

Abstract
Osteoclasts are multinuclear bone-resorbing cells that differentiate from hematopoietic precursor cells. Prostate cancer cells frequently spread to bone and secrete soluble signaling factors to accelerate osteoclast differentiation and bone resorption. However, processes and mechanisms that govern the expression of osteoclastogenic soluble factors secreted by prostate cancer cells are largely unknown. MacroH2A (mH2A) is a histone variant that replaces canonical H2A at designated genomic loci and establishes functionally distinct chromatin regions. Here we report that mH2A1.2, one of the mH2A isoforms, attenuates prostate cancer-induced osteoclastogenesis by maintaining the inactive state of genes encoding soluble factors in prostate cancer cells. Our functional analyses of soluble factors identify lymphotoxin beta (LTβ) as a major stimulator of osteoclastogenesis and an essential mH2A1.2 target for its anti-osteoclastogenic activity. Mechanistically, mH2A1.2 directly interacts with HP1α and H1.2 and requires them to inactivate LTβ gene in prostate cancer cells. Consistently, HP1α and H1.2 have an intrinsic ability to inhibit osteoclast differentiation in a mH2A1.2-dependent manner. Together, our data uncover a new and specific role for mH2A1.2 in modulating osteoclastogenic potential of prostate cancer cells and demonstrate how this signaling pathway can be exploited to treat osteolytic bone metastases at the molecular level.
Keywords: macroH2A, Prostate cancer, Bone, Osteoclast, Lymphotoxin
INTRODUCTION
Bone is a dynamic tissue that is constantly renewed through a series of highly regulated steps that depend on the coordinated action of two cell lineages, osteoblasts and osteoclasts.1, 2 Osteoblasts are cells with single nuclei that form bone matrix, whereas osteoclasts are large multinuclear cells whose function is to resorb bone matrix.3, 4 Osteoclast differentiation is triggered by receptor activator of NF-κB ligand (RANKL), which is expressed as a membrane-bound TNF-related factor in osteoblasts and provides osteoclast-specific differentiation signals in the presence of macrophage colony stimulating factor (M-CSF).5–7 The binding of RANKL to its cognate receptor RANK on pre-osteoclast cell membrane triggers a cascade of intracellular events, resulting in activation and translocation of NF-κB to the nucleus, which in turn induces the initial expression of key determinants of osteoclastogenesis such as NFATc1.8 Then, these transcription factors trigger major signaling pathways to turn on multiple downstream genes for osteoclast formation, activation, and survival. Given the fact that deregulated osteoclastogenic activity in pathologic conditions leads to different types of bone diseases including osteoporosis, rheumatoid arthritis, and bone metastases,9–12 it seems of importance to unravel molecular mechanisms and signaling pathways regulating the expression of osteoclastogenesis-associated genes.
Prostate cancer has the second highest incidence in men and tends to metastasize to bone and interrupt the normal bone remodeling process.13 While initially thought to be primarily osteoblastic, it is now well recognized that prostate cancer bone metastases have extensive bone resorptive components that are caused primarily by osteoclasts.14 The osteolytic nature of prostate cancer bone metastases relies on the ability of cancer cells to secrete soluble factors that stimulate osteoclast differentiation.15–17 The unbalanced generation of osteoclasts leads to a massive bone resorption and releases the growth factors stored in the bone matrix, promoting metastatic tumor growth. This feed-forward vicious cycle creates a fertile microenvironment for cancer cell growth in bone to drive the devastating effects of bone destruction. Thus, the secretion and function of factors capable of promoting osteoclast formation are crucial for initiating osteolytic bone metastases of prostate cancer. However, the molecular components and mechanisms that regulate cancer cell production of secreted osteoclastogenic factors in the bone microenvironment are still largely unknown.
Histone variants are non-allelic isoforms of canonical histones and emerge as key players in creating unique chromatin domain and regulating specific transcription programs.18, 19 As all genes encoding secreted factors are expressed in the context of chromatin, a fundamental mechanism underlying prostate cancer-induced osteoclastogenesis might involve histone variant-mediated regulatory pathways. Of special relevance to the current study, macroH2A (mH2A) is one of the H2A variants and is defined by the presence of a large C-terminal macrodomain connected to the H2A-like domain.20 In mice and humans, there are two genes encoding mH2A1 and mH2A2, with mH2A1 being more abundant variant.19, 21 mH2A1 has two isoforms, mH2A1.1 and mH2A1.2, which are generated by the alternative splicing in the macrodomain.22 Whereas mH2A was originally implicated in X-chromosome inactivation and autosomal gene silencing, recent studies revealed that mH2A exerts both positive and negative effects on gene transcription in a context-dependent manner.23–25 Physical and functional interactions with other chromatin regulators may be attributed in part to the unique functional property of mH2A, as exemplified by PARP1, HDAC1/2, and Pbx1 whose recruitment is mediated by mH2A1 and necessary for mH2A1 function in establishing distinct transcription states of genes.26–29 Although several lines of evidence suggest that at least some of histone modifications may influence bone metastases and remodeling, nothing is currently known about the effects of histone variants on these processes. As such, the primary goal of this study was to assess the role of mH2A as a regulator of the expression and secretion of soluble factors influencing osteoclast differentiation and function in the process of prostate cancer bone metastases. Our second goal was to determine whether mH2A collaborates with other factors to enhance its capacity to create functionally distinct chromatin domain and regulate the expression of osteoclastogenic genes in prostate cancer cells.
RESULTS
mH2A1.2 impairs osteoclastogenic potential of soluble factors secreted by prostate cancer cells.
In osteolytic bone metastases, prostate cancer cells produce and secrete soluble factors that stimulate osteoclast differentiation and bone resorption.14, 16, 17 However, it remains elusive how the expression of these secreted factors is regulated in prostate cancer cells and whether any of histone variants participates in this process. To address these issues, we first set up an assay system, where osteoclast precursor (OCP) cells are synchronously differentiated into TRAP-positive multinuclear osteoclasts in α-minimal essential medium (α-MEM) following treatment with RANKL and conditioned media (CMs) (Supplementary Figure S1a).30 In this setting, enhanced osteoclastogenesis was evident, when OCP cells were cultured for 7 days in a 1:1 mixture of α-MEM and CMs prepared from DU145 prostate cancer cells with moderate metastatic potential (Supplementary Figure S1b). For the purpose of analyzing possible roles played by histone variants in regulating CM-induced osteoclastogenesis, we prepared CMs from DU145 cells depleted of each of histone variants. Our infection of DU145 cells with lentiviruses expressing shRNAs designed against histone variants showed that each shRNA specifically and potently ablated the expression of target histone variant (Supplementary Figures S2a and b). As indicated by TRAP staining, depletion of H2AX, H2A.Z, mH2A2, H2ABbd, H3.3 or CENP-A in DU145 cells had only minor effects on the formation of mature osteoclasts in differentiation assays with DU145 CMs (Figure 1a). On the other hand, when OCP cells were treated with CMs from mH2A1-depleted DU145 cells, the number of differentiated osteoclasts was increased significantly (Figure 1a).
Figure 1.
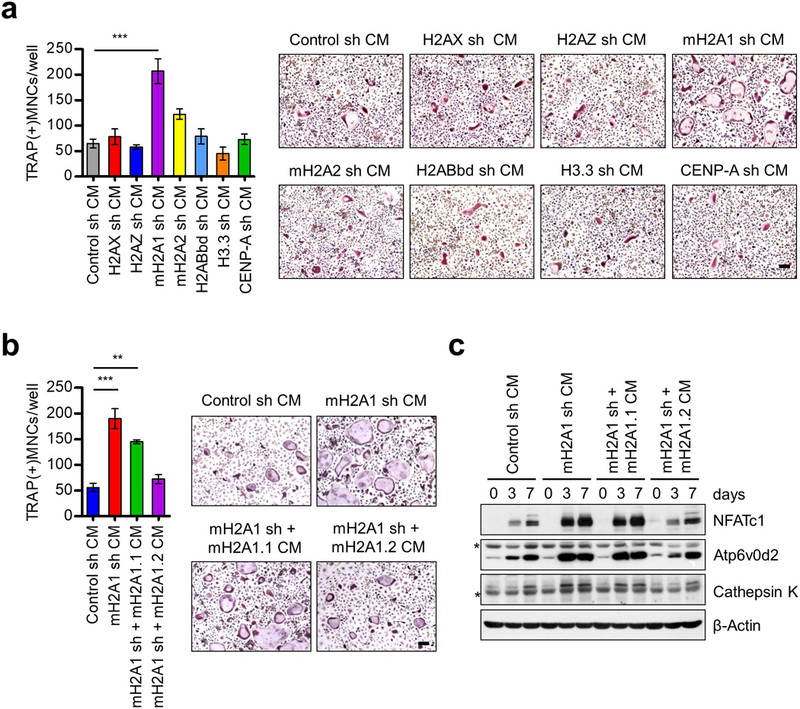
Osteoclastogenic activity of CMs from mH2A1.2-depleted prostate cancer cells. (a) Primary OCP cells derived from adult mouse long bone were cultured with CMs collected from DU145 prostate cancer cells depleted of each of the indicated histone variants in the presence of RANKL. At the end of day 7, OCP-induced cells were fixed, stained for TRAP, and photographed under a light microscope (10×) (right). Representative images of osteoclasts are shown (scale bar, 100 μm). TRAP positive cells containing three or more nuclei and a full actin ring were counted as osteoclasts (left). Cells exposed to CMs from mock-depleted DU145 cells served as control. (b) mH2A1-depleted DU145 cells were infected with lentiviruses expressing shRNA-resistant mH2A1.1 or mH2A1.2, and their CMs were prepared and analyzed for osteoclastogenic activity as in (a). Scale bar, 100 μm. (c) OCP cells were treated with CMs for the indicated time periods, and their lysates were collected and analyzed for the expression of three osteoclast marker genes (NFATc1, Atp6v0d2, and Cathepsin K) by Western blot. β-Actin was used as a loading control. Asterisks indicate non-specific bands. Shown in (a) and (b) are the means ± s.d. (n = 3) of three independent experiments; **P<0.01, ***P<0.001 (ANOVA analysis).
