

Abstract
Autosomal dominant polycystic kidney disease is characterized by multiple cysts in both kidneys manifesting in adult life. In general, the disorder is caused by a pathogenic variant in one allele of PKD1 or PKD2 genes, while the other allele is normal. Pathogenic variants in both the alleles are rare and have variable phenotypes, from lethal or perinatal presentation to a mild form in later adulthood, depending on the type of variant. Here, we describe a proband with two variants (p.Thr1773Ile and p.Ala1871Thr in trans) in PKD1 gene, who presented with disease at age 24 years. Both the parents and one brother had a variant in one allele, the other being wild type only and had normal ultrasound findings. Segregation studies suggest that both the variants may act as “hypomorphic” or “incompletely penetrant” alleles and acting together resulted in haploinsufficiency of protein PC1 in renal cells, leading to cystogenesis in the proband. The consequences of the presence of two hypomorphic variants have been poorly documented in literature. We reviewed the few published cases having two hypomorphic variants and the data conform to the conclusions that we reached by study of the family described. It is emphasized that to resolve the significance of suspected hypomorphic variants, segregation studies in the parents and siblings are essential.
Keywords: Autosomal dominant polycystic kidney disease, genetic counseling, hypomorphic alleles, PKD1
Introduction
Autosomal dominant polycystic kidney disease (ADPKD), OMIM 173900, is a monogenic disorder caused by a variant in either PKD1 (~ 85% of cases) or PKD2 (~ 15%) gene. ADPKD patients have wide intra- and interfamilial phenotypic variability, attributable to the role of genes, alleles, modifier genes, or the environment. Pathogenic variants in PKD1 gene result in a more severe phenotype than those in PKD2 gene.[1] Majority of the cases result from variant in one allele, the other allele being normal. Cases with variants in both alleles can present as a lethal or perinatal form to a mild one manifesting in adulthood. The phenotype is determined by the type of variants. Protein-truncating variants (deletions, insertions, nonsense, and canonical splice-site variants) are definitely pathogenic. These variants in combination with nontruncating variants (hypomorphic variants) give a variable phenotype.[2,3,4] We describe a family with two hypomorphic variants that presented as adult-onset PKD.
Case Report
The proband II-1 [pedigree in Figure 1a] was a 34-year-old male, diagnosed with ADPKD using Ravine's criteria,[5,6] at the age of 24 years and had progressed to renal failure within 7 years of diagnosis. He was on dialysis for a year and underwent transplant at the age of 32 years, the mother being the donor. At the time of the transplant, both the kidneys of the proband had prominent lobulated outline and a heterogeneous echotexture. Corticomedullary differentiation was absent in both kidneys. His left and right kidneys measured 10.2 cm × 4.68 cm and 10.06 cm × 4.08 cm, respectively. He had multiple cysts in both the kidneys, largest measuring 4.1 cm in the right kidney and 2.5 cm in the left kidney. There were no extrarenal manifestations. There was no history of renal disease in the family, and both the parents (aged 65 and 60 years) and one brother (aged 30 years) did not have any cysts in their kidneys on ultrasound examination. However, the mother of the proband had a positive history of hypertension in her family. She has remained well without any renal symptoms or signs 7 years after the transplant (last contacted in September 2017). The proband was the eldest of three siblings. Informed consent was obtained from all participants. Blood samples were collected from the family members and the proband, and extracted DNA was analyzed using Sanger sequencing of PKD1 (RefSeq: NM_001009944.2) and PKD2 (RefSeq: NM_000297.3) genes.[7]
Figure 1.
