

Abstract
With the advent of new cancer therapies in the last few years, the goals of reducing disease burden and improving quality of life are frequently achieved. Yet despite the advances seen with numerous monotherapies, a multimodality approach that targets different aspects of tumor biology may yield the greatest clinical benefit for patients with late-stage disease. Many such strategies have been employed with varying degrees of success. The addition of immunotherapy to standard-of-care radiation therapy has shown evidence of efficacy in some preclinical models and in the clinical setting. However, exploiting these two modalities safely and effectively remains an ongoing challenge. It is feasible that the addition of another therapeutic modality could further enhance the antitumor effects of these treatments. The recent addition of angiogenesis inhibitors to the cancer treatment armamentarium represents an attractive option, especially since these agents have been shown to be most effective when combined with other therapies. This review examines preclinical and clinical data on the interaction between immunotherapy and radiation, and discusses the potential synergy between these two modalities and angiogenesis inhibitors.
Radiation, immunotherapy, and angiogenesis inhibitors: ‘‘all for one’’
Radiotherapy is a major cancer treatment modality, and efforts to improve its efficacy will likely impact significant numbers of people. In North America in 2009, radiation therapy will be used at some point in 75% of the estimated million patients who will be diagnosed with a noncutaneous form of cancer.1 Some progress has been made combining radiation with standard cytotoxic chemotherapy, and chemo- radiation is the standard of care for multiple cancer types, including carcinomas of the lung, head and neck, gastrointestinal tract, cervix, bladder, and brain. In addition, targeted therapies designed to exploit the molecular basis of cancer and radiation- induced cellular changes are actively being investigated. Angiogenesis inhibitors are one example of targeted agents that can improve the therapeutic ratio of radiation therapy. Based on promising preclinical results with the combination of radiation and angiogenesis inhibitors, the Radiation Therapy Oncology Group has recently opened a phase III trial investigating the potential benefit of bevacizumab, an antivascular endothelial growth factor (VEGF) antibody, concurrent with radiation and temozolomide in patients with newly diagnosed glioblastoma.2
Immunotherapy is another targeted therapy that modifies the tumor microenvironment. It has had limited success as a monotherapy, but has shown more promising results when used in combination with other treatment modalities, including radiation. The immune system is an important regulator of cellular responses to radiation, which can be used to modulate immune-mediated responses to tumors.
Despite these advances, altering a single facet of tumor biology appears unlikely to have a meaningful impact on tumor control. Solid tumor cells are extensively heterogeneous; thus, inhibiting one target will affect some, but not all, tumor cells. One way to potentially overcome this is to develop treatment strategies that inhibit or affect multiple aspects of tumor growth, such as combining radiation, angiogenesis inhibitors, and immunotherapy. Angiogenesis inhibitors can normalize tumor vasculature and enhance the effects of radiation while favorably altering the immunologic tumor microenvironment. But while joining all three modalities in common cause may seem like an ideal ‘‘Three Musketeers’’ approach to cancer treatment, we must cautiously consider whether this is just another quixotic strategy. Two recently reported phase III trials showed decreased progression-free survival in metastatic colorectal cancer patients given the combination of epidermal growth factor inhibitors with bevacizumab and standard chemotherapy.3,4
Previous reports have focused on the combination of various modalities to improve cancer treatment. This review summarizes current preclinical and clinical research data on radiation and immune system interactions, and offers a rationale for combining radiation, immunotherapy, and angiogenesis inhibitors for enhanced tumor control.
‘‘Athos’’: combining radiation with immunotherapy
Effects of radiation on the immune system
Radiation can induce cancer cell death through a number of mechanisms, including initiation of apoptotic and cell-cycle arrest pathways as well as interfering with the repair of damaged DNA. Furthermore, radiation-induced cell death is also an immunogenic process that can potentially be exploited to stimulate tumor-specific immune responses. While the immunogenic process of radiation-induced cancer cell death is not fully understood, certain key mechanisms are known. Cells undergoing immunologic cell death develop distinctive changes on their plasma membrane which can act as signals to promote phagocytosis by antigen-presenting cells (APCs) (Fig. 1). One set of proteins known to move from the intracellular to the extracellular compartment during immunologic cell death following radiation are heat shock proteins.5 Plasma membrane expression of these proteins helps mark damaged cells for elimination by the immune system and facilitates antigen cross-presentation,6 dendritic cell (DC) maturation,7 and natural killer (NK) cell activation.8 Taken together, these effects help to induce and amplify antitumor responses.
Fig. 1.

Antigen release from dying tumor cells can activate immune responses—irradiation induces death of cancer cells. As these cells die, they are taken up by professional antigen presenting cells. These antigen presenting cells then travel to regional lymph nodes where they present antigen to T cells, initiating or potentiating an antitumor immune response. Activated tumor-specific T cells can then traffic to areas of tumor to participate in immune-mediated tumor killing.
Another protein that translocates to the cell surface following radiation therapy prior to tumor cell death is calreticulin (CRT).9 CRT is a Ca2+ binding protein located mainly in the lumen of the endoplasmic reticulum and is involved in Ca2+ homeostasis. The presence of CRT is a crucial determinant of whether dying tumor cells are phagocytosed by macro- phages and DCs.9,10 However, CRT translocation alone is not sufficient to provoke an immune response, which appears to require other ‘‘danger signals.’’ High-mobility group box 1, a nonhistone chromatin-binding protein released by dying tumor cells in response to radiation, has recently been shown to be one of these critical ‘‘danger signals’’ (Fig. 1).11,12
Immune-mediated tumor eradication has also been linked with radiation-induced DNA damage pathways via ataxia telangiectasia mutated (ATM) protein and p53. In an ATM-dependent fashion, DNA-damaged tumor cells can upregulate ligands such as NKG2D, making them more effciently recognized for elimination by NK and activated CD8+ T cells.13 In a study using an in vivo hepatocellular carcinoma model, Xue et al. showed that restoring p53 tumor expression in p53-deficient tumors could produce complete tumor regression. These changes were not mediated by apoptosis, but rather involved p53-mediated cellular senescence, followed by upregulation of inflammatory cytokines and induction of tumor-specific innate immune responses.14
Modulating radiation response with immunotherapy
Because radiation-induced cell death is an immunologically active process, many studies have tried to enhance the effects of radiation by combining it with various immunotherapeutic strategies. For instance, preclinical studies combining DCs with radiation therapy have shown increased tumor-specific CD8+ T cells and improved antitumor responses compared with radiation alone.15,16 Other preclinical studies have shown improved tumor control and/or survival by combining radiation with immune modulators such as anti-CTLA-4 antibodies,17 IL-2 gene transfer,18 IL-12,19,20 IL-3,21 Flt3 ligand,17 and TNF-a in combination with temozolomide.22 While these results are promising, the interactions between radiation and the immune system are complex and influenced by multiple cell types. Still, this is an active area of research with many potential clinical applications.
