

Abstract
Arboviruses such as Zika virus (ZIKV, Flaviviridae; Flavivirus) must replicate in both mammalian and insect hosts possessing strong immune defenses. Accordingly, transmission between and replication within hosts involves genetic bottlenecks, during which viral population size and genetic diversity may be significantly reduced. To help quantify these bottlenecks and their effects, we constructed 4 “barcoded” ZIKV populations that theoretically contain thousands of barcodes each. After identifying the most diverse barcoded virus, we passaged this virus 3 times in 2 mammalian and mosquito cell lines and characterized the population using deep sequencing of the barcoded region of the genome. C6/36 maintain higher barcode diversity, even after 3 passages, than Vero. Additionally, field-caught mosquitoes exposed to the virus to assess bottlenecks in a natural host. A progressive reduction in barcode diversity occurred throughout systemic infection of these mosquitoes. Differences in bottlenecks during systemic spread were observed between different populations of Aedes aegypti.
Keywords: Zika virus, Flavivirus, arbovirus, virus evolution, mosquito-borne virus
INTRODUCTION
Zika virus (ZIKV, Flaviviridae; Flavivirus) was responsible for a pandemic first noted on the Pacific Island of Yap in 2007 that expanded throughout the Americas beginning in 2013–14 [1,2]. ZIKV produces either an asymptomatic infection or mild febrile disease in most infected humans. However, the ongoing outbreak has been characterized by a high incidence of malformations in developing fetuses and neurological complications in adults resulting in massive public concern [3,4]. In addition to mosquito-borne transmission, ZIKV is capable of being spread sexually [5], via direct contact [6], and vertically in both humans and mosquitoes [7–9]. Understanding the dynamics of transmission is essential for developing strategies to prevent future pandemics caused by ZIKV or other arboviruses.
Transmission bottlenecks are a major stochastic force in virus evolution that occur when conditions prevent a significant proportion of the viral population from establishing infection in a new host or after some barrier. When the effective population size (Ne) is reduced following a population bottleneck, the effect of genetic drift is increased, which can have either negative or positive effects on viral fitness. In particular, arthropod-borne viruses (arboviruses) must replicate efficiently in two vastly different hosts in order to perpetuate in nature. Several bottlenecks may exist within each host. For example, for mosquito transmission to occur, arboviruses must first infect and replicate within the insect’s midgut epithelial cells and disseminate to secondary tissues such as the fat body, hemocytes, and the nervous system [10]. Following infection of these tissues, the virus must then productively infect the salivary glands to eventually be expectorated from the saliva during feeding. Any of these steps may impose genetic bottlenecks that reduce the genetic diversity of arboviruses including West Nile virus (WNV) [11], Powassan virus [12], and Venezuelan equine encephalitis virus [13]. Recovery of lost genetic diversity is dependent on error-prone virus replication that is vector species-specific and depends on virus population size and the time elapsed since the bottleneck event. Previously, we demonstrated that WNV genetic diversity is increased in Culex mosquitoes during systemic infection. However, in Aedes aegypti, this diversity becomes progressively lower as the virus passes through several anatomical barriers, ultimately to the saliva [11]. The degree to which replication in mosquitoes imposes bottlenecks on ZIKV populations has not been described. Repeated bottlenecks are known to reduce virus fitness [14] and therefore represent targets for interrupting virus transmission.
In this study we describe the construction and characterization of a set of ZIKV constructs that contain degenerate nucleotides that result in random “barcodes” within a small region of the genome. Barcoded viruses were characterized in vitro and then used to assess bottlenecks in mammalian and mosquito cell lines and in Ae. aegypti mosquitoes. We then used an amplicon-based next-generation sequencing technique to quantify the barcode frequencies and measure bottlenecks. These viruses represent a useful tool to study ZIKV population biology within ecologically relevant hosts.
RESULTS
Barcoded ZIKV viruses replicate at similar levels to wild-type ZIKV
Barcoded viruses have been used to track bottlenecks in different virus systems [13,15–18]. Using an approach similar to Fennessey et al., we introduced genetic barcodes consisting of 8 or 9 degenerate nucleotides into the ZIKV genome (strain PRVABC59, GenBank accession number KU501215). Two barcoded viruses were constructed in the coding sequence (bc1 in the NS2a protein (nucleotide position 4008–4031) and bc2 in the NS4a protein (nucleotide position 6639–6665), and two were constructed in the 3′ UTR (bc3 after position 10388 and bc4 after position 10475) (Fig. 1A). In order to do this, we used a novel technique called “bacteria-free cloning” or BFC (Fig. 1B). This technique has the advantage of facilitating the easy propagation of constructs that are toxic to E. coli and maintaining the degeneracy of the barcode sequences. Since no bacteria are used, the potential negative effects of lipopolysaccharide during transfection and recovery of virus are also mitigated. Virus replication for all four barcoded viruses and the wild-type clone-derived virus (wt ZIKV-IC) was assessed on four cell lines. Two mammalian cell lines, Vero and LLC-MK2, both primate kidney epithelial cells, were chosen because the former lacks a functional type-1 interferon (IFN) response while the latter does not [19]. The two insect cell lines, C6/36 and Aag2, derived from Aedes embryonic cells, were selected because the former lacks a functional RNAi response and the latter does not [20,21]. Upon infection at a multiplicity of infection (MOI) of 0.01 PFU/cell, all four cell lines supported replication of all four barcoded viruses and the wild-type derived infectious clone (Fig. 2A–D). Few differences were observed in multi-step growth curves between the viruses with the exception of bc4 replicating to higher levels in LLC-MK2 cells. As expected, all viruses replicated faster and to higher peak titers in cells lacking IFN or RNAi responses. Replication in Aag2 cells (Fig. 2D) was poor for all viruses, but titers on days 5 and 6 were significantly higher (p<0.05 by two-way ANOVA) as compared to day 0 for all viruses except for barcode 2 (bc2), indicative of replication.
Figure 1. Insertion of genetic barcodes into the ZIKV genome.

A) Insertion site of degenerate nucleotide barcodes into the genome of ZIKV. Four barcode viruses were constructed, two in the coding sequence and two in the 3′UTR. The coding changes are a consecutive series of degenerate synonymous nucleotide changes at the third codon position. B) Schematic of construction of barcode viruses using a bacteria-free cloning (BFC) approach. This approach uses Gibson assembly and rolling circle amplification in place of bacteria. Virus is then rescued by transfection in Vero cells.
Figure 2.

ZIKV barcode viruses replicate similar to wild-type clone derived virus and have differences in barcode diversity.
(A–D) The four barcode viruses have similar replication in two mammalian (Vero (A) and LLC-MK2 (B)) and two insect-derived (C6/36 (C) and Aag2 (D)) cell lines. Cells were infected at MOI 0.01. Titers were compared using a two-way ANOVA. E) The number of unique barcodes present in each barcoded virus after transfection (passage 0) or one passage at MOI 0.01 on Vero cells (n=3). Values were compared using a one-way ANOVA for each passage. F) Average genetic complexity at the barcode positions for each barcoded virus. Calculated as . Where ni is the frequency of each nucleotide at each barcode position and N is the total number of barcode calls at that position. Values were compared using a one-way ANOVA for each passage and between passages. **** p<0.0001. G) Frequency of individual barcodes. Each color represents a unique barcode. More colors indicate increased barcode diversity.
