the pharmacodynamic response monitored to minimize the risk of ADRs from DDIs (48, 104). Therapeutic drug monitoring (TDM) is another method to minimize ADRs from DDIs (104). In the method, the drug plasma concentration is measured directly in the blood or indirectly through biological fluids and correlated to a pharmacodynamic endpoint such as blood pressure (104). Although less common, another approach is to give a digitalizing dose, which is a series of small doses to control and achieve a therapeutic concentration and avoid ADRs (48).
Cardiovascular ion channel inhibitor DDIs and their observed pharmacokinetics discussed in the review are summarized in Table 3. The first column shows the name of the drug, while the next columns identify substrates, inhibitors and inducers of Pgp. Since many ion channel inhibitors have DDIs with digoxin (e.g. 61), their effects on digoxin PK parameters are shown in the next column. The penultimate column shows other observed DDIs and the last column are the corresponding references.
Table 3 shows that cardiovascular ion channel inhibitors range from being non-ligands to good substrates for Pgp. Although digoxin is often considered the gold standard for measuring Pgp-mediated DDIs (40), drugs with very similar molecular structures can have dramatically different inhibitory potency to digoxin transport such as nifedipine and amlodipine (61, 64, 66). Some of the drugs are also Pgp inducers (70, 78). In the clinic, cardiovascular ion channel inhibitors show a large range of effects on digoxin pharmacokinetics. Large increases in digoxin exposure and drug plasma concentration are observed in the presence of dronedarone (59), while decreases in digoxin plasma concentration are observed in the presence of phenytoin (96). Unfortunately, in vitro Pgp-mediated inhibition of digoxin transport and the observed pharmacokinetics are not well correlated. For example, despite being potent inhibitors of digoxin in vitro, nicardipine and nifedipine had relatively modest effects of digoxin pharmacokinetics in vivo (61, 64). The most studied cardiovascular ion channel inhibitors, digoxin, amiodarone and verapamil, are known to exhibit DDIs with several Pgp ligands. On the other hand, only a few Pgp-mediated DDIs have been noted in the literature with the other drugs, and this reflects a significant gap in our understanding of Pgp-mediated DDIs. Bridging this knowledge gap will require additional Pgp DDI studies in the future.
7. CONCLUSIONS AND FUTURE PERSPECTIVES
This review was focused on DDIs-mediated by Pgp, but, in reality, clinically-observed DDIs are multifactorial and reflects the complex interplay between drug metabolizing enzymes and transporters (105). For example, the pharmacokinetics profile from verapamil-quinidine DDIs reflects the combined inhibition of Pgp-mediated transport and drug metabolism by cytochromes P450 (106). Drug metabolites can also contribute significant Pgp-mediated DDIs (105) such as is the case with amiodarone and its metabolite DEA (55, 56). Alternative transporters such as MRP2 can potentially mitigate Pgp-mediated DDIs at the BBB and the placenta by effluxing the same drugs (76, 77, 107, 108). Influx transporters that actively transport Pgp inhibitors, and are found in specific tissues can potentially increase Pgp’s sensitivity to inhibitors skewing the pharmacokinetics. For example, amiodarone is a substrate of the organic anionic transporting polypeptide 2B1 (OAT2B1) influx transporter that is found in relatively high concentrations in hepatocytes and intestinal cells, but relatively low concentrations in the kidneys (109, 110). Under these conditions, Pgp will be more sensitive to amiodarone inhibition in hepatocytes and intestinal cells than kidney cells because of the higher intracellular amiodarone concentration mediated by OAT2B1. Under these conditions, we anticipate relatively high intestinal absorption and decreased extrarenal clearance as a result of Pgp inhibition and relatively little effect on renal clearance of Pgp substrates. This is exactly what we observe pharmacokinetically with amiodarone and digoxin (58).
Because of the involvement of alternate transporters and drug metabolizing enzymes in cardiovascular ion channel inhibitor disposition, extrapolating clinically observed DDIs to Pgp-mediated DDIs observed in vitro remains a significant challenge (111). To overcome this challenge, future in vitro studies with cardiovascular ion channel inhibitors will need to consider contributions from alternate transporters and drug metabolizing enzymes in addition to Pgp.