Modulating immune response with radiation therapy
Preclinical rationale.
The most significant clinical issue in radiation therapy for cancer is how to safely increase the dose of radiation to enhance tumor cell killing. However, radiation can also be regarded as a targeted therapy that can be used to improve tumor-specific immune responses (Fig. 1). Sublethal doses of radiation bring about significant changes in cell surface protein expression. Garnett et al. treated human cancer cell lines (12 colon, 7 lung, and 4 prostate) with sublethal doses of radiation. They found that 72 hours post-treatment, 91% of the cell lines upregulated one or more cell surface molecules, including Fas, ICAM-1, MUC-1, CEA, and MHC class I,23 all of which have been shown to render tumor cells more amenable to immune recognition and to potentiate antitumor immune responses. Furthermore, in a preclinical in vivo model, 8 Gy of radiation in combination with a recombinant poxvirus vaccine expressing CEA and three T-cell costimulatory molecules (CEA-TRICOM) showed dramatic tumor reduction and significant cure rates of established tumors. Notably, neither radiation nor vaccine alone was capable of inhibiting tumor growth.24 These studies show that sublethal doses of radiation can alter the expression of cell surface molecules on tumor cells, making them more susceptible to immune-mediated attack.
Radiation therapy not only enhances MHC class I expression, but also modulates the peptide repertoire expressed by MHC class I peptides through antigen cascade.25 The mechanisms involved in this process include degradation of proteins damaged by radiation, increased peptide production, and improved antigen presentation and cytotoxic T lymphocyte recognition of radiated cells.
More recent studies have investigated the use of much higher doses of radiation to alter the immune response. Using a mouse lung carcinoma model, Crittenden et al. radiated tumors with 20 Gy fractions three times.26 There were significant decreases in CD4+ and CD8+ tumor-infiltrating cells within 48 h of radiation; however, by day 7 a significantly higher fraction of CD8+ cells in the tumor had an activated effector phenotype. A study by Lee et al. used an in vivo mouse model to treat mouse melanoma tumors with 20 Gy of radiation in immune-competent and T cell-deficient mice and found significant tumor regression only in the immune- competent mice.27 Additional studies showed that this effect was largely mediated by CD8+ T cells, and that the mechanisms involved in this process included increased T-cell priming and expansion of effector T cells to both low- and high-affinity antigens following radiation treatment.
The studies described above focused on changes in the immune system following external beam radiation therapy (EBRT). Other preclinical studies have demonstrated that delivering radiation in different forms can also modulate tumor phenotype and enhance T cell-mediated killing. In one study, human tumor cell lines exposed to 153samarium (Quadramet), a chelated, bone-seeking radionuclide, upregulated critical molecules involved in initiating antitumor immune responses, including Fas, CEA, MUC-1, and MHC class I.26 These phenotypic changes subsequently rendered the tumor cells more susceptible to cytotoxic T lymphocyte cell lysis. A second study employed yttrium-90-labeled anti-CEA antibody, a radiolabeled monoclonal antibody, in combination with vaccine therapy and showed statistically significant increases in survival in tumor-bearing mice compared to vaccine or yttrium-90 alone.28 This study also reported that the combination treatment resulted in an increase in the percentage of tumor-infiltrating antigen-specific T cells. Interestingly, these T cells conferred a measure of resistance to radiation-induced damage, a finding consistent with a preclinical study by Grayson et al. that found that murine memory T cells were more resistant to apoptosis than naive T cells after whole-body irradiation.29
These studies demonstrate the ability of radiation therapy and immunotherapy to enhance tumor-specific immune responses and immune-mediated tumor killing, and have provided a rationale for their combined use in the clinic.
Clinical studies.
A recent clinical trial combined a recombinant cancer vaccine with standard definitive radiation therapy in patients with localized prostate cancer.30 Patients were assigned to receive definitive radiotherapy with or with- out vaccine to determine if the vaccine could induce an immune response in the presence of tumor irradiation. The study was designed as a randomized phase II trial with a primary endpoint of immunologic response, as determined by the generation of PSA-specific T cells, with secondary endpoints of safety and clinical response. Of 30 total patients, 19 received radiation plus a priming vaccine of recombinant vaccinia (rV) expressing PSA (rV-PSA) admixed with rV expressing the costimulatory molecule B7–1 (rV-PSA-B7–1), followed by monthly booster vaccines with recombinant fowlpox (rF) expressing PSA (rF-PSA). The vaccines were administered with low-dose systemic IL-2 and local granulocyte-macrophage colony-stimulating factor (GM-CSF, Leukine) to augment the immune response. Patients then received EBRT at a dose of Z70 Gy in 1.8 to 2.0 Gy fractions between the fourth and sixth vaccinations.30
Of 17 patients in the combination arm who completed all eight vaccinations, 13 (76%) had at least three fold increases in circulating PSA-specific T cells.30,31 Patients receiving radiation only had no detectable increases in PSA-specific T cells (P o 0.0005). Patients in the combination arm also showed evidence of de novo generation of T cells to prostate-associated antigens other than PSA,30 confirming the phenomenon of antigen cascade and providing indirect evidence of immune-mediated tumor killing.
Based on preclinical studies showing that Quadramet induced immunomodulating phenotypic changes in tumor cells,24 a clinical trial currently enrolling patients at the National Cancer Institute (NCI) is evaluating the combination of Quadramet and vaccine in castration-resistant prostate cancer with bone metastases.32 Here, the vaccine is composed of poxviral vectors expressing PSA with three T-cell costimulatory molecules (B7–1, ICAM-1, and LFA-3), designated PROSTVAC or PSA-TRICOM.31 Another recent trial at the NCI, the results of which have not yet been reported, used the same vaccine platform and regimen, but with CEA instead of PSA as the tumor antigen. In this phase I study, patients with CEA-expressing tumors metastatic to the liver received radiation therapy and CEA-TRICOM vaccine to evaluate the clinical safety and tolerability of the combined treatment.33
An alternative approach to using a vaccine platform that expresses a prominent tumor antigen to enhance tumor-specific immune responses is to utilize dying tumor cells as a rich environment in which to prime DCs. Chi et al. conducted a phase I clinical trial in which patients with refractory hepatoma received 8 Gy of EBRT, followed by intratumoral injection of immature DCs on days 2 and 24.34 It has been reported that cells exposed to this dose of radiation are killed predominantly through the apoptotic pathway, thereby allowing for effcient and proper antigen processing and presentation by DCs.35 Of the 14 patients enrolled in the study, four had minor responses and two had partial responses, including one patient whose alpha-fetoprotein (AFP) levels decreased from 128 to 1.6 ng mL—1.34 In addition, of 10 patients evaluated for induction of AFP-specific immune responses, eight showed an increased response as measured by cytokine-release assay.
