

Abstract
Small-molecule inhibitor selectivity may be influenced by variation in dynamics among members of a protein family. Regulator of G-protein Signaling (RGS) proteins are a family that plays a key role in G-Protein Coupled Receptor (GPCR) signaling by binding to active Gα subunits and accelerating GTP hydrolysis, thereby terminating activity. Thiadiazolidinones (TDZDs) inhibit the RGS-Gα interaction by covalent modification of cysteine residues in RGS proteins. Some differences in specificity may be explained by differences in the complement of cysteines among RGS proteins. However, key cysteines shared by RGS proteins inhibited by TDZDs are not exposed on the protein surface, and differences in potency exist among RGS proteins containing only buried cysteines. We hypothesize that differential exposure of buried cysteine residues among RGS proteins partially drives TDZD selectivity. Hydrogen-deuterium exchange (HDX) studies and molecular dynamics (MD) simulations were used to probe the dynamics of RGS4, RGS8, and RGS19, three RGS proteins inhibited at a range of potencies by TDZDs. When these proteins were mutated to contain a single, shared cysteine, RGS19 was found to be most potently inhibited. HDX studies revealed differences in α4 and α6 helix flexibility among RGS isoforms, with particularly high flexibility in RGS19. This could cause differences in cysteine exposure and lead to differences in potency of TDZD inhibition. MD simulations of RGS proteins revealed motions that correspond to solvent exposure observed in HDX, providing further evidence for a role of protein dynamics in TDZD selectivity.
Graphical Abstract
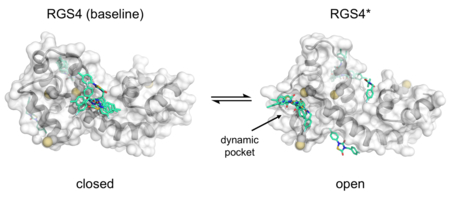
INTRODUCTION
Protein-protein interactions (PPIs) remain a poorly tapped pool of potential targets for small-molecule inhibitors. Targeting PPIs has been challenging because many protein-protein interfaces are flat and lack a dedicated small-molecule binding pocket.1–3 However, it may be possible to interrupt PPIs by binding to transiently exposed pockets,4,5 either at the protein-protein interface6 or at allosteric sites.7,8 Targeting of allosteric sites, as they are less evolutionarily conserved, may confer better specificity than directly targeting interfaces.9 In addition, there may be variation in dynamic exposure of allosteric pockets among members of a protein family. Such differences in protein dynamics could drive inhibitor specificity.10
G-protein signaling is critical in pharmacology. Approximately thirty percent of marketed drugs target G-Protein Coupled Receptors (GPCRs), and more target related pathways.11 Regulators of G-protein Signaling (RGS) proteins control GPCR signaling by binding to active, GTP-bound Gα subunits and accelerating GTP hydrolysis. This terminates G-protein signaling. By inhibiting an RGS protein, signaling through a GPCR may be amplified. We previously identified thiadiazolidinone (TDZD) inhibitors of the RGS-Gα interaction in a high-throughput screen.12 They allosterically inhibit RGS proteins by covalent modification of cysteine residues at sites distant from the RGS-Gα interface. The TDZD inhibitor CCG-50014 is most potent against RGS4, followed by RGS19 and distantly by RGS8.13 RGS4 inhibitors may be valuable as therapeutics for Parkinson’s disease. RGS4 is highly expressed in the striatum,14,15 where it regulates synaptic plasticity in response to dopamine signaling.16,17 A TDZD inhibitor with enhanced specificity for RGS4, CCG-203769, reduces bradykinesia in a raclopride model of certain Parkinson’s-like motor deficits in mice.18
The RGS homology domain, which is responsible for the GTP-ase accelerating activity of RGS proteins, is a 120-amino acid domain consisting of nine α-helices (Fig 1A).19,20 Differences in TDZD potency may be due to different locations or numbers of cysteines among RGS isoforms, or due to differential transient cysteine exposure. RGS4, RGS8, and RGS19 all share an α4 helix cysteine, while RGS4 and RGS8 share one on the α6-α7 interhelical loop (Fig 1A). Notably, these cysteines are buried beneath the protein surface in crystal structures.21,22 Therefore, it may be necessary for dynamic pockets to open to expose these cysteines for TDZD interaction. Understanding of dynamic pockets will be beneficial, as such a pocket may be exploited in rational design of novel non-covalent inhibitors using a docking based virtual screen. We previously showed that the α5-α6 helical pair is flexible using enhanced sampling MD simulations.23 Covalent modification by TDZD inhibitors could lock the α5-α6 interhelical loop in a position that prevents RGS interaction with Gα proteins. We hypothesize that differential transient exposure of buried cysteine residues drives TDZD selectivity. Here, we used hydrogen/deuterium exchange with mass spectroscopy (HDX-MS) and long time-scale classical unbiased molecular dynamics (MD) studies to examine differences in dynamics between RGS4, RGS8, and RGS19. These RGS protein isoforms represent a range of potencies of inhibition by TDZD inhibitors (RGS4>RGS19>RGS8). HDX-MS and MD studies make a powerful combined experimental and computational approach for evaluating protein dynamics.24,25 These revealed a dual role of protein dynamics and cysteine complement in selectivity of TDZDs against RGS proteins.
