

Abstract
Patients with cancer who develop sepsis have a markedly higher mortality than patients who were healthy prior to the onset of sepsis. Potential mechanisms underlying this difference have previously been examined in two preclinical models of cancer followed by sepsis. Both pancreatic cancer/pneumonia and lung cancer/cecal ligation and puncture (CLP) increase murine mortality, associated with alterations in lymphocyte apoptosis and intestinal integrity. However, pancreatic cancer/pneumonia decreases lymphocyte apoptosis and increases gut apoptosis while lung cancer/CLP increases lymphocyte apoptosis and decreases intestinal proliferation. These results cannot distinguish the individual roles of cancer versus sepsis since different models of each were used. We therefore created a new cancer/sepsis model to standardize each variable. Mice were injected with a pancreatic cancer cell line and three weeks later cancer mice and healthy mice were subjected to CLP. Cancer septic mice had a significantly higher 10-day mortality than previously healthy septic mice. Cancer septic mice had increased CD4+ T cells and CD8+ T cells, associated with decreased CD4+ T cell apoptosis 24 hours after CLP. Further, splenic CD8+ T cell activation was decreased in cancer septic mice. In contrast, no differences were noted in intestinal apoptosis, proliferation or permeability, nor were changes noted in local bacterial burden, renal, liver or pulmonary injury. Cancer septic mice thus have consistently reduced survival compared to previously healthy septic mice, independent of the cancer or sepsis model utilized. Changes in lymphocyte apoptosis are common to cancer model and independent of sepsis model whereas gut apoptosis is common to sepsis model and independent of cancer model. The host response to the combination of cancer and sepsis is dependent, at least in part, on both chronic co-morbidity and acute illness.
Keywords: Sepsis, CD8 T cell, CD4 T cell, cell death, intestine, gut, mortality
INTRODUCTION
The global burden of sepsis is enormous, with over 31.5 million cases of sepsis and at least 5 million deaths annually (1). Outside of antibiotics, treatment for sepsis is non-specific, leading to intense efforts to better understand the mechanisms behind mortality from sepsis (2–4). However, not all patients have the same risk of developing sepsis or of having a poor outcome if they become septic (5). Patients with cancer are nearly ten times more likely to develop sepsis than the general population (6). Further, cancer represents the most common co-morbidity in septic patients, and sepsis is the leading cause of ICU admission in patients with malignancy (7–9). Notably, cancer is the co-morbidity associated with the highest risk of death in sepsis, and hospital mortality can exceed 50% in patients with cancer and sepsis (9–12).
The etiology behind the increased mortality seen in cancer septic patients is multifactorial. Some deaths are attributable to secondary effects of cancer treatment such as chemotherapy. However, other deaths are likely related to the host’s inability to develop an adaptive response to infection in the setting of pre-existing immunosuppression as well as immune cell exhaustion related to the underlying malignancy (13).
In an effort to better understand how cancer impacts the pathophysiology of sepsis, we have previously published two preclinical studies on mice that received injections of transplantable cancer cell lines prior to the onset of sepsis (14;15). Both demonstrated increased mortality in cancer septic mice compared to previously healthy septic mice, mirroring outcomes seen in patients. However, findings outside of mortality varied significantly between the models. Mice with pancreatic cancer followed by Pseudomonas aeruginosa pneumonia had decreased T cell apoptosis and increased intestinal epithelial apoptosis, without changes in local infection control, renal function or liver function (14). In contrast, mice with lung cancer followed by CLP had increased CD4+ T cell apoptosis and no change in intestinal epithelial apoptosis but a decrease in crypt proliferation, decreased local infection, increased BUN and creatinine, and increased BAL protein (15). It is notable, however, that both the cancer model (pancreas, lung) and sepsis model (pneumonia, CLP) differed in these sets of experiments making it difficult to ascertain the relative contributions of cancer and sepsis towards these outcomes. We therefore developed a new model (pancreatic cancer followed by CLP) using a common variable from each of these prior publications to determine the relative importance of pre-existing comorbidity and type of sepsis on the physiologic abnormalities induced by the combination of cancer and sepsis.
METHODS
Mice
Six week old male C57Bl6 mice were obtained from Jackson Laboratories (Bar Harbor, Maine) and housed in an approved animal facility at Emory University. Male mice were used in light of a desire to compare to our original publication on cancer and sepsis (14), which was published prior to the current NIH policy on rigor and reproducibility. Mice were allowed 1 week to acclimatize to the animal facility before any interventions were performed. Animals were then randomized to either receive cancer or not and were followed for an additional three weeks. All animals were then subjected to CLP at ten weeks of age, and they were either sacrificed 24 hours later or followed ten days for survival. Mice had free access to food and water throughout. All mice that were injected with tumor cells were monitored according to the Emory IACUC guidelines for tumor burden and were sacrificed if the tumor ulcerated, caused difficulties in ambulation or exceeded acceptable tumor size. In addition, all animals were checked twice daily after the induction of CLP, and moribund animals were sacrificed. All experiments were performed in accordance with the National Institutes of Health Guidelines for the Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Emory University School of Medicine (Protocol DAR-2003199-071415N).
Cancer models
Mice randomized to the cancer group received a 200 µl subcutaneous injection into the right hind-limb containing 375,000 live cells from the mouse pancreatic adenocarcinoma cell line Pan02 (16). Cancer cells were cultured in RMPI medium with 10% fetal bovine serum, 1% penicillin-streptomycin, 1% glutamine, and 1% 4-(2-hydroxyethyl)-1-piperazeneethanesulfonic acid (Cellgro, Herndon, VA). Prior to injection, cells were lysed from culture using 0.25% trypsin, washed, and re-suspended in PBS. Control mice (subsequently referred to as previously healthy) did not receive any injections and thus had no intervention prior to CLP. To verify that the presence of vehicle alone did not impact CD4+ and CD8+ cell numbers, apoptosis and activation, mice injected with vehicle alone and mice without injection were compared by flow cytometry three weeks after injection and no difference was noted in any group (Supplemental figure 1). Cancer mice and previously health mice were monitored for the identical length of time after arrival to the animal facility and thus were aged matched throughout all experiments. Of note, body weights were similar between previously healthy animals and cancer mice three weeks after injection of cancer cells (i.e. just prior to the induction of sepsis): 22.7 ± 1.6 grams vs. 23.1 ± 1.6 grams, p=0.68.