Barcodes in the coding sequence maintain more diversity than those in the 3′ UTR
A high level of nucleotide diversity in the barcode region is necessary to assess population size changes. Therefore, we sought to characterize the initial barcode composition of all four viruses using next-generation sequencing. By sequencing the wild-type clone stock, which should be predominantly wild-type sequence, we were able to generate a cutoff to differentiate between likely valid and possibly artifactual barcodes present in the samples. We tested three independent approaches to define which barcodes to consider ‘authentic.’ In the first approach (Approach ‘A’), we identified all the distinct non-WT barcodes detected in the wt ZIKV-IC and the ZIKV-bc1 stocks in the region encompassing the barcode. We subsequently calculated the arithmetic mean plus 3 times the standard deviation of the frequency of all the non-WT barcodes present in three replicates of the wt ZIKV-IC stock, even if the frequency of a specific barcode in the stock was 0%. This threshold frequency was 0.01% and resulted in 146 ‘authentic’ barcodes called in the ZIKV-bc1 stock. For the second approach (Approach ‘B’), we calculated the arithmetic mean plus 3 times the standard deviation of the frequency of all the non-WT barcodes present only in the wt ZIKV-IC stock. This threshold frequency was 0.10% and resulted in 37 ‘authentic’ barcodes called in the ZIKV-bc1 stock. For the final method (Approach ‘C’), we identified the highest frequency of the most common non-WT barcode present in any of the three replicates of the wt ZIKV-IC stock. This threshold frequency was 0.42% and resulted in 18 ‘authentic’ barcodes called in the ZIKV-bc1 stock. As this third calculation was the most conservative, we used 0.42% to be the minimum threshold to consider a barcode in ZIKV-bc1 as ‘authentic.’ Using this value, we included 18 sequences in our list of authentic barcodes, and followed these throughout the study. The same analysis was performed for the three other barcoded viruses. A list of the ‘authentic’ barcodes used for ZIKV-bc1 and their sequence is presented in Table 1. The number of unique barcodes present in the two coding sequence barcoded viruses (barcoded viruses 1 (bc1) and bc2) was significantly higher than the number of unique barcodes present in viruses with the 3′ UTR insertions (Fig. 2E, by one-way ANOVA; bc1 vs bc3 p=0.0051, bc1 vs bc4 p=0.0016, bc2 vs bc3 p=0.0107, bc2 vs bc4 p=0.0027). This was true in both the transfection-derived stock and after one passage in Vero cells (by one-way ANOVA; bc1 vs bc3 p=0.0045, bc1 vs bc4 p=0.0007, bc2 vs bc3 p=0.0088, bc2 vs bc4 p=0.0011). Diversity in the barcode region was also assessed by calculating the complexity present at the degenerate positions that were inserted in the barcode. In order to do this, we used Shannon’s index, which takes into account the frequency of each nucleotide present in the barcode sequence and the total number of barcode sequences present [22]. With greater complexity, the likelihood of encountering the same nucleotide (i.e. the same barcode) becomes lower, indicating greater diversity. Levels of complexity were similar among all barcodes in the initial stock following transfection (Fig. 2F). However, following a passage on Vero cells at MOI 0.01, the barcode 4 (bc4) virus (3′ UTR insertion) had a significant reduction in complexity in the barcode region (by one-way ANOVA; p<0.0001). The complexity of the other barcoded viruses remained relatively stable following a single passage (all p>0.05 by one-way ANOVA). Consistent with this result, we performed Kolmogorov-Smirnov tests (herein called KS tests) to compare the average (n=2) barcode distributions between two populations and found that only the bc4 virus barcode distribution was significantly different after one passage on Vero cells (p-values, bc1: .794, bc2: .5262, bc3: .954, bc4: .037). The proportion of barcodes present in the population during this passage is presented in Fig. 2G. In the stock viruses derived directly from transfection (passage 0), the diversity of barcoded viruses appears to be greater in bc1 virus than the others, consistent with data in Fig. 2E–F. Given that barcode diversity was highest and replication differences compared to the wild-type infectious clone were lowest in bc1 virus, we chose this for all future studies in vitro and in vivo.
Table 1.
List of ‘authentic’ barcodes in ZIKV-bc1 virus.
| Barcode Name | Barcode Sequence | Average Percentage in Stock±95% CI |
|---|---|---|
| BC_1 | CTCGCAGCACTGACTCCTCTTGCG | 16.18±0.19 |
| BC_2 | CTGGCCGCGCTGACTCCTCTCGCT | 10.19±0.22 |
| BC_3 | CTCGCTGCCCTCACACCTCTTGCA | 10.21±0.25 |
| BC_4 | CTGGCTGCACTAACTCCGCTGGCG | 5.42±0.04 |
| BC_5 | CTTGCAGCTCTAACCCCCCTAGCA | 4.16±0.05 |
| BC_6 | CTCGCTGCTCTGACTCCTCTCGCC | 3.88±0.07 |
| BC_7 | CTGGCTGCACTGACTCCCCTAGCC | 3.62±0.1 |
| BC_8 | CTAGCCGCACTAACGCCGCTAGCC | 3.79±0.02 |
| BC_9 | CTCGCGGCACTAACGCCGCTGGCG | 2.47±0.07 |
| BC_10 | CTAGCCGCCCTAACCCCGCTAGCG | 2.21±0.2 |
| BC_11 | CTGGCCGCGCTGACGCCGCTGGCG | 2.01±0.06 |
| BC_12 | CTTGCGGCCCTGACTCCTCTAGCG | 1.48±0.1 |
| BC_13 | CTCGCGGCGCTTACGCCTCTTGCC | 0.77±0.09 |
| BC_14 | CTTGCAGCGCTGACGCCTCTAGCC | 0.77±0.02 |
| BC_15 | CTAGCCGCTCTGACTCCGCTAGCG | 0.8±0.04 |
| BC_16 | CTTGCCGCTCTAACGCCCCTTGCC | 0.64±0.01 |
| BC_17 | CTCGCTGCCCTCACGCCGCTCGCT | 0.55±0.01 |
| BC_18 | CTAGCTGCTCTAACACCTCTAGCT | 0.43±0.05 |
| BC_WT | CTGGCTGCTCTGACACCACTGGCC | 0.08±0 |
| other BC | n/a | 30.33±0.55 |
| Degenerate pattern | CTNGCNGCNCTNACNCCNCTNGCN |
Reduced bottlenecks in mosquito cells lacking functional RNAi
In-vitro passaging experiments have been instrumental in dissecting basic evolutionary mechanisms and assessing host differences in arboviruses [23–25]. Therefore, we used four cell lines, Vero, LLC-MK2, C6/36, and Aag2, to analyze bottleneck forces in vitro after three passages. While all cells were infected at an MOI of 0.01 for the first passage, due to lower replication rates on LLC-MK2 and Aag2, passaging was performed at MOI 0.001 on these cells while an MOI of 0.01 was used on Vero and C6/36 cells. Following the three passages, the number of unique barcodes present in each of the samples was reduced significantly in all cell lines except C6/36, which lacks an RNAi response (Fig. 3A, by one-way ANOVA, all as compared to the input; Vero p=0.0133, C6/36 p=0.9893, LLC-MK2 p<0.0001, Aag2 p<0.0001). In addition, the barcode populations present in virus after passage in both Vero and C6/36 were not significantly different to the input population by the KS test (Table 2). Finally, as expected, the number of genomes that was estimated to initiate infection in C6/36 cells was higher than any of the other cell lines, consistent with the result that the bottleneck force in these cells is lower than the other 3 cell lines tested (Table 3). Virus passaged in C6/36 cells similarly maintained high levels of complexity and the distribution of barcodes after the three passages, as compared to the input (Fig. 3B–C).