References
- 1.Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the united states from 1999–2012. JAMA. 2015;314(17):1818–30. doi: 10.1001/jama.2015.13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaidenstein R, Eyal S, Efrati S, Akivison L, Michowitz MK, Nagornov V, et al. Adverse drug events in hospitalized patients treated with cardiovascular drugs and anticoagulants. Pharmacoepidemiol Drug Saf. 2002;11(3):235–8. doi: 10.1002/pds.693. [DOI] [PubMed] [Google Scholar]
- 3.Tabrizchi R. Molecular mechanisms of adverse drug reactions in cardiac tissue. Handb Exp Pharmacol. 2010;(196):77–109. doi: 10.1007/978-3-642-00663-0_4. [DOI] [PubMed] [Google Scholar]
- 4.Gholami K, Ziaie S, Shalviri G. Adverse drug reactions induced by cardiovascular drugs in outpatients. Pharm Pract (Granada) 2008;6(1):51–5. doi: 10.4321/s1886-36552008000100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mowry JB, Spyker DA, Brooks DE, McMillan N, Schauben JL. 2014 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 32nd Annual Report. Clin Toxicol (Phila) 2015;53(10):962–1147. doi: 10.3109/15563650.2015.1102927. [DOI] [PubMed] [Google Scholar]
- 6.Karimzadeh I, Namazi S, Shalviri G, Gholami K. Cardiovascular drug adverse reactions in hospitalized patients in cardiac care unit. African Journal of Pharmacy and Pharmacology. 2011;5(4):493–9. [Google Scholar]
- 7.Mohebbi N, Shalviri G, Salarifar M, Salamzadeh J, Gholami K. Adverse drug reactions induced by cardiovascular drugs in cardiovascular care unit patients. Pharmacoepidemiol Drug Saf. 2010;19(9):889–94. doi: 10.1002/pds.1916. [DOI] [PubMed] [Google Scholar]
- 8.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glaeser H. Importance of P-glycoprotein for drug-drug interactions. Handb Exp Pharmacol. 2011;(201):285–97. doi: 10.1007/978-3-642-14541-4_7. [DOI] [PubMed] [Google Scholar]
- 10.Seelig A. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem. 1998;251:1–2. 252–61. doi: 10.1046/j.1432-1327.1998.2510252.x. [DOI] [PubMed] [Google Scholar]
- 11.Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61(25):2495–502. doi: 10.1016/j.jacc.2013.02.058. [DOI] [PubMed] [Google Scholar]
- 12.Luepker RV. Cardiovascular disease: rise, fall, and future prospects. Annu Rev Public Health. 2011;32:1–3. doi: 10.1146/annurev-publhealth-112810-151726. [DOI] [PubMed] [Google Scholar]
- 13.Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm. 2007;13(8 Suppl B):9–20. doi: 10.18553/jmcp.2007.13.s8-b.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helfand M, Peterson K, Christensen V, Dana T, Thakurta S. Drug Class Review: Beta Adrenergic Blockers: Final Report Update 4. Portland (OR): 2009. [PubMed] [Google Scholar]
- 15.Jensen BC, O’Connell TD, Simpson PC. Alpha-1-adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2011;51(4):518–28. doi: 10.1016/j.yjmcc.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giovannitti JA, Jr, Thoms SM, Crawford JJ. Alpha-2 adrenergic receptor agonists: a review of current clinical applications. Anesth Prog. 2015;62(1):31–9. doi: 10.2344/0003-3006-62.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harter K, Levine M, Henderson SO. Anticoagulation drug therapy: a review. West J Emerg Med. 2015;16(1):11–7. doi: 10.5811/westjem.2014.12.22933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith MEB, Lee NJ, Haney E, Carson S. Drug Class Review: HMG-CoA Reductase Inhibitors (Statins) and Fixed-dose Combination Products Containing a Statin: Final Report Update 5. Portland (OR): 2009. [PubMed] [Google Scholar]
- 19.Grant AO. Cardiac ion channels. Circ Arrhythm Electrophysiol. 2009;2(2):185–94. doi: 10.1161/CIRCEP.108.789081. [DOI] [PubMed] [Google Scholar]