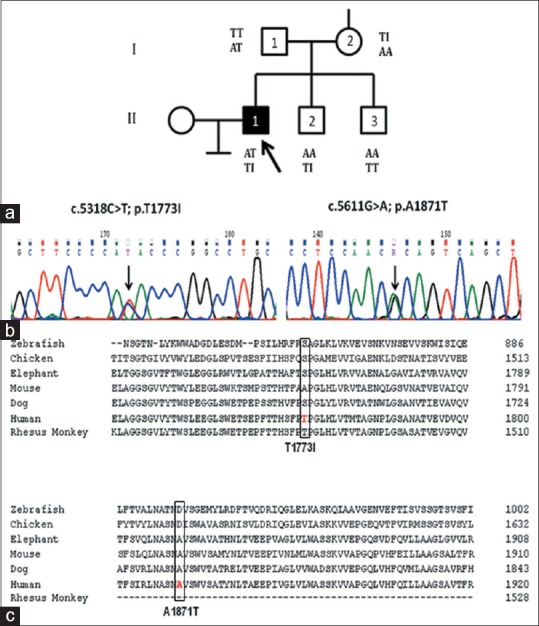
Family PKDF9 (a) Pedigree showing proband (II-1) and his siblings, unaffected parents (I-1 and I-2), and unaffected siblings (II-2 and II-3). The p.Thr1773Ile and p.Ala1871Thr variants are shown below the symbol of the individual. (b) Sequence data for variants showing the nucleotide and amino acid changes. (c) Sequence alignment of PC1 orthologues
Bidirectional sequencing of PKD1 and PKD2 genes revealed that the proband was compound heterozygote for two missense variants in exon 15 of PKD1 gene-c.5318C>T; p. Thr1773Ile (rs140162759) and c.5611G>A; p. Ala1871Thr (rs144137200) [Figure 1b]. No variants were observed in PKD2 gene. The presence of large deletions or duplications of PKD1 or PKD2 were excluded using multiple ligation-dependent probe amplification assays, as per the manufacturer's protocol (MRC, Holland). Segregation was tested by sequence analysis of the relevant genomic fragments in all the family members. It was observed that the proband's mother (I-2) and father (I-1) were heterozygous for p. Thr1773Ile and p. Ala1871Thr, respectively. The asymptomatic family members had one or the other variant but not both. One of the brothers (II-2) aged 30 years carried only one variant – p. Thr1773Ile. He was thus heterozygous for the variant, the other allele being normal. The younger brother (II-3), aged 28 years, was negative for both the variants and had a normal ultrasound study. The parents and one brother having one variant only did not have any renal cysts or renal complications. The variants p. Thr1773Ile and p. Ala1871Thr are within the PKD repeat domains 13 and 14 of polycystin 1 (PC1) protein, respectively. The two missense substitutions were predicted to be nonpathogenic by various online bioinformatic tools (SIFT, PolyPhen2, Mutation Taster, AlignGVGD). The variants p. Thr1773Ile and p. Ala1871Thr are conservative changes (Grantham distance:[8] 69 and 7, respectively) at a poorly conserved site (Grantham variation: 104 and 227, respectively) [Figure 1c].
Discussion
In the present family, ADPKD in the proband was caused by the coinheritance of two variants in trans from two unaffected parents, who had variant in one allele only. This was suggestive of autosomal recessive inheritance. The majority of PKD1 and PKD2 variants are likely fully inactivating although recent studies indicate that some alleles retain partial activity (hypomorphic alleles). Homozygotes for such alleles are viable, and in combination with an inactivating allele can result in early-onset disease.[3,4,9] Hypomorphism of both alleles leading to adult-onset polycystic kidney disease has been rarely described. Rossetti et al.[3] reported two consanguineous families, with children having homozygous variants in PKD1 gene. In the first family, two siblings were homozygous for p. Arg3277Cys variant, had multiple, relatively uniform-sized renal cysts (atypical of ADPKD), and had ESRD at the age of 75 and 62 years, respectively. The heterozygous parents and four heterozygous children had few cysts with normal renal function. In the second family, the father and two children with moderate-to-severe disease were homozygous for p. Asn3188Ser variant. Children developed clinical manifestations of ADPKD in the first decade of life. However, mother and siblings, who were heterozygotes for p. Asn3188Ser variant, had normal kidneys on ultrasound studies. In these two families, the affected homozygotes (those carrying variants in both alleles) had an early onset of renal failure. In third family, the proband, 22 years old, presented with normal-sized kidneys and was diagnosed with medullary sponge kidney with bilateral renal cysts at the age of 11 years. The proband was compound heterozygous for p. Arg3105Trp and p. Arg2765Cys variants in the PKD1 gene. The heterozygous parents had a normal renal phenotype, and the siblings, with an occasional renal cyst, had one of the variants but not both. One of the siblings (aged 42 years) had a single renal cyst, whereas the other sibling had one cyst each in kidney and liver at the age of 45 years.