Adoptive immunotherapy, an alternate approach to immunotherapy that has been used for over 30 years by Rosenberg et al., involves the isolation, expansion, and reinfusion of tumor-infiltrating lymphocytes.36 In a pilot study, 28 patients with advanced cancer who had failed all standard treatments were enrolled in a trial designed to evaluate the benefit of combining total body radiation therapy with adoptive immunotherapy.37 Four patients had significant decreases in tumor size at the irradiated site, and one achieved a transient complete response. This pilot trial showed the feasibility of administering radiation with adoptive immunotherapy and the potential benefit of the combination of these two therapies.
A different vaccine strategy currently being employed in the clinic is a biological response modifier prepared from heat-killed Lactobacillus casei YTTT018, designated LC-9018. Okawa et al. conducted a phase II randomized clinical trial to determine the clinical efficacy of this vaccine in combination with radiation in 61 patients with stage IIB or III carcinoma of the uterine cervix.38 The combination therapy demonstrated significant tumor reduction, compared with radiation therapy alone (P <0.05). Furthermore, statistically significant objective responses of 31% and 73% were seen in the cohorts receiving cumulative doses of 15 and 30 Gy, respectively.
There are several scheduled and ongoing clinical trials involving the combination of immunotherapy and radiotherapy. One study that will be recruiting patients in the near future is investigating the potential benefit of combining EBRT with anti-CTLA-4 antibody (ipilimumab) in patients with advanced prostate cancer.39
The studies described above demonstrate that the combination of immunotherapy and radiotherapy can effectively enhance tumor-specific immune responses, and may augment tumor cell killing compared to the use of either therapy alone. Future studies should further investigate whether this clinical benefit is due to synergistic or additive effects.
‘‘Porthos’’: combining radiation with antiangiogenic modalities
Preclinical rationale
Multiple preclinical studies suggest that combining radiation and angiogenesis inhibitors such as sunitinib40–42 enhances the therapeutic ratio of radiation therapy. Abnormal vasculature is associated with an uncharacteristic micro- environment, including uneven perfusion, hypoxia, and increased interstitial fluid pressure. This malignant micro- environment desensitizes malignant cells to radiation.43 While the molecular mechanisms responsible for radiation enhancement with antiangiogenic agents are not entirely understood, they appear to be related to tumor vasculature normalization, which consequently improves tumor oxygenation and ultimately enhances the efficacy of radiation therapy (Fig. 2).44 The optimal timing and duration of antiangiogenic therapies to maximize radiation’s therapeutic ratio has not been determined. However, because vasculature normalization is most frequently hypothesized as the mechanism responsible for increased radiosensitivity, identifying this ‘‘normalization window’’ would be an essential first step in initiating combination therapy. In vivo studies in mouse models of intracranial human glioblastoma multiforme (GBM) tumors treated with a monoclonal antibody to VEGF receptor-2 suggest that the vascular normalization window may be approximately seven days.45 This finding differs from clinical data on patients with recurrent GBM treated with AZD2171, a pan-VEGF tyrosine kinase inhibitor, that demonstrated features of vascular normalization as early as one day after treatment, with a reversal of this normalization starting around day 28.46 Either way, it appears that there is a relatively small window of opportunity for maximizing the effect of combined radiation and antiangiogenic agents.
Fig. 2.

Anti-angiogenic agents can increase radiation sensitivity—the use of antiangiogenic agents can normalize tumor vasculature which increases tumor perfusion while decreasing interstitial pressure. The increased oxygen content in the formerly hypoxic tumor gives rise to elevated reactive oxygen species (ROS) upon irradiation, leading to increased DNA damage, mitotic catastrophe, and ultimately, cell death.
Clinical studies
Refuting the notion that angiogenesis inhibitors impair the effects of radiation therapy by inducing tumor hypoxia, several clinical studies have shown that these two modalities can be combined safely and effectively.
Interfering with epidermal growth factor receptor (EGFR) signaling can indirectly inhibit angiogenesis by decreasing production of VEGF by tumor cells, and can in some cases directly target angiogenesis by inducing apoptosis of endothelial cells that express and activate EGFR.47–50 In a phase III clinical trial enrolling 424 patients with locoregionally advanced head and neck cancer, 211 patients were treated with weekly cetuximab, a monoclonal antibody against EGFR, and one of three high-dose radiotherapy fractionation regimens. The other 213 patients were treated with radiation alone.51 Median survival for the combined modality cohort was 49 months compared to 29 months for the radiation alone group (P = 0.03), with a median follow-up of 54 months.
Celecoxib, a selective cyclo-oxygenase-2 inhibitor, has exhibited antiangiogenic activities through direct effects on endothelial cells and by interfering with mitogen-activated protein kinase (ERK2) activity.52 In a recent phase I trial, celecoxib was administered concurrently with thoracic radiotherapy to patients with inoperable or unresectable non-small cell lung cancer (NSCLC).53 The combined treatment resulted in actuarial local progression-free survival of 66% at one year and 42.2% at two years. Furthermore, of 37 patients evaluable for tumor response, 27 had complete or partial responses.53 Other antiangiogenic agents are currently being investigated for their potential clinical benefit in this patient population. In a phase II single-agent trial in patients with relapsed NSCLC, sorafenib, a tyrosine kinase inhibitor of VEGF, was well tolerated and active against NSCLC, as evidenced by one patient who experienced a 41% tumor reduction.54 Similarly, a multicenter phase II trial evaluating the activity of sunitinib, a tyrosine kinase inhibitor of VEGF receptors 1, 2, and 3, platelet-derived growth factor, KIT (stem cell factor receptor), RET, and Flt-3 in refractory NSCLC reported a median progression-free survival of 12 weeks and median overall survival of 23.4 weeks, with an acceptable toxicity profile.55
The combination of antiangiogenic factors with chemoradiotherapy has been increasingly employed in recent years. In an ongoing phase II trial, bevacizumab was added to temozolomide and radiation therapy in patients who had undergone surgical resection of GBM.56 All patients received standard EBRT and concurrent temozolomide, with bevacizumab given every two weeks at 10 mg kg—1 starting the first day of chemoradiotherapy. At the completion of radiation therapy, bevacizumab and temozolomide were continued. Preliminary efficacy analysis shows an encouraging mean progression-free survival of >8.8 months and an acceptable toxicity profile.56 However, as in other clinical trials that have reported increased toxicity with bevacizumab and chemoradiotherapy,57–60 healing difficulties and thromboembolic events were observed and attributed, in part, to the addition of bevacizumab to the regimen.