- 20.Padwal R, Straus SE, McAlister FA. Evidence based management of hypertension. Cardiovascular risk factors and their effects on the decision to treat hypertension: evidence based review. BMJ. 2001;322(7292):977–80. doi: 10.1136/bmj.322.7292.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roush GC, Sica DA. Diuretics for Hypertension: A Review and Update. Am J Hypertens. 2016 doi: 10.1093/ajh/hpw030. [DOI] [PubMed] [Google Scholar]
- 22.Dimmeler S. Cardiovascular disease review series. EMBO Mol Med. 2011;3(12):697. doi: 10.1002/emmm.201100182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amin AS, Tan HL, Wilde AA. Cardiac ion channels in health and disease. Heart Rhythm. 2010;7(1):117–26. doi: 10.1016/j.hrthm.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Martens JR, Gelband CH. Ion channels in vascular smooth muscle: alterations in essential hypertension. Proc Soc Exp Biol Med. 1998;218(3):192–203. doi: 10.3181/00379727-218-44286. [DOI] [PubMed] [Google Scholar]
- 25.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3(8):a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JB. Channelopathies. Korean J Pediatr. 2014;57(1):1–18. doi: 10.3345/kjp.2014.57.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giudicessi JR, Ackerman MJ. Potassium-channel mutations and cardiac arrhythmias–diagnosis and therapy. Nat Rev Cardiol. 2012;9(6):319–32. doi: 10.1038/nrcardio.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28(5):287–94. doi: 10.1111/j.1755-5922.2010.00210.x. [DOI] [PubMed] [Google Scholar]
- 29.Ho WS, Davis AJ, Chadha PS, Greenwood IA. Effective contractile response to voltage-gated Na+ channels revealed by a channel activator. Am J Physiol Cell Physiol. 2013;304(8):C739–47. doi: 10.1152/ajpcell.00164.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suhail M. Na, K-ATPase: Ubiquitous Multifunctional Transmembrane Protein and its Relevance to Various Pathophysiological Conditions. J Clin Med Res. 2010;2(1):1–17. doi: 10.4021/jocmr2010.02.263w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roden DM, Darbar D, Kannankeril PJ. Antiarrhythmic Drugs. In: Willerson JT, Wellens HJJ, Cohn JN, Holmes DR, editors. Cardiovascular Medicine. London: Springer London; 2007. pp. 2085–102. [Google Scholar]
- 32.McDonagh MS, Eden KB, Peterson K. Drug Class Review: Calcium Channel Blockers: Final Report. Portland, OR: 2005. [PubMed] [Google Scholar]
- 33.Buckley N, Dawson A, Whyte I. Calcium channel blockers. Medicine. 2007;35(11):599–602. doi: 10.1016/j.mpmed.2007.08.025. [DOI] [Google Scholar]
- 34.Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8(12):982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siddoway LA. Amiodarone: guidelines for use and monitoring. Am Fam Physician. 2003;68(11):2189–96. [PubMed] [Google Scholar]
- 36.Fuerstenwerth H. On the differences between ouabain and digitalis glycosides. Am J Ther. 2014;21(1):35–42. doi: 10.1097/MJT.0b013e318217a609. [DOI] [PubMed] [Google Scholar]
- 37.Hauptman PJ, Kelly RA. Digitalis. Circulation. 1999;99(9):1265–70. doi: 10.1161/01.cir.99.9.1265. [DOI] [PubMed] [Google Scholar]
- 38.Ziff OJ, Kotecha D. Digoxin: The good and the bad. Trends Cardiovasc Med. 2016 doi: 10.1016/j.tcm.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Sharom FJ. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Front Oncol. 2014(4):41. doi: 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staud F, Cerveny L, Ceckova M. Pharmacotherapy in pregnancy; effect of ABC and SLC transporters on drug transport across the placenta and fetal drug exposure. J Drug Target. 2012;20(9):736–63. doi: 10.3109/1061186X.2012.716847. [DOI] [PubMed] [Google Scholar]
- 42.Couture L, Nash JA, Turgeon J. The ATP-binding cassette transporters and their implication in drug disposition: a special look at the heart. Pharmacol Rev. 2006;58(2):244–58. doi: 10.1124/pr.58.2.7. [DOI] [PubMed] [Google Scholar]