Rossetti et al.[3] also screened three families with an in utero presentation of PKD. These severely affected babies were reported to have one protein-truncating variant besides a hypomorphic variant. In one family, the in utero case had a truncating variant (p. Gln2158Ter) in addition to a hypomorphic variant p. Arg3277Cys in PKD1 gene. The affected father had typical ADPKD and was heterozygous for truncating variant only. The other in utero case had a truncating variant p. Tyr3819Ter and hypomorphic variant p. Arg2765Cys. Affected father had only truncating variant. In both these cases, the probands manifested hypertension with bilaterally enlarged, multicystic kidneys in the 1st year of life. The third in utero case died perinatally of pulmonary hypoplasia with massively enlarged cystic kidneys. The fetus was found to have p. Arg2765Cys as hypomorphic variant in addition to a truncating variant c.7915dup20, whereas affected mother was heterozygous only for c.7915dup20 variant. Cases with just the p. Arg2765Cys variant did not have renal cysts by ultrasound examination.
Vujic et al.[4] studied two families with in utero onset of polycystic kidney disease due to the inheritance of hypomorphic alleles. In one of the families, the individual with compound heterozygote for p. Arg3277Cys and p. Arg2220Trp variants had in utero presentation of PKD, whereas the heterozygous parents had normal renal ultrasound. In their second family, the individual who was homozygous for both p. Val1045Met and p. Thr1570Met variants in trans had in utero ADPKD presentation and developed renal failure by 8 years of age with renal transplant being done at 9 years of age. The parents and sibling, heterozygous for two variants on the same haplotype (same chromosome), had normal kidneys.
In the consanguineous family described by Bergmann et al.,[2] the inheritance of homozygous PKD1 variant p. Val1274Met in cis with p. Gly2906Ser on the maternal allele resulted in development of bilateral multiple renal cysts and mild hypertension during the first decade of life. Both the parents were heterozygous for the variant p. Val1274Met and mother also had variant p. Gly2906Ser in cis (on the same chromosome). They were in early twenties and had normal kidneys on ultrasound. Corroboration of these studies is also provided by studies on mouse models with hypomorphic variants in the PKD1 gene, suggesting that level of polycystins below a critical threshold (due to two hypomorphic alleles) results in cyst formation in renal epithelia.[9,10]
In the light of above data, we propose that in the family described, both the variants (p. Thr1773Ile and p. Ala1871Thr) were “hypomorphic,” that had a combined effect resulting in reduced level of protein PC1 in renal cells and hence caused cystogenesis in the proband. Thus, the phenotype of individuals with alterations in both alleles of the ADPKD gene is variable, depending on the functional consequences of the variants. However, family studies are essential in determining the pathogenicity and clinical significance of such hypomorphic alleles.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors would like to thank the family for their cooperation for the study. This work was supported by Indian Council of Medical Research, New Delhi, India and partly by Ganga Ram Institute of Postgraduate Medical Education and Research, New Delhi, India.
References
- 1.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease Type I. Cell. 1996;87:979–87. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 2.Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol. 2011;22:2047–56. doi: 10.1681/ASN.2010101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–55. doi: 10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 2010;21:1097–102. doi: 10.1681/ASN.2009101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–7. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 6.Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205–12. doi: 10.1681/ASN.2008050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossetti S, Chauveau D, Walker D, Saggar-Malik A, Winearls CG, Torres VE, et al. A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int. 2002;61:1588–99. doi: 10.1046/j.1523-1755.2002.00326.x. [DOI] [PubMed] [Google Scholar]
- 8.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 9.Reiterová J, Štekrová J, Merta M, Kotlas J, Elišáková V, Lněnička P, et al. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol. 2013;14:59. doi: 10.1186/1471-2369-14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, et al. Lowering of PKD1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13:3069–77. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]