Figure 1.
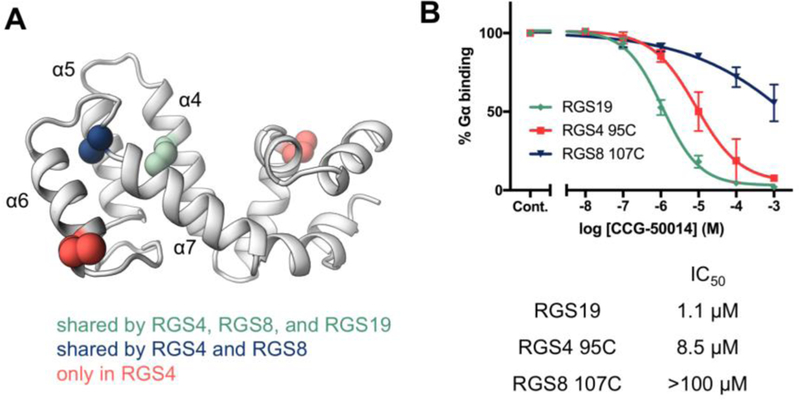
(a) Locations of cysteines in RGS4, RGS8, and RGS19. (b) Potency of CCG-50014 against RGS19, which has only one cysteine, and mutant RGS4 and RGS8 containing only the shared α4 helix cysteine. n=3.
RESULTS
Previous work has demonstrated a role for the number and position of cysteine residues in the potency of RGS inhibitors.26 To eliminate this confounding variable and allow better assessment of the role of protein dynamics, the potency of CCG-50014 was compared among RGS19 and mutants of RGS4 and RGS8 containing only the shared α4 cysteine. These mutants are termed RGS4 95C and RGS8 107C respectively. While removal of additional cysteines reduced potency in both RGS4 and RGS8, dramatic differences in TDZD potency still exist among single-cysteine proteins. RGS19 was most potently inhibited at 1.1 μM, while RGS4 95C was inhibited at 8.5 μM, and RGS8 107C at >100 μM (Fig 1B).
To compare solvent exposure kinetics on the α4 helix, we performed HDX-MS on RGS4, RGS8, and RGS19 apo-proteins. A map of pepsin cleavage fragments observed in each protein is shown in Fig S1. Consistent with the higher potency of inhibition by the TDZD, the cysteine-containing fragment from α4 (residues 92–97) in RGS4 shows significantly higher exchange than that from RGS8 (residues 86–91). After a 1000 minute incubation in D2O, the 92–97 fragment of RGS4 had 35% deuterium incorporation, while the analogous fragment in RGS8 had only 8% deuterium incorporation. Further strengthening the correlation of dynamics with selectivity, RGS19 had much faster exchange than RGS4 or RGS8 in the α4 helix. It reached 48% deuterium incorporation by only 100 minutes, while RGS4 and RGS8 had 9% and 1% respectively (Fig 2A). A similar trend was observed in the α5 helix. RGS8 had the least exchange after 1000 minutes (24% deuterium incorporation), followed by RGS4 and RGS19 (38% and 49% deuterium incorporation, respectively, Fig 2B). One pattern consistent among all three isoforms is high exchange in the α5-α6 interhelical loop, indicating that RGS proteins are flexible in this region. Those fragments in all three proteins exceeded 50% deuterium incorporation by 100 minutes (Fig 2C). This was not surprising, as the α5-α6 loop is the longest unstructured region within the RGS domain. In the α6 helix, RGS19 again had higher exchange than RGS8 and RGS4. RGS8 was particularly protected in the residue 126–136 fragment, reaching only 7% deuterium incorporation after 1000 minutes. However, higher exchange was observed in the residue 130–140 fragment of RGS8, likely because this fragment also contains residues that are a part of the α6-α7 loop (Fig 2D). A similar effect was seen in RGS4 near the α7 helix, in which a fragment wholly within α7 (residues 150–159) had much slower exchange than a fragment partially overlapping the α6-α7 loop (residues 143–151) (Fig 2E).