Since mH2A1 shRNA used in this assay causes knockdown of both mH2A1.1 and mH2A1.2 isoforms, we tried to develop isoform specific shRNAs. However, our attempts to selectively knockdown mH2A1.1 or mH2A1.2 were not successful due to the limited amount of sequence that could be targeted discriminatively. Therefore, we took an alternative approach in which we attempted to block osteoclastogenic effects of CMs by individually transfecting RNAi-resistant, FLAG-tagged mH2A1.1 and mH2A1.2 in mH2A1-depleted DU145 cells (Supplementary Figure S2c). Expression of ectopic mH2A1.2 in mH2A1-depleted DU145 cells reduced osteoclastogenic property of CMs quantitatively similar to those observed for mock-depleted control cells (Figure 1b). However, despite its high sequence homology with mH2A1.2, mH2A1.1 was unable to substitute for the anti-osteoclastogenic function of mH2A1.2 within the scope of this analysis (Figure 1b). Western blot analysis of lysates from CM-treated OCP cells revealed higher expression of NFATc1 and its downstream target genes Atp6v0d2 and Cathepsin K, suggesting that mH2A1.2 suppresses gene sets for osteoclast differentiation pathway (Figure 1c).
mH2A1.2 represses the expression and secretion of key regulators of osteoclast differentiation in prostate cancer cells.
We next wanted to examine whether the above-described effects of mH2A1.2 reflect its functional property leading to transcriptional inactivation of genes encoding secreted factors. Toward this end, we isolated total RNA from mock-depleted or mH2A1-depleted DU145 prostate cancer cells and conducted microarray-based screening for mH2A1-regulated secretome. Although recent genome-wide studies developed several databases summarizing known or predicted secreted proteins, these resources are incomplete and in an inconsistent format because of their use of different protein and gene identifiers. Therefore, we collected public databases as well as published papers providing lists of secreted proteins, trans-membrane/membrane proteins and signal peptides, and derived a prostate cancer secretome through in silico approaches. As described in Methods, we used a bioinformatics approach and GRCh37 genome in each database to curate the secreted proteins associated with prostate cancer (Supplementary Table S1). A total of 4,050 secreted genes were mapped to Illumina Human HT-12 V4 ChIP.
Most common approach for selecting differentially expressed genes is to order genes based on their fold changes in expression levels and use this short list of genes to determine whether any gene set is overrepresented as compared with the whole list. This approach is reasonable to certain extent, but has several limitations as recently detailed.31 Thus, we used Gene Set Enrichment Analysis (GSEA) method to identify enriched pathways and prioritize candidate genes in each pathway (see Materials and Methods for details).32 GSEA of 4,050 genes showed an overall significant enrichment of prostate cancer specific, metastatic, and RB1 target pathways (Supplementary Table S2). A constellation map of gene set enrichment classified both replicates into distinct groups between control and mH2A1-depleted cells (Figure 2a). One of the key pathways significantly enriched in this analysis was bone remodeling (normalized enrichment score=3.92, p=0.0002) (Figure 2b). Also of note, when 15 core genes in bone remodeling category were ranked in the order of their enrichment score, LTβ gene showed the highest score (Figure 2c). The data sets resulting from microarray-based secretome analysis were further confirmed by qRT-PCR (Figure 2d). Because our microarray data were generated by using DU145 cells depleted of both mH2A1.1 and mH2A1.2, we again expressed mH2A1.1 or mH2A1.2 in mH2A1-depleted DU145 cells to distinguish mH2A1.1-specifc versus mH2A1.2-specific target genes. Ectopic expression of mH2A1.2 led to a strong suppression of eight out of fifteen genes encoding secreted bone remodeling factors that were most highly expressed in mH2A1-depleted DU145 cells (Figure 2d). Among the remaining genes, six genes were inactivated by ectopic mH2A1.1, while one gene was slightly affected by both ectopic mH2A1.1 and mH2A1.2 (Figure 2d).
Figure 2.
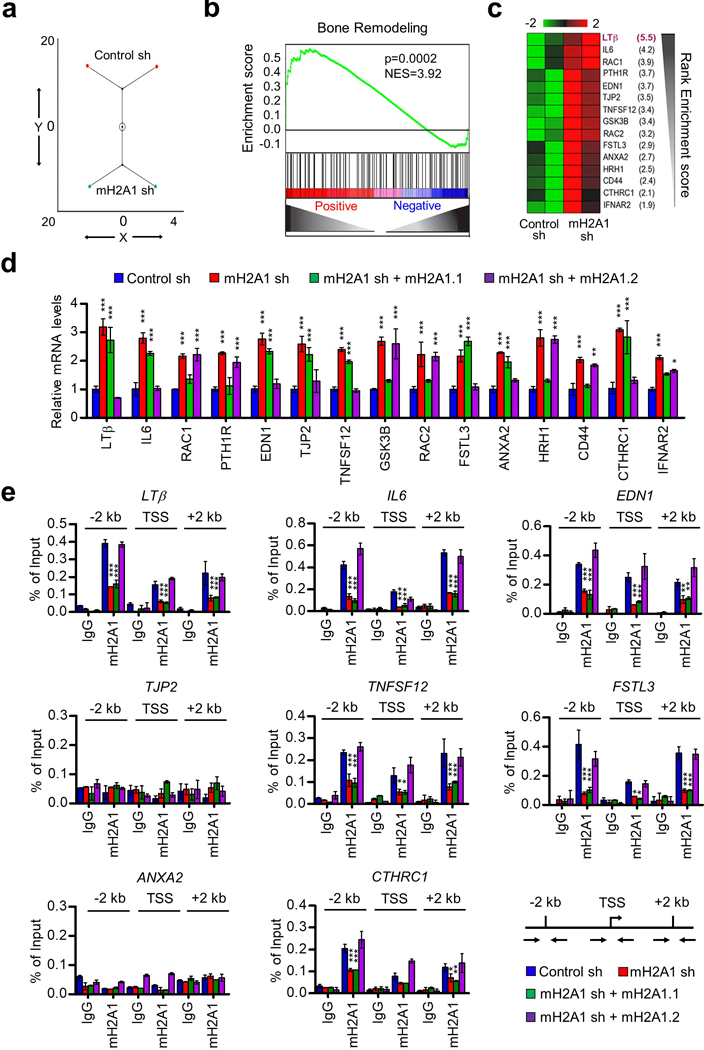
mH2A1.2-dependent repression of osteoclastogenic genes in prostate cancer cells. (a) A constellation map of the genes that were identified by GSEA analysis and significantly enriched in different pathways (Supplementary Table S1) can be classified in two distinct groups corresponding to control and mH2A1-depleted cells. (b) GSEA of 4050 genes representing prostate secretome shows the enrichment of genes associated with bone remodeling. Y-axis plots the enrichment score, and X-axis is the rank of genes differentially expressed in mH2A1-depleted cells. The genes were ranked by increasing expression differences (green curve). (c) Ranking 15 core enriched genes based on their enrichment scores in GSEA of bone remodeling pathway suggests that LTβ is the most significantly enriched gene. (d) mH2A1-depleted DU145 cells were complemented with mH2A1.1 or mH2A1.2, and relative mRNA levels of the top 15 genes were quantified by qRT-PCR using primers listed in Supplementary Table S3. (e) ChIP assays of the eight mH2A1.2-repressed genes were performed in mock-depleted (Control sh) or mH2A1-depleted (mH2A1 sh) DU145 cells using mH2A1 antibody. To rescue the effects of mH2A1 knockdown, mH2A1-depleted cells were transfected with shRNA-resistant mH2A1.1 or mH2A1.2. Precipitation efficiencies were determined for the regions 2 kb upstream and 2 kb downstream of TSS by qPCR with primers depicted in the lower right panel and listed in Supplementary Table S4. Percent input was determined as the amount of immunoprecipitated DNA relative to input DNA. Error bars in (d) and (e) denote the means ± s.d. obtained from triplicate real-time PCR reactions; *P<0.05, **P<0.01, ***P<0.001 versus Control sh (ANOVA analysis).
Although the results suggested that mH2A1.2 inactivates the genes involved in bone-remodeling pathways, the possible indirect effects of mH2A1.2 knockdown on target genes could not be distinguished by transcription analysis. We therefore investigated the ability of mH2A1.2 to localize along the eight potential mH2A1.2 target genes by chromatin immunoprecipitation (ChIP). Due to the unavailability of ChIP-grade mH2A1.2-specific antibody, we used in these experiments mH2A1 antibody that recognizes both mH2A1.1 and mH2A1.2. Crosslinked chromatin was isolated from control or mH2A1-depleted DU145 cells, and the precipitated DNA was amplified by quantitative PCR (qPCR) with three primer pairs targeting transcription start site (TSS), +2 kb region, and −2 kb region. In mock-depleted cells, we were able to detect mH2A1 occupancy in six out of the eight genes we analyzed (Figure 2e). We observed higher levels of enrichments for mH2A1 at the promoter and coding regions compared to TSS, although the precipitation patterns were slightly different among the target genes (Figure 2e). The two genes that failed to generate mH2A1 ChIP signals in our assay may represent genes inactivated by mH2A1 through an indirect mechanism such as the ability of free mH2A1 to interact with specific gene regulatory factors. Expectedly, when ChIP experiments were performed after mH2A1 knockdown, almost complete loss of mH2A1 ChIP signals was detected at the six target genes (Figure 2e). Expressing mH2A1.1 in mH2A1-depleted DU145 cells could not recover mH2A1 ChIP signals at the target genes (Figure 2e). However, ectopic mH2A1.2 largely overrides the ChIP signal defects caused by mH2A1 knockdown (Figure 2e), indicating that mH2A1.2 is responsible for mH2A1 ChIP enrichment detected at the six target genes. An analogous ChIP experiment was also carried out in DU145 cells transfected with mH2A1.1 or mH2A1.2, and ectopic mH2A1.2 was detected at the target genes while ectopic mH2A1.1 was not (Supplementary Figure S3), again reinforcing the conclusion that mH2A1.2, not mH2A1.1, is capable of localizing at the target genes.
LTβ is osteoclastogenic and a crucial target for mH2A1.2 activity.