A different group of cancer mice received a 200 µl subcutaneous injection into the right hind-limb of 250,000 live cells from the murine lung carcinoma cell line (LLC1, American Type Culture Collection, Manassas, VA) as previously described (15).
Sepsis models
Animals were subjected to CLP, a well-established murine model of polymicrobial intra-abdominal sepsis (17). Mice were anesthetized via inhaled isoflurane and a midline abdominal incision was made. The cecum was identified, exteriorized and ligated near its base with silk suture. The cecum was then punctured twice with a 25-gauge needle, and a small amount of stool was extruded. The bowel was then returned to the abdominal cavity, and the incision was closed. To minimize animal suffering, all mice received pre-operative analgesia with Buprenex (0.1 mg/kg, McKesson Medical, San Francisco, CA). Mice received a 1ml subcutaneous injection of sterile saline immediately after surgery, and antibiotics (50 mg/kg of ceftriaxone, Sigma-Aldrich, St. Louis, MO and 35 mg/kg metronidazole, Apotex Corp, Weston, FL) were injected subcutaneously at 12, 24, and 36 hours after surgery. Animals were either followed for survival for ten days or sacrificed either 24 hours or 5 days after CLP. Data shown represents 1–3 replicates/experiment and the number of mice used for each experiment in presented in each figure legend.
A different subset of mice were subjected to pneumonia. Under isoflurane anesthesia, mice were given an intratracheal injection of P. aeruginosa. A 1 cm. midline neck incision was created, and after separation of the strap muscles, the trachea was identified and 40uL of P. aeruginosa (Strain ATCC 27853, Manassas, VA) diluted in PBS (2 × 108 CFU/mL) and injected via a 28-gauge syringe. Animals were then held vertically for 10 seconds to improve bacterial delivery to the lungs. Sham animals underwent the identical procedure, except they received an intratracheal injection of sterile saline instead of bacteria.
Flow cytometry
Spleens were removed and processed to single-cell suspension by grinding with a plunger from a small syringe in a 70 µm filter placed over a 50 ml conical tube. Samples were rinsed with cold PBS, spun down in a centrifuge and re-suspended in PBS. CountBright Absolute Counting Beads (Thermo Fisher, Waltham, MA) were added, and surface marker stains were incubated for 30 minutes on ice before being analyzed using color flow cytometry. Two panels were used: Panel 1 - FITC:CD25 (Clone: PC61, BD Pharmingen, San Jose, CA), PE:CD69 (Clone: H.2F3, Biolegend, San Diego, CA), PB:CD4 (Clone: PM4–5, BD Pharmingen), PO:CD8 (Clone: 53-6.7, Thermo Fisher), APC-Cy7:CD3 (Clone: 17A2, Biolegend); Panel 2 - FITC:Annexin V (Biolegend), PE:empty, PerCP:7-AAD (Biolegend), PB:CD4 (Clone: RPA-T4, BD Pharmingen), PO:CD8 (Clone 53-6.7, Thermo Fisher), AF700:CD3 (Clone 500A2, eBioscience). Flow cytometry was also performed on mesenteric lymph nodes which were collected, homogenized and stained. In addition, blood was collected via cardio-puncture at time of sacrifice, treated with High-yield Lyse (Life Technologies, Waltham, MA) and then stained as above.
Serum and BAL studies
At time of sacrifice, whole blood was collected and centrifuged for 10 minutes at 10,000 RPM. The supernatant was collected and analyzed using a microplate creatinine assay (Oxford Biomedical Research, Rochester Hills, MI). A nitrogen colorimetric kit (Arbor Assays, Ann Arbor, MI) was used to measure blood urea nitrogen (BUN). Liver enzymes asparagine aminotransferase (AST) and alanine aminotransferase (ALT) were measured with a Beckman AU480 chemistry analyzer machine (Beckman Diagnostics, LaBrea, CA).
To determine protein levels in BAL fluid, the trachea of sacrificed animals was lavaged with 1 ml of PBS, withdrawn, and centrifuged for 10 minutes at 10,000 RPM. The concentration of protein was then determined by treating samples with protein assay reagent (Thermo Scientific, Rockford, IL) and analyzing absorbance in a microplate reader at 660 nm in comparison to a standard curve of bovine serum albumin.
Intestinal integrity
Apoptotic cells were quantified in the jejunum of 100 consecutive well-oriented crypts by two complementary techniques: active caspase 3 staining to identify cells with activation of the common caspase executioner and H&E staining to identify cells with characteristic morphologic changes (18). For active caspase-3 staining, slides were deparaffinized, rehydrated, incubated in 3% hydrogen peroxide for 15 minutes, and treated with antigen decloaker (Biocare Medical, Concord, CA) for 45 minutes in a pressure cooker. Slides were then treated with protein block (Dako, Carpinteria, CA) for 30 minutes at room temperature and then incubated overnight at 4°C with rabbit anti-caspase 3 (1:100; Cell Signaling, Beverly, MA), followed by goat anti-rabbit biotinylated antibody (1:500; Vector Laboratories) and streptavidin horseradish peroxidase (1:500; Dako) each for one hour at room temperature. Slides were developed in DAB and then counterstained with hematoxylin.
Intestinal proliferation was measured by staining S-phase cells with 5-bromo-2-deoxyuridine (BrdU, 5mg/ml in 0.9% saline, Sigma-Aldrich), which was injected into the peritoneum of mice 90 minutes prior to sacrifice. Small intestinal tissue was then fixed, deparaffinized, and rehydrated as above, before being incubated in 1% hydrogen peroxide for 15 minutes at room temperature. Slides were treated with antigen decloaker, protein block, and then incubated overnight at 4°C with rat monoclonal anti-BrdU (1:500; Accurate Chemical and Scientific, Westbury, NJ). Slides were then treated with goat anti-rat antibody (1:500; Accurate Chemical & Scientific) and streptavidin horseradish peroxidase (1:500; Dako), for one hour each at room temperature before being developed in DAB. Positive cells were quantified in 20 contiguous well-oriented crypts.