Figure 2.
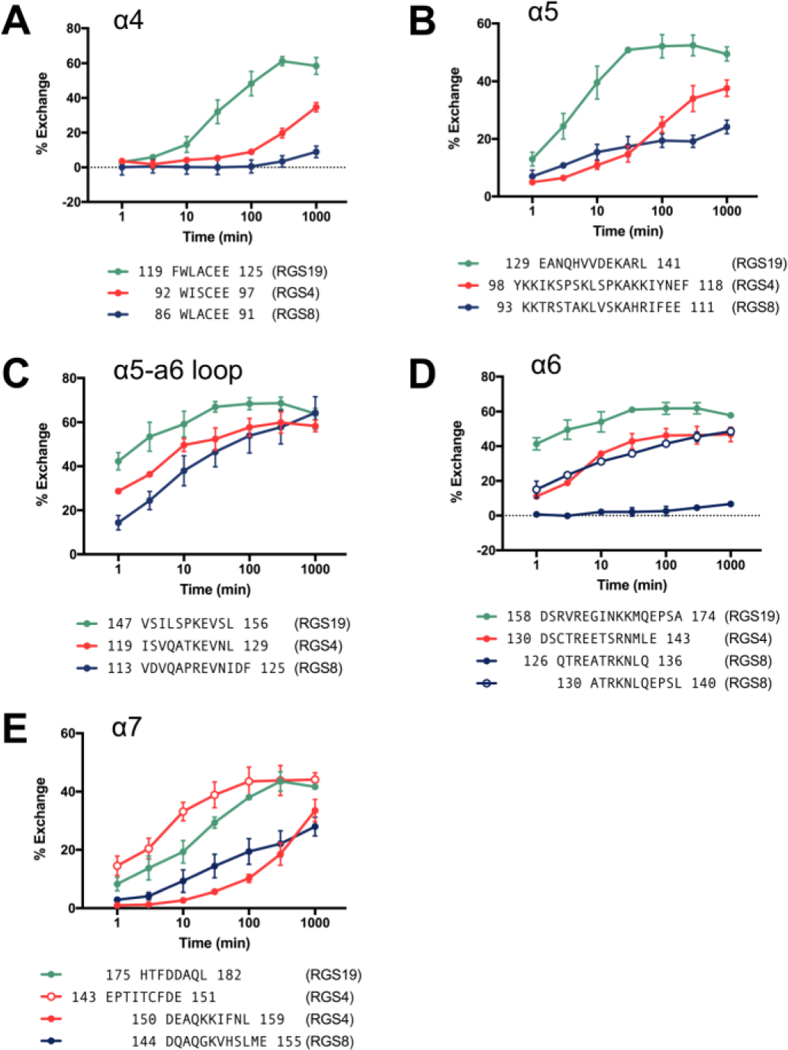
(a-e) Kinetics of deuterium exchange in selected protein fragments from (a) α4, (b) α5, (c) α5-α6 interhelical region, (d) α6 and (e) α7. Sequences of observed fragments are aligned and residue numbers of each fragment indicated. n=3.