As an extension of the above-described studies that established a repressive function for mH2A1.2 on the genes involved in bone remodeling, it was also important to investigate the relative contribution of the target genes to osteoclastogenesis, which is the initial process of bone remodeling. For this objective, we suppressed the expression of the six mH2A1.2 target genes that were shown to be directly bound and repressed by mH2A1.2 in DU145 prostate cancer cells (Figures 2d and e). Note that shRNA-mediated downregulation of one target gene did not affect the expression of other target genes, enabling us to functionally evaluate each of the target genes (Supplementary Figure S4). When OCP cells were treated with CMs from DU145 cells depleted of IL6, EDN1, TNFSF12, FSTL3 or CTHRC1, only minimal changes were observed in the average number of osteoclasts formed in our differentiation assays (Figure 3a). On the contrary, specific knockdown of lymphotoxin beta (LTβ) in DU145 cells significantly attenuated the effects of CMs on RANKL-induced osteoclast differentiation as well as osteoclast-specific gene expression (Figures 3a-c and Supplementary Figures S5a and b). Consistent with this finding, overexpression of LTβ in DU145 cells markedly enhanced osteoclast formation (Supplementary Figures S6a-c). These results exclude the possibility that the observed effects of LTβ shRNA are generated by off-target activity, and indicate that osteoclastogenic function of LTβ is indeed accurately established in our assays. Importantly, knockdown of mH2A1.2 in LTβ-deficient DU145 cells was unable to augment osteoclastogenic potential of CMs (Figures 3b and c), strongly suggesting that anti-osteoclastogenic function of mH2A1.2 is dependent on its inhibitory activity against LTβ expression. Given that osteoclast differentiation involves several distinct stages, we also analyzed the impact of altering LTβ levels on OCP cell migration and proliferation. Our analysis clearly showed that knockdown and overexpression of LTβ decreased and increased, respectively, the capacity of OCP cells to migrate (Supplementary Figures S5c and S6d). Since we detected no significant effects of LTβ knockdown/overexpression on OCP cell proliferation (data not shown), these findings indicate that LTβ has the main impact on the migration of OCP cells during osteoclast formation.
Figure 3.
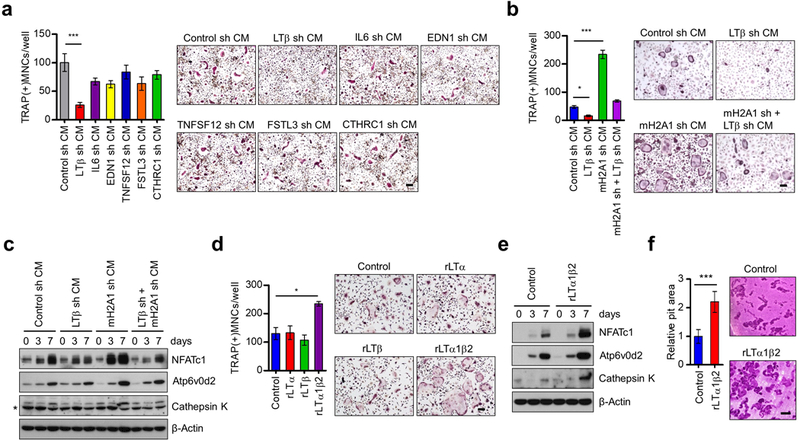
Identification of LTβ gene as a major target for macroH2A1.2 anti-osteoclastogenic activity. (a) OCP cells were treated with CMs from DU145 cells depleted of each of the six mH2A1.2 target genes identified in Figure 2e, and stained for TRAP expression (right). Multinuclear osteoclasts were counted (left). Scale bar, 100 μm. (b) Osteoclast differentiation assays were carried out as in (a), but using CMs from DU145 cells depleted of mH2A1 and LTβ individually or together. Scale bar, 100 μm. (c) After treating OCP cells with CMs as in (b), expression changes of osteoclast marker genes NFATc1, Atp6v0d2, and Cathepsin K at the indicated time points were analyzed by Western blot. β-Actin was used as a loading control. (d) OCP cells were treated with M-CSF/RANKL and rLTα, rLTβ, or rLTα1β2 for 7 days, and TRAP-positive multinucleated osteoclasts were stained (right) and counted (left). Scale bar, 100 μm. (e) Western blot analyses of three osteoclast marker genes after treating OCP cells with rLTα1β2 for 0, 3 and 7 days. β-Actin was used as a loading control. (f) Osteoclasts were seeded on dentin slices and treated with vehicle (control) or rLTα1β2 (100 ng/ml) in the presence of M-CSF and RANKL for 2 days. After ultrasonication, dentin slices were stained with hematoxylin solution and photographed (right). Representative images are shown for each condition (scale bar, 100 μm). The pit areas (Bone resorption areas) were analyzed using Image-Pro Plus program. Error bars in (a, b, d, f) represent the means ± s.d. (n = 3) of three independent experiments; *P<0.05, ***P<0.001 (ANOVA analysis or Student’s t-test).
The above data provide the first experimental evidence for an osteoclastogenic activity of LTβ, because most previous studies focused on its function in the development and maintenance of peripheral lymphoid organs as a heterotrimer with LTα.33, 34 To further validate the role of LTβ in osteoclastogenesis, we treated OCP cells with recombinant LTα (rLTα), rLTβ, or rLTα1β2 in the presence of M-CSF and RANKL. When treated with rLTα1β2 for 7 days, OCP cells were differentiated into osteoclasts and expressed osteoclast marker genes (NFATc1, TRAP, Cathepsin K, OSCAR, Integrin β3, Atp6v0d2, DC-STAMP, and OC-STAMP) at much higher levels compared with mock-treated control cells (Figures 3d and e and Supplementary Figure S7). However, no changes in osteoclast differentiation were detected in case of treatments with rLTβ (Figure 3d). This is an unexpected result based on the fact that LTβ overexpression drives increased osteoclast differentiation (Supplementary Figure S6c). Although the reasons for these contrasting results are under investigation, we favor a model where rLTβ requires rLTα for its transport into OCP cells and osteoclastogenic function. Thus, unlike ectopic LTβ that can functionally couple with endogenous LTα upon expression, rLTβ should be used together with rLTα to get imported efficiently into OCP cells. In agreement with the overexpression data (Supplementary Figure S6), treatment with rLTα1β2 also potentiates transwell movements of OCP cells, again linking LTβ to OCP cell migration (Supplementary Figure S5d).
Next, we plated osteoclasts on dentin slices and evaluated the effect of rLTα1β2 treatment on their resorbing ability. As expected, rLTα1β2-enhanced osteoclast formation increased the rate of bone resorption, stimulating the formation of resorption pits (Figure 3f). To further investigate whether the rLTα1β2 treatment is also effective in vivo, we injected rLTα1β2 into the periosteal regions of male mouse calvaria at 1-day intervals for 10 days. Micro-computed tomography (micro-CT) analysis of the calvaria showed that the bone mineral density (BMD) was higher in rLTα1β2-injected mice compared to control mice (Figures 4a and b). Additional analysis revealed that treating mice with rLTα1β2 generated an apparent decrease in the bone volume/tissue volume ratio (BV/TV) (Figure 4c). The observed changes in BMD and BV/TV were due to enhanced osteoclastogenesis, because higher numbers of TRAP-positive osteoclasts were detected in rLTα1β2-injected mice compared to control mice in our histomorphometric analysis (Figure 4d). Moreover and as shown in Figure 4e, the fact that mice treated with rLTα1β2 display larger cavities in bone strongly argues that rLTα1β2 promotes bone resorption, thereby enhancing bone destruction. Consistent with these findings, our qRT-PCR data showed that osteoclast differentiation marker genes were more highly expressed in rLTα1β2-injected mice compared to control mice (Figure 4f).
Figure 4.
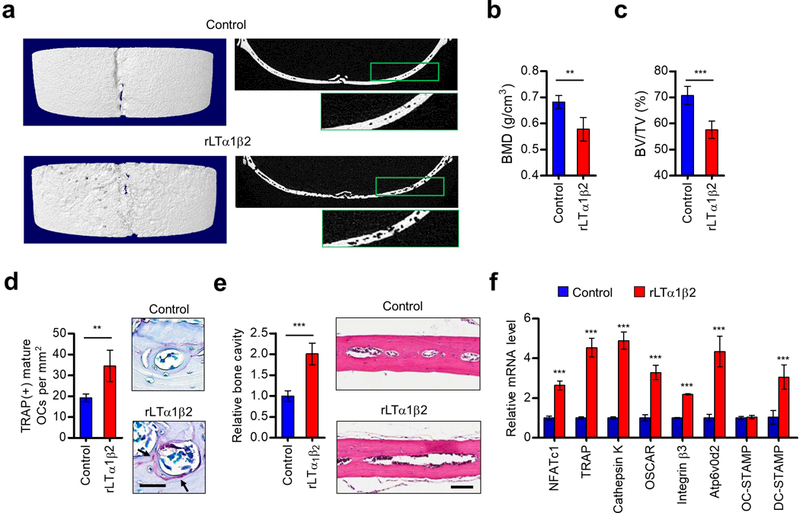
Effects of LTβ on osteoclast formation and bone resorption. (a) Seven-week-old male C57BL/6 mice were injected with rLTα1β2 (0.2 mg/kg in 100 μl PBS) or PBS alone (control) every other day for 10 days. Mice were then sacrificed, and calvarial bones were subjected to micro-computed tomography (micro-CT) analysis. Representative three-dimensional micro-CT (left) and cross sectional micro-CT (right) images are shown. Right panels also show higher magnification images of boxed area of low magnification views. (b, c) Bone mineral density (b) and bone volume per tissue volume (c) were calculated using three-dimensional micro-CT images. (d) Calvarial bones were fixed, embedded, and subjected to sectioning, TRAP staining, and histological analysis. Representative images of TRAP staining are shown on the right (scale bar, 100 μm). Arrows point to TRAP-positive mature osteoclasts. (e) Bone sections from C57BL/6 mice treated with vehicle (control) or rLTα1β2 were stained for hematoxylin-eosin (H&E) and analyzed for osteolytic legion formation. Representative H&E stained images are shown on the right (scale bar, 100 μm). (f) Total RNAs were isolated from calvarial bones, and the expression levels of osteoclast differentiation markers were quantified by qRT-PCR. Error bars in (b-f) represent the means ± s.d. (n = 5); **P<0.01, ***P<0.001 versus Control (Student’s t-test or ANOVA analysis).