Figure 3. ZIKV barcode viruses undergo cell-type specific reduction in barcode diversity.
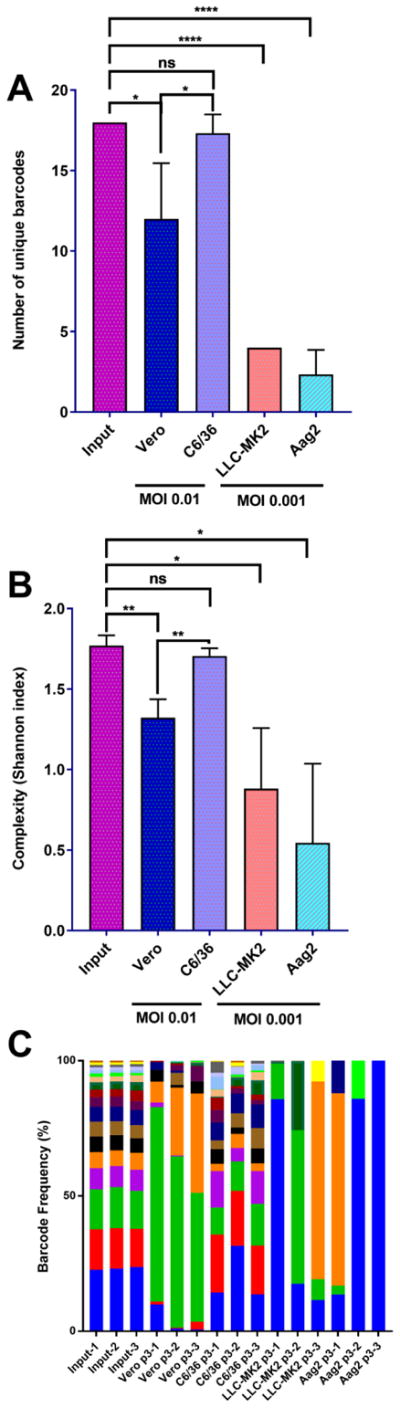
The four barcode viruses were subjected to 3 three serial passages in two mammalian (Vero and LLC-MK2) and two insect-derived (C6/36 and Aag2) cell lines. Passages were performed at MOI 0.01 for Vero and C6/36 and 0.001 for LLC-MK2 and Aag2. A) The number of unique barcodes present in each barcode virus iteration. Values were compared statistically using a one-way ANOVA with Tukey’s multiple comparisons test, * p<0.05 **** p<0.0001 ns p>0.05 B) Average genetic complexity at the barcode positions measured by Shannon’s index. Calculated as . Where ni is the frequency of each nucleotide at each barcode position and N is the total number of barcode calls at that position. Values were compared statistically using a one-way ANOVA with Tukey’s multiple comparisons test, * p<0.05 ** p<0.01 ns p>0.05 C) Frequency of individual barcodes. Each color represents a unique barcode. More bars indicate more barcode diversity. n=3 for all treatments. The number following the group label on the x-axis refers to the biological replicate.
Table 2.
Results of the Kolmogorov-Smirnov test to compare distribution probabilities after three passages in different cell lines as compared to unpassaged virus.
| Cell Line | p-value (as compared to input) | Significant ? |
|---|---|---|
| Vero | 0.0571 | No |
| C6/36 | 0.9639 | No |
| LLC-MK2 | 0.0002 | Yes |
| Aag2 | 0.0002 | Yes |
Bold indicates distributions are significantly different.
Table 3.
The estimated number of Zika virus genomes that founded the population in different cell lines, estimated using Fst.
| Cell Line | |||||
|---|---|---|---|---|---|
|
| |||||
| Input | Vero | C6/36 | LLCMK2 | Aag2 | |
| HTa | - | 0.8686 | 0.9258 | 0.8823 | 0.8886 |
| Hsa | 0.9421 | 0.6769 | 0.9051 | 0.7713 | 0.7730 |
| Nfa | - | 3.5525 | 25.4954 | 5.5163 | 5.5717 |
HT, Hs and Nf were calculated according to Monsion et al. 2008. Nf is the number of ZIKV-bc1 genomes that founded the population from one site to another.
A sequential reduction of barcode diversity occurs during infection and dissemination in mosquitoes characterized by stochastic forces
We exposed three different recently field-derived Ae. aegypti populations to an infectious bloodmeal containing 1.5e06 PFU/mL of either wild-type clone-derived or bc1 virus. Two of the 3 populations tested (Merida and Poza Rica, herein called Merida and PR) had relatively high rates of infectious virus expectorated in saliva, while one (Coatzacoalcos) had a lower transmission rate for wild-type PRVABC59 (Fig. 4A, by two-tailed Fisher’s exact test; p<0.0001 for Coatzacoalcos compared to Merida and PR). However, no difference in transmission rate for infectious virus was observed between the three groups for bc1 virus (Two-tailed Fisher’s exact test; Merida vs Coatzacoalcos p=1, Merida vs PR p=0.76, Coatzacoalcos vs PR p=0.57). In contrast, the percentage of mosquitoes with ZIKV RNA positive saliva was significantly higher for Merida and PR than Coatzacoalcos for the both wild-type PRVABC59 and bc1 virus (Supp. Fig. 1A, by two-tailed Fisher’s exact test; for wild-type PRVABC59 p<0.0001 for Coatzacoalcos compared to Merida and PR; for bc1 virus, Merida vs Coatzacoalcos p=.02, Merida vs PR p=0.34, Coatzacoalcos vs PR p<0.0001). Despite the difference in transmission rate as tested by PCR, there was no difference in average saliva titer between the three groups (Supp. Fig. 1B, one-way ANOVA with Tukey’s correction, all p>0.05). Five samples (bloodmeal, midguts, legs, salivary glands (sg) and saliva) were collected from three mosquitoes (for three biological replicates per population) of each population to represent possible anatomical barriers to virus transmission and sequenced using next-generation sequencing (NGS). For the Merida and PR populations, the drop in barcode complexity and number of unique barcodes from the bloodmeal to the midgut was not significant (Fig. 4B). In contrast, in comparing the bloodmeal and the midgut for the Coatzacoalcos population, the difference in the number of unique barcodes and the barcode complexity was significant (p=0.001) and approaching significance (p=0.051, both by one-way ANOVA), respectively. In all populations, both the complexity and number of unique barcodes decreased as the virus spread through the mosquito body to the saliva (Fig. 4B–C). This is clearly visualized in Fig. 4D–F, where in the majority of mosquitoes a single barcode population took over in the legs and predominated through to the saliva. In most cases, the barcode that was predominant in the saliva was different for each individual mosquito tested from each population. Additionally, the highest frequency barcode in the saliva was not necessarily the highest in the bloodmeal, as even the fifth and tenth most abundant barcode in the bloodmeal was able to take over after midgut infection (Fig. 4F). Finally, it was possible for a barcode that was not the most frequent or even absent in previous tissues to either take over in the saliva (See Fig. 4, panel E, sample 3; blue color in replicate 1 or brown in replicate 2) or re-emerge. The titer of bc1 virus in the saliva in both PFU/mL and genome equivalents/mL of the mosquitoes that were used for sequencing is presented in supplementary table 1.