According to these results, RGS8 had low deuterium exchange relative to other RGS proteins throughout the helices surrounding its cysteines. This is indicative of rigidity of these helices in RGS8, which likely prevents exposure of cysteines to solvent. This observation also could explain the low potency of TDZDs against RGS8 relative to other RGS isoforms. The α6 helix of RGS4 has more deuterium exchange than the α4, α5, and α7 helices (Fig 3A and B). Rapid exchange in the α6 helix may be due to movement away from neighboring helices or unfolding of the helix itself. Such a movement could increase solvent exposure of the otherwise buried cysteine 148 on the α6-α7 loop. This would allow access by TDZD inhibitors. Because the higher exchange on α6 compared to other nearby helices is unique to RGS4, this potentially contributes to the increased potency of TDZDs against wild type RGS4 versus RGS8. In the α4, α5, and α6 helices, RGS19 shows higher deuterium exchange than RGS4 or RGS8, indicating that RGS19 is highly dynamic. For example, in a fragment of the α5 helix, RGS19 had 51% deuterium incorporation after 30 minutes, while similar fragments in RGS4 and RGS8 had 15% and 17% incorporation, respectively (Fig 2B). This fits with functional data showing that RGS19 is more potently inhibited by CCG-50014 than single-cysteine RGS4 and RGS8 (Fig 1B). Although RGS19 lacks cysteines on the α6 helix and α6-α7 loop which may contribute to potency of inhibition of RGS4 by TDZDs (Cys 132 and Cys 148 in RGS4), it has the highest potency of inhibition among single-cysteine RGS proteins. This may be due to a pronounced movement of the α4, α5, and α7 helices, allowing TDZDs to access RGS19’s cysteine on the α4 helix.
Figure 3.
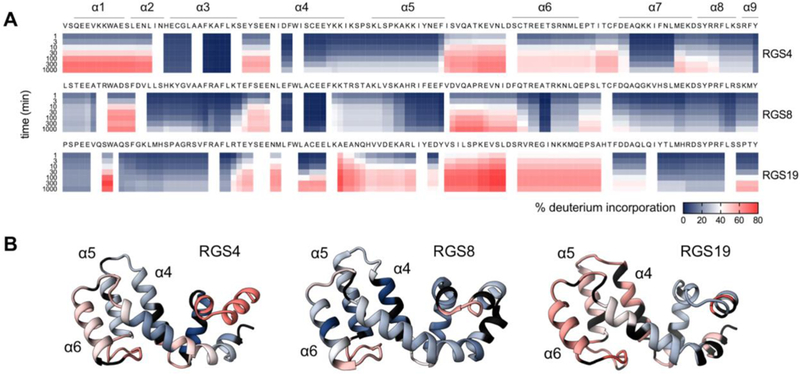
(a) Global kinetics of deuterium exchange. Deuterium incorporation is expressed as a percent of exchangeable amide hydrogen positions. (b) Degree of deuterium incorporation at 300 minutes in 90% D2O is mapped onto protein structure of RGS4, RGS8, and RGS19. n=3.
To probe the molecular details of dynamic motions in RGS4, RGS8, and RGS19 that underlie the flexibility differences observed in HDX-MS as well as to evaluate possible routes of access to cysteines by TDZDs, we performed long time-scale classical MD simulations in explicit-solvent (see supplemental methods). Our previous short time-scale classical MD simulations did not show any major conformational changes; but enhanced sampling simulations did show changes23. Here, we conducted microsecond time-scale classical MD simulations, through which the flexibility in key helices became apparent.
The first set of simulations that were 2 µs long (set 1 in Table S1) showed regions of pronounced movement in all three proteins. RGS4 showed unique motions within the α6 helix (Fig 4A), while in RGS8 and RGS19, movement was primarily within the α6-α7 interhelical loop (Fig 4B and C). A second independent set of simulations that were 3 µs-long (set 2 in Table S1) showed the largest movement in RGS19, again particularly prominent in the α6-α7 interhelical loop, with the α6 helix and α5-α6 interhelical loop also relatively flexible (Fig S2). However, RGS4 and RGS8 were relatively stable. Taken together, these simulation sets indicate highest flexibility in RGS19, with potential for flexibility in distinct regions in RGS8 and RGS4. In all simulations for each protein, pronounced movements also occurred in the residues located in terminal helices. This is likely an effect of free terminal ends; residues outside of the RGS homology domains were not included in the simulations.
Figure 4.