- 43.Meissner K, Sperker B, Karsten C, Meyer Zu Schwabedissen H, Seeland U, Bohm M, et al. Expression and localization of P-glycoprotein in human heart: effects of cardiomyopathy. J Histochem Cytochem. 2002;50(10):1351–6. doi: 10.1177/002215540205001008. [DOI] [PubMed] [Google Scholar]
- 44.Cascorbi I, Paul M, Kroemer HK. Pharmacogenomics of heart failure – focus on drug disposition and action. Cardiovasc Res. 2004;64(1):32–9. doi: 10.1016/j.cardiores.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Chung FS, Eyal S, Muzi M, Link JM, Mankoff DA, Kaddoumi A, et al. Positron emission tomography imaging of tissue P-glycoprotein activity during pregnancy in the non-human primate. Br J Pharmacol. 2010;159(2):394–404. doi: 10.1111/j.1476-5381.2009.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42(1):59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- 47.Ehle M, Patel C, Giugliano RP. Digoxin: clinical highlights: a review of digoxin and its use in contemporary medicine. Crit Pathw Cardiol. 2011;10(2):93–8. doi: 10.1097/HPC.0b013e318221e7dd. [DOI] [PubMed] [Google Scholar]
- 48.Rowland M, Tozer TN. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications Fourth ed Philadelphia, PA: Lipponcott. Williams & Wilkins; 2010. [Google Scholar]
- 49.Wegener FT, Ehrlich JR, Hohnloser SH. Dronedarone: an emerging agent with rhythm- and rate-controlling effects. J Cardiovasc Electrophysiol. 2006;17(Suppl 2):S17–20. doi: 10.1111/j.1540-8167.2006.00583.x. [DOI] [PubMed] [Google Scholar]
- 50.Elsherbiny ME, El-Kadi AO, Brocks DR. The metabolism of amiodarone by various CYP isoenzymes of human and rat, and the inhibitory influence of ketoconazole. J Pharm Pharm Sci. 2008;11(1):147–59. doi: 10.18433/j3sg66. [DOI] [PubMed] [Google Scholar]
- 51.Kimoto E, Seki S, Itagaki S, Matsuura M, Kobayashi M, Hirano T, et al. Efflux transport of N-monodesethylamiodarone by the human intestinal cell-line Caco-2 cells. Drug Metab Pharmacokinet. 2007;22(4):307–12. doi: 10.2133/dmpk.22.307. [DOI] [PubMed] [Google Scholar]
- 52.Latini R, Tognoni G, Kates RE. Clinical pharmacokinetics of amiodarone. Clin Pharmacokinet. 1984;9(2):136–56. doi: 10.2165/00003088-198409020-00002. [DOI] [PubMed] [Google Scholar]
- 53.Patel C, Yan GX, Kowey PR. Dronedarone. Circulation. 2009;120(7):636–44. doi: 10.1161/CIRCULATIONAHA.109.858027. [DOI] [PubMed] [Google Scholar]
- 54.Lafuente-Lafuente C, Alvarez JC, Leenhardt A, Mouly S, Extramiana F, Caulin C, et al. Amiodarone concentrations in plasma and fat tissue during chronic treatment and related toxicity. Br J Clin Pharmacol. 2009;67(5):511–9. doi: 10.1111/j.1365-2125.2009.03381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katoh M, Nakajima M, Yamazaki H, Yokoi T. Inhibitory effects of CYP3A4 substrates and their metabolites on P-glycoprotein-mediated transport. Eur J Pharm Sci. 2001;12(4):505–13. doi: 10.1016/S0928-0987(00)00215-3. [DOI] [PubMed] [Google Scholar]
- 56.Kakumoto M, Takara K, Sakaeda T, Tanigawara Y, Kita T, Okumura K. MDR1-mediated interaction of digoxin with antiarrhythmic or antianginal drugs. Biol Pharm Bull. 2002;25(12):1604–7. doi: 10.1248/bpb.25.1604. [DOI] [PubMed] [Google Scholar]
- 57.Horie A, Ishida K, Shibata K, Taguchi M, Ozawa A, Hirono K, et al. Pharmacokinetic variability of flecainide in younger Japanese patients and mechanisms for renal excretion and intestinal absorption. Biopharm Drug Dispos. 2014;35(3):145–53. doi: 10.1002/bdd.1877. [DOI] [PubMed] [Google Scholar]
- 58.Robinson K, Johnston A, Walker S, Mulrow JP, McKenna WJ, Holt DW. The digoxin-amiodarone interaction. Cardiovasc Drugs Ther. 1989;3(1):25–8. doi: 10.1007/BF01881526. [DOI] [PubMed] [Google Scholar]
- 59.Vallakati A, Chandra PA, Pednekar M, Frankel R, Shani J. Dronedarone-induced digoxin toxicity: new drug, new interactions. Am J Ther. 2013;20(6):e717–9. doi: 10.1097/MJT.0b013e31821106c9. [DOI] [PubMed] [Google Scholar]