Figure 4.

Stochastic forces dominate the sequential reduction in barcode diversity during replication in Aedes aegypti mosquitoes.
Mosquitoes were given infectious ZIKV bloodmeals and dissected for midguts, legs, salivary glands and saliva 14 days later. A) Percent of mosquitoes with infectious virus in the saliva 14 days post-exposure. Comparison between wild-type clone derived ZIKV and barcode 1 virus. n=16–32 for each group. Statistical comparisons were made using a two-tailed Fisher’s exact test. B) Average genetic complexity at the barcode positions measured by Shannon’s index. Calculated as . Where ni is the frequency of each nucleotide at each barcode position and N is the total number of barcode calls at that position. This was measured in the three different mosquito populations across tissue type. Statistical comparisons were made using a one-way ANOVA with Tukey’s correction for multiple comparisons. C) The number of unique barcodes present in each barcode virus iteration. Statistical comparisons were made using a one-way ANOVA with Tukey’s correction for multiple comparisons. D–F) Frequency of individual barcodes from different tissues from the three mosquito populations; Merida (D), Coatzacoalcos (E), and Poza Rica (F). Each color represents a unique barcode. The light green portion at the top of each bar represents the remaining proportion of barcodes that were not considered “authentic”. More bars indicate more barcode diversity. n=3 for all treatments.
Further analyses to compare distributions of barcodes between tissues using the KS test showed that the population of barcodes present in the midgut samples was not significantly different in the Merida and Poza Rica mosquitoes, as compared to the bloodmeal control (Table 4). The population in the legs was significantly different for both than in the midgut, however. In contrast, the population in the midgut was significantly different from that of the bloodmeal in Coatzacoalcos mosquitoes, while the legs were not significantly different than the midguts. Finally, we estimated effective population size (Nf) using Fst statistics with previously published statistical methods [26]. In the midguts, Nf was highest in the Merida and Poza Rica populations and lowest in Coatzacoalcos, consistent with complexity and KS test results (Table 5). Also consistent with KS test results, Nf for the population infecting the legs from the midguts was highest in Coatzacoalcos. Generally, Nf continued to increase or stay stable after dissemination from the midgut.
Table 4.
Results of the Kolmogorov-Smirnov test to compare distribution probabilities between tissues compared between three distinct mosquito populations.
| p-value (as compared to previous tissue) | ||||
|---|---|---|---|---|
|
| ||||
| Mosquito Population | Midgut | Legs | Salivary Glands | Saliva |
| Merida | 0.27 | 0.0222 | 0.7658 | 0.7658 |
| Coatzacoalcos | 0.0077 | 0.1314 | 0.9999 | >0.9999 |
| Poza Rica | 0.7658 | 0.0024 | 0.9999 | 0.9999 |
Bold indicates distributions are significantly different.
Table 5.
The estimated number of Zika virus genomes that founded the population in different tissues as compared between three distinct mosquito populations, estimated using Fst.
| NF (estimated pop. size founding infection in this tissue | ||||
|---|---|---|---|---|
|
| ||||
| Mosquito Population | Midgut | Legs | Salivary Glands | Saliva |
| Merida | 169.94 | 2.96 | 8.84 | 23.46 |
| Coatzacoalcos | 5.23 | 59.6 | 33.17 | 51.17 |
| Poza Rica | 43.72 | 8.26 | 17.18 | 731.11 |
DISCUSSION
We developed a barcoded ZIKV to measure population bottlenecks both in vitro and in vivo. Using a novel approach called bacteria-free cloning, we constructed four distinct barcoded viruses and characterized their in vitro growth characteristics and barcode composition. Using one of the barcoded viruses, called bc1 virus, we characterized bottlenecks in vitro in several cell lines of both mammalian and mosquito origin. C6/36 cells, RNAi deficient Ae. albopictus cells, were shown to maintain the most barcode diversity after three passages. In order to characterize bottlenecks in vivo, three distinct populations of recently field-derived Ae. aegypti mosquitoes were exposed to bc1 virus. Multiple populations were employed as it is known that large vector competence and therefore possibly bottleneck differences can exist between different collections of Ae. aegypti [28]). Bottleneck strength differed between different mosquito populations of the same species, particularly as the virus infected the midgut. Sequencing of the barcode region in different tissues representing different points for potential bottlenecks emphasized the stochastic nature of replication and transmission of ZIKV in mosquitoes. The barcodes present at a high frequency (>10%) in the bloodmeal stock virus commonly were the most abundant in the saliva, although this was not always the case, suggesting that the input virus population structure is a critical determinant of what goes on to be transmitted.
Barcoded viruses have become important tools to investigate virus population dynamics. Several technical strategies have been used to construct these viruses [13, 15–18]. While some approaches used a mixture of known tags, approaches that are more recent have incorporated degenerate nucleotides that increase the theoretical number of barcodes present in a population [18]. We sought to use a similar approach and introduced 8 or 9 degenerate nucleotides in either the coding sequence (CDS) or 3′UTR of an infectious clone of ZIKV strain PRVABC59, which was originally derived from an infected individual in Puerto Rico in 2015. We found two areas in the region of the genome encoding the non-structural proteins that allowed for the introduction of either 8 or 9 synonymous changes. For the 3′UTR, we used the RNA secondary structure of ZIKV to identify two regions that would allow insertion of an 8-nucleotide barcode sequence [29].
The flavivirus 3′UTR is highly structured and known to be sensitive to indels that disrupt native RNA folding. Several deletions in the 3′UTR of dengue virus (DENV) have been shown to result in almost complete abrogation of RNA replication in different cell lines [30]. In addition, motifs within the 3′UTR are responsible for genome circularization as well as production of sfRNA, both of which are required for efficient replication [31, 32]. This may have led to decreased maintenance of diversity observed in the two barcoded viruses with 3′UTR insertions. While RNA structure in the coding region can be important for function [33–35], those sequences in the barcoded viruses in the CDS that disrupted this likely would have been quickly removed. Since this was not an insertion, but rather substitutions at 8 nucleotide sites, it is less likely that RNA structure would have a comparable effect on the maintenance of diversity of barcodes as might be expected in the highly structured UTRs. It may be that although degenerate nts in the CDS are translationally “silent,” they could influence virus fitness through codon- and codon-pair biases or translational selection, a theory that suggests that highly expressed genes will favor codons that are more efficiently translated in a given system. Our results on virus replication in various cell lines and mosquitoes demonstrated that the barcoded viruses replicated to similar levels compared to wild-type clone-derived virus and had similar transmission rates in Ae. aegypti mosquitoes, suggesting that the impact of these alterations to the viral genome carried minimal fitness cost. It’s possible that individual barcodes within a population have fitness advantages through codon usage or other mechanisms and certain sequences could have been selected for/against. This is supported by the fact that several barcodes were present at high frequency in the virus stock and further increased in frequency during additional passage. If individual barcode sequences have much higher fitness than others, this would be a major drawback to this approach. This initial appearance of high frequency barcodes may have been due to low transfection efficiency in Vero cells, which would result in a significant bottleneck during virus recovery that would not be due to selection. Virus rescue in more readily transfectable cells has validated this hypothesis and we are using this more complex starting population for future studies. Distribution analyses suggested that selection for individual barcodes was not particularly strong in vitro or in vivo, as even after three passages in Vero or C6/36 cells the composition of barcodes did not significantly change as compared to the stock virus. This was further supported by results in mosquitoes, where two of the three populations showed no difference in barcode composition in the midgut as compared to the bloodmeal input, suggesting little selection during infection of this preliminary tissue for specific barcodes. These observations allow us to conclude that ZIKV possessing barcodes in the CDS are appropriate tools to measure virus population dynamics.