To measure intestinal permeability, mice were gavaged with fluorescein isothiocyanate-dextran (FD4, 0.5 ml of 22 mg/ml, Sigma-Aldrich) 5 hours before sacrifice. Serum was analyzed for FD4 concentration on a microplate reader (BioTek Synergy HT, Winooski, VT) using fluorospectrometry with 485/520 nm excitation/emission wavelengths against a standard curve of serial dilutions.
Peritoneal cultures
A total of 3 ml of PBS was injected into the abdomen of mice at time of sacrifice, gently agitated for 5 seconds and then withdrawn. Quantitative peritoneal fluid cultures were prepared using 10-fold serial dilutions of samples with sterile 0.9% saline. Each dilution and a 100 µl aliquot of raw sample were plated on blood agar plates (Remel, Lenexa, KS) and incubated for 24 hours (35°C in 5% CO2 atmosphere). Plates containing fewer than 300 colonies were analyzed for colony counts, multiplying the number of counted colonies by the reciprocal of the dilution counted, adjusting for the volume of plated sample.
Tissue histology
Sections of whole lung, liver, and kidney tissue were fixed overnight in 10% formalin before being fixed in paraffin and sectioned. Tissues were stained with H&E and assessed for injury and percent inflammatory infiltrate by a pathologist blinded to section identity (ABF).
Statistics
All data were analyzed using the software program Prism 6.0 (GraphPad, San Diego, CA) and are presented as mean ± SD. Survival data were analyzed using the log-rank test. Data were tested for normality using the D'Agostino-Pearson omnibus normality test. Comparisons on data with a Gaussian distribution were performed using the Student’s t-test while comparisons on data that did not have a Gaussian distribution were performed using the Mann Whitney test. Data were considered to be statistically significant for a p value of <0.05.
RESULTS
The presence of pancreatic cancer worsens survival following sepsis
All mice that received subcutaneous injections of pancreatic cancer cells developed non-metastatic solitary tumors at the site of injection prior to the onset of sepsis. Mice that had cancer prior to the onset of sepsis had a significantly lower 10-day survival than mice that were healthy prior to the onset of sepsis (16% vs 50%, Fig. 1).
FIG. 1. Effect of cancer and sepsis on survival.
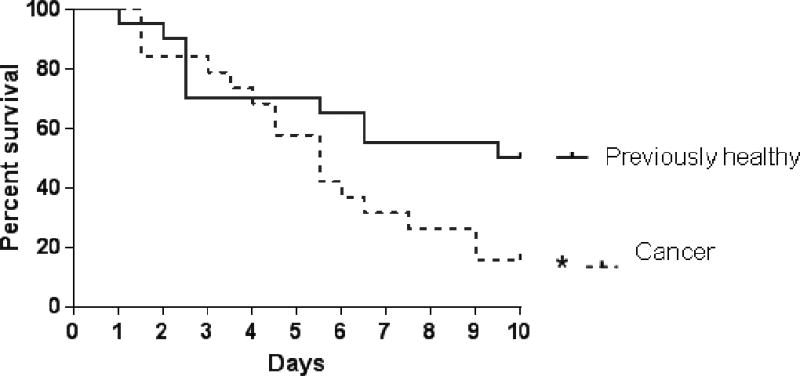
Cancer septic mice had significantly worse 10-day survival compared to previously healthy septic mice (p=0.048, n=19–20/group).
The presence of pancreatic cancer increases CD4+ and CD8+ T cell counts, decreases septic CD4+ T cell apoptosis and decreases CD8+T cell activation following sepsis
Cancer septic mice had an increase in the absolute number of CD4+ T cells in the spleen compared to previously healthy septic mice (Fig. 2A). This was associated with a decrease in Annexin+7AAD+ CD4+ apoptotic cells (Fig. 2B). No difference was noted in the frequency of activated CD4+ T cells, whether measured by the percentage of CD25+ or CD69+ within the CD4+ T cell compartment (Fig. 2 C, D). Cancer septic mice also had an increase in the absolute number of CD8+ T cells compared to previously healthy septic mice (Fig. 2E), although this was not associated with a change in the frequency of Annexin+7AAD+ CD8+ apoptotic cells (Fig. 2F). Activated CD8+ T cells were decreased in cancer septic mice compared to previously healthy septic mice, as evidenced by decreased percentage of both CD25+ T cells and CD69+ cells within the CD8+ T cell compartment (Fig. 2 G, H).
FIG. 2. Effect of cancer and sepsis on splenocyte T cell number, apoptosis and activation.
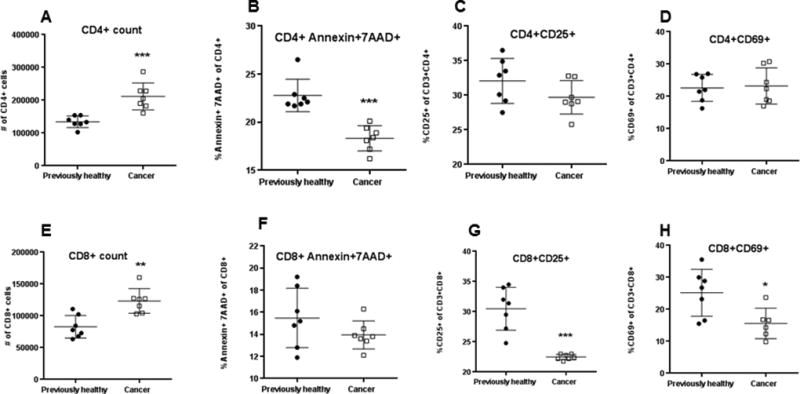
Cancer septic mice had an increase in the absolute number of CD4+ T cells 24 hours after CLP compared to previously healthy mice (A, p=0.0006) associated with decreased Annexin+7AAD+ CD4+ apoptotic cells (B, p=0.0006). This was not associated with a change in activation as measured by the percentage of CD25+ CD4+ (C, p=0.10) or CD69+ CD4+ cells (D, p=0.87). Cancer septic mice also had an increase in the absolute number of CD8+ T cells (E, p=0.002) without a statistically significant change in Annexin+7AAD+ CD8+ apoptotic cells (F, p=0.31). Activation was decreased in CD8+ cells in cancer septic mice as measured by the percentage of CD25+ (G, p=0.0006) or CD69+ cells (H, p=0.05), n=7 for all groups except n=6 for cancer septic CD69.