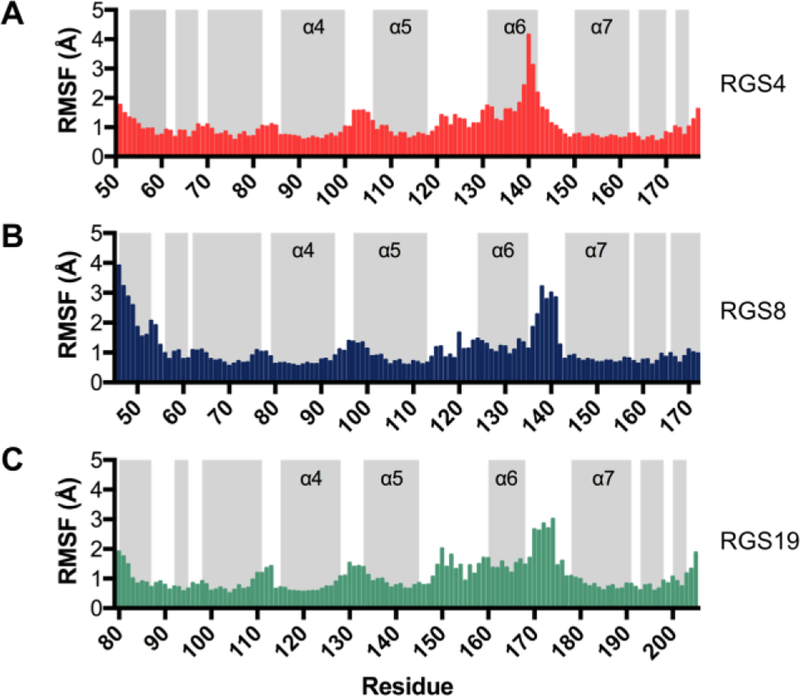
Root mean squared fluctuations (RMSF) per residue during 2-μs MD simulations of (a) RGS4 (PDB: 1AGR), (b) RGS8 (PDB: 2ODE), and (c) RGS19 (PDB: 1CMZ). The RMSF trends for each protein for the simulation set 2 are shown in Fig S2. Gray bars indicate helical regions.
Analysis of solvent exposure of sulfur atoms reveals exposure of initially buried cysteines. (Fig 5). Cys 123 in RGS19 is more exposed than analogous cysteines in RGS4 and RGS8 in the 2 μs simulation set (Fig 5A) and again in the 3 μs simulation set (Fig 5B). This may explain the potency of RGS19 relative to the analogous single-cysteine RGS4 and RGS8. Pronounced exposure of the α6-α7 interhelical Cys 160 in RGS8 was observed in both sets of simulations (Fig 5C and 5D).
Figure 5.
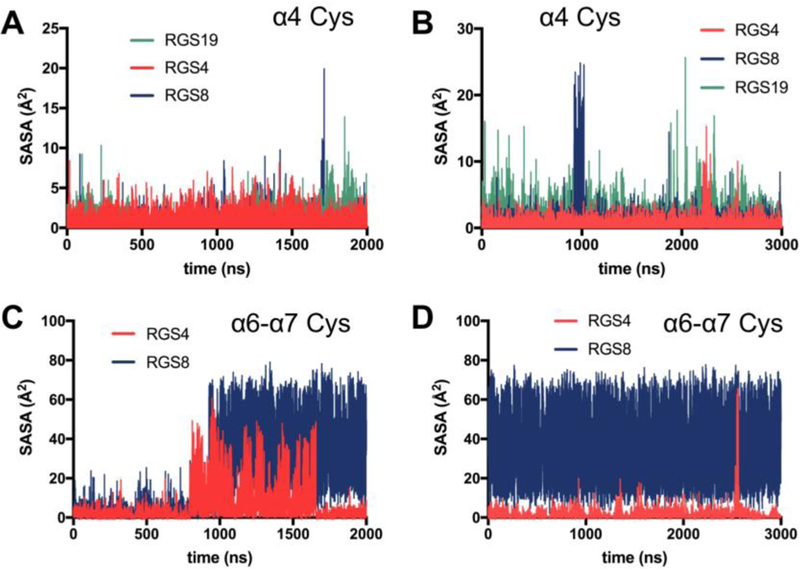
Solvent-accessible surface areas (SASA) are shown for sulfur atoms in shared cysteines on α4 helix for simulation set 1 (a) and set 2 (b) in RGS4, RGS8, and RGS19, and for shared cysteines on α6-α7 interhelical loop in simulation set 1 (c) and set 2 (d) in RGS4 and RGS8.