Using the most diverse barcoded ZIKV, we quantified bottlenecks in several different cell lines in vitro and in Ae. aegypti mosquitoes in vivo. Our results demonstrated that passaging in C6/36 cells places a weaker bottleneck on the virus population, especially when compared to Aag2 cells and even compared to both Vero and LLC-MK2 cell lines. Similarly, virus passaged on Vero cells maintained barcode diversity and distribution when compared to LLC-MK2. The fact that different MOI were used for passaging in C6/36 and Vero (MOI 0.01) and LLC-MK2 and Aag2 (MOI 0.001) should temper the conclusions here. However, this was experimentally necessary due to low replication in the latter cell lines. It’s possible that the lack of functional RNAi response in C6/36 and IFN response in Vero were responsible for the observed phenotypes as compared to both Aag2 and LLC-MK2, respectively, although further studies are required to confirm this. The reason for differences in population size reduction between C6/36 and Vero are unclear but may be due to differences in susceptibility to infection between the two cell lines [36], effectively making the starting MOI higher for the invertebrate cells. ZIKV has been shown to induce an RNAi response in mosquitoes and thus it is likely playing a role during infection of mosquito cells as well [37]. Vero cells are also known to produce type III interferon which likely plays a role in reducing replication of ZIKV, thereby reducing barcode diversity [38]. Since we did not specifically silence the RNAi or IFN system, we cannot definitively attribute this result to the lack of functional RNAi or IFN response. Additionally, C6/36 and Aag2 cells are known to harbor persistently infecting viruses [39, 40] and also flavivirus endogenous elements [41], in addition to being derived from different species. Future work should address the role of RNAi and IFN, along with endogenous viral elements in relation to bottlenecks using barcoded ZIKV. It should be noted that all virus titrations were performed on Vero cells and that these titers were used to calculate MOI. Therefore, initial infectivity may have been increased for C6/36 as compared to other cell lines, making the effective population size larger. This was confirmed by using Fst based estimations of population size.
We observed differences in population size after infection of different anatomical barriers in different populations of Ae. aegypti mosquitoes by using both the Kolmogorov-Smirnov test and Fst based estimations the measure population size. This appeared to be related to the transmission rate (TR) of the mosquito population, as the population with the biggest reduction in barcode complexity, significant difference in barcode distribution and smallest NF between the bloodmeal and midgut also had the lowest TR. This suggested the midgut barrier is a major determinant of vector competence in this population. As previously observed with a marked population of VEEV, diversity progressively decreased as the virus passed through anatomical barriers to transmission [13]. Previous work showed that WNV haplotypes changed considerably in Culex mosquitoes as the virus moved from the midgut to the saliva [11]. However, in Ae. aegypti mosquitoes, WNV haplotypes remained relatively stable during the same progression. A similar trend was observed here, as the barcodes present in the midgut of one mosquito rarely differed from the virus in the saliva. However, in contrast to that study, we observed a sharp initial drop in virus diversity from the bloodmeal to the midgut and then the legs in infected mosquitoes. This was presumably due to a high amount of complexity (present in the barcoded region) in our starting virus stocks, whereas the cloned virus used in the previous study had very little initial genetic diversity. Thus, although we started with low overall population diversity on a whole genome level, the barcode sequence provided a high level of starting complexity to assess bottlenecks. One potential drawback of this approach is that it is difficult to measure virus diversification after a bottleneck due to the massive loss of barcode complexity. Combined sequencing of the barcode region along with the whole genome may allow for further analyses after a bottleneck event occurs. The reason for the observed differences in bottleneck strength between the different mosquito populations is unclear at present. However, many factors have been shown to impact vector competence; including RNAi [42], the microbiome [43], vector genotype and virus genotype interactions [44], among others.
Many of the mosquitoes tested here had one barcode predominate in the legs, salivary glands and saliva, indicating that a severe population bottleneck occurs as virus either infects or disseminates from the midgut. This was confirmed using the KS test and measuring effective population size. However, the virus that predominated in the saliva was not always the highest frequency barcode in the midgut, reflecting the stochastic nature of virus population dynamics, and the importance of genetic drift during mosquito transmission. Visual inspection of the barcode sequences during spread throughout individual mosquitoes (Fig. 4D–F) illustrates this: the vast majority of mosquitoes in each population has a different barcode that predominates in the saliva with significantly reduced total numbers of barcodes and complexity. This result is entirely consistent with previous reports showing that mosquitoes significantly constrain arbovirus evolution [11, 23, 45]. It is interesting to note that the smallest NF and tissue where the KS test was found significant always occurred at either infection of the midgut or dissemination from this tissue. This strongly suggests that in these mosquito populations, the predominant bottleneck occurs early in infection with ZIKV. Another caveat with this approach is that changes in barcode frequencies could occur by dilution of the stock virus, effectively resulting in a different population being used during every experiment. By using a conservative approach to select ‘authentic’ barcodes we mitigated this problem, as the top barcodes were the same in the undiluted stock as in the diluted infection inocula or bloodmeal. Future studies with these viruses should always account for the potential stochastics effects of using a highly diverse viral population.
In conclusion, barcoded ZIKV populations are a promising tool to use for experimental evolution in vitro and in vivo. These viruses are currently being tested in monkeys and in mice to track viral replication dynamics and bottlenecks in mammalian species. By sequencing the stock virus in replicate for every experiment, it is possible to track the barcode population from starting virus to final outcome, even with new virus stocks. In addition, although the barcoded viruses described here had high complexity, the number of barcodes present did not approach the theoretical limit. With 8 degenerate nucleotides, the theoretical number of barcodes present in a “perfect” stock is ~65,000, considerably higher than what we achieved here. By improving our rescue technique, we have now improved the number of unique barcodes present in the stock virus to a level of almost perfect complexity, which will be used in future studies in pregnant monkeys, mice and mosquitoes.
MATERIALS AND METHODS
Cells and mosquitoes
Vero (ATCC CCL-81) and LLC-MK2 (ATCC CCL-7) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 50 μg/mL gentamicin at 37°C with 5% CO2. C6/36 cells (ATCC CRL-1660) were maintained in MEM with 10% FBS and 50 μg/mL gentamicin at 28°C with 5% CO2. Aag2 cells (obtained from Dr. Aaron Brault) were maintained in Schneider’s insect medium with 10% FBS at 28°C.
Aedes aegypti [L.] mosquitoes were collected from wild populations in different locations of Mexico (Merida, Coatzacoalcos and Poza Rica) (García-Luna et al., in submission). The mosquitoes used in this study were between the F2 and F4 generation and were maintained on citrated sheep blood and given 10% sucrose ad libitum. Following emergence, adults were maintained under controlled conditions of temperature (28°C), humidity (70% RH), and light (14:10 L:D diurnal cycle). Experiments involving infectious ZIKV in mosquitoes were performed under BSL3 conditions.
