

Summary:
The discovery of the Autoimmune Regulator (AIRE) protein and the delineation of its critical contributions in the establishment of central immune tolerance has significantly expanded our understanding of the immunological mechanisms that protect from the development of autoimmune disease. The parallel identification and characterization of patient cohorts with the monogenic disorder Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED), which is typically caused by biallelic AIRE mutations, has underscored the critical contribution of AIRE in fungal immune surveillance at mucosal surfaces and in prevention of multiorgan autoimmunity in humans. In this review, we synthesize the current clinical, genetic, molecular and immunological knowledge derived from basic studies in Aire-deficient animals and from APECED patient cohorts. We also outline major advances and research endeavors that show promise for informing improved diagnostic and therapeutic approaches for patients with APECED.
Keywords: AIRE, APECED, APS-1, thymic medullary epithelial cells, central tolerance, immunomodulation
Introduction
Immunological recognition of self and non-self is of paramount importance to the body’s ability to harness the appropriate defense against invading pathogens while simultaneously avoiding a misdirected autoimmune response. The Autoimmune Regulator (AIRE) protein is a transcriptional regulator that plays a critical role in self-tolerance through orchestrating the promiscuous gene expression of tissue-specific antigens (TSAs) in a subset of medullary thymic epithelial cells (mTECs).1 Naïve thymocytes that recognize these TSAs with high affinity undergo apoptosis in a process referred to as negative selection.2–4 Dysfunction of this process allows self-reactive T cells to escape into the periphery resulting in autoimmune destruction of certain tissues.5 Consonant with the crucial role of AIRE in mediating central immune tolerance, loss-of-function mutations in the AIRE gene result in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED; Online Mendelian Inheritance in Man [OMIM] number, 240300), also known as autoimmune polyglandular syndrome type 1 (APS-1) or polyglandular autoimmune (PGA) syndrome type-1.
APECED/APS-1 is a rare monogenic disorder characterized by development of multiorgan autoimmunity that targets several endocrine and non-endocrine tissues, and susceptibility to a “signature” infectious disease, chronic mucocutaneous candidiasis (CMC), which manifests with chronic, recurrent and severe infections of the mucous membranes, skin and/or nails by the commensal yeast fungus Candida.6–9 Since the initial clinical descriptions of APECED/APS-1 in the medical literature in between 1929 and 194310–12 and the subsequent discovery of its genetic etiology via heroic positional cloning efforts by the Finnish-German Consortium and Dr. Shimizu’s research group in 199713,14, there have been significant advances in our understanding of this rare disease. In this review, we discuss the broad clinical implications of AIRE deficiency on autoimmunity, our understanding of the molecular and genetic mechanisms that play a role in the pathogenesis of APECED, and how these findings may guide therapeutic strategies based on the evaluation and treatment of ~120 APECED patients at the NIH Clinical Center in the past five years.
Prevalence
APECED exhibits its highest prevalence amongst certain historically isolated populations such as Persian Jews (1:9,000), Sardinians (1:14,000) and Finns (1:25,000)14–18, which display enrichment for corresponding founder “signature” AIRE mutations; indeed, the missense mutation Y85C, the nonsense mutation R139X, and the nonsense mutation R257X are commonly observed and are somewhat specific for these patient populations, respectively. The prevalence of APECED has also been estimated in other countries such as Slovenia (1:43,000), Norway (1:80,000) and Poland (1:129,000).19–21 The estimated prevalence of APECED in most other countries is thought to be in the range 1:80,000–200,000, although a systematic effort to accurately calculate country-specific prevalence rates is missing thus far.