To determine whether these results were compartment specific, activation status was assessed on CD4+ T and CD8+ T cells from blood (Fig. 3A–D) and mesenteric lymph nodes (Fig. 3E–H). In contrast to the spleen, activation as measured by CD25+ or CD69+ was similar in both CD4+ T and CD8+ T cells in both blood and mesenteric lymph node except for an increase in CD69+ of CD4+ T cells from cancer/septic mice in mesenteric lymph nodes.
FIG. 3. Effect of cancer and sepsis on blood and mesenteric T cell activation.
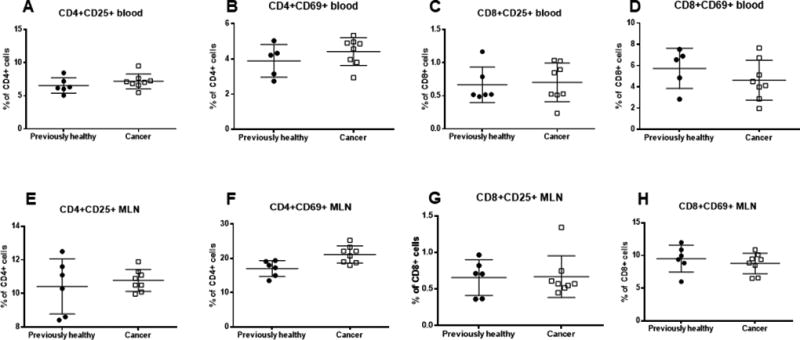
CD4+ activation was similar in the blood between cancer septic mice and previously healthy septic mice as measured by CD25+ (A, p=0.28) and CD69+ (B, p=0.43) cells. Similarly blood CD8+ activation was similar in the blood as measured by CD25+ (C, p=0.63) and CD69+ (D, p=0.43) cells. CD4+CD25+ cells were similar in the mesenteric lymph nodes between cancer septic mice and previously healthy septic mice (E, p=0.87) although CD4+CD69+ cells were higher in cancer/septic mice (F, p=0.02). No difference was detected in activation of CD8+ cells in the mesenteric lymph nodes whether measured by CD25+ (G, p=0.82) or CD69+ (H, p=0.43) cells, n=6–8/group for all panels.
To determine whether cancer sepsis altered cell numbers in other immune cells, B cells, NK cells, macrophages, neutrophils and dendritic cells were also examined (Fig 4). All were similar between cancer septic mice and previously healthy septic mice except dendritic cells which were lower in cancer septic mice.
FIG. 4. Effect of cancer and sepsis on non-T cell immune numbers.
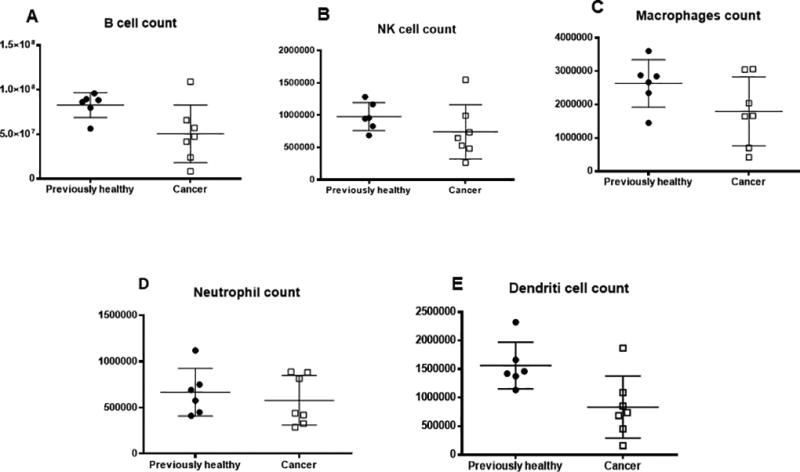
Cancer septic mice and previously healthy mice had similar numbers of B cells (A, p=0.07), NK cells (B, p=0.18), macrophages (C, p=0.29) and neutrophils (D, p=0.62) 24 hours after CLP. Dendritic cell numbers were lower in cancer septic mice (E, p=0.02), n=6–8 for all groups.
The presence of pancreatic cancer does not impact local infection control following sepsis
Bacterial burden in the peritoneal cavity was similar in both cancer septic and previously healthy septic mice 24 hours after CLP (Fig. 5).
FIG. 5. Effect of cancer and sepsis on peritoneal bacterial burden.
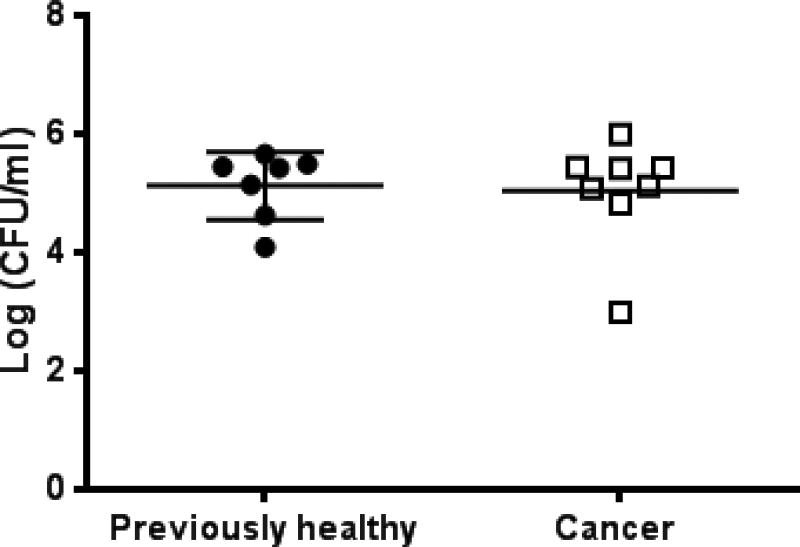
Cancer septic mice had similar numbers of bacteria in peritoneal fluid as previously healthy septic mice 24 hours after the onset of sepsis (p=0.63, n=7–8/group).
The presence of pancreatic cancer does not impact gut integrity following sepsis
Gut epithelial apoptosis was similar in both cancer septic mice and previously healthy septic mice (Fig. 6A, B). In addition, crypt proliferation (Fig. 6C) and intestinal permeability (Fig. 6D) were similar between cancer septic mice and previously healthy septic mice.
FIG. 6. Effect of cancer and sepsis on intestinal integrity.