Construction of Barcoded ZIKV
A total of four barcodes consisting of 8 or 9 degenerate nucleotides were constructed, 2 present in the coding sequence at consecutive wobble positions and 2 in the 3′ untranslated region (Figure 1A). Coding-sequence barcodes were selected by searching for consecutive codons in which inserting a degenerate nucleotide in the third position would result in a synonymous change. Barcoded ZIKV was constructed using BFC. First the genome of ZIKV was amplified in two overlapping pieces from the two-part plasmid system previously described [46]. The CMV promoter was amplified from pcDNA3.1 (Invitrogen). The barcode region was then introduced in the form of an overlapping PCR-amplified oligo (for the coding sequence) or gBlock (for the 3′ UTR) (IDT, Iowa, USA). Primers used in the study are presented in Table 1. All PCR amplifications were performed with Q5 DNA polymerase (NEB, MA, USA). The amplified pieces were then excised from a gel using crystal violet for DNA visualization [47], thus avoiding the need for exposure to ultraviolet light, and purified with a gel extraction kit (Macherey-Nagel). The purified overlapping pieces were then assembled using Gibson assembly with the HiFi DNA assembly master mix (NEB) and incubated at 50°C for four hours. The Gibson assembly reaction was then digested with Exonuclease I to digest ssDNA, lambda exonuclease to remove non-circular dsDNA, and DpnI to remove any original bacteria derived plasmid DNA at 37°C for 30 minutes followed by heat inactivation at 80°C for 20 minutes. 2 μl of this reaction was then used for rolling circle amplification (RCA) using the Qiagen repli-g mini kit (Qiagen). RCA was performed as instructed except that 2M trehalose was used in place of water for the reaction mix to reduce secondary amplification products [48]. Reactions were incubated at 30°C for 4 hours and then inactivated at 65°C for 3 minutes. Correct banding pattern was confirmed by restriction digestion and sequence was confirmed by Sanger sequencing. A schematic depicting the construction and rescue of the barcoded viruses can be seen in Figure 1B.
Rescue of Infectious Clones
Following completion, the RCA reactions were digested with NruI at 37°C for 1 hour in order to linearize the product and remove the branched structure of the RCA reaction. Generation of an authentic 3′UTR was assured due to the presence of the hepatitis-delta ribozyme immediately following the viral genome. The digested RCA reaction was then purified using a PCR purification kit (Macherey-Nagel) and eluted in molecular grade water. Purified and digested RCAs were transfected into 80–90% confluent T75 flasks of Vero cells using Xfect transfection reagent (Clontech) following the recommended protocol. Infectious virus was harvested upon seeing 50–75% cytopathic effect (CPE), which was 6 days-post transfection. Viral supernatant was then clarified and supplemented to a final concentration of 20% fetal bovine serum (FBS) and 10 mM HEPES before being frozen in single-use aliquots. The titer was measured by plaque assay on Vero cells.
In vitro replication
Multi-step growth curves were performed on Vero, LLC-MK2, C6/36, and Aag2 cells at an MOI of 0.01. The day before infection, cells were seeded in 12-well plates. Following infection, the virus was allowed to absorb for two hours, at which point virus inoculum was removed, cells were washed with PBS and fresh culture media was added. To monitor replication dynamics, a 100 μL supernatant was harvested and frozen for later titration every day (including day 0) for a total of 6 days. Cells were then supplemented with 100 μL fresh media at each time point. Viral titers were determined by plaque assay on Vero cells.
In vitro passaging experiments
Passaging experiments were performed on Vero, LLC-MK2, C6/36, and Aag2 cells in 12-well plates. Passages on Vero and C6/36 cells were performed at a MOI of 0.01. Due to low replication levels, passages were performed at a MOI of 0.001 in LLC-MK2 and Aag2 cells. Cell culture supernatant was harvested at day 3 post-infection for Vero and C6/36 and day 6 post-infection for LLC-MK2 and Aag2. Infectious virus was then quantified by plaque assay on Vero cells and used to initiate another passage on the corresponding cell line. The original inoculum of diluted virus stock and the passage 3 supernatant was used for NGS to analyze barcode populations.
Vector competence studies
To assess bottlenecks in vivo, adult females from the 3 different populations were exposed to an infectious bloodmeal containing 1.5e06 PFU/mL of either ZIKV wild-type clone virus or barcoded virus. Following each infectious bloodmeal, back-titration was performed to ensure that virus titers were comparable. The titers after back-titration were as follows; for Coatzacoalcos and Merida, wild-type 3.87e06 PFU/mL and bc1 2.93e06 PFU/mL, for Poza Rica, wild-type 2e06 PFU/mL and bc1 2.5e06 PFU/mL. On day 14, mosquitoes were dissected for midguts, legs/wings, salivary glands and saliva in the same manner as we have previously described [49]. Presence of virus was assessed in the saliva using both qRT-PCR [50] and plaque assay. The bloodmeal containing diluted virus stock and mosquito tissues were subsequently used for NGS to analyze barcode populations.
Library Prep and Data Analysis
RNA was extracted from all samples using the Mag-Bind Viral DNA/RNA 96 kit (Omega Bio-Tek) on the KingFisher Flex Magnetic Particle Processor (Thermo Fisher Scientific). RNA was eluted in 30 μl nuclease-free water. RNA was then reverse transcribed into cDNA using Protoscript II reverse transcriptase (NEB) random dodecamer primers (IDT) at 23°C for ten minutes, 50°C for one hour and 85°C for 5 minutes. Regions around the barcode were amplified using primers tagged with Illumina compatible adapters using the NEBNext Ultra II Q5 Master Mix (NEB) adapted for qPCR by adding 5 μM Syto 9 green fluorescent nucleic acid stain (primers listed in supplementary table 2). Conditions used for the PCR were as follows; initial denaturation at 98°C for 30 seconds, followed by an initial touchdown PCR for 14 cycles of 98°C for 10 seconds, 72–65°C (down 0.5 C/cycle) for 15 seconds and 72°C for 30 seconds. The PCR was continued for an additional 6–20 cycles at a constant melting temperature of 65°C. Amplicons were then purified with AMPure XP beads (Beckman-Coulter) at a 1.0x ratio. A second indexing PCR was performed using homemade Illumina indexing primers (ordered from IDT) in the same manner as the first PCR, for 10 cycles, followed by a bead purification. Libraries were then pooled by volume and sequenced following manufacturer’s instructions on the Nextseq 500 (Illumina) via paired-end sequencing (2x150bp).
Sequencing data was demultiplexed, and fastq files were trimmed of adapter and index sequences. The paired-end reads were then merged using BBMerge, aligned to the ZIKV genome using BBMap, trimmed to the barcode region using Reformat.sh, and barcode sequences were counted and provided in fasta format using kmercountexact.sh. All of the programs listed are a part of the BBTools suite of software (Brian Bushnell, sourceforge.net/projects/bbmap/).