Having established the importance of LTβ gene silencing in mH2A1.2 function, we then performed qPCR to measure mH2A1.2 and LTβ levels in a cDNA array of 39 prostate tumor and 9 normal samples. In these experiments, we found that about 90% of tumor samples showed lower expression of mH2A1.2 compared with normal samples (Supplementary Figure S8). By contrast, a substantial increase of LTβ expression was observed in about 70% of the tumor samples. When the two expression data were compared, the majority of samples demonstrated an inverse correlation between mH2A1.2 and LTβ expression. It is also noteworthy that mH2A1.2 and LTβ show more significant expression changes in advanced stages of prostate cancer (Supplementary Figure S8). Together these data suggest that dysregulated expression of mH2A1.2 and LTβ is a common occurrence in prostate cancer and might be used to measure the potential for prostate cancer-induced bone loss.
LTβ promotes nuclear localization and transactivation function of NF-κB during osteoclastogenesis.
The NF-κB p50/p65 heterodimer plays a key role in osteoclast formation, and NFATc1 is an essential downstream target for its osteoclastogenic function.35–38 Considering that IκBα is the most abundant inhibitory protein for NF-κB and that LTα1β2 can activate NF-κB signaling pathways,39 we suspected that the stimulatory effects of LTα1β2 observed in our osteoclastogenic reactions are derived from its ability to control the level of IκBα in OCP cells. Indeed, the treatment of OCP cells with rLTα1β2 in the presence of RANKL induced the higher levels of IκBα degradation over a 30 min time period, as compared with those observed in mock-treated control cells (Figure 5a). Since IκBα was shown to govern stability and subcellular localization of NF-κB,35, 38 we also measured NF-κB p65 levels in OCP cells after rLTα1β2 treatment. Western blot analysis of total cell lysates did not show any effects of rLTα1β2 treatment on cellular levels of p65 in RANKL-stimulated OCP cells (Figure 5b). However, further analysis with cytoplasmic and nuclear extracts showed that the nuclear content of p65 was specifically elevated in response to rLTα1β2 treatment (Figure 5c). The involvement of LTα1β2 in NF-κB function was further confirmed by the reporter gene assays showing that NF-κB-mediated transcription from the reporter construct in RANKL-stimulated RAW264.7 macrophage cells was enhanced after rLTα1β2 treatment (Figure 5d).
Figure 5.
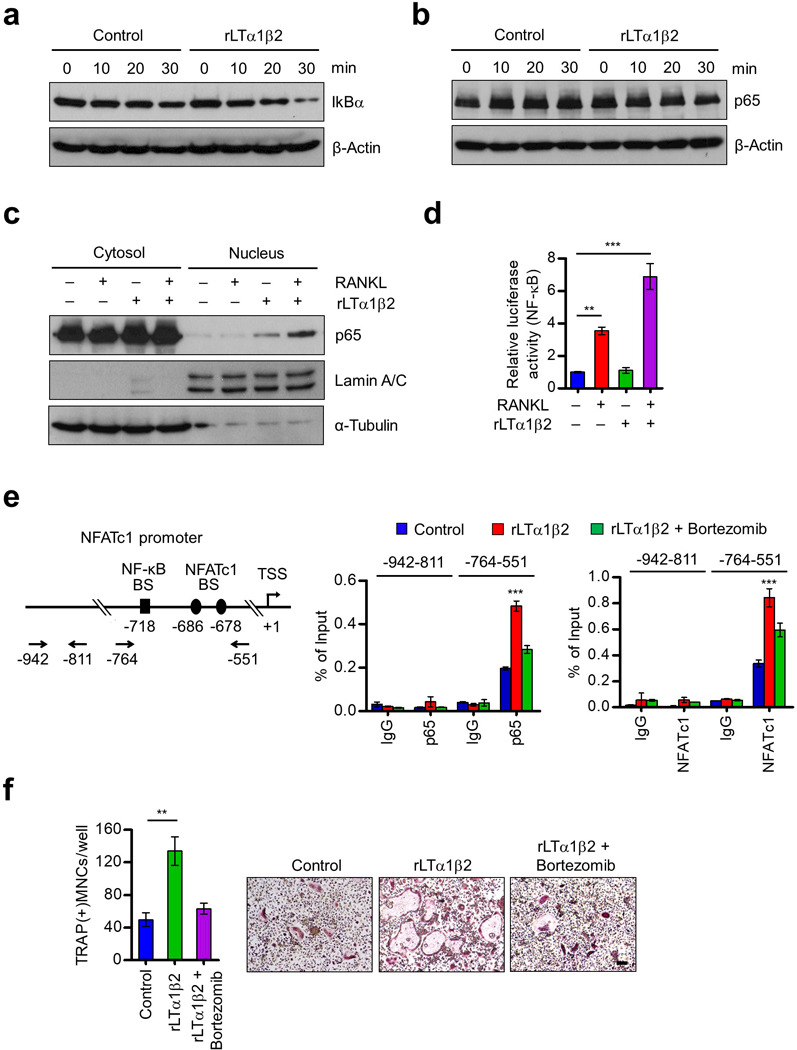
Stimulation of p65 nuclear translocation and NFATc1 expression by LTβ. (a) OCP cells were cultured with M-CSF and RANKL for 3 days, deprived of M-CSF/RANKL for 6 h, and treated with RANKL only (control) or RANKL plus rLTα1β2 (100 ng/ml) for 0, 10, 20 or 30 min as indicated. Cell lysates were prepared, and IkBα levels were assessed by Western blot. β-Actin was used as a loading control. (b) OCP cells were treated as in (a), and subjected to Western blot analysis for p65 expression. (c) OCP cells were cultured with M-CSF and RANKL for 3 days, deprived of M-CSF/RANKL for 6 h, and then stimulated with RANKL or RANKL plus rLTα1β2 for 30 min. Nuclear and cytosolic fractions were prepared and analyzed by Western blot using anti-p65, anti-Lamin, or anti-α-tubulin antibodies. Lamin and α-tubulin were used as markers for nuclear and cytoplasmic fractions, respectively. (d) RAW264.7 cells were cultured with RANKL for 3 days, transfected with the NF-κB-TA-Luc reporter gene, and then further cultured with RANKL, rLTα1β2, or RANKL plus rLTα1β2. Luciferase activity in the lysates was determined after 24 h of RANKL/rLTα1β2 stimulation. (e) OCP cells were cultured with M-CSF and RANKL for 3 days, deprived of M-CSF/RANKL for 6 h, and then sequentially treated with Bortezomib (100 nM) for 2 h and RANKL only (Control) or RANKL plus rLTα1β2 for 1 h (p65) or 6 h (NFATc1). ChIP assays were performed using p65 (middle) and NFATc1 (right) antibodies with qPCR amplicons specific for p65 and NFATc1 binding sites and TSS of NFATc1 gene as indicated on the left. (f) OCP cells were cultured with rLTα1β2 alone or together with Bortezomib (1 nM) in the presence of M-CSF and RANKL, and stained for TRAP. Scale bar, 100 μm. Each bar in (d-f) represents the mean ± s.d. from triplicate wells; **P<0.01, ***P<0.001 versus Control (ANOVA analysis).
Upon RANKL induction, NF-κB is recruited to the NFATc1 gene to activate transcription, and the newly generated NFATc1 can autoamplify its own transcription. Thus, to gain support for the above results, we also tested whether increasing NF-κB and NFATc1 levels in the NFATc1 gene is attributable to the observed effects of rLTα1β2 treatment. Expectedly, we found that NF-κB and NFATc1 are enriched at their binding sites of the NFATc1 promoter in RANKL-stimulated OCP cells (Figure 5e). Importantly, rLTα1β2 treatment of RANKL-stimulated cells generated a distinct increase in the promoter occupancy of NF-κB and NFATc1 (Figure 5e). Under these conditions, positive effects of rLTα1β2 treatment on RANKL-induced osteoclastogenesis were also easily detectable (Figure 5f and Supplementary Figure S9). The effects were specific, since rLTα1β2 treatment together with Bortezomib, which blocks NF-κB activation through proteasome inhibition,40 failed to yield any enhancement of NF-κB/NFATc1 recruitment and RANKL-induced osteoclast differentiation (Figures 5e and f and Supplementary Figure S9). These results constitute a powerful argument that LTα1β2 is both necessary and sufficient for nuclear localization and transactive function of NF-kB in RANKL-stimulated OCP cells.
HP1α and H1.2 interact with mH2A1.2 and are necessary for LTβ gene silencing in prostate cancer cells.
Although our data clearly implicate mH2A1.2 in LTβ gene silencing in prostate cancer cells, they left open the molecular basis of the action of mH2A1.2. Recent studies, including ours, suggested that specific factors are required for mH2A-mediated transcriptional regulation in a chromatin context.26–28 Thus we sought to determine whether other factors participate and play a role, if any, in the repressive activity of mH2A1.2 in prostate cancer cells. Toward this end, we generated a DU145 cell line stably expressing a FLAG- and HA-tagged version of mH2A1.2 to identify factors which can physically associate with mH2A1.2. After confirming that ectopic mH2A1.2 was mainly localized in the nucleus (data not shown), ectopic mH2A1.2 and its associated factors were purified from nuclear-soluble extracts by sequential immunoprecipitations with anti-FLAG antibody, followed by anti-HA antibody under stringent conditions (300 mM KCl, 0.1% Nonidet P-40). Coomassie blue staining of the mH2A1.2-associated factors fractionated on SDS-PAGE showed several distinct bands, which were not detectable in a control sample similarly purified from DU145 cells that do not express ectopic mH2A1.2 (Figure 6a). Mass spectrometry analysis of the mH2A1.2-specific bands identified a collection of fifteen different proteins, which were further validated by Western blotting (Supplementary Figure S10).
Figure 6.