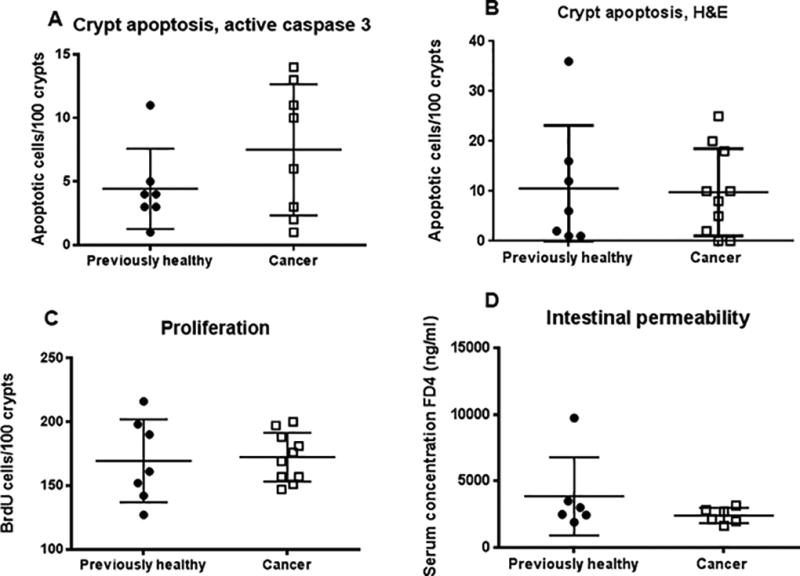
Intestinal epithelial apoptosis whether measured by active caspase 3 staining (A, p=0.38, n=7–8/group) or H&E staining (B, p=0.98, n=7–10/group) was similar between cancer septic mice and previously healthy septic mice. Crypt proliferation (C, p=0.87, n=7–10/group) and intestinal permeability (D, p=0.39, n=6/group) were also similar between the groups
The presence of pancreatic cancer does not impact organ dysfunction following sepsis
Renal function as measured by BUN and creatinine was similar in both cancer septic mice and previously healthy septic mice (Fig. 7A, B). Liver function as measured by levels of AST or ALT was also similar (Fig. 7C, D). Levels of BUN, creatinine, AST and ALT were also similar between cancer septic mice and previously healthy mice 5 days after CLP (Supplemental figure 2). Protein concentration in BAL fluid was also similar between cancer septic and previously healthy septic mice (Fig. 7E). Kidney, liver and lung histology also did not demonstrate differences in organ injury or inflammation between cancer septic and previously healthy septic mice (data not shown).
FIG. 7. Effect of cancer and sepsis on organ dysfunction.
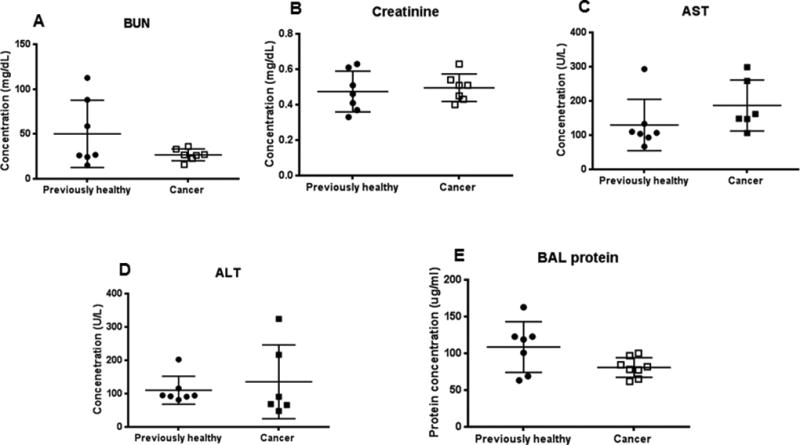
Renal function as measured by BUN (A, p=0.59, n=7/group), and creatinine (B, p=0.64, n=7/group), liver function as measured by AST (C, p= 0.07, n=6–7/group) and ALT (D, p=0.51, n=6–7/group) and lung injury as measured by BAL protein (E, p=0.09, n=7–8/group) were all similar between cancer septic mice and previously healthy septic mice.
The presence of lung cancer does not alter lymphocyte or gut apoptosis or renal function following pneumonia-induced sepsis
Previous publications have examined pancreatic cancer/pneumonia and lung cancer/CLP while this manuscript examines pancreatic cancer/CLP. The fourth possible combination of cancer/sepsis is lung cancer/pneumonia. A new model was therefore created by injecting mice lung cancer cells followed three weeks later by P. aeruginosa pneumonia. There were no differences in CD4+, CD8+ or gut apoptosis between cancer septic mice and previously healthy septic mice (Fig. 8). Similarly no difference was noted in renal function between cancer septic mice and previously healthy septic mice (Fig. 8).
FIG. 8. Effect of lung cancer/pneumonia on lymphocyte and gut apoptosis and renal function.
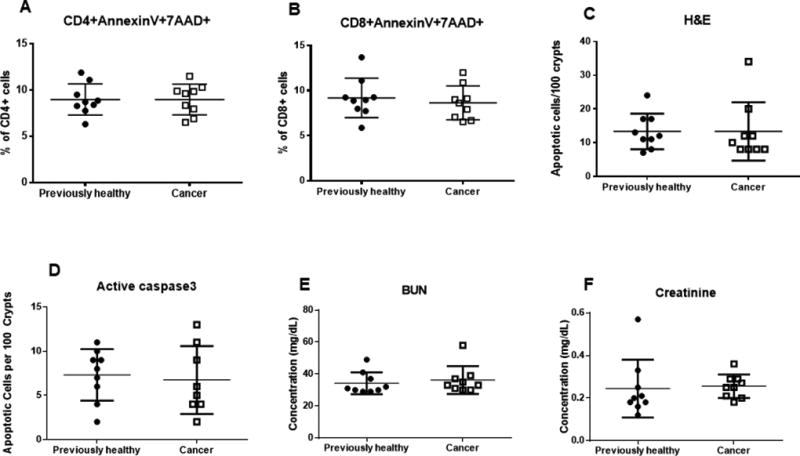
No differences were noted between cancer septic mice and previously healthy septic mice in CD4+ apoptosis (A, p=0.99), CD8+ apoptosis (B, p=0.87), or gut apoptosis as measured by H&E (C, p=0.58) or active caspase 3 (D, p=0.73). No differences were noted in renal function as measured by BUN (E, p=0.37) or Cr (F, p=0.21), n=9 for all groups.