Recent studies by Willett et al. combining bevacizumab with chemoradiation have resulted in an ongoing phase II trial for patients with rectal cancer. In one of their prior phase I trials, the combination of bevacizumab with 5-fluorouracil and radiation was shown to be safe and effective, as all six patients completed the combined treatment without dose-limiting toxicity and underwent surgery without perioperative or postoperative complications.61 In a second phase I trial, bevacizumab at 10 mg kg—1 combined with chemoradiotherapy induced complete responses in two of five patients with locally advanced rectal cancer.59 This investigation supports the antivascular effects of bevacizumab and its ability to normalize tumor vasculature.
Taken together, these studies demonstrate that antiangiogenic therapy can improve the antitumor effects of radiation. Nevertheless, in order to avoid the toxicities reported in the above trials, additional studies should further investigate the precise timing, duration, and dose of antiangiogenic agents when used in combination with radiotherapy.
‘‘Aramis’’: combining immunotherapy with antiangiogenic modalities
Preclinical/translational studies
DCs are considered the most powerful APCs and, thus, a critical component of the adaptive immune response. Their function is highly influenced by the tumor microenvironment, and tumor-secreted VEGF negatively affects DC maturation.62 VEGF was initially shown to influence DC maturation in both in vivo and in vitro settings when administration of VEGF-specific neutralizing antibodies normalized DC differentiation.63
VEGF is also involved in promoting myeloid-derived stem cell (MDSC) expansion. MDSCs, which are immature myeloid cells that are precursors of DCs, macrophages, and/or granulocytes, are driven by multiple tumor-secreted factors.64 MSDCs are critical because they can induce T regulatory cells (Tregs) that can in turn downregulate cell-mediated immunity. Multiple preclinical studies have tested various antiangiogenic agents to see if they can improve DC maturation and inhibit MDSC expansion. For instance, bevacizumab and matrix metalloproteinase-9 inhibition can decrease the MDSC population in the peripheral blood in mouse tumor models.65,66 In vivo studies in a colon cancer mouse model showed that treatment with sunitinib decreased the number of MDSCs and Tregs through KIT ligand inhibition.67 This resulted in enhanced IFN-g expression and decreased expression of negative costimulatory molecules such as CTLA-4 and programmed death ligand 1. This study is significant because it demonstrates that the interplay between antiangiogenic agents and the immune system goes beyond VEGF and tumor vasculature normalization to include ‘‘off targets’’ that can mediate helpful changes.
Some investigators have reached different conclusions regarding the effects of sunitinib on the immune system. In one in vivo study, sorafenib, but not sunitinib, had inhibitory effects on DC function, expression of costimulatory molecules, migration ability, and T-cell stimulatory capacity.68 This was in contrast to the in vitro study by Alfaro et al., which found that only sunitinib, and not sorafenib or bevacizumab, significantly reduced IL-12 expression when added to differentiated DCs.69 It may be that antiangiogenic agents have a different effect on myeloid immature precursors to DCs than they have on fully differentiated DCs, thus explaining the different results of these studies. Clearly, much more research is needed to determine the optimal timing of immunotherapy with other treatment modalities.
Translational studies have also provided insight into the effects of sunitinib on the immune system. In one study, metastatic renal cell cancer patients treated with sunitinib had significant reductions in MDSCs, which correlated with a reversal in type 1 T-cell suppression and decreased Tregs.70 Another study in the same patient population showed an increase in a myeloid dendritic cell subset after sunitinib treatment, which correlated with decreased tumor size and an increase in progression-free survival.71
Overall, preclinical and translational studies demonstrate that different angiogenesis inhibitors have different effects on the immune system, and that exposure during different phases of immune cell maturation can have different effects. While data are limited, combining sunitinib with immunotherapy seems promising, as it may lead to enhanced DC function and decreased MDSCs and Tregs (Fig. 3). As previously mentioned, numerous studies have shown that angiogenesis inhibitors cause regression of existing vessels, prevent new vessel formation, and normalize tumor vasculature.72–76 This improvement in blood flow and oxygenation is postulated not only to improve delivery of drugs to the tumor, but also to result in higher levels of glucose and oxygen, and thus increased availability of amino acids (Fig. 3).77 Because T cells require adequate levels of glucose, oxygen, and amino acids to retain optimal activity at the tumor site, the combination of immunotherapy and antiangiogenic agents in the clinic is reasonable and timely. Finally, it has been demonstrated that increased levels of VEGF can lead to endothelial cell ‘‘anergy’’ characterized by the downregulation of adhesion molecules such as ICAM-1. This downregulation can make it difficult for leukocytes to attach and extravasate from the endothelium into the stroma and ultimately to tumors. The use of antiangiogenesis inhibitors has shown that they can reverse this endothelial cell anergy and show increased lymphocyte infiltration into tumors.78
Fig. 3.
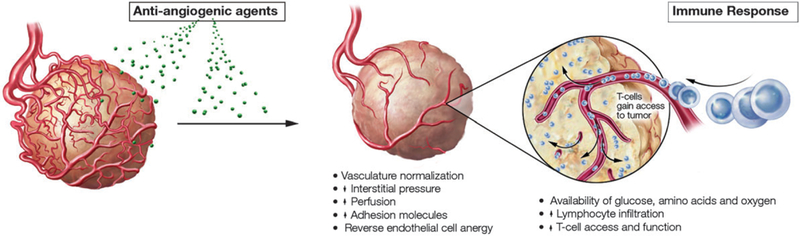
Angiogenesis inhibitors can increase tumor responses to immunotherapy—angiogenesis inhibitors cause regression of existing vessels, prevent new vessel formation, and normalize tumor vasculature. This results in increased blood flow, oxygenation, and glucose required by infiltrating lymphocytes.