In addition, the conformations observed during movements of the α6 helix and α6-α7 loop show distinct routes of cysteine exposure among the three RGS proteins. In the RGS4 crystal structure (PDB: 1AGR),21 Asn 140 occludes Cys 148 from exposure to the protein surface (Fig 6D). In the MD simulation set 1 using 1AGR as initial coordinates, a transient movement of the α6 helix was observed, reaching 15.1 Å between α-carbons at 1.24 μs (Fig 6G), versus 5.9 Å at baseline. This movement coincided with a high solvent exposure of Cys 148 (Fig 5C). In MD simulation set 1 of RGS8 (using PDB code 2ODE27 as initial coordinates), helices α4, α5, α6, and α7 were stable relative to the same helices in other proteins tested. However, the α6-α7 interhelical region, which includes cysteine 160, underwent a pronounced movement (Fig 6B). Cys 160 rotated toward the protein surface at 1 μs, and remained exposed to solvent for the remainder of the trajectory (Fig 5B and 6H). This cysteine exposure was observed again for the duration of simulation set 2 (Fig 5D). RGS19 lacks the cysteine in the α6-α7 interhelical loop, having only Cys123 on α4. Both MD simulations of RGS19 (starting with the PDB code 1CMZ28) revealed a movement of the α6-α7 interhelical loop away from the α4 and α5 helices, resulting in an open groove in the protein surface (arrow in Fig 6I and S3). This observation likely explains the higher observed deuterium incorporation of α4 and α5 helices in RGS19 compared to RGS4 and RGS8, but additional changes, perhaps induced by compound binding, may be required for full exposure of Cys 123.
Figure 6.
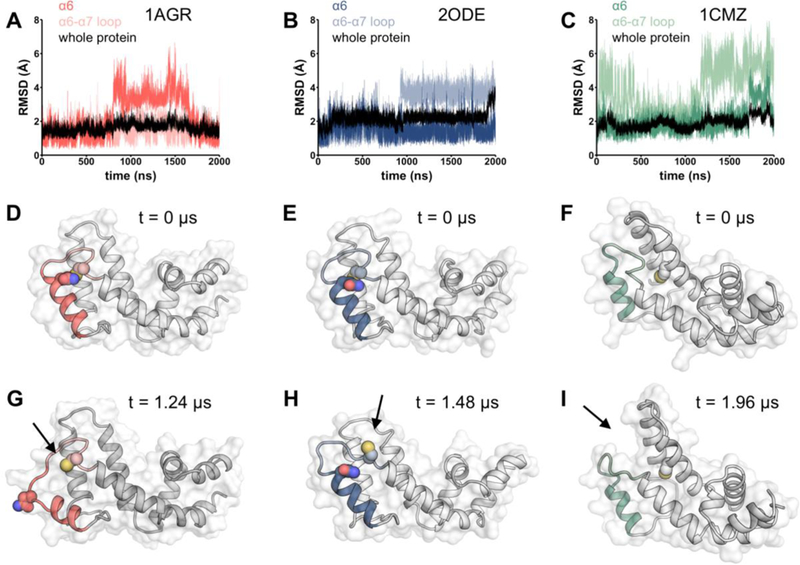
Conformational changes during molecular dynamics simulations. Root mean square deviations of α6 helix and α6-α7 loop, starting conformation, and a snapshot conformation during MD simulation are shown for (a, d, g) RGS4, (b, e, h) RGS8, and (c, f, i) RGS19. Protein regions plotted in MD trajectories are depicted in color in protein structures. Arrows indicate locations of notable solvent exposure during simulation.
DISCUSSION
RGS protein flexibility, as measured both by deuterium incorporation and solvent exposure of the α4 cysteine in MD simulations, is correlated with potency of inhibition of TDZDs against proteins containing only a single shared cysteine. RGS19 had the most pronounced deuterium incorporation throughout the α4-α7 helix bundle, and it was more potently inhibited by CCG-50014 than single-cysteine RGS4 or RGS8. Such flexibility could result in increased likelihood of binding of TDZDs at the α4 cysteine. This may lead to perturbation of residues involved in G-protein binding, as suggested by previous NMR experiments.23
There was also good concordance between regional protein flexibility in the HDX-MS studies and in MD simulations. In RGS8, helices α4, α5, α6, and α7 were protected from deuterium exchange and were also stable during MD simulations. The dramatic movement of the RGS4 α6 helix in simulation set 1 mirrors its high solvent exposure in HDX studies. This suggests that movement of the α6 helix is likely responsible for solvent exposure of Cys 148 in RGS4, providing a plausible route of access by TDZD inhibitors. Indeed, cysteine 148 was the most important single cysteine for inhibition of RGS4 by our other cys-linking inhibitor, CCG-4986.26
Deuterium exchange was measured on a much longer timescale than MD simulations. In order for exchange to occur, amide hydrogens must be in a conformation amenable to exchange, requiring both interruption of H-bonds and proximity of solvent waters. These exchange-competent states are short lived, often existing on a 10–100 picosecond timescale.25 They are frequent enough to be readily observed in microsecond timescale simulations; however, the rate of intrinsic hydrogen exchange is much slower than the rate of hydrogen solvent exposure. This is termed EX2 kinetics, in which an amide hydrogen may make multiple visits to a solvent-exposed state before an exchange event occurs.29 While exchange is still representative of the time spent in an open state, this allows observation of exchange on much longer timescales than those of dynamic motions.