DISCUSSION
Mice with pancreatic cancer followed by CLP had increased mortality compared to previously healthy septic mice. This was accompanied by a decrease in apoptotic CD4+ T cells and by a decrease in activated CD8+ T cells in cancer septic mice. In contrast, no differences were detected in any element of intestinal integrity, local infection control or renal, liver or lung function or histology.
These results are best interpreted in the context of two previous publications on cancer and sepsis. All three studies (pancreatic cancer/CLP herein and previously pancreatic cancer/pneumonia and lung cancer/CLP) demonstrate increased mortality in cancer/sepsis, consistent with the human condition. However, there was minimal overlap in abnormalities associated with increased mortality in our prior studies. Specifically, the dominant findings in pancreatic cancer followed by pneumonia were decreased T cell apoptosis and increased intestinal epithelial apoptosis, without changes in local bacterial burden or renal, liver of lung function or histology (14). In contrast, mice with lung cancer followed by CLP had increased CD4+ T cell apoptosis, no change in intestinal epithelial apoptosis but a decrease in crypt proliferation, decreased local bacterial burden, increased BUN and creatinine, increased BAL protein and no change in AST, ALT (15). This study took a single variable that was common to our two previous publications (cancer model, sepsis model) to determine whether the specific type of cancer or specific type of sepsis played a dominant role in the abnormalities seen in cancer/sepsis. This is conceptually important since if findings were similar in lung cancer/CLP and pancreatic cancer/CLP but different from pancreatic cancer/pneumonia, this would suggest that type of sepsis is more important than type of cancer in the host response to cancer/sepsis. Alternatively, if findings were similar in pancreatic cancer/pneumonia and pancreatic cancer/CLP but different from lung cancer/CLP, this would suggest that type of cancer is more important than type of sepsis. The reality appears to be more nuanced than either of these extremes. In fact, while there is minimal overlap between pancreatic cancer/pneumonia and lung cancer/CLP, there is significant overlap of the pancreatic cancer/CLP with both. This suggests that both the chronic inflammatory state induced by cancer and the acute illness induced by sepsis play important roles in determining the host response.
It is notable that T cell apoptosis is altered in all models of sepsis and cancer. Multiple studies have demonstrated that CD4+ and CD8+ T cell apoptosis is increased in both septic patients and preclinical models of sepsis. Further, preventing sepsis-induced lymphocyte apoptosis via multiple pathways improves survival in murine sepsis (19–23). However, preventing sepsis-induced apoptosis by either lymphocyte-specific overexpression of Bcl-2 or genetic deletion of Bim increases mortality in pancreatic cancer followed by pneumonia (24). This suggests that the presence of cancer may fundamentally alter T cell behavior during sepsis. To study this further, we examined CD25 and CD69 expression in T cell subsets following pancreatic cancer/CLP. Activation was decreased in splenic CD8+ cells in cancer septic mice compared to previously healthy septic mice. Notably, this was compartment specific since activation was not altered in either blood or mesenteric lymph nodes. T cell activation has not previously been examined in models of cancer and sepsis, so we cannot draw any conclusions about the generalizability of this finding. However, it is plausible that decreased splenic T cell activation leads to an ineffective immune response that, coupled with immune exhaustion that occurs in both cancer and sepsis (13;25–28) independently represents a potential mechanism for the increased mortality seen when these two deadly entities are combined. It is also important to note that despite differences in the immune system, there was no difference in the amount of bacteremia between cancer septic mice and previously healthy septic mice. This is consistent with recent studies of the co-inhibitory marker 2B4, where genetic deletion or pharmacologic inhibition leads to improved mortality due to altered signaling and function of CD4+ cells, yet there is no difference in bacterial burden regardless of immune function (29). This may potentially be explained by “disease tolerance,” a relatively new biological concept in which the host can protect against infectious disease by reducing the negative impact of infections on its overall fitness (30). The mechanisms underlying “disease tolerance” are complex and multifactorial, but include damage control allowing for preservation of functional output of parenchymal tissues, thus maintaining homeostatic parameters within a dynamic range (31).
An unexpected negative finding in this study was the observation that all components of gut integrity examined (apoptosis, proliferation, permeability) were similar between cancer septic mice and previously healthy septic mice. Given previous alterations in intestinal integrity in both pancreatic cancer/pneumonia and lung cancer/CLP, as well as the gut’s role as “the motor” of sepsis (32–34), it might have been predicted that multiple components of intestinal integrity would be altered by pancreatic cancer/CLP. The absence of a difference between cancer septic and previously healthy septic mice argues suggests that the gut epithelium plays a less important role (or at a minimum, a less consistent role) in mediating mortality in cancer sepsis. It should be noted, however, that we did not examine the microbiome in this study. Considering the role of the microbiome in mediating outcomes in both cancer and sepsis (35;36) and the role of the microbiome in modulating interaction between immune and cancer cells (37;38), the microbiome assuredly deserves further investigation in cancer and sepsis.
This study has a number of limitations. First, only male mice were used. The rationale behind this decision was that we wanted to be able to directly compare results to our study on pancreatic cancer and pneumonia (14), which was published prior to current NIH guidelines on rigor and reproducibility. However, the impact of gender on outcomes from sepsis is well known (39), and we acknowledge the downside to the current experimental design is that the results presented herein are not generalizable to half the population and should be repeated in female mice in future experiments. Next, the majority of endpoints other than survival were assayed 24 hours after CLP, and this likely misses insights that could be gained by examining other timepoints. Further, we chose to examine endpoints for which we had previously published data in pancreatic cancer/pneumonia and lung cancer/CLP. While this had the advantage of allowing direct comparisons to answer our fundamental question of the relative importance of cancer model vs. sepsis model, multiple other endpoints could potentially have yielded new insights, and the absence of molecular data in this manuscript limits conclusions that may be drawn. Further, the absolute CD4+ T cell and CD8+ T cell counts are lower than might have been expected in the literature, even in the setting of sepsis for which we do not have a clear explanation. To highlight this limitation, splenic CD8+ T cell activation -- the sole endpoint in this study that we have not previously examined in cancer/sepsis -- demonstrated new findings herein. Next, between the experiments presented herein and prior publications, we now have extensive data on three of the four combinations of cancer/sepsis -- pancreatic cancer/CLP, pancreatic cancer/pneumonia and lung cancer/CLP. However, a fourth unexplored combination is lung cancer/pneumonia. We performed a limited analysis on lymphocyte and gut apoptosis and renal function in these mice and found no difference between cancer septic and previously healthy septic mice. These results should be interpreted with caution, however, as we did not perform a dose/response curve to examine the mortality of this model, instead using our previously published models of lung cancer and pneumonia. There is no way to know if the combination examined replicates the mortality in cancer/sepsis without performing extensive additional experiments outside the scope of this manuscript. Finally, the transplantable model of cancer in which mice develop solitary tumors 3 weeks after injection may not accurately reflect the more gradual development of naturally occurring malignancies, while the development of tumors in hind-limbs as opposed to in-situ pancreatic tissue may alter how the tumor is perceived by immune cell populations.