Clinical studies
The ability to improve DC maturation through the use of angiogenesis inhibitors has been tested in the clinic. In one study, 41 patients with lung, breast, or colorectal cancer treated with bevacizumab had enhanced DC and T-cell allostimulatory capacity against recall antigens and decreased immature progenitor cells.79 In another study, Almand et al. examined DC populations in a group of metastatic lung cancer patients before and after treatment with carboplatin, taxol, and bevacizumab followed by bevacizumab maintenance and found a shift in immature to competent DCs from baseline to post-three months of bevacizumab treatment.80 Taken together, these data suggest that bevacizumab treatment can affect DC maturation in patients, but do not indicate whether these changes are clinically meaningful.
Interferon-alpha (IFN-α), an immunotherapeutic cytokine, has become a standard initial therapy in patients with metastatic renal cell carcinoma (RCC).81–83 Recent studies of the pathogenesis of RCC have demonstrated that silencing the Von Hippel-Lindau tumor suppressor gene induces the transcription of several pro-angiogenic genes, including VEGF.81,84–88 Based on the biology of RCC, further studies evaluated the clinical benefit of adding sorafenib to IFN monotherapy in patients with advanced RCC. In a phase II trial conducted by the Southwest Oncology Group, nearly 20% of evaluable patients achieved an objective confirmed response, and an additional 50% had unconfirmed partial responses or stable disease after receiving the combination of sorafenib and IFN.89 The results from this study suggest that the combination treatment induces a higher incidence of predefined clinical responses than either agent alone.89 Gollob et al. reported similar results in a phase II trial of patients with metastatic RCC, where the combination of sorafenib and IFN induced a response rate of 33%, including 28% partial responses and 5% complete responses.90 More recently, a randomized phase II trial combining the same agents in patients with metastatic RCC reported greater rates of tumor reduction, better quality of life, and improved tolerability when sorafenib was added to the standard regimen.91
Based on the results reported in these phase II trials, the addition of the anti-VEGF monoclonal antibody bevacizumab to IFN monotherapy was investigated in two randomized phase III trials.81,92 The first reported superior progression- free survival in the bevacizumab plus IFN cohort compared to patients receiving IFN alone (8.5 months versus 5.2 months, P o 0.001). The combination treatment also resulted in significantly higher objective response rates.81 Similarly, the second study reported significantly longer progression-free survival in the combination group (10.2 months) compared to IFN alone (5.4 months).92 While overall toxicity was greater for bevacizumab plus IFN in both studies, based on the above results, the addition of an antiangiogenic agent to the standard initial therapy of IFN may represent an important advance in the treatment of metastatic RCC.
Bevacizumab has been studied as a component of combination strategies in the treatment of various carcinomas. One such study combined a cellular prostate cancer vaccine with bevacizumab in patients with serologic progression of prostate cancer after definitive local therapy.93 The vaccine contained autologous APCs loaded with a recombinant prostatic acid phosphatase/GM-CSF fusion protein as the immunogen. Patients received both the vaccine and bevacizumab on weeks 0, 2, and 4, then bevacizumab alone every two weeks thereafter until toxicity or disease progression. Of the 22 patients treated in this study, one achieved a Z 50% decrease in PSA, nine had decreases in PSA from baseline ranging from 6% to 72%, and three experienced a decrease of Z 25%.93 In addition, the median PSA doubling time pretreatment was 6.9 months compared to 12.7 months post-treatment. Because this was a small, single-arm study without a concurrent control arm of vaccine or bevacizumab alone, the results, while promising, did not allow for an estimation of the potential additive or synergistic clinical effects of the combination.
The clinical data outlined above demonstrate that targeting antivascular pathways and normalizing tumor vasculature with antiangiogenics can improve antitumor immune responses elicited by immunomodulators or vaccine approaches. Despite the increased toxicity reported in some studies, the results are encouraging, and the ability of the combination to enhance immune function warrants further investigation.
Radiation, immunotherapy, and angiogenesis inhibitors: ‘‘one for all’’
While a multimodal approach to cancer treatment is no longer a novel concept, the combination of immunotherapy, radiation, and angiogenesis inhibitors represents an innovative approach with potential clinical benefits. The ability of antiangiogenic agents to normalize tumor vasculature, increase lymphocyte infiltration, limit hypoxia, improve DC maturation, reduce MDSCs and Tregs, and transiently increase perfusion may produce an optimal tumor microenvironment for radiotherapy and immunotherapy to attack tumor cells. By normalizing blood flow and lowering intratumoral pressure, angiogenesis inhibitors enable T cells to gain efficient access to the tumor, while increasing the necessary ingredients for effective T-cell function at the tumor site.72–77 Furthermore, by reducing hypoxia, antiangiogenic agents allow for optimization of radiotherapy, thereby providing the immune system with a rich supply of tumor antigens while modulating tumor-cell phenotype for enhanced T-cell killing (Fig. 4).
Fig. 4.

The combined use of radiation, immunotherapy, and angiogenesis inhibitors—by normalizing blood flow and lowering intratumoral pressure, angiogenesis inhibitors enable T cells to gain efficient access to the tumor, while increasing the necessary components for effective T-cell function at the tumor site. Furthermore, by reducing hypoxia, antiangiogenic agents allow for optimization of radiotherapy, thereby providing the immune system with a rich supply of tumor antigens while modulating tumor-cell phenotype for enhanced T-cell killing.
Taken together, the results of the preclinical and clinical studies described above offer a rationale for, and outline the potential advantages of, combining antiangiogenic agents with radiation and immunotherapy in the management of cancer patients. Each modality affects a different aspect of tumor biology and microenvironment, which can subsequently facilitate and enhance the action of the other two. Therefore, for certain cancer patients, the triplet of radiation therapy, immunotherapy, and angiogenesis inhibitors could represent the optimal prescription.
Before these three modalities can be successfully incorporated into clinical strategies, however, particular attention must be given to the design of clinical trials involving their concurrent administration. As previously described, the administration of antiangiogenic therapy with radiation and immunotherapy can potentially increase toxicity. Therefore, the timing, dose, and duration of these therapies are crucial to determining the potential efficacy of the combination. Choosing the optimal patient population poses another challenge, as some types of cancer have been shown to be relatively resistant to certain treatment modalities.
Despite these obstacles, the combination of antiangiogenic agents with radiotherapy and immunotherapy warrants further investigation. Future studies should focus on enhancing the antitumor responses induced by these three therapies, while limiting potential adverse effects of the combination.
References
- 1.National Cancer Institute/NIH Available from: http://www.cancer.gov/.