Diagnosis of the Suspected APECED Patient
The diagnosis of APECED is established by the presence of any two clinical manifestations from the classical triad of CMC, hypoparathyroidism and adrenal insufficiency (Addison’s disease), although the diagnosis can be based on the development of only one of the classic triad manifestations in a child whose sibling already carries the diagnosis of APECED.6,22,23 Nonetheless, recent work from our group and others has underscored the much broader and deeper clinical phenotype of APECED patients compared to that previously recognized.7–9,18,24 Indeed, highlighting the vast phenotypic diversity of the disease, greater than 30 distinct manifestations can be seen, with more than 25 of those involving non-endocrine organs (Lionakis, unpublished data).8,9 Specifically, beyond the classical triad components of APECED, patients may also exhibit varying frequencies of urticarial eruption, enamel hypoplasia, intestinal malabsorption, autoimmune hepatitis, autoimmune pneumonitis, autoimmune gastritis, Sjogren’s-like syndrome, B12 deficiency, vitiligo, keratoconjuctivits, alopecia, type-1 diabetes, tubulointerstitial nephritis and asplenia, amongst others (Table 1, Fig. 1).7–9,18–20,25–30 Of interest, some of the above clinical manifestations, particularly those that target the skin, the lungs, the salivary glands, the stomach, the intestine and the liver appear to be significantly enriched among American APECED patients compared to their frequency reported in European patient cohorts. Whether this phenotypic discrepancy is explained by genetic, environmental, microbiome and/or other differences among the various patient cohorts or it is the result of the deeper systematic phenotyping that was recently performed in American APECED patients remains to be elucidated by future studies.
Table 1.
Prevalence (%) of Disease Manifestations in American APECED with Comparison International Cohorts
| Disease manifestations | American (n=35) |
Russian (n=112) |
Finnish (n=91) |
Norwegian (n=52) |
Italian (n=41) |
Irish (n=31) |
Sardinian (n=22) |
Iranian Jewish (n=23) |
Turkish (n=23) |
Indian (n=22) |
French (n=19) |
British (n=18) |
Polish (n=16) |
Sicilian (n=15) |
Slovakian (n=4) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Classical triad | |||||||||||||||
| CMC | 86 | 75 | 100 | 77 | 83 | 100 | 95 | 18 | 82.6 | 96 | 89 | 100 | 87.5 | 80 | 0 |
| Hypoparathyroidism | 91 | 78 | 72 | 73 | 93 | 84 | 77 | 96 | 86 | 91 | 63 | 89 | 100 | 100 | 50 |
| Adrenal insufficiency | 83 | 67 | 72 | 63 | 73 | 68 | 68 | 22 | 56.5 | 55 | 79 | 83 | 50 | 67 | 100 |
| Other endocrine | |||||||||||||||