Interestingly, dynamic cysteine exposure varied among protein isoforms. In RGS4, movement of helix 6 exposed the α6-α7 cysteine, while in RGS8, helix 6 was stable and that cysteine rotated toward solvent in during a movement of the α6-α7 loop. RGS19 lacks a cysteine on the α6-α7 loop, but opens a cleft toward a deeply buried α4 helix cysteine. These results suggest that the route of modification by covalent inhibitors varies among RGS isoforms, even at shared cysteine locations.
These differences in dynamic motions among RGS isoforms may contribute to differences in potency of TDZD inhibition by two ways. First, differences in the rate of covalent modification or the magnitude of effect on Gα binding may be driven by differences between RGS isoforms in the direction of cysteine solvent exposure. Second, distinct transient conformations occurring more frequently in certain RGS isoforms may permit unique non-covalent docking to drive covalent modification. In such a scenario, the open state could be taken advantage of in a docking-based virtual screen, permitting the discovery of non-covalent RGS inhibitors. Although additional future work is required to fully understand the inhibitor access routes and mechanisms (e.g. conformational selection versus induced fit), we have previously shown23 using nuclear magnetic resonance (NMR) and MD simulation analyses that an open conformation of RGS4 facilitates covalent docking of CCG-50014 and leads to significant perturbations in residues near the binding pocket and at the protein-protein interface. This is because inhibitor binding only allows a partial recovery of the open conformation to an apo-like conformation as opposed to a significant recovery in the absence of the inhibitor. Because conformational changes induced by compound binding may be a factor in inhibition, we aim to undertake studies involving docking of other TDZD and non-TDZD analogs12 using conformations of RGS proteins reported in this work. These possibilities remain an object of future investigations.
CONCLUSION
The application of HDX-MS and MD methods reveal that RGS isoforms differ in their mechanism of transient cysteine exposure, suggesting distinct routes of access by covalent inhibitors. These differences are potentially responsible for the selective potency of TDZD inhibitors among RGS isoforms. Importantly, the conformations of RGS proteins in which cysteine residues are transiently exposed could be potentially useful for designing the next generation of inhibitory small-molecules.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge Sundari Chodavarapu in the lab of Dr. Jon Kaguni and Anthony Schilmiller and Daniel Jones in the RTSF Mass Spectrometry and Metabolomics core (Michigan State University) for assistance with hydrogen/deuterium exchange. We are grateful for computational support from Trillian, a Cray XE6m-200 supercomputer supported by the NSF MRI program under grant PHY-1229408, and the NSF-supported (ACI-1053575) Extreme Science and Engineering Discovery Environment (XSEDE) under grant TG-MCB140029/TG-MCB160183 (HV).
Funding Sources
This work was supported by NSF grants 1507588 (RRN) and 1508595 (HV) and NIH training grant T32 GM092715 (RRN).
ABBREVIATIONS
- HDX-MS
hydrogen-deuterium exchange with mass spectrometry
- GPCR, G
protein coupled receptor
- MD
molecular dynamics
- RGS
regulator of G-protein signaling
- TDZD
thiadiazolidinone
Footnotes
Notes
No competing financial interests have been declared.