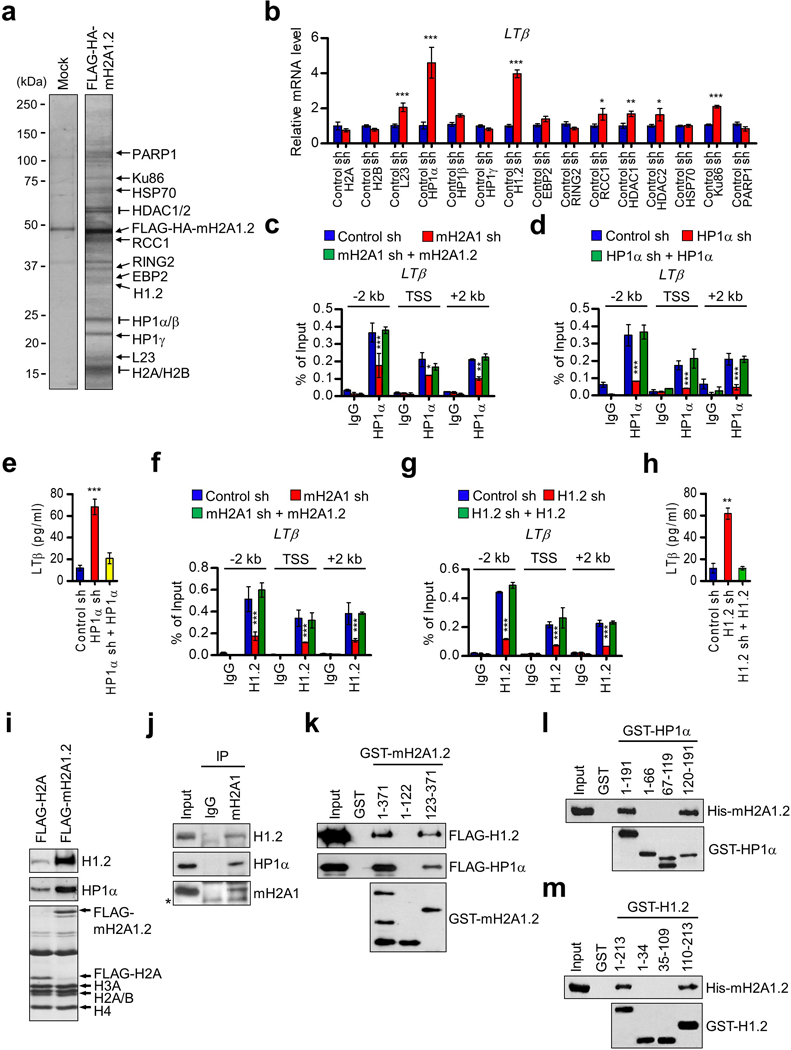
mH2A1.2-dependent recruitments of HP1α and H1.2 at osteoclastogenic genes. (a) Nuclear extracts derived from mock-transduced and FLAG-HA-mH2A1.2 expressing DU145 cells were subjected to sequential immunoprecipitations using anti-FLAG and anti-HA beads. After the co-purified proteins were resolved by 4–20% SDS-PAGE, major bands were excised and subjected to mass spectrometric analysis. The positions of the molecular mass markers are indicated on the left. (b) DU145 prostate cancer cells were transfected with nontargeting control shRNA (Control) or shRNAs targeting genes encoding mH2A1.2-associated factors for 48 h. mRNA levels of LTβ gene were quantified by qRT-PCR using primers listed in Supplementary Table S5. (c) The levels of HP1α at the LTβ gene were assessed in mock-depleted or mH2A1-depleted DU145 cells complemented with shRNA-resistant mH2A1.2 by ChIP assays. Primer sets locating at TSS, 2 kb upstream, and 2 kb downstream regions of the LTβ gene were used. (d) ChIP assays were carried out as in (c), but using mock-depleted control or HP1α-depleted DU145 cells complemented with HP1α. (e) LTβ expression levels in mock-depleted control or HP1α-depleted DU145 cells were measured by ELISA. The rescue effects of HP1α expression in HP1α-depleted cells were also analyzed. (f) ChIP assays were performed to quantitate the recruitment of H1.2 to the LTβ gene using mock-depleted (Control sh) or mH2A1-depleted DU145 cells complemented with mH2A1.2. (g) ChIP assays were carried out as in (f), but using mock-depleted (Control sh) or H1.2-depleted (H1.2 sh) cells complemented with H1.2. (h) LTβ expression was assessed in mock-depleted or H1.2-depleted DU145 cells complemented with H1.2 by ELISA. (i) DU145 cells were transfected with H2A or mH2A1.2 expression vectors, and mononucleosomes were prepared by MNase digestion as recently described. Mononucleosomes containing ectopic H2A or mH2A1.2 were immunoprecipitated from total mononucleosomes with anti-FLAG antibody. Histone compositions of the purified nucleosomes were analyzed by 15% SDS-PAGE followed by Coomassie brilliant blue staining (lower). The relative levels of H1.2 and HP1α bound to the purified nucleosomes were determined by Western blot (upper and middle). (j) Endogenous mH2A1 was immunoprecipited from cell lysates prepared from DU145 cells. The presence of H1.2 and HP1α in the precipitate was confirmed by Western blot. Asterisk indicates non-specific bands. (k) GST alone or GST fusions containing indicated mH2A1.2 domains were immobilized on glutathione Sepharose beads and incubated with FLAG-H1.2 and FLAG-HP1α. After extensive washing, bound H1.2 and HP1α proteins were fractionated by 10% SDS-PAGE, and probed with anti-FLAG antibody. Input lanes represent 5% of H1.2 and HP1α used in the binding reactions. (l) His-tagged mH2A1.2 was incubated with immobilized GST or the indicated GST-H1.2 fusions, and mH2A1.2 binding was analyzed by Western blot with anti-His antibody. Of the input proteins, 5% were examined by Western blot. (m) GST pull-down assays were conducted as in (k) but using the indicated GST-HP1α fusions. Error bars in (b-h) denote the means ± s.d. from triplicate reactions; *P<0.05, **P<0.01, ***P<0.001 versus Control sh (ANOVA analysis).
To examine whether some of the mH2A1.2-associated factors act as a functional partner for mH2A1.2 in inactivating LTβ gene, we transfected DU145 cells with shRNAs targeting the genes encoding the fifteen associated factors, and then evaluated the effects on LTβ expression. Confirmatory qRT-PCR analysis verified that each of these shRNAs generates a selective and effective silencing of the expression of target genes (Supplementary Figure S11). Under these conditions, we detected only minor changes in LTβ expression after knockdown of H2A, H2B, L23, HP1β/γ, EBP2, RING2, RCC1, HDAC1/2, HSP70, Ku86 or PARP1 in DU145 prostate cancer cells (Figure 6b). In sharp contrast, however, individual knockdown of HP1α and H1.2 caused a significant increase in LTβ expression in the cells (Figure 6b), strongly supporting that HP1α and H1.2 are required for mH2A1.2 function in repressing LTβ expression.
HP1α and H1.2 localize to the LTβ promoter in mH2A1.2-dependent manner.
To check whether the observed requirements for HP1α and H1.2 reflect their targeted recruitment, we next investigated their localization at the LTβ locus by ChIP assays. Our assays in DU145 prostate cancer cells revealed that HP1α was indeed strongly enriched in the −2 kb to +2 kb regions surrounding the TSS of LTβ gene (Figure 6c). These results were further supported by the finding that ectopic HP1α was able to localize at the LTβ locus and mediate transrepression in HP1α-depleted DU145 cells (Figures 6d and e and Supplementary Figure S12a). In parallel assays, we observed H1.2 occupancy at the LTβ locus, and confirmed its specific localization and function from H1.2 knockdown and ectopic expression studies (Figures 6f-h and Supplementary Figure S12b). It was also apparent in our additional ChIP experiments that loss and rescue of HP1α or H1.2 have no effects on the localization of mH2A1.2 at the LTβ locus (Supplementary Figure S13). Furthermore, the levels of HP1α and H1.2 in mH2A1-depleted DU145 cells were comparable to those in control cells (Supplementary Figure S14). These results suggest that HP1α and H1.2 act primarily as major downstream effectors of mH2A1.2-dependent suppression of LTβ expression and osteoclast differentiation.
The fact that HP1α and H1.2 are recruited to osteoclastogenic LTβ gene in a mH2A1.2-dependent manner prompted us to ask whether they have an ability to recognize and bind mH2A1.2 nucleosomes in DU145 prostate cancer cells. To this end, we transfected cells with plasmids expressing FLAG-H2A or FLAG-mH2A1.2, and purified mononucleosomes containing ectopic H2A/mH2A1.2 and their bound proteins following the procedure described.41 We confirmed that similar levels (~60%) of ectopic H2A and mH2A1.2 are present in the purified nucleosomes by Coomassie blue staining (Figure 6i). In Western blot analysis using HP1α and H1.2 antibodies, we detected substantial binding of both HP1α and H1.2 to mH2A1.2 nucleosomes (Figure 6i). However, the observed binding was dramatically reduced in H2A nucleosomes. These binding data were further supported by our finding that mH2A1 antibody is able to coimmunoprecipitate endogenous HP1α and H1.2 from lysates of DU145 prostate cancer cells (Figure 6j).
To determine more precise nature of the observed interactions, we also performed a series of GST pull down assays. rHP1α and rH1.2 formed a stable complex with glutathione S-transferase (GST)-mH2A1.2 immobilized on glutathione-Sepharose beads (Figure 6k). The specificity of these interactions was confirmed by the failure of HP1α and H1.2 to bind GST alone. In similar binding experiments with truncated versions of mH2A1.2, mH2A1.2 deletion mutant (residues 123–371) lacking N-terminal histone domain still showed strong affinity for both HP1α and H1.2 (Figure 6k). However, another deletion mutant (residues 1–122) lacking C-terminal macrodomain failed to interact with HP1α and H1.2, indicating that the binding of mH2A1.2 to HP1α and H1.2 is dependent upon its macrodomain. In additional binding assays, the C-terminal region of HP1α (residues 119–191) showed a direct interaction with mH2A1.2, while no apparent interaction was observed with the N-terminal (residues 1–66) and central (residues 67–119) regions of HP1α (Figure 6l). In mapping mH2A1.2-interacting region of H1.2, the binding of the C-terminal tail domain (residues 110–213) of H1.2 was readily detectable, but its N-terminal tail and central globular domains (residues 1–34 and 35–109) failed to show any interaction with mH2A1.2 (Figure 6m). These results reinforce the conclusion that the primary mH2A1.2-binding capacity of HP1α and H1.2 resides in their C-terminal regions.