- 60.Stollberger C, Finsterer J. Relevance of P-glycoprotein in stroke prevention with dabigatran, rivaroxaban, and apixaban. Herz. 2015;40(Suppl 2):140–5. doi: 10.1007/s00059-014-4188-9. [DOI] [PubMed] [Google Scholar]
- 61.Fenner KS, Troutman MD, Kempshall S, Cook JA, Ware JA, Smith DA, et al. Drug–Drug interactions mediated through P-glycoprotein: Clinical relevance and in vitro–in vivo correlation using digoxin as a probe drug. Clin Pharmacol Ther. 2009;85(2):173–81. doi: 10.1038/clpt.2008.195. [DOI] [PubMed] [Google Scholar]
- 62.Laer S, Scholz H, Buschmann I, Thoenes M, Meinertz T. Digitoxin intoxication during concomitant use of amiodarone. Eur J Clin Pharmacol. 1998;54(1):95–6. doi: 10.1007/s002280050427. [DOI] [PubMed] [Google Scholar]
- 63.Liesenfeld KH, Lehr T, Dansirikul C, Reilly PA, Connolly SJ, Ezekowitz MD, et al. Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non-valvular atrial fibrillation from the RE-LY trial. J Thromb Haemost. 2011;9(11):2168–75. doi: 10.1111/j.1538-7836.2011.04498.x. [DOI] [PubMed] [Google Scholar]
- 64.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, et al. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299(2):620–8. [PubMed] [Google Scholar]
- 65.Rausl D, Fotaki N, Zanoski R, Vertzoni M, Cetina-Cizmek B, Khan MZ, et al. Intestinal permeability and excretion into bile control the arrival of amlodipine into the systemic circulation after oral administration. J Pharm Pharmacol. 2006;58(6):827–36. doi: 10.1211/jpp.58.6.0013. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz JB. Effects of amlodipine on steady-state digoxin concentrations and renal digoxin clearance. J Cardiovasc Pharmacol. 1988;12(1):1–5. doi: 10.1097/00005344-198807000-00001. [DOI] [PubMed] [Google Scholar]
- 67.Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, et al. Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res. 2004;21(9):1686–91. doi: 10.1023/b:pham.0000041466.84653.8c. [DOI] [PubMed] [Google Scholar]
- 68.Nishio S, Watanabe H, Kosuge K, Uchida S, Hayashi H, Ohashi K. Interaction between amlodipine and simvastatin in patients with hypercholesterolemia and hypertension. Hypertens Res. 2005;28(3):223–7. doi: 10.1291/hypres.28.223. [DOI] [PubMed] [Google Scholar]
- 69.Elsby R, Surry DD, Smith VN, Gray AJ. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica. 2008;38:7–8. 1140–64. doi: 10.1080/00498250802050880. [DOI] [PubMed] [Google Scholar]
- 70.IS Haslam, K Jones, T Coleman, NL Simmons. Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84) Biochem Pharmacol. 2008;76(7):850–61. doi: 10.1016/j.bcp.2008.07.020. [DOI] [PubMed] [Google Scholar]
- 71.ME Cavet, M West, NL Simmons. Transport and epithelial secretion of the cardiac glycoside, digoxin, by human intestinal epithelial (Caco-2) cells. Br J Pharmacol. 1996;118(6):1389–96. doi: 10.1111/j.1476-5381.1996.tb15550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, et al. Drug-drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13(5):331–42. doi: 10.1007/s40256-013-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rameis H. Quinidine-digoxin interaction: are the pharmacokinetics of both drugs altered? . Int J Clin Pharmacol Ther Toxicol. 1985;23(3):145–53. [PubMed] [Google Scholar]
- 74.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94(8):4028–33. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pedersen KE, Dorph-Pedersen A, Hvidt S, Klitgaard NA, Nielsen-Kudsk F. Digoxin-verapamil interaction. Clin Pharmacol Ther. 1981;30(3):311–6. doi: 10.1038/clpt.1981.165. [DOI] [PubMed] [Google Scholar]
- 76.Eyal S, Hsiao P, Unadkat JD. Drug interactions at the blood-brain barrier: fact or fantasy? . Pharmacol Ther. 2009;123(1):80–104. doi: 10.1016/j.pharmthera.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.S Lowes, ME Cavet, NL Simmons. Evidence for a non-MDR1 component in digoxin secretion by human intestinal Caco-2 epithelial layers. Eur J Pharmacol. 2003;458:1–2. 49–56. doi: 10.1016/s0014-2999(02)02764-4. [DOI] [PubMed] [Google Scholar]