Despite these limitations, our results demonstrate that both cancer and sepsis play distinct roles in the host response when the two conditions are combined. Considering the marked increase in both incidence and mortality of cancer and sepsis in patients compared to previously healthy patients, this has significant implications for studying this common and deadly clinical scenario. When more detailed studies of cancer and sepsis are performed in the future, it is insufficient to assume there is a common response to either variable, which should help guide experimental design and interpretation.
Supplementary Material
Supplemental FIG. 1. Effect of vehicle injection on T cell number, activation and apoptosis. Three weeks after injection of vehicle or no injection of vehicle, no differences were noted in absolute number of CD4+ T cells (A, p=0.09) 7AAD+ CD4+ apoptotic cells (B, p=0.84) or activation as measured by the percentage of CD25+ CD4+ (C, p=0.72) or CD69+ CD4+ cells (D, p=0.34). Similarly, no differences were noted in absolute number of CD8+ T cells (E, p=0.85) 7AAD+ CD8+ apoptotic cells (F, p=0.46) or activation as measured by the percentage of CD25+ CD8+ (G, p=0.89) or CD69+ CD8+ cells (H, p=0.98). n=10 for all groups.
Supplemental FIG. 2 Effect of cancer and sepsis on organ dysfunction 5 days following CLP. Renal function as measured by BUN (A, p=0.29), creatinine (B, p=0.87), and liver function as measured by AST (C, p= 0.71) and ALT (D, p=0.94) were all similar between cancer septic mice and previously healthy septic mice, n=4–10/group.
Acknowledgments
Conflicts of Interest and Source of Funding: This work was supported by funding from the National Institutes of Health (GM072808, GM095442, GM104323, GM109779, GM113228, GM117895)
References
- 1.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am J Respir Crit Care Med 1. 2016;193(3):259–272. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 2.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit Care Med. 2017;45(3):486–552. doi: 10.1097/CCM.0000000000002255. [DOI] [PubMed] [Google Scholar]
- 3.Leisman DE, Doerfler ME, Ward MF, Masick KD, Wie BJ, Gribben JL, Hamilton E, Klein Z, Bianculli AR, Akerman MB, et al. Survival Benefit and Cost Savings From Compliance With a Simplified 3-Hour Sepsis Bundle in a Series of Prospective, Multisite, Observational Cohorts. Crit Care Med. 2017;45(3):395–406. doi: 10.1097/CCM.0000000000002184. [DOI] [PubMed] [Google Scholar]
- 4.Seymour CW, Gesten F, Prescott HC, Friedrich ME, Iwashyna TJ, Phillips GS, Lemeshow S, Osborn T, Terry KM, Levy MM. Time to Treatment and Mortality during Mandated Emergency Care for Sepsis. N Engl J Med. 2017;376(23):2235–2244. doi: 10.1056/NEJMoa1703058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esper AM, Martin GS. The impact of cormorbid conditions on critical illness. Crit Care Med. 2011;39(12):2728–2735. doi: 10.1097/CCM.0b013e318236f27e. [DOI] [PubMed] [Google Scholar]
- 6.Danai PA, Moss M, Mannino DM, Martin GS. The epidemiology of sepsis in patients with malignancy. Chest. 2006;129(6):1432–1440. doi: 10.1378/chest.129.6.1432. [DOI] [PubMed] [Google Scholar]
- 7.Rosolem MM, Rabello LS, Lisboa T, Caruso P, Costa RT, Leal JV, Salluh JI, Soares M. Critically ill patients with cancer and sepsis: clinical course and prognostic factors. J Crit Care. 2012;27(3):301–307. doi: 10.1016/j.jcrc.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Williams MD, Braun LA, Cooper LM, Johnston J, Weiss RV, Qualy RL, Linde-Zwirble W. Hospitalized cancer patients with severe sepsis: analysis of incidence, mortality, and associated costs of care. Crit Care. 2004;8(5):R291–R298. doi: 10.1186/cc2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melamed A, Sorvillo FJ. The burden of sepsis-associated mortality in the United States from 1999 to 2005: an analysis of multiple-cause-of-death data. Crit Care. 2009;13(1):R28. doi: 10.1186/cc7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de ME, Tandjaoui-Lambiotte Y, Legrand M, Lambert J, Mokart D, Kouatchet A, Lemiale V, Pene F, Bruneel F, Vincent F, et al. Outcomes in critically ill cancer patients with septic shock of pulmonary origin. Shock. 2013;39(3):250–254. doi: 10.1097/SHK.0b013e3182866d32. [DOI] [PubMed] [Google Scholar]
- 11.Soares M, Caruso P, Silva E, Teles JM, Lobo SM, Friedman G, Dal PF, Mello PV, Bozza FA, Silva UV, et al. Characteristics and outcomes of patients with cancer requiring admission to intensive care units: a prospective multicenter study. Crit Care Med. 2010;38(1):9–15. doi: 10.1097/CCM.0b013e3181c0349e. [DOI] [PubMed] [Google Scholar]
- 12.Torres VB, Azevedo LC, Silva UV, Caruso P, Torelly AP, Silva E, Carvalho FB, Vianna A, Souza PC, Godoy MM, et al. Sepsis-Associated Outcomes in Critically Ill Patients with Malignancies. Ann Am Thorac Soc. 2015;12(8):1185–1192. doi: 10.1513/AnnalsATS.201501-046OC. [DOI] [PubMed] [Google Scholar]
- 13.Mittal R, Wagener M, Breed ER, Liang Z, Yoseph BP, Burd EM, Farris AB, III, Coopersmith CM, Ford ML. Phenotypic T cell exhaustion in a murine model of bacterial infection in the setting of pre-existing malignancy. PLoS ONE. 2014;9(5):e93523. doi: 10.1371/journal.pone.0093523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox AC, Robertson CM, Belt B, Clark AT, Chang KC, Leathersich AM, Dominguez JA, Perrone EE, Dunne WM, Hotchkiss RS, et al. Cancer causes increased mortality and is associated with altered apoptosis in murine sepsis. Crit Care Med. 2010;38(3):886–893. doi: 10.1097/CCM.0b013e3181c8fdb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyons JD, Mittal R, Fay KT, Chen CW, Liang Z, Margoles LM, Burd EM, Farris AB, Ford ML, Coopersmith CM. Murine Lung Cancer Increases CD4+ T Cell Apoptosis and Decreases Gut Proliferative Capacity in Sepsis. PLoS ONE. 2016;11(3):e0149069. doi: 10.1371/journal.pone.0149069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP, Jr, Schabel FM., Jr Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44(2):717–726. [PubMed] [Google Scholar]