- 2.Radiation Therapy Oncology Group (RTOG). Phase III double-blind placebo-controlled trial of conventional concurrent chemoradiation and adjuvant temozolomide plus bevacizumab versus conventional concurrent chemoradiation and adjuvant temozolomide in patients with newly diagnosed glioblastoma Available from: http://www.rtog.org/members/protocols/0825/0825.pdf.
- 3.Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Borger ME, Antonini NF, Mol L, van Krieken JH, Dalesio O and Punt CJ, N. Engl. J. Med, 2009, 360, 563–572. [DOI] [PubMed] [Google Scholar]
- 4.Hecht JR, Mitchell E, Chidiac T, Scroggin C, Hagenstad C, Spigel D, Marshall J, Cohn A, McCollum D, Stella P, Deeter R, Shahin S and Amado RG, J. Clin. Oncol, 2009, 27, 672–680. [DOI] [PubMed] [Google Scholar]
- 5.Gehrmann M, Marienhagen J, Eichholtz-Wirth H, Fritz E, Ellwart J, Jaattela M, Zilch T and Multhoff G, Cell Death Differ, 2005, 12, 38–51. [DOI] [PubMed] [Google Scholar]
- 6.Binder RJ, Blachere NE and Srivastava PK, J. Biol. Chem, 2001, 276, 17163–17171. [DOI] [PubMed] [Google Scholar]
- 7.Singh-Jasuja H, Toes RE, Spee P, Munz C, Hilf N, Schoenberger SP, Ricciardi-Castagnoli P, Neefjes J, Rammensee HG, Arnold-Schild D and Schild H, J. Exp. Med, 2000, 191, 1965–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickman-Miller HD and Hildebrand WH, Trends Immunol, 2004, 25, 427–433. [DOI] [PubMed] [Google Scholar]
- 9.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L and Kroemer G, Nat. Med. (N. Y.), 2007, 13, 54–61. [DOI] [PubMed] [Google Scholar]
- 10.Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L and Kroemer G, Cell Death Differ, 2008, 15, 3–12. [DOI] [PubMed] [Google Scholar]
- 11.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F,Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer F and Zitvogel L, Nat. Med. (N. Y.), 2007, 13, 1050–1059. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi ME, Beltrame M and Paonessa G, Science, 1989, 243, 1056–1059. [DOI] [PubMed] [Google Scholar]
- 13.Gasser S, Orsulic S, Brown EJ and Raulet DH, Nature, 2005, 436, 1186–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C and Lowe SW, Nature, 2007, 445, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikitina EY and Gabrilovich DI, Int. J. Cancer, 2001, 94, 825–833. [DOI] [PubMed] [Google Scholar]
- 16.Teitz-Tennenbaum S, Li Q, Rynkiewicz S, Ito F, Davis MA, McGinn CJ and Chang AE, Cancer Res, 2003, 63, 8466–8475. [PubMed] [Google Scholar]
- 17.Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N,Liebes L and Formenti SC, Int. J. Radiat. Oncol. Biol. Phys, 2004, 58, 862–870. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Moran JP, Fenton BM, Koch CJ, Frelinger JG, Keng PC and Lord EM, Br. J. Cancer, 2000, 82, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lohr F, Hu K, Haroon Z, Samulski TV, Huang Q, Beaty J, Dewhirst MW and Li CY, Mol. Ther, 2000, 2, 195–203. [DOI] [PubMed] [Google Scholar]
- 20.Seetharam S, Staba MJ, Schumm LP, Schreiber K, Schreiber H, Kufe DW and Weichselbaum RR, Int. J. Oncol, 1999, 15, 769–773. [DOI] [PubMed] [Google Scholar]
- 21.Chiang CS, Syljuasen RG, Hong JH, Wallis A, Dougherty GJ and McBride WH, Cancer Res, 1997, 57, 3899–3903. [PubMed] [Google Scholar]
- 22.Yamini B, Yu X, Pytel P, Galanopoulos N, Rawlani V, Veerapong J, Bickenbach K and Weichselbaum RR, Clin. Cancer Res, 2007, 13, 6217–6223. [DOI] [PubMed] [Google Scholar]
- 23.Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J and Hodge JW, Cancer Res, 2004, 64, 7985–7994. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J and Hodge JW, Cancer Res, 2004, 64, 4328–4337. 25 [DOI] [PubMed] [Google Scholar]
- 25.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, Griekspoor A, Mesman E, Verreck FA, Spits H, Schlom J, van Veelen P and Neefjes JJ, J. Exp. Med, 2006, 203, 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crittenden M, Gough M, Seung S, Pang P, Thomas C and Hu H, American Society for Therapeutic Radiology and Oncology, Annual Meeting, 2008, p. 1113. [Google Scholar]
- 27.Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T, Weichselbaum RR and Fu YX, Blood, 2009, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakraborty M, Gelbard A, Carrasquillo JA, Yu S, Mamede M, Paik CH, Camphausen K, Schlom J and Hodge JW, Cancer Immunol. Immunother, 2008, 57, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grayson JM,Harrington LE, Lanier JG, Wherry EJ and Ahmed R, J. Immunol, 2002, 169, 3760–3770 [DOI] [PubMed] [Google Scholar]
- 30.Gulley JL, Arlen PM, Bastian A, Morin S, Marte J, Beetham P, Tsang KY, Yokokawa J, Hodge JW, Menard C, Camphausen K, Coleman CN, Sullivan F, Steinberg SM, Schlom J and Dahut W, Clin. Cancer Res, 2005, 11, 3353–3362. [DOI] [PubMed] [Google Scholar]
- 31.Hodge JW, Guha C, Neefjes J and Gulley JL, Oncology, 2008, 22, 1064–1070, discussion 1075, 1080–1061, 1084. [PMC free article] [PubMed] [Google Scholar]
- 32.National Institutes of Health Clinical Trials Database. Samarium Sm 153 Lexidronam Pentasodium with or without vaccine therapy in treating patients with prostate cancer and bone metastases Available from: http://www.clinicaltrials.gov/ct2/show/NCT00450619?term=NCT00450619&rank=1.
- 33.National Institutes of Health Clinical Trials Database. Vaccine therapy and radiation therapy in treating patients with carcino- embryonic antigen-positive solid tumors that have metastasized to the liver Available from: http://www.clinicaltrials.gov/ct2/show/NCT00085241?term=NCT00085241&rank=1.