- 78.Riganti C, Campia I, Polimeni M, Pescarmona G, Ghigo D, Bosia A. Digoxin and ouabain induce P-glycoprotein by activating calmodulin kinase II and hypoxia-inducible factor-1alpha in human colon cancer cells. Toxicol Appl Pharmacol. 2009;240(3):385–92. doi: 10.1016/j.taap.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 79.Pedersen KE, Dorph-Pedersen A, Hvidt S, Klitgaard NA, Pedersen KK. The long-term effect of verapamil on plasma digoxin concentration and renal digoxin clearance in healthy subjects. Eur J Clin Pharmacol. 1982;22(2):123–7. doi: 10.1007/BF00542456. [DOI] [PubMed] [Google Scholar]
- 80.Gozalpour E, Wilmer MJ, Bilos A, Masereeuw R, Russel FG, Koenderink JB. Heterogeneous transport of digitalis-like compounds by P-glycoprotein in vesicular and cellular assays. Toxicol In Vitro. 2016;32:138–45. doi: 10.1016/j.tiv.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 81.Pauli-Magnus C, Murdter T, Godel A, Mettang T, Eichelbaum M, Klotz U, et al. P-glycoprotein-mediated transport of digitoxin, alpha-methyldigoxin and beta-acetyldigoxin. Naunyn Schmiedebergs. Arch Pharmacol. 2001;363(3):337–43. doi: 10.1007/s002100000354. [DOI] [PubMed] [Google Scholar]
- 82.Emi Y, Tsunashima D, Ogawara K-I, Higaki K, Kimura T. Role of P-glycoprotein as a secretory mechanism in quinidine absorption from rat small intestine. J Pharm Sci. 1998;87(3):295–9. doi: 10.1021/js970294v. [DOI] [PubMed] [Google Scholar]
- 83.Ramos E, O’Leary ME. State-dependent trapping of flecainide in the cardiac sodium channel. J Physiol. 2004;560(Pt 1):37–49. doi: 10.1113/jphysiol.2004.065003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lewis GP, Holtzman JL. Interaction of flecainide with digoxin and propranolol. Am J Cardiol. 1984;53(5):52B–7B. doi: 10.1016/0002-9149(84)90502-2. [DOI] [PubMed] [Google Scholar]
- 85.Leuranguer V, Mangoni ME, Nargeot J, Richard S. Inhibition of T-type and L-type calcium channels by mibefradil: physiologic and pharmacologic bases of cardiovascular effects. J Cardiovasc Pharmacol. 2001;37(6):649–61. doi: 10.1097/00005344-200106000-00002. [DOI] [PubMed] [Google Scholar]
- 86.Schwab D, Fischer H, Tabatabaei A, Poli S, Huwyler J. Comparison of in vitro P-glycoprotein screening assays: recommendations for their use in drug discovery. J Med Chem. 2003;46(9):1716–25. doi: 10.1021/jm021012t. [DOI] [PubMed] [Google Scholar]
- 87.Holtzman CW, Wiggins BS, Spinler SA. Role of P-glycoprotein in statin drug interactions. Pharmacotherapy. 2006;26(11):1601–7. doi: 10.1592/phco.26.11.1601. [DOI] [PubMed] [Google Scholar]
- 88.Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol. 2004;94(9):1140–6. doi: 10.1016/j.amjcard.2004.07.080. [DOI] [PubMed] [Google Scholar]
- 89.Eijkelkamp N, Linley JE, Baker MD, Minett MS, Cregg R, Werdehausen R, et al. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135(Pt 9):2585–612. doi: 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang C, Kwan P, Zuo Z, Baum L. In vitro concentration dependent transport of phenytoin and phenobarbital, but not ethosuximide, by human P-glycoprotein. Life Sci. 2010;86:23–24. 899–905. doi: 10.1016/j.lfs.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 91.Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Loscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007;52(2):333–46. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 92.Potschka H, Loscher W. In vivo evidence for P-glycoprotein-mediated transport of phenytoin at the blood-brain barrier of rats. Epilepsia. 2001;42(10):1231–40. doi: 10.1046/j.1528-1157.2001.01901.x. [DOI] [PubMed] [Google Scholar]