REFERENCES
- (1).Whitty A; Kumaravel G Nat. Chem. Biol 2006, 2 (3), 112–118. [DOI] [PubMed] [Google Scholar]
- (2).Jin L; Wang W; Fang G Annu. Rev. Pharmacol. Toxicol 2014, 54 (1), 435–456. [DOI] [PubMed] [Google Scholar]
- (3).Arkin MR; Tang Y; Wells JA Chem. Biol 2014, 21 (9), 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Stank A; Kokh DB; Fuller JC; Wade RC Acc. Chem. Res 2016, 49 (5), 809–815. [DOI] [PubMed] [Google Scholar]
- (5).Oleinikovas V; Saladino G; Cossins BP; Gervasio FL J. Am. Chem. Soc 2016, 138 (43), 14257–14263. [DOI] [PubMed] [Google Scholar]
- (6).Eyrisch S; Helms VJ Med. Chem 2007, 50 (15), 3457–3464. [DOI] [PubMed] [Google Scholar]
- (7).Gunasekaran K; Ma B; Nussinov R Proteins: Struct., Funct., Bioinf 2004, 57 (3), 433–443. [DOI] [PubMed] [Google Scholar]
- (8).Kunze J; Todoroff N; Schneider P; Rodrigues T; Geppert T; Reisen F; Schreuder H; Saas J; Hessler G; Baringhaus K-H; Schneider GJ Chem. Inf. Model 2014, 54 (3), 987–991. [DOI] [PubMed] [Google Scholar]
- (9).Christopoulos A Nat. Rev. Drug Discov 2002, 1 (3), 198–210. [DOI] [PubMed] [Google Scholar]
- (10).Mittal A; Johnson ME J. Mol. Graph. Model 2015, 55, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hopkins AL; Groom CR Nat. Rev. Drug Discov 2002, 1 (9), 727–730. [DOI] [PubMed] [Google Scholar]
- (12).Turner EM; Blazer LL; Neubig RR; Husbands SM ACS Med. Chem. Lett 2011, 3 (2), 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Blazer LL; Zhang H; Casey EM; Husbands SM; Neubig RR Biochemistry 2011, 50 (15), 3181–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gold SJ; Ni YG; Dohlman HG; Nestler EJJ Neurosci 1997, 17 (20), 8024–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Larminie C; Murdock P; Walhin J-P; Duckworth M; Blumer KJ; Scheideler MA; Garnier M Mol. Brain Res 2004, 122 (1), 24–34. [DOI] [PubMed] [Google Scholar]
- (16).Lerner TN; Kreitzer AC Neuron 2012, 73 (2), 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shen W; Plotkin JL; Francardo V; Ko WKD; Xie Z; Li Q; Fieblinger T; Wess J; Neubig RR; Lindsley CW; Conn JP; Greengard P; Bezard E; Cenci MA; Surmeier DJ Neuron 2015, 88 (4), 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Blazer LL; Storaska AJ; Jutkiewicz EM; Turner EM; Calcagno M; Wade SM; Wang Q; Huang X-P; Traynor JR; Husbands SM; Morari M; Neubig RR ACS Chem. Neurosci 2015, 6 (6), 911–919. [DOI] [PubMed] [Google Scholar]
- (19).Neubig RR; Siderovski DP Nat. Rev. Drug Discov 2002, 1 (3), 187–197. [DOI] [PubMed] [Google Scholar]
- (20).Tesmer JJG Prog. Mol. Biol. Transl. Sci 2009, 86, 75–113. [DOI] [PubMed] [Google Scholar]
- (21).Tesmer JJG; Berman DM; Gilman AG; Sprang SR Cell 1997, 89 (2), 251–261. [DOI] [PubMed] [Google Scholar]
- (22).Taylor VG; Bommarito PA; Tesmer JJG J. Biol. Chem 2016, 291 (10), 5138–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vashisth H; Storaska AJ; Neubig RR; Brooks CL 3rd ACS Chem. Biol 2013, 8 (12), 2778–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Park I-H; Venable JD; Steckler C; Cellitti SE; Lesley SA; Spraggon G; Brock AJ Chem. Inf. Model 2015, 55 (9), 1914–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Persson F; Halle B Proc. Natl. Acad. Sci. U.S.A 2015, 112 (33), 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Roman DL; Blazer LL; Monroy CA; Neubig RR Mol. Pharmacol 2010, 78 (3), 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Soundararajan M; Willard FS; Kimple AJ; Turnbull AP; Ball LJ; Schoch GA; Gileadi C; Fedorov OY; Dowler EF; Higman VA; Hutsell SQ; Sundström M; Doyle DA; Siderovski DP Proc. Natl. Acad. Sci. U.S.A 2008, 105 (17), 6457–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).de Alba E; De Vries L; Farquhar MG; Tjandra NJ Mol. Biol 1999, 291 (4), 927–939. [DOI] [PubMed] [Google Scholar]
- (29).Weis DD; Wales TE; Engen JR; Hotchko M; Ten Eyck LF J. Am. Soc. Mass Spectrom 2006, 17 (11), 1498–1509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.