- 34.Chi KH, Liu SJ, Li CP, Kuo HP, Wang YS, Chao Y and Hsieh SL, J. Immunother, 2005, 28, 129–135. [DOI] [PubMed] [Google Scholar]
- 35.Shinomiya N, Kuno Y, Yamamoto F, Fukasawa M, Okumura A, Uefuji M and Rokutanda M, Int. J. Radiat. Oncol., Biol., Phys, 2000, 47, 767–777. [DOI] [PubMed] [Google Scholar]
- 36.Topalian SL, Muul LM, Solomon D and Rosenberg SA, J. Immunol. Methods, 1987, 102, 127–141. [DOI] [PubMed] [Google Scholar]
- 37.Lange JR, Raubitschek AA, Pockaj BA, Spencer WF, Lotze MT, Topalian SL, Yang JC and Rosenberg SA, J. Immunother, 1992, 12, 265–271. [DOI] [PubMed] [Google Scholar]
- 38.Okawa T, Kita M, Arai T, Iida K, Dokiya T, Takegawa Y, Hirokawa Y, Yamazaki K and Hashimoto S, Cancer, 1989, 64, 1769–1776. [DOI] [PubMed] [Google Scholar]
- 39.National Institutes of Health Clinical Trials Database. A randomized, double-blind, phase 3 trial comparing ipilimumab vs. placebo following radiotherapy in subjects with castration resistant prostate cancer that have received prior treatment with docetaxel Available from: http://www.clinicaltrials.gov/ct2/show/NCT00861614?term=NCT00861614&rank=1.
- 40.Cuneo KC, Geng L, Fu A, Orton D, Hallahan DE and Chakravarthy AB, Int. J. Radiat. Oncol. Biol. Phys, 2008, 71, 873–879. [DOI] [PubMed] [Google Scholar]
- 41.Schueneman AJ, Himmelfarb E, Geng L, Tan J, Donnelly E, Mendel D, McMahon G and Hallahan DE, Cancer Res, 2003, 63, 4009–4016. [PubMed] [Google Scholar]
- 42.Zwolak P, Jasinski P, Terai K, Gallus NJ, Ericson ME, Clohisy DR and Dudek AZ, Eur. J. Cancer, 2008, 44, 2506–2517. [DOI] [PubMed] [Google Scholar]
- 43.Huang G and Chen L, Cancer Biother. Radiopharm, 2008, 23, 661–667. [DOI] [PubMed] [Google Scholar]
- 44.Hansen-Algenstaedt N, Stoll BR, Padera TP, Dolmans DE, Hicklin DJ, Fukumura D and Jain RK, Cancer Res, 2000, 60, 4556–4560. [PubMed] [Google Scholar]
- 45.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev L, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL and Jain RK, Cancer Cell, 2004, 6, 553–563. [DOI] [PubMed] [Google Scholar]
- 46.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY and Jain RK, Cancer Cell, 2007, 11, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu W, O’Reilly MS, Langley RR, Tsan RZ, Baker CH, Bekele N, Tang XM, Onn A, Fidler IJ and Herbst RS, Mol. Cancer Ther, 2007, 6, 2652–2663. [DOI] [PubMed] [Google Scholar]
- 48.Baker CH, Kedar D, McCarty MF, Tsan R, Weber KL, Bucana CD and Fidler IJ, Am. J. Pathol, 2002, 161, 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey CJ, Evans DB, Abbruzzese JL, Hicklin DJ and Radinsky R, Clin. Cancer Res, 2000, 6, 1936–1948. [PubMed] [Google Scholar]
- 50.Yokoi K, Thaker PH, Yazici S, Rebhun RR, Nam DH, He J, Kim SJ, Abbruzzese JL, Hamilton SR and Fidler IJ, Cancer Res, 2005, 65, 3716–3725. [DOI] [PubMed] [Google Scholar]
- 51.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK and Ang KK, N. Engl. J. Med, 2006, 354, 567–578. [DOI] [PubMed] [Google Scholar]
- 52.Jones MK, Wang H, Peskar BM, Levin E, Itani RM, Sarfeh LJ and Tarnawski AS, Nat. Med. (N. Y.), 1999, 5, 1418–1423. [DOI] [PubMed] [Google Scholar]
- 53.Liao Z, Komaki R, Milas L, Yuan C, Kies M, Chang JY, Jeter M, Guerrero T, Blumenschien G, Smith CM, Fossella F, Brown B and Cox JD, Clin. Cancer Res, 2005, 11, 3342–3348. [DOI] [PubMed] [Google Scholar]
- 54.Liu B, Barrett T, Choyke P, Maynard K, Wright J, Kummar S, Murgo A, Doroshow J and Gutierrez M, ASCO Meeting Abstracts, 2006, 24, 17119. [Google Scholar]
- 55.Socinski MA, Novello S, Brahmer JR, Rosell R, Sanchez JM, Belani CP, Govindan R, Atkins JN, Gillenwater HH, Pallares C, Tye L, Selaru P, Chao RC and Scagliotti GV, J. Clin. Oncol, 2008, 26, 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai A, Filka E, McGibbon B, Nghiemphu PL, Graham C, Yong WH, Mischel P, Liau LM, Bergsneider M, Pope W, Selch M and Cloughesy T, Int. J. Radiat. Oncol. Biol. Phys, 2008, 71, 1372–1380. [DOI] [PubMed] [Google Scholar]
- 57.Seiwert TY, Haraf DJ, Cohen EE, Stenson K, Witt ME, Dekker A, Kocherginsky M, Weichselbaum RR, Chen HX and Vokes EE, J. Clin. Oncol, 2008, 26, 1732–1741. [DOI] [PubMed] [Google Scholar]
- 58.Cohen MH, Gootenberg J, Keegan P and Pazdur R, Oncologist, 2007, 12, 713–718. [DOI] [PubMed] [Google Scholar]
- 59.Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT, Kozin SV, Petit L, Jain RK, Chung DC, Sahani DV, Kalva SP, Cohen KS, Scadden DT, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Shellito PC, Mino-Kenudson M and Lauwers GY, J. Clin. Oncol, 2005, 23, 8136–8139. [DOI] [PubMed] [Google Scholar]
- 60.Crane CH, Ellis LM, Abbruzzese JL, Amos C, Xiong HQ, Ho L, Evans DB, Tamm EP, Ng C, Pisters PW, Charnsangavej C, Delclos ME, O’Reilly M, Lee JE and Wolff RA, J. Clin. Oncol, 2006, 24, 1145–1151. [DOI] [PubMed] [Google Scholar]