- 93.Maines LW, Antonetti DA, Wolpert EB, Smith CD. Evaluation of the role of P-glycoprotein in the uptake of paroxetine, clozapine, phenytoin and carbamazapine by bovine retinal endothelial cells. Neuropharmacology. 2005;49(5):610–7. doi: 10.1016/j.neuropharm.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 94.Baker AF, Dorr RT. Drug interactions with the taxanes: clinical implications. Cancer Treat Rev. 2001;27(4):221–33. doi: 10.1053/ctrv.2001.0228. [DOI] [PubMed] [Google Scholar]
- 95.Johannessen SI, Landmark CJ. Antiepileptic drug interactions -principles and clinical implications. Curr Neuropharmacol. 2010;8(3):254–67. doi: 10.2174/157015910792246254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rameis H. On the interaction between phenytoin and digoxin. Eur J Clin Pharmacol. 1985;29(1):49–53. doi: 10.1007/BF00547368. [DOI] [PubMed] [Google Scholar]
- 97.Fromm MF, Kim RB, Stein CM, Wilkinson GR, Roden DM. Inhibition of P-glycoprotein-mediated drug transport: A unifying mechanism to explain the interaction between digoxin and quinidine. Circulation. 1999;99(4):552–7. doi: 10.1161/01.cir.99.4.552. [DOI] [PubMed] [Google Scholar]
- 98.Schenck-Gustafsson K, Dahlqvist R. Pharmacokinetics of digoxin in patients subjected to the quinidine-digoxin interaction. Br J Clin Pharmacol. 1981;11(2):181–6. doi: 10.1111/j.1365-2125.1981.tb01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pedersen KE, Christiansen BD, Klitgaard NA, Nielsen-Kudsk F. Effect of quinidine on digoxin bioavailability. Eur J Clin Pharmacol. 1983;24(1):41–7. doi: 10.1007/BF00613925. [DOI] [PubMed] [Google Scholar]
- 100.Munafo A, Buclin T, Tuto D, Biollaz J. The effect of a low dose of quinidine on the disposition of flecainide in healthy volunteers. Eur J Clin Pharmacol. 1992;43(4):441–3. doi: 10.1007/BF02220625. [DOI] [PubMed] [Google Scholar]
- 101.Kharasch ED, Hoffer C, Altuntas TG, Whittington D. Quinidine as a probe for the role of P-glycoprotein in the intestinal absorption and clinical effects of fentanyl. J Clin Pharmacol. 2004;44(3):224–33. doi: 10.1177/0091270003262075. [DOI] [PubMed] [Google Scholar]
- 102.Kharasch ED, Hoffer C, Whittington D. The effect of quinidine, used as a probe for the involvement of P-glycoprotein, on the intestinal absorption and pharmacodynamics of methadone. Br J Clin Pharmacol. 2004;57(5):600–10. doi: 10.1111/j.1365-2125.2003.02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Becquemont L, Funck-Brentano C, Jaillon P. Mibefradil, a potent CYP3A inhibitor, does not alter pravastatin pharmacokinetics. Fundam Clin Pharmacol. 1999;13(2):232–6. doi: 10.1111/j.1472-8206.1999.tb00344.x. [DOI] [PubMed] [Google Scholar]
- 104.Shargel L, Yu ABC, Wu-Pong S. Applied biopharmaceutics & pharmacokinetics. 6th. New York: McGraw-Hill; 2012. [Google Scholar]
- 105.Pang KS, Rodrigues AD, Peter RM. Enzyme- and transporter-based drug-drug Interactions: progress and future challenges. New York: Springer; 2010. [Google Scholar]
- 106.Edwards DJ, Lavoie R, Beckman H, Blevins R, Rubenfire M. The effect of coadministration of verapamil on the pharmacokinetics and metabolism of quinidine. Clin Pharmacol Ther. 1987;41(1):68–73. doi: 10.1038/clpt.1987.11. [DOI] [PubMed] [Google Scholar]
- 107.Vahakangas K, Myllynen P. Drug transporters in the human blood-placental barrier. Br J Pharmacol. 2009;158(3):665–78. doi: 10.1111/j.1476-5381.2009.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holcberg G, Sapir O, Tsadkin M, Huleihel M, Lazer S, Katz M, et al. Lack of interaction of digoxin and P-glycoprotein inhibitors, quinidine and verapamil in human placenta in vitro. Eur J Obstet Gynecol Reprod Biol. 2003;109(2):133–7. doi: 10.1016/s0301-2115(02)00513-4. [DOI] [PubMed] [Google Scholar]
- 109.Segawa M, Ogura J, Seki S, Itagaki S, Takahashi N, Kobayashi M, et al. Rapid stimulating effect of the antiarrhythmic agent amiodarone on absorption of organic anion compounds. Drug Metab Pharmacokinet. 2013;28(3):178–86. doi: 10.2133/dmpk.dmpk-12-rg-010. [DOI] [PubMed] [Google Scholar]