HP1α and H1.2 are anti-osteoclastogenic factors.
The above data suggested that HP1α and H1.2 directly assist mH2A1.2 in establishing and maintaining a repressive chromatin environment over the LTβ locus. We hypothesized that this LTβ gene-targeted activity confers anti-osteoclastogenic properties to HP1α and H1.2. To test this in a direct fashion, we prepared CMs from DU145 prostate cancer cells depleted of HP1α and examine its effects on OCP cell differentiation after seven days of RANKL treatment. It was apparent in these experiments that selective depletion of endogenous HP1α in DU145 cells endowed CMs with the ability to stimulate RANKL-induced osteoclast differentiation (Figure 7a). In our attempt to limit the differentiation rate provoked by HP1α loss by ectopic expression of HP1α in DU145 cells, exogenous HP1α almost completely restored the original incapacity of CMs (Figure 7a). shRNA-mediated downregulation of H1.2 in DU145 cells also increased the number of osteoclasts formed in the presence of CMs (Figure 7c). The observed changes were specific, because ectopic expression of H1.2 in H1.2-depleted DU145 cells restored OCP cell differentiation to levels nearly identical to those generated by control cell CMs (Figure 7c). The inhibitory effects of HP1α and H1.2 on osteoclastogenesis were also verified by monitoring changes in the expression of NFATc1, Atp6v0d2 and Cathepsin K genes in response to knockdown and rescue of HP1α and H1.2 in DU145 cells (Figures 7b and d). Since LTβ activates NF-κB signaling pathways to enhance osteoclastogenic reactions (Figure 5), it is possible that treating prostate cancer cells with NF-kappaB inhibitors would affect osteoclastogenic activities of CMs from mH2A1/HP1α/H1.2-depleted cancer cells. To investigate these possibilities, we repeated differentiation assays in the presence of NF-kappaB inhibitor bortezomib. Consistent with this idea, osteoclastogenic effects of CMs from DU145 cells depleted of mH2A1, HP1α, or H1.2 were largely abolished after treatment with bortezomib (Supplementary Figure S15). Beside CMs from DU145 cells, we also examined the effects of CMs from PC3 prostate cancer cells depleted of mH2A1, HP1α, or H1.2. Western blots confirmed the activation of LTβ expression after individual knockdown of mH2A1, HP1α and H1.2 in PC3 cells (Supplementary Figure S16a). The higher level of LTβ was accompanied by increases in OCP cell differentiation (Supplementary Figure S16b), indicating that HP1α and H1.2 participate in mH2A1-induced anti-differentiation program. Together, these data provide insights into the specific effector mechanisms whereby HP1α and H1.2 influence anti-osteoclastogenic function of mH2A1.2 in metastatic prostate cancer cells.
Figure 7.
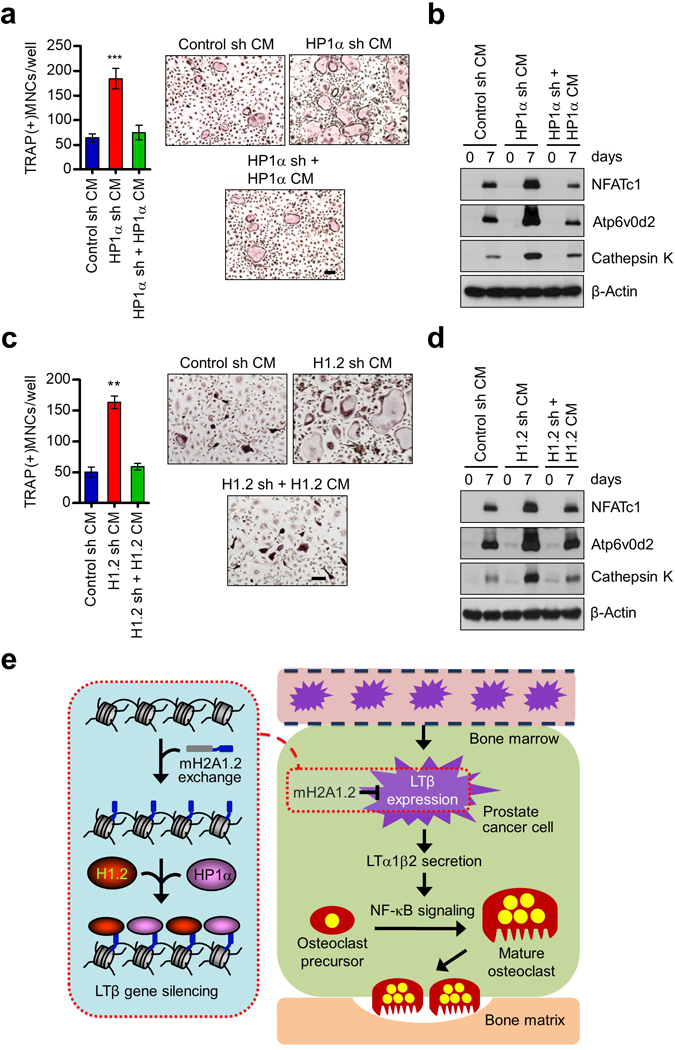
Inhibition of osteoclastogenesis by HP1α and H1.2. (a) OCP cells were treated with CMs from HP1α-depleted DU145 prostate cancer cells in the presence of RANKL, and stained for TRAP. HP1α-depleted cells were infected with lentiviruses expressing HP1α, and their CMs were also analyzed for osteoclastogenic activity. Scale bar, 100 μm. (b) Expression levels of osteoclast marker genes (NFATc1, Atp6v0d2 and Cathepsin K) in OCP cells treated with indicated CMs were analyzed by Western blot. β-Actin was used as a loading control. (c) CMs were harvested from mock-depleted or H1.2-depleted DU145 cells expressing shRNA-resistant H1.2 and analyzed for RANKL-induced osteoclastogenesis. Scale bar, 100 μm. (d) Western blot analyses of three osteoclast marker genes after treating OCP cells with indicated CMs for 0 and 7 days. β-Actin was used as a loading control. (e) A model for how mH2A1.2 might affect prostate cancer-induced osteoclast differentiation. See Discussion for details. Error bars in (a, c) denote the means ± s.d. from triplicate wells; **P<0.01, ***P<0.001 versus Control sh CM (ANOVA analysis).
DISCUSSION
In the present study, we investigated a potential role of histone variants in regulating the expression of soluble factors secreted by prostate cancer cells and their effects on osteoclast differentiation. Our initial studies indicate that mH2A1.2 constitutes a repressive barrier to the osteoclastogenic activity of soluble factors derived from prostate cancer cells, while the two other mH2A isoforms (mH2A1.1 and mH2A2) and five other histone variants (H2AX, H2AZ, H2ABbd, H3.3 and CENP-A) do not serve as major regulators in the context of OCP differentiation. In agreement with these observations, our microarray-based secretome analysis demonstrates that mH2A1.2 is essential for repressing the genes encoding the factors that influence bone development, growth, and repair. The observed function of mH2A1.2 is specific, since modulation of two other isoforms, mH2A1.1 and mH2A2, either through knockdown or ectopic expression does not significantly affect the expression of the target genes bound and inactivated by mH2A1.2. While these data strongly argue for the suppressive role played by mH2A1.2 in prostate cancer-derived osteoclastogenesis, we cannot rule out a possible role for mH2A1.1 and mH2A2, since their depletion/re-expression leads to a mildly altered osteoclast differentiation rate. However, unlike mH2A1.2, mH2A1.1 and mH2A2 do not appear to play any crucial role in inactivating osteoclastogensis-associated genes, and they cannot compensate for mH2A1.2 following depletion of this protein. Currently it is not clear if there is a biological explanation for why only mH2A1.2 serves as a major regulator of osteoclastogenic activity of prostate cancer cells, or if its localization at target genes involves an exchange factor recognizing distinct features of mH2A1.2. Nevertheless, the discovery that a group of genes related to bone remodeling are specific for mH2A1.2, with respect to both deposition and transcriptional regulation, strongly supports our conclusion that mH2A1.2 is required for OCP differentiation, since this group contains osteoclast-specific mH2A1.2 targets involved in differentiation.
Among the genes that encode regulators of bone remodeling and are inactivated by mH2A1.2 in prostate cancer cells, we focused on LTβ gene based on the fact that our secretome analysis of microarray data ranked LTβ gene highest and that LTβ has not been implicated in osteoclast differentiation. In support of an important role for LTβ in prostate cancer-triggered osteoclastogenesis, our in vitro studies identified its stimulatory functions in OCP differentiation. In addition to its effect in RANKL-treated OCP cells, administration of rLTβ together with its interacting partner LTα to animal models resulted in osteoclastic bone resorption. Our data further suggest that LTβ plays a role in controlling nuclear import of NF-κB which is necessary for the expression of NFATc1, a pivotal component for osteoclast differentiation.36 This idea is supported by our demonstration that treatment of OCP cells with rLTα/β complex significantly increases NF-κB p65 nuclear translocation and binding to the NFATc1 promoter, which in turn induces NFATc1 expression and osteoclast differentiation. This is the first study to report that LTβ is a key molecule in determining the differentiation rate of OCP cells as an upstream effector of NFATc1 signaling pathway. This finding also underscores the importance of mH2A1.2-mediated suppression of LTβ expression as an early step necessary for the inhibition of prostate cancer-induced osteoclastogenesis and bone resorption. We failed to detect LTβ occupancy at the gene encoding c-Fos (data not shown), which is another master regulator of osteoclastogenesis,42 suggesting that LTβ is mainly targeting NF-κB to fulfil its osteoclastogenic function. Our results, however, do not rule out the possibility that other signal transduction pathways operating in osteoclastogenesis may also offer a molecular basis of LTβ-triggered activation of osteoclast differentiation. Thus, to better understand how much the mH2A1.2-mediated regulatory mechanism contributes to the inhibition of osteoclast formation, it will be important to characterize other osteoclast-specific genes whose transcription is particularly sensitive to LTβ expression in future investigations.