- 61.Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, Chung DC, Sahani DV, Kalva SP, Kozin SV, Mino M, Cohen KS, Scadden DT, Hartford AC, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Chen HX, Shellito PC, Lauwers GY and Jain RK, Nat. Med. (N. Y.), 2004, 10, 145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fricke I and Gabrilovich DI, Immunol. Invest, 2006, 35, 459–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S and Carbone DP, Blood, 1998, 92, 4150–4166. [PubMed] [Google Scholar]
- 64.Gabrilovich DI and Nagaraj S, Nat. Rev. Immunol, 2009, 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kusmartsev S, Eruslanov E, Kubler H, Tseng T, Sakai Y, Su Z, Kaliberov S, Heiser A, Rosser C, Dahm P, Siemann D and Vieweg J, J. Immunol, 2008, 181, 346–353. [DOI] [PubMed] [Google Scholar]
- 66.Melani C, Sangaletti S, Barazzetta FM, Werb Z and Colombo MP, Cancer Res, 2007, 67, 11438–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY and Chen SH, Cancer Res, 2009, 69, 2514–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, Weinschenk T, Singh-Jasuja H and Brossart P, Blood, 2008, 111, 5610–5620. [DOI] [PubMed] [Google Scholar]
- 69.Alfaro C, Suarez N, Gonzalez A, Solano S, Erro L, Dubrot J, Palazon A, Hervas-Stubbs S, Gurpide A, Lopez-Picazo JM, Grande-Pulido E, Melero I and Perez-Gracia JL, Br. J. Cancer, 2009, 100, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R and Finke JH, Clin. Cancer Res, 2009, 15, 2148–2157. [DOI] [PubMed] [Google Scholar]
- 71.van Cruijsen H, van der Veldt AA, Vroling L, Oosterhoff D, Broxterman HJ, Scheper RJ, Giaccone G, Haanen JB, van den Eertwegh AJ, Boven E, Hoekman K and de Gruijl TD, Clin. Cancer Res, 2008, 14, 5884–5892. [DOI] [PubMed] [Google Scholar]
- 72.Ferrara N, Gerber HP and LeCouter J, Nat. Med. (N. Y.), 2003, 9, 669–676. [DOI] [PubMed] [Google Scholar]
- 73.Jain RK, Science, 2005, 307, 58–62. [DOI] [PubMed] [Google Scholar]
- 74.Jain RK, Duda DG, Clark JW and Loeffler JS, Nat. Clin. Pract. Oncol, 2006, 3, 24–40. [DOI] [PubMed] [Google Scholar]
- 75.Lee TH, Avraham HK, Jiang S and Avraham S, J. Biol. Chem, 2003, 278, 5277–5284. [DOI] [PubMed] [Google Scholar]
- 76.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ and Jain RK, Cancer Res, 2004, 64, 3731–3736. [DOI] [PubMed] [Google Scholar]
- 77.Lee CG, Heijn M, di Tomaso E, Griffon-Etienne G, Ancukiewicz M, Koike C, Park KR, Ferrara N, Jain RK, Suit HD and Boucher Y, Cancer Res, 2000, 60, 5565–5570. [PubMed] [Google Scholar]
- 78.Griffioen AW, Cancer Immunol. Immunother, 2008, 57, 1553–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, Hurwitz HI, Dev I, Nixon AB, Lyerly HK, Clay T and Morse MA, Cancer Immunol. Immunother, 2008, 57, 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP and Gabrilovich DI, Clin. Cancer Res, 2000, 6, 1755–1766. [PubMed] [Google Scholar]
- 81.Rini BI, Halabi S, Taylor J, Small EJ and Schilsky RL, Clin. Cancer Res, 2004, 10, 2584–2586. [DOI] [PubMed] [Google Scholar]
- 82.Pyrhonen S, Salminen E, Ruutu M, Lehtonen T, Nurmi M, Tammela T, Juusela H, Rintala E, Hietanen P and Kellokumpu-Lehtinen PL, J. Clin. Oncol, 1999, 17, 2859–2867. [DOI] [PubMed] [Google Scholar]
- 83.Motzer RJ, Bacik J, Murphy BA, Russo P and Mazumdar M, J. Clin. Oncol, 2002, 20, 289–296. [DOI] [PubMed] [Google Scholar]
- 84.Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, Lubensky I, Duan DR, Florence C, Pozzatti R, Walther MM, Bander NH, Grossman HB, Brauch H, Pomer S, Brooks JD, Isaacs WB, Lerman MI, Zbar B and Linehan WM, Nat. Genet, 1994, 7, 85–90. [DOI] [PubMed] [Google Scholar]
- 85.Shuin T, Kondo K, Torigoe S, Kishida T, Kubota Y, Hosaka M, Nagashima Y, Kitamura H, Latif F, Zbar B, Lerman MI and Yao M, Cancer Res, 1994, 54, 2852–2855. [PubMed] [Google Scholar]
- 86.Wiesener MS, Munchenhagen PM, Berger I, Morgan NV, Roigas J, Schwiertz A, Jurgensen JS, Gruber G, Maxwell PH, Loning SA, Frei U, Maher ER, Grone HJ and Eckardt KU, Cancer Res, 2001, 61, 5215–5222. [PubMed] [Google Scholar]
- 87.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR and Linehan WM, Proc. Natl. Acad. Sci. U. S. A, 1994, 91, 9700–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dvorak HF, J. Clin. Oncol, 2002, 20, 4368–4380. [DOI] [PubMed] [Google Scholar]
- 89.Ryan CW, Goldman BH, Lara PN Jr., Mack PC, Beer TM, Tangen CM, Lemmon D, Pan CX, Drabkin HA and Crawford ED, J. Clin. Oncol, 2007, 25, 3296–3301. [DOI] [PubMed] [Google Scholar]
- 90.Gollob JA, Rathmell WK, Richmond TM, Marino CB, Miller EK, Grigson G, Watkins C, Gu L, Peterson BL and Wright JJ, J. Clin. Oncol, 2007, 25, 3288–3295. [DOI] [PubMed] [Google Scholar]
- 91.Escudier B, Szczylik C, Hutson TE, Demkow T, Staehler M, Rolland E, Negrier S, Laferriere N, Scheuring UJ, Cella D, Shah S and Bukowski RM, J. Clin. Oncol, 2009, 27, 1280–1289. [DOI] [PubMed] [Google Scholar]
- 92.Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, Chevreau C, Filipek M, Melichar B, Bajetta E, Gorbunova V, Bay JO, Bodrogi I, Jagiello-Gruszfeld A and Moore N, Lancet, 2007, 370, 2103–2111. [DOI] [PubMed] [Google Scholar]
- 93.Rini BI, Weinberg V, Fong L, Conry S, Hershberg RM and Small EJ, Cancer, 2006, 107, 67–74. [DOI] [PubMed] [Google Scholar]