- 110.Pan AZ, Dong XA, Zhang SJ, Xiang T, Chen ZX, Lin YW. [Study on mRNA and protein expressions of organic anion transporting polypeptide (oatp2b1) in rats with high fat diet and overstrain induced Pi deficiency syndrome] Zhongguo Zhong Xi Yi Jie He Za Zhi. 2013;33(7):953–7. [PubMed] [Google Scholar]
- 111.Prueksaritanont T, Chu X, Gibson C, Cui D, Yee KL, Ballard J, et al. Drug-drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 2013;15(3):629–45. doi: 10.1208/s12248-013-9470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hartter S, Sennewald R, Nehmiz G, Reilly P. Oral bioavailability of dabigatran etexilate (Pradaxa((R))) after co-medication with verapamil in healthy subjects. Br J Clin Pharmacol. 2013;75(4):1053–62. doi: 10.1111/j.1365-2125.2012.04453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ledwitch KV, Barnes RW, Roberts AG. Unraveling the complex drug-drug Interactions of the cardiovascular drugs, verapamil and digoxin, with P-glycoprotein. Biosci Rep. 2016;36(2) doi: 10.1042/BSR20150317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, et al. In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos. 2006;34(5):786–92. doi: 10.1124/dmd.105.008615. [DOI] [PubMed] [Google Scholar]
- 115.Feng Q, Wilke RA, Baye TM. Individualized risk for statin-induced myopathy: current knowledge, emerging challenges and potential solutions. Pharmacogenomics. 2012;13(5):579–94. doi: 10.2217/pgs.12.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moss AJ, Davis HT, Conard DL, DeCamilla JJ, Odoroff CL. Digitalis-associated cardiac mortality after myocardial infarction. Circulation. 1981;64(6):1150–6. doi: 10.1161/01.cir.64.6.1150. [DOI] [PubMed] [Google Scholar]
- 117.Ranger S, Sheldon R, Fermini B, Nattel S. Modulation of flecainide’s cardiac sodium channel blocking actions by extracellular sodium: a possible cellular mechanism for the action of sodium salts in flecainide cardiotoxicity. J Pharmacol Exp Ther. 1993;264(3):1160–7. [PubMed] [Google Scholar]
- 118.Keefe DL. Anthracycline-induced cardiomyopathy. Semin Oncol. 2001;28(4 Suppl 12):2–7. [PubMed] [Google Scholar]
- 119.Piazza G, Nguyen TN, Cios D, Labreche M, Hohlfelder B, Fanikos J, et al. Anticoagulation-associated adverse drug events. Am J Med. 2011;124(12):1136–42. doi: 10.1016/j.amjmed.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lefkowitz JM, Shapiro M. Quinine-induced thrombocytopenia. Can Fam Physician. 1986;32:1949–53. [PMC free article] [PubMed] [Google Scholar]
- 121.Ruiz-Casado A, Calzas J, Garcia J, Soria A, Guerra J. Life-threatening adverse drug reaction to paclitaxel. Postmarketing surveillance. Clin Transl Oncol. 2006;8(1):60–1. doi: 10.1007/s12094-006-0098-5. [DOI] [PubMed] [Google Scholar]
- 122.Hunt G, Bruera E. Respiratory depression in a patient receiving oral methadone for cancer pain. J Pain Symptom Manage. 1995;10(5):401–4. doi: 10.1016/0885-3924(95)00021-p. [DOI] [PubMed] [Google Scholar]
- 123.Smydo J. Delayed respiratory depression with fentanyl. Anesth Prog. 1979;26(2):47–8. [PMC free article] [PubMed] [Google Scholar]
- 124.Dixon AJ, Wall GC. Probable colchicine-induced neutropenia not related to intentional overdose. Ann Pharmacother. 2001;35(2):192–5. doi: 10.1345/aph.10184. [DOI] [PubMed] [Google Scholar]
- 125.Makinson A, Martelli N, Peyriere H, Turriere C, Le Moing V, Reynes J. Profound neutropenia resulting from interaction between antiretroviral therapy and vinblastine in a patient with HIV-associated Hodgkin’s disease. Eur J Haematol. 2007;78(4):358–60. doi: 10.1111/j.1600-0609.2007.00827.x. [DOI] [PubMed] [Google Scholar]
- 126.Thein T, Koene RA, Wijdeveld PG. Orthostatic hypotension due to prazosin. Lancet. 1977;1(8007):363. doi: 10.1016/s0140-6736(77)91163-1. [DOI] [PubMed] [Google Scholar]