In this report, we also examined whether other factors contribute to the observed transrepression potential of mH2A1.2 by purifying mH2A1.2-interacting factors from prostate cancer cells. In accordance with previous studies,26–28 we found that mH2A1.2 can stably associate with multiple chromatin and gene regulatory factors. Importantly, our RNAi-based screening of the interacting factors led to the conclusion that H1.2 and HP1α act as major functional partners of mH2A1.2 in attenuating LTβ expression in prostate cancer cells. We observed that HP1β and HP1γ are unable to bind LTβ gene, and ectopically expressed HP1β and HP1γ cannot precisely mirror the binding and inactivation normally seen with endogenous HP1α (data not sown). This suggests that HP1β and HP1γ cannot compensate for the absence of HP1α. Similarly, no prominent compensation for H1.2 activity by other H1 subtypes seems to occur, as mH2A1.2 target genes were not affected upon the expression of four other linker histone subtypes (H1.1, H1.3, H1.4, and 1.5) in H1.2-depleted prostate cancer cells (data not shown), and a repressive environment over LTβ gene was restored solely upon re-expression of H1.2 (Figure 6h and Supplementary Figure S12b). Currently, we do not know the exact mechanism behind this selectivity. The C-terminal domains of H1.2 and HP1α have several unique amino acids and are proposed to interact with other factors.43–45 Thus, mH2A1.2-dependent behaviors of H1.2 and HP1α may be derived from their distinct structural features conferred by these unique amino acids existing in H1.2 and HP1α. This is consistent with our observation that the macrodomain of mH2A1.2 directly interacts with the C-terminal domains of H1.2 and HP1α. This also supports a model where H1.2 and HP1α are non-redundant and function in concert to ensure correct action of mH2A1.2 toward silencing pro-osteoclastogenic genes in prostate cancer cells. Although we have linked H1.2 and HP1α to mH2A1.2-mediated LTβ gene silencing, we have not shown that mH2A1.2-dependent recruitments of H1.2 and HP1α to LTβ gene alter local chromatin structure. In addition, changes in DNA inaccessibility surrounding LTβ gene in response to knockdown of H1.2 and HP1α have not yet been investigated. However, since H1.2 and HP1α are important modulators of chromatin higher order folding, it is tempting to speculate that mH2A1.2-dependent recruitment of H1.2 and HP1α will promote the formation and maintenance of higher order chromatin structure to achieve sustained silencing of LTβ gene. Data presented here only indicate that mH2A1.2 plays a role in localizing H1.2 and HP1α at LTβ chromatin locus that was analyzed. It remains to be determined whether these observations also hold true on other mH2A1.2-regulated genes in prostate cancer cells.
Based on the data presented here and previously known function of mH2A, we propose a model wherein mH2A1.2 serves as a negative regulator of prostate cancer-mediated osteoclastogenesis (Figure 7e). In prostate cancer cells in the bone microenvironment, mH2A1.2 is incorporated into the LTβ locus and recruits H1.2 and HP1α through direct interaction. Subsequent localization of H1.2 and HP1α at mH2A1.2-rich LTβ locus leads to the local compaction of chromatin around the promoter and early coding regions, with decreased accessibility of transcription factors to their target sites. In this state, LTβ is only minimally expressed, and its effect on osteoclast differentiation is significantly attenuated. In this scenario, mH1.2-mediated recruitment of H1.2 and HP1α can be viewed as a critical regulatory process to modulate LTβ expression in a specific manner.
MATERIALS AND METHODS
Constructs and antibodies, cDNA array analysis, generation of prostate cancer secretome, microarray data analysis, candidate gene prioritization via pathway-based approach, reporter gene assay, migration assay, bone resorption assay, and statistical analysis are detailed in the Supplementary Materials and Methods.
Cell culture and osteoclast differentiation
Human prostate cancer cell line DU145 was obtained from American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). To prepare CMs, media was changed to α-minimum essential medium (α-MEM) containing 0.5% FBS when cells were approximately 80% confluent. After 24 h incubation, CMs were collected, centrifuged at 1200 rpm for 10 min, concentrated, and filtered. For in vitro osteoclastogenesis assays, bone marrow cells were collected from the femurs and tibia of 6-week-old C57BL/6 mice (The Jackson Laboratory, ME, USA) as previously described30 and in accordance with the University of Southern California guidelines. The cells were centrifuged and removed by a treatment with red blood cell lysis buffer (Sigma-Aldrich, St Louis, MO). The harvested cells were cultured with α-MEM containing 10% FBS and macrophage colony stimulating factor (M-CSF, 5 ng/ml) for 16 h. Non-adherent mononuclear cells were collected, seeded on 100-mm culture dishes, and further incubated for 3 days with α-MEM containing M-CSF (30 ng/ml) to generate osteoclast precursor (OCP) cells. OCP cells were seed into a 48-well plate (2 × 104 cells/well) and grown in a 1:1 mixture of α-MEM culture medium and CMs in the presence of RANKL (50 ng/ml) for 7 days with a change of CMs every 2 days. On day 7, the cells were stained with tartrate-resistant acid phosphatase (TRAP) using a leukocyte acid phosphatase staining kit (Sigma-Aldrich). TRAP-positive multinuclear cells containing three or more nuclei and a full actin ring were counted as osteoclasts under a light microscope. In certain instances, CM treatments were terminated at days 0, 3 and 7, and expression levels of osteoclastogenic genes were analyzed.
RNA Interference
DNA oligonucleotides encoding shRNAs specific for the coding regions of histone variants and mH2A1.2 target genes (listed in Supplementary Table S3) were annealed and ligated into the lentiviral expression vector pLKO.1 (Addgene, Cambridge, MA). To prepare lentiviral particles, 293T cells were transfected with plasmids encoding VSV-G, NL-BH and the shRNAs using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Cells stably expressing the shRNA were generated by lentivirus infection and subsequent selection for puromycin resistance for two weeks. For shRNA-mediated depletion of the genes encoding mH2A1.2-associated factors, shRNAs targeting each of the associated factors and non-targeted control shRNA (listed in Supplementary Table S3) were generated as described above. DU145 cells were transiently transfected with these shRNAs, and changes in the target gene mRNA levels were measured using qRT-PCR.
Purification and identification of mH2A1.2-associated factors
DU145 prostate cancer cells continuously expressing mH2A1.2 were generated by stable transfection of pLenti-FLAG-HA-mH2A1.2 vector. Nuclear extracts were prepared as described,41 and ectopic mH2A1.2 and its interacting proteins were purified by sequential immunoprecipitation using anti-FLAG M2 and anti-HA antibodies in the precipitation buffer (20 mM Tris-HCl, pH 7.3, 300 mM KCl, 0.2 mM EDTA, 20% Glycerol, and 0.1% Nonidet P-40). The purified proteins were resolved by 4–20% gradient SDS-PAGE, and stained with Coomassie blue. Major protein bands were excised and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Chromatin immunoprecipitation
ChIP assays with DU145 prostate cancer cells were performed using the ChIP assay kit (Millipore, Bedford, MA) as recently described.46 Antibodies specific for mH2A1, H1.2, HP1α, HP1β and HP1γ were used for immunoprecipitation. Immunoprecipitated DNA was analyzed by qPCR using the primers that amplify the TSSs, +2 kb regions, and −2 kb regions of the LTβ, IL6, EDN1, TNFSF12, FSTL3, ANXA2, GSK3β, HRH1 and CTHRC1 genes. The primers used for qPCR are listed in Supplementary Table S4. Specificity of amplification was determined by melting curve analysis and all samples were run in triplicate with results averaged.
qRT-PCR
Total RNA (2 μg) was isolated from DU145 cell pellets using an RNeasy mini kit (Qiagen, Germany). RNA was converted to first strand cDNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time RT-PCR was performed for the selected genes (listed in Supplementary Table S5) using one-step QuantiTect SYBR Green RT-PCR kit (Qiagen) according to the manufacturer’s protocol.
Recombinant LT treatment
Seven-week-old male C57BL/6 mice were injected with rLTα1β2 (0.2 mg/kg in 100 μl PBS) or PBS alone (Control) subcutaneously over the calvaria every other day. Five male mice were injected for each group. After 10 days, mice were sacrificed, and calvaria was collected and fixed with 3.7% formaldehyde. Paraffin-embedded sections (6 μm) of calvaria were stained for hematoxylin/eosin and TRAP for histological analysis. Images were acquired with an Aperio ScanScope Model T3 and were analyzed with ImageScope software (Aperio Technologies, Vista, CA). All animal studies were conducted according to protocols approved by the Institutional Animal Care and Use Committee. OCP cells (2 × 104 cells/well) were also treated with rLTα, rLTβ, or rLTα1β2 (100 ng/ml) along with RANKL (15 ng/ml) and M-CSF (30 ng/ml) and used to evaluate in vitro response to rLT proteins.
Micro-CT analysis
Calvarial bones were excised from euthanized mice, and fixed with 4% paraformaldehyde. The micro-CT scanning was performed SkyScan 1172 scanner (Bruker, Belgium) using a set at 67 kV and 147 μA for 705 milliseconds. The bone volume (mm3) over tissue volume and bone mineral density in the region of interest were measured directly with 2D/3D Image analysis CT-analyser (CTAn) software (Bruker). We defined the regions of interest as the areas between 2.066 mm and 2.754 mm rearward of coronal suture in calvaria. For visualization, the segmented data were imported and reconstructed as a three-dimensional image displayed in CT-volume (CTVol) software (Bruker).
Protein-protein interactions
GST and GST-tagged proteins were produced from E. coli Rosetta 2 (DE3) pLysS cells (Novagen, Madison, WI), purified on Glutathione-Agarose beads (Sigma-Aldrich), and quantified by Coomassie staining as described previously.46 In vitro interaction assays were performed with equal amounts of GST or GST fusion proteins in affinity buffer (20 mM HEPES-KOH, pH 7.9, 0.5 mM EDTA, 200 mM NaCl, 10% glycerol, and 0.1% Nonidet P-40) supplemented with PMSF, dithiothreitol, and protease inhibitor mixture. Bound proteins were washed four times and resuspended in Laemmli buffer for Western blot analysis. Ten percent of the interaction mix was loaded as an input fraction.