The number of ‘authentic’ barcodes was determined statistically by identifying the highest frequency of the most common non-WT barcode present in any of the three replicates of the wt ZIKV-IC stock. For ZIKV-bc1, this frequency was found to be 0.42% and was used as the minimum threshold to consider a barcode in ZIKV-bc1 as ‘authentic.’ Using this value, we included 18 sequences in our list of authentic barcodes for ZIKV-bc1, and these were followed throughout the study. The number of unique barcodes was defined as the number of barcodes called at any frequency higher than the cutoff. The percent of unique barcodes is defined as the number of unique barcodes divided by the total number of called barcode sequences. Genetic complexity was calculated using Shannon’s index using the following equation -
Where ni is the frequency of each nucleotide at each barcode position and N is the total number of barcode calls at that position. A perfectly complex viral population (a barcode sequence with 25% of each nucleotide) would equal 2.
Measurement of effective population size
The effective population size (NF) was measured in the same manner as previously described by Monsion et al [26]. Briefly, Fst [51] is expressed as Fst = (HT - Hs)/HT where HT is the average gene diversity for all samples, assuming they form a single population and HS is the within population average gene diversity. The gene diversity was calculated with the formula where pi is the frequency for each barcode. The frequency from all 18 barcodes was used for all calculations. Since we could not conclusively determine that barcodes called 0 times in any given sample were truly negative or due to sampling, barcodes that were not called in a given sample were assigned a value of the cutoff divided by 2 (0.21%). For our application, HT was the gene diversity of both the sample and input populations combined and HS was the gene diversity for the individual group. Monsion et al. previously showed that from this equation it is possible to calculate NF with the following equation
where Fst and F′st are from the input and sample populations, respectively.
Statistical analysis
Comparisons of virus titers in growth curves were performed using two-way ANOVA with Tukey’s correction at each time point. One-way ANOVA with Tukey’s correction was used for all other data sets. To compare barcode distributions, we used the Kolmogorov-Smirnov test (KS test) which compares probability distributions. A p-value of < 0.05 was taken as an indication that the two distributions were significantly different from one another. Transmission rates were compared using a two-tailed Fisher’s exact test. GraphPad Prism 7.0 (La Jolla, CA) was used for all statistical tests, and significance was defined as p<0.05.
Supplementary Material
{kind=link}
Supplementary Table 1. PFU/mL and GE/mL of bc1 Zika virus for saliva samples that were used for next-generation sequencing to characterize the barcode populations.
Supplementary Table 2. Primers used to prepare libraries for next-generation sequencing.
Acknowledgments
We gratefully thank Brian Geiss for discussions involving infectious clones. We would also like to thank Jeff Kieft and Benjamin Akiyama for insight into potential insertion sites into the 3′ UTR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zanluca C, de Melo VCA, Mosimann ALP, Santos GIVD, Santos CNDD, Luz K. First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz. 2015;110:569–572. doi: 10.1590/0074-02760150192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffy MR, Chen T-H, Hancock WT, Powers AM, Kool JL, Lanciotti RS, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med. 2009;360:2536–2543. doi: 10.1056/NEJMoa0805715. [DOI] [PubMed] [Google Scholar]
- 3.Oehler E, Watrin L, Larre P, Leparc-Goffart I, Lastere S, Valour F, et al. Zika virus infection complicated by Guillain-Barre syndrome--case report, French Polynesia, December 2013. Euro Surveill. 2014:19. doi: 10.2807/1560-7917.es2014.19.9.20720. Available: https://www.ncbi.nlm.nih.gov/pubmed/24626205. [DOI] [PubMed]
- 4.Schuler-Faccini L, Ribeiro EM, Feitosa IML, Horovitz DDG, Cavalcanti DP, Pessoa A, et al. Possible Association Between Zika Virus Infection and Microcephaly - Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:59–62. doi: 10.15585/mmwr.mm6503e2. [DOI] [PubMed] [Google Scholar]
- 5.Foy BD, Kobylinski KC, Chilson Foy JL, Blitvich BJ, Travassos da Rosa A, Haddow AD, et al. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis. 2011;17:880–882. doi: 10.3201/eid1705.101939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaminathan S, Schlaberg R, Lewis J, Hanson KE, Couturier MR. Fatal Zika Virus Infection with Secondary Nonsexual Transmission. N Engl J Med. 2016;375:1907–1909. doi: 10.1056/NEJMc1610613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thangamani S, Huang J, Hart CE, Guzman H, Tesh RB. Vertical Transmission of Zika Virus in Aedes aegypti Mosquitoes. Am J Trop Med Hyg. 2016;95:1169–1173. doi: 10.4269/ajtmh.16-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miner JJ. Congenital Zika virus infection: More than just microcephaly. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aan8195. [DOI] [PubMed] [Google Scholar]
- 9.Ciota AT, Bialosuknia SM, Ehrbar DJ, Kramer LD. Vertical Transmission of Zika Virus by Aedes aegypti and Ae. albopictus Mosquitoes. Emerg Infect Dis. 2017;23:880–882. doi: 10.3201/eid2305.162041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salazar MI, Richardson JH, Sánchez-Vargas I, Olson KE, Beaty BJ. Dengue virus type 2: replication and tropisms in orally infected Aedes aegypti mosquitoes. BMC Microbiol. 2007;7:9. doi: 10.1186/1471-2180-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grubaugh ND, Weger-Lucarelli J, Murrieta RA, Fauver JR, Garcia-Luna SM, Prasad AN, et al. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe. 2016;19:481–492. doi: 10.1016/j.chom.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grubaugh ND, Rückert C, Armstrong PM, Bransfield A, Anderson JF, Ebel GD, et al. Transmission bottlenecks and RNAi collectively influence tick-borne flavivirus evolution. Virus Evol. 2016;2:vew033. doi: 10.1093/ve/vew033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forrester NL, Guerbois M, Seymour RL, Spratt H, Weaver SC. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog. 2012;8:e1002897. doi: 10.1371/journal.ppat.1002897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duarte E, Clarke D, Moya A, Domingo E, Holland J. Rapid fitness losses in mammalian RNA virus clones due to Muller’s ratchet. Proc Natl Acad Sci U S A. 1992;89:6015–6019. doi: 10.1073/pnas.89.13.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lauring AS, Andino R. Exploring the fitness landscape of an RNA virus by using a universal barcode microarray. J Virol. 2011;85:3780–3791. doi: 10.1128/JVI.02217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfeiffer JK, Kirkegaard K. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A. 2006;103:5520–5525. doi: 10.1073/pnas.0600834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, Sachs D, et al. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe. 2014;16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fennessey CM, Pinkevych M, Immonen TT, Reynaldi A, Venturi V, Nadella P, et al. Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLoS Pathog. 2017;13:e1006359. doi: 10.1371/journal.ppat.1006359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emeny JM, Morgan MJ. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol. 1979;43:247–252. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- 20.Brackney DE, Scott JC, Sagawa F, Woodward JE, Miller NA, Schilkey FD, et al. C6/36 Aedes albopictus cells have a dysfunctional antiviral RNA interference response. PLoS Negl Trop Dis. 2010;4:e856. doi: 10.1371/journal.pntd.0000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott JC, Brackney DE, Campbell CL, Bondu-Hawkins V, Hjelle B, Ebel GD, et al. Comparison of dengue virus type 2-specific small RNAs from RNA interference-competent and -incompetent mosquito cells. PLoS Negl Trop Dis. 2010;4:e848. doi: 10.1371/journal.pntd.0000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.SHANNON EC. A mathematical theory of communication, Part I, Part II. Bell Syst Tech J. 1948;27:623–656. [Google Scholar]