- 17.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94(2):331–335. [PubMed] [Google Scholar]
- 18.Vyas D, Robertson CM, Stromberg PE, Martin JR, Dunne WM, Houchen CW, Barrett TA, Ayala A, Perl M, Buchman TG, et al. Epithelial apoptosis in mechanistically distinct methods of injury in the murine small intestine. Histol Histopathol. 2007;22(6):623–630. doi: 10.14670/hh-22.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96(25):14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, Robbins P, Ulbrandt N, Suzich J, Green J, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care. 2014;18(1):R3. doi: 10.1186/cc13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bommhardt U, Chang KC, Swanson PE, Wagner TH, Tinsley KW, Karl IE, Hotchkiss RS. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2004;2172(12):7583–75891. doi: 10.4049/jimmunol.172.12.7583. [DOI] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;5(5):496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick ASS, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306(23):2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fox AC, Breed ER, Liang Z, Clark AT, Zee-Cheng BR, Chang KC, Dominguez JA, Jung E, Dunne WM, Burd EM, et al. Prevention of lymphocyte apoptosis in septic mice with cancer increases mortality. J Immunol. 2011;187(4):1950–1956. doi: 10.4049/jimmunol.1003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mittal R, Chen CW, Lyons JD, Margoles LM, Liang Z, Coopersmith CM, Ford ML. Murine lung cancer induces generalized T-cell exhaustion. J Surg Res. 2015;195(2):541–549. doi: 10.1016/j.jss.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol. 2016;100(6):1239–1254. doi: 10.1189/jlb.4HI0616-255R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res. 2017;208:33–39. doi: 10.1016/j.jss.2016.08.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Olive D, Kuchroo V, Zarour HM. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72(4):887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CW, Mittal R, Klingensmith NJ, Burd EM, Terhorst C, Martin GS, Coopersmith CM, Ford ML. Cutting Edge: 2B4-Mediated Coinhibition of CD4(+) T Cells Underlies Mortality in Experimental Sepsis. J Immunol. 2017;199(6):1961–1966. doi: 10.4049/jimmunol.1700375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;35(6071):936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, Larsen R, Rebelo S, Schauble S, Del BL, et al. Metabolic Adaptation Establishes Disease Tolerance to Sepsis. Cell. 2017;169(7):1263–1275. doi: 10.1016/j.cell.2017.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fay KT, Ford ML, Coopersmith CM. The intestinal microenvironment in sepsis. Biochim Biophys Acta. 2017;1863(10 PtB):2574–2583. doi: 10.1016/j.bbadis.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klingensmith NJ, Coopersmith CM. The Gut as the Motor of Multiple Organ Dysfunction in Critical Illness. Crit Care Clin. 2016;32(2):203–212. doi: 10.1016/j.ccc.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng M, Klingensmith NJ, Coopersmith CM. New insights into the gut as the driver of critical illness and organ failure. Curr Opin Crit Care. 2017;23(2):143–148. doi: 10.1097/MCC.0000000000000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alverdy JC, Krezalek MA. Collapse of the Microbiome, Emergence of the Pathobiome, and the Immunopathology of Sepsis. Crit Care Med. 2017;45(2):337–347. doi: 10.1097/CCM.0000000000002172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krezalek MA, Defazio J, Zaborina O, Zaborin A, Alverdy JC. The Shift of an Intestinal "Microbiome" to a "Pathobiome" Governs the Course and Outcome of Sepsis Following Surgical Injury. Shock. 2016;45(5):475–482. doi: 10.1097/SHK.0000000000000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, Kroemer G. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2018;7(271):271ps1. doi: 10.1126/scitranslmed.3010473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weniger M, D'Haese JG, Angele MK, Chaudry IH. Potential therapeutic targets for sepsis in women. Expert Opin Ther Targets. 2105;19(11):1531–1543. doi: 10.1517/14728222.2015.1057570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental FIG. 1. Effect of vehicle injection on T cell number, activation and apoptosis. Three weeks after injection of vehicle or no injection of vehicle, no differences were noted in absolute number of CD4+ T cells (A, p=0.09) 7AAD+ CD4+ apoptotic cells (B, p=0.84) or activation as measured by the percentage of CD25+ CD4+ (C, p=0.72) or CD69+ CD4+ cells (D, p=0.34). Similarly, no differences were noted in absolute number of CD8+ T cells (E, p=0.85) 7AAD+ CD8+ apoptotic cells (F, p=0.46) or activation as measured by the percentage of CD25+ CD8+ (G, p=0.89) or CD69+ CD8+ cells (H, p=0.98). n=10 for all groups.
Supplemental FIG. 2 Effect of cancer and sepsis on organ dysfunction 5 days following CLP. Renal function as measured by BUN (A, p=0.29), creatinine (B, p=0.87), and liver function as measured by AST (C, p= 0.71) and ALT (D, p=0.94) were all similar between cancer septic mice and previously healthy septic mice, n=4–10/group.