Subcellular fractionation of osteoclasts
Nuclear and cytosolic fractions were prepared as described previously30 with minor modifications. Briefly, mock-treated/rLTα1β2-treated osteoclasts were lysed in buffer A (10 mM HEPES-NaOH, pH 7.9, 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA, 1 mM dithiothreitol, 1 mM PMSF, and 1x Roche protease inhibitor cocktail) on ice for 30 min with occasional mixing by gently tapping the tube. NP-40 was then added to a final concentration of 0.3%. Cell lysates were spun at 5000 rpm for 5 min to obtain nuclear pellets. Supernatants were collected as cytoplasmic fractions. Nuclear pellets were resuspended in buffer B (20 mM HEPES-NaOH, pH 7.9, 0.5 mM EGTA, 0.5 mM EDTA, 420 mM NaCl, 1 mM dithiothreitol, 1 mM PMSF, and 1x Roche protease inhibitor cocktail) and incubated on ice for 30 min with occasional mixing to extract nuclear proteins. Nuclear extracts were cleared by centrifugation at 12,000 rpm for 5 min and supernatants were collected as nuclear fraction.
Nucleosome purification
DU145 prostate cancer cells were transfected with expression vectors for FLAG-tagged H2A and mH2A1.2. 48 h post-transfection, nuclei were prepared and digested with micrococcal nuclease (MNase) to produce predominantly mononucleosomes. Digested nuclei were collected and incubated in nuclear extraction buffer (20 mM HEPES, pH 7.4, 500 mM NaCl, 1.5 mM MgCl2, 0.2 mM EGTA, and 1x Roche protease inhibitor cocktail) for 1 h, and centrifuged to remove nuclear debris. After adjusting the salt concentration of the extract to 300 mM NaCl, ectopic H2A/mH2A1.2-containing nucleosomes were isolated by immunoprecipitation using anti-FLAG antibody in washing buffer (20 mM HEPES, pH 7.8, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EGTA, 10% Glycerol, 0.2% Triton X-100, and 1x Roche protease inhibitor cocktail). Bead-bound nucleosomes and H1.2/HP1α were analyzed by Western blot analysis.
LTβ ELISA
ELISA assay kit (Antibodies-online Inc., Atlanta, GA) was used to measure LTβ levels in CMs. Before use, an aliquot of conditioned medium was appropriately diluted with buffers recommended by the manufacturer. Optical density (OD) was measured on a Plate Chameleon V plate reader (Hidex, Finland) at an absorbance of 450 nm. Protein concentration was determined after correcting for optical imperfections in the plate by subtracting OD values at 540/570 nm from OD values obtained at 450 nm.
Co-immunoprecipitation
DU145 prostate cancer cells were washed with cold PBS and lysed in cell lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, and 1x Roche protease inhibitor cocktail). Whole cell lysates were incubated with mH2A1 antibody for overnight followed by incubation with Protein A/G agarose beads (Santa Cruz Biotechnology, Santacruz, CA). Beads were washed four times with lysis buffer, and the binding of endogenous mH2A1 and H1.2/HP1α was analyzed by Western blot.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Yongwon Choi for the gift of RANKL plasmid and M-CSF protein. This work was supported by NIH Grant CA201561 awarded to W.A. The study was also funded in part by pilot project grants from Keck School of Medicine of USC.
Footnotes
Conflict of Interest
The authors declare no competing financial interests.
REFERENCES
- 1.Matsuo K, Irie N. Osteoclast-osteoblast communication. Arch Biochem Biophys 2008; 473: 201–209. [DOI] [PubMed] [Google Scholar]
- 2.Edwards CM, Mundy GR. Eph receptors and ephrin signaling pathways: a role in bone homeostasis. Int J Med Sci 2008; 5: 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–147. [DOI] [PubMed] [Google Scholar]
- 4.Ash P, Loutit JF, Townsend KM. Osteoclasts derived from haematopoietic stem cells. Nature 1980; 283: 669–670. [DOI] [PubMed] [Google Scholar]
- 5.Baron R Arming the osteoclast. Nat Med 2004; 10: 458–460. [DOI] [PubMed] [Google Scholar]
- 6.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93: 165–176. [DOI] [PubMed] [Google Scholar]
- 7.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 1998; 95: 3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teitelbaum SL. Bone resorption by osteoclasts. Science 2000; 289: 1504–1508. [DOI] [PubMed] [Google Scholar]
- 9.Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol 2008; 3: 457–484. [DOI] [PubMed] [Google Scholar]
- 10.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet 2003; 4: 638–649. [DOI] [PubMed] [Google Scholar]
- 11.Zaidi M Skeletal remodeling in health and disease. Nat Med 2007; 13: 791–801. [DOI] [PubMed] [Google Scholar]
- 12.Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature 2003; 423: 343–348. [DOI] [PubMed] [Google Scholar]
- 13.Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1998. CA Cancer J Clin 1998; 48: 6–29. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest 2001; 107: 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell Res 2005; 15: 57–62. [DOI] [PubMed] [Google Scholar]
- 16.Armstrong AP, Miller RE, Jones JC, Zhang J, Keller ET, Dougall WC. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008; 68: 92–104. [DOI] [PubMed] [Google Scholar]
- 17.Mori K, Le Goff B, Charrier C, Battaglia S, Heymann D, Redini F. DU145 human prostate cancer cells express functional receptor activator of NFkappaB: new insights in the prostate cancer bone metastasis process. Bone 2007; 40: 981–990. [DOI] [PubMed] [Google Scholar]
- 18.Maze I, Noh KM, Soshnev AA, Allis CD. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat Rev Genet 2014; 15: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 2010; 11: 264–275. [DOI] [PubMed] [Google Scholar]
- 20.Pehrson JR, Fried VA. MacroH2A, a core histone containing a large nonhistone region. Science 1992; 257: 1398–1400. [DOI] [PubMed] [Google Scholar]
- 21.Melters DP, Nye J, Zhao H, Dalal Y. Chromatin Dynamics in Vivo: A Game of Musical Chairs. Genes (Basel) 2015; 6: 751–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasmussen TP, Huang T, Mastrangelo MA, Loring J, Panning B, Jaenisch R. Messenger RNAs encoding mouse histone macroH2A1 isoforms are expressed at similar levels in male and female cells and result from alternative splicing. Nucleic Acids Res 1999; 27: 3685–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T et al. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009; 28: 3423–3428. [DOI] [PubMed] [Google Scholar]
- 24.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010; 468: 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, Kraus WL. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev 2010; 24: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol 2009; 16: 923–929. [DOI] [PubMed] [Google Scholar]
- 27.Dell’Orso S, Wang AH, Shih HY, Saso K, Berghella L, Gutierrez-Cruz G et al. The Histone Variant MacroH2A1.2 Is Necessary for the Activation of Muscle Enhancers and Recruitment of the Transcription Factor Pbx1. Cell Rep 2016; 14: 1156–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JM, Heo K, Choi J, Kim K, An W. The histone variant MacroH2A regulates Ca(2+) influx through TRPC3 and TRPC6 channels. Oncogenesis 2013; 2: e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, Gamble MJ. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat Struct Mol Biol 2014; 21: 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim K, Punj V, Kim JM, Lee S, Ulmer TS, Lu W et al. MMP-9 facilitates selective proteolysis of the histone H3 tail at genes necessary for proficient osteoclastogenesis. Genes Dev 2016; 30: 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Punj V, Matta H, Chaudhary PM. A computational profiling of changes in gene expression and transcription factors induced by vFLIP K13 in primary effusion lymphoma. PLoS One 2012; 7: e37498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–273. [DOI] [PubMed] [Google Scholar]
- 33.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity 1997; 6: 491–500. [DOI] [PubMed] [Google Scholar]
- 34.Liepinsh DJ, Grivennikov SI, Klarmann KD, Lagarkova MA, Drutskaya MS, Lockett SJ et al. Novel lymphotoxin alpha (LTalpha) knockout mice with unperturbed tumor necrosis factor expression: reassessing LTalpha biological functions. Mol Cell Biol 2006; 26: 4214–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyce BF, Yao Z, Xing L. Functions of nuclear factor kappaB in bone. Ann N Y Acad Sci 2010; 1192: 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med 2005; 202: 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med 2004; 10: 617–624. [DOI] [PubMed] [Google Scholar]
- 38.Jimi E, Ghosh S. Role of nuclear factor-kappaB in the immune system and bone. Immunol Rev 2005; 208: 80–87. [DOI] [PubMed] [Google Scholar]
- 39.Madge LA, Kluger MS, Orange JS, May MJ. Lymphotoxin-alpha 1 beta 2 and LIGHT induce classical and noncanonical NF-kappa B-dependent proinflammatory gene expression in vascular endothelial cells. J Immunol 2008; 180: 3467–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61: 3071–3076. [PubMed] [Google Scholar]
- 41.Heo K, Kim H, Choi SH, Choi J, Kim K, Gu J et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell 2008; 30: 86–97. [DOI] [PubMed] [Google Scholar]
- 42.Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994; 266: 443–448. [DOI] [PubMed] [Google Scholar]
- 43.Kim K, Lee B, Kim J, Choi J, Kim JM, Xiong Y et al. Linker Histone H1.2 cooperates with Cul4A and PAF1 to drive H4K31 ubiquitylation-mediated transactivation. Cell Rep 2013; 5: 1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smothers JF, Henikoff S. The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr Biol 2000; 10: 27–30. [DOI] [PubMed] [Google Scholar]
- 45.Richart AN, Brunner CI, Stott K, Murzina NV, Thomas JO. Characterization of chromoshadow domain-mediated binding of heterochromatin protein 1alpha (HP1alpha) to histone H3. J Biol Chem 2012; 287: 18730–18737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JM, Kim K, Schmidt T, Punj V, Tucker H, Rice JC et al. Cooperation between SMYD3 and PC4 drives a distinct transcriptional program in cancer cells. Nucleic Acids Res 2015; 43: 8868–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.