- 23.Vasilakis N, Deardorff ER, Kenney JL, Rossi SL, Hanley KA, Weaver SC. Mosquitoes put the brake on arbovirus evolution: experimental evolution reveals slower mutation accumulation in mosquito than vertebrate cells. PLoS Pathog. 2009;5:e1000467. doi: 10.1371/journal.ppat.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene IP, Wang E, Deardorff ER, Milleron R, Domingo E, Weaver SC. Effect of alternating passage on adaptation of sindbis virus to vertebrate and invertebrate cells. J Virol. 2005;79:14253–14260. doi: 10.1128/JVI.79.22.14253-14260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jerzak GVS, Brown I, Shi P-Y, Kramer LD, Ebel GD. Genetic diversity and purifying selection in West Nile virus populations are maintained during host switching. Virology. 2008;374:256–260. doi: 10.1016/j.virol.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monsion B, Froissart R, Michalakis Y, Blanc S. Large bottleneck size in Cauliflower Mosaic Virus populations during host plant colonization. PLoS Pathog. 2008;4:e1000174. doi: 10.1371/journal.ppat.1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lequime S, Fontaine A, Ar Gouilh M, Moltini-Conclois I, Lambrechts L. Genetic Drift, Purifying Selection and Vector Genotype Shape Dengue Virus Intra-host Genetic Diversity in Mosquitoes. PLoS Genet. 2016;12:e1006111. doi: 10.1371/journal.pgen.1006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett KE, Olson KE, de Muñoz ML, Fernandez-Salas I, Farfan-Ale JA, Higgs S, et al. Variation in vector competence for dengue 2 virus among 24 collections of Aedes aegypti from Mexico and the United States. Am J Trop Med Hyg. 2002;67:85–92. doi: 10.4269/ajtmh.2002.67.85. [DOI] [PubMed] [Google Scholar]
- 29.Akiyama BM, Laurence HM, Massey AR, Costantino DA, Xie X, Yang Y, et al. Crystal structure of an exonuclease resistant RNA from Zika virus [Internet] 2016 doi: 10.2210/pdb5tpy/pdb. [DOI] [Google Scholar]
- 30.Alvarez DE, De Lella Ezcurra AL, Fucito S, Gamarnik AV. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology. 2005;339:200–212. doi: 10.1016/j.virol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 31.You S, Falgout B, Markoff L, Padmanabhan R. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5′-and 3′-terminal regions that influence RNA structure. J Biol Chem ASBMB. 2001;276:15581–15591. doi: 10.1074/jbc.M010923200. [DOI] [PubMed] [Google Scholar]
- 32.Moon SL, Anderson JR, Kumagai Y, Wilusz CJ, Akira S, Khromykh AA, et al. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA. 2012;18:2029–2040. doi: 10.1261/rna.034330.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lobert PE, Escriou N, Ruelle J, Michiels T. A coding RNA sequence acts as a replication signal in cardioviruses. Proc Natl Acad Sci U S A. 1999;96:11560–11565. doi: 10.1073/pnas.96.20.11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linger BR, Kunovska L, Kuhn RJ, Golden BL. Sindbis virus nucleocapsid assembly: RNA folding promotes capsid protein dimerization. RNA. 2004;10:128–138. doi: 10.1261/rna.5127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lulla V, Kim DY, Frolova EI, Frolov I. The amino-terminal domain of alphavirus capsid protein is dispensable for viral particle assembly but regulates RNA encapsidation through cooperative functions of its subdomains. J Virol. 2013;87:12003–12019. doi: 10.1128/JVI.01960-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan JF-W, Yip CC-Y, Tsang JO-L, Tee K-M, Cai J-P, Chik KK-H, et al. Differential cell line susceptibility to the emerging Zika virus: implications for disease pathogenesis, non-vector-borne human transmission and animal reservoirs. Emerg Microbes Infect. 2016;5:e93. doi: 10.1038/emi.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saldaña MA, Etebari K, Hart CE, Widen SG, Wood TG, Thangamani S, et al. Zika virus alters the microRNA expression profile and elicits an RNAi response in Aedes aegypti mosquitoes. PLoS Negl Trop Dis. 2017;11:e0005760. doi: 10.1371/journal.pntd.0005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stoltz M, Klingström J. Alpha/beta interferon (IFN-alpha/beta)-independent induction of IFN-lambda1 (interleukin-29) in response to Hantaan virus infection. J Virol. 2010;84:9140–9148. doi: 10.1128/JVI.00717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paterson A, Robinson E, Suchman E, Afanasiev B, Carlson J. Mosquito densonucleosis viruses cause dramatically different infection phenotypes in the C6/36 Aedes albopictus cell line. Virology. 2005;337:253–261. doi: 10.1016/j.virol.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 40.Zhang G, Asad S, Khromykh AA, Asgari S. Cell fusing agent virus and dengue virus mutually interact in Aedes aegypti cell lines. Sci Rep. 2017;7:6935. doi: 10.1038/s41598-017-07279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki Y, Frangeul L, Dickson LB, Blanc H, Verdier Y, Vinh J, et al. Uncovering the Repertoire of Endogenous Flaviviral Elements in Aedes Mosquito Genomes. J Virol. 2017:91. doi: 10.1128/JVI.00571-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blair CD. Mosquito RNAi is the major innate immune pathway controlling arbovirus infection and transmission. Future Microbiol. 2011;6:265–277. doi: 10.2217/fmb.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dennison NJ, Jupatanakul N, Dimopoulos G. The mosquito microbiota influences vector competence for human pathogens. Curr Opin Insect Sci. 2014;3:6–13. doi: 10.1016/j.cois.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambrechts L, Chevillon C, Albright RG, Thaisomboonsuk B, Richardson JH, Jarman RG, et al. Genetic specificity and potential for local adaptation between dengue viruses and mosquito vectors. BMC Evol Biol. 2009;9:160. doi: 10.1186/1471-2148-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC. Arbovirus evolution in vivo is constrained by host alternation. Proc Natl Acad Sci U S A. 2008;105:6970–6975. doi: 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weger-Lucarelli J, Duggal NK, Bullard-Feibelman K, Veselinovic M, Romo H, Nguyen C, et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. J Virol. 2017:91. doi: 10.1128/JVI.01765-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rand KN. Crystal violet can be used to visualize DNA bands during gel electrophoresis and to improve cloning efficiency. Tech Tips Online. 1996;1:23–24. [Google Scholar]
- 48.Pan X, Urban AE, Palejev D, Schulz V, Grubert F, Hu Y, et al. A procedure for highly specific, sensitive, and unbiased whole-genome amplification. Proc Natl Acad Sci U S A. 2008;105:15499–15504. doi: 10.1073/pnas.0808028105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weger-Lucarelli J, Rückert C, Chotiwan N, Nguyen C, Garcia Luna SM, Fauver JR, et al. Vector Competence of American Mosquitoes for Three Strains of Zika Virus. PLoS Negl Trop Dis. 2016;10:e0005101. doi: 10.1371/journal.pntd.0005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis. 2008;14:1232–1239. doi: 10.3201/eid1408.080287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright S. Evolution in Mendelian Populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. PFU/mL and GE/mL of bc1 Zika virus for saliva samples that were used for next-generation sequencing to characterize the barcode populations.
Supplementary Table 2. Primers used to prepare libraries for next-generation sequencing.