| Hypothyroidism | 23 | 13 | 4 | 19 | 10 | NR | 0 | 4 | 21.7 | 4.5 | 5 | NR | NR | 13 | 50 |
| Type 1 diabetes | 11 | 9 | 16.5 | 8 | 2 | 10 | 4.5 | 4 | 8.7 | 4.5 | 5 | 28 | NR | 7 | 0 |
| GH deficiency | 17 | NR | 5.5 | NR | NR | 6 | 22 | NR | 4.3 | NR | 5 | 33 | 6.25 | NR | 0 |
| Ovarian failure | 38 | 48 | 25 | 33 | 36.5 | 61 | 61 | 38 | 8.6 | 60 | 71 | NR | 25 | 20 | 0 |
| Testicular failure | 21 | NR | 7.7 | 0 | 14.6 | NR | 0 | 33 | 13 | 0 | 0 | NR | NR | NR | 50 |
| Intra-abdominal | |||||||||||||||
| Intestinal dysfunction | 80* | 25 | 5 | 23 | 15 | 32 | 50 | NR | 8.7 | 27 | 26 | NR | 12.5 | 33 | NR |
| Autoimmune hepatitis | 43* | 11 | 17.5 | 4 | 20 | 19 | 27 | NR | 13 | 9 | 11 | 11 | 37.5 | 20 | NR |
| Autoimmune gastritis | 49* | NR | 19 | NR | 15 | NR | 32 | 9 | 8.7 | NR | 31.5 | NR | NR | 20 | NR |
| B12 deficiency | 29 | 8 | 31 | 15 | 15 | 10 | 32 | 9 | 17 | 21 | 21 | 22 | 6.25 | 20 | NR |
| Interstitial nephritis | 6 | NR | 9 | 3.8 | NR | NR | NR | NR | NR | 0 | NR | NR | NR | NR | NR |
| Asplenia | 9 | 2 | 19 | 16 | 11 | NR | 4.5 | NR | 0 | 4.5 | 0.5 | NR | NR | NR | NR |
| Skin/Nail/Dental | |||||||||||||||
| Urticarial eruption | 66* | NR | 14.3 | NR | NR | NR | NR | NR | 4.3 | 9 | NR | NR | NR | NR | NR |
| Alopecia | 17 | 34 | 35 | 31 | 37 | 19 | 45 | 13 | 39 | 27 | 53 | 22 | 18.75 | 14 | 25 |
| Vitiligo | 37 | 9 | 2 | 15 | 15 | NR | 22.7 | NR | 26 | 27 | 21 | NR | NR | 20 | NR |
| Nail dystrophy | 17 | NR | 84 | 13 | NR | NR | 55 | NR | 13 | 22 | NR | NR | NR | 27 | NR |
| Enamel hypoplasia | 86 | 28 | 77 | 72 | NR | NR | 45 | NR | 4.3 | 61 | 0 | NR | NR | 20 | 25 |
| Other | |||||||||||||||
| Pneumonitis | 40* | NR | 1 | NR | NR | NR | NR | NR | 0 | 4.5 | NR | NR | NR | 7 | NR |
| Sjogren’s-like syndrome | 43* | NR | 11 | NR | 12 | NR | NR | NR | 4.3 | 9 | NR | NR | NR | NR | NR |
| Keratoconjunctivitis | 29 | NR | 9 | 12 | 12 | NR | 36 | 0 | 4.3 | 9 | 31.5 | 11 | 25 | 13 | NR |
| Early onset HTN | 17 | NR | 15 | NR | NR | NR | 9 | NR | NR | 0 | NR | NR | NR | NR | NR |
Shown are approximate percentages of corresponding APECED manifestations pooled from various published studies comparing data from the American8 or other international cohorts 9, 15, 18, 20, 24, 27–32, 40, 146, 147
Disease manifestations that appear more prevalent in American APECED patients relative to reported frequencies in European APECED patients.
CMC, chronic mucocutaneus candidiasis; GH, growth hormone; HTN, hypertension; NR, not reported.
Fig. 1. Diagram depicting organ systems affected by APECED.
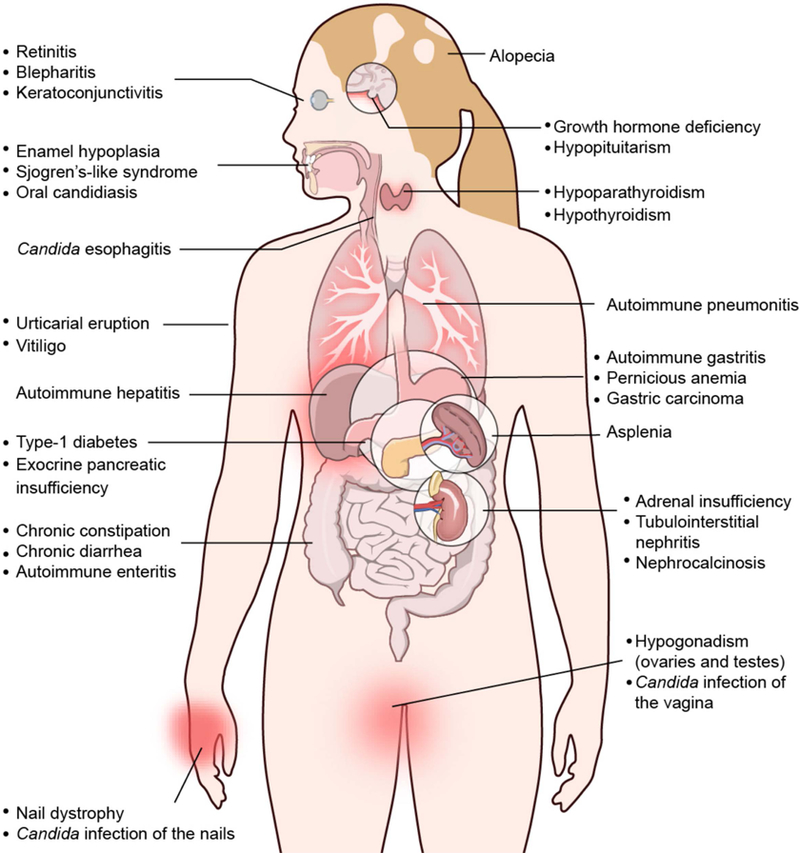
The presented organ-specific autoimmune manifestations affect APECED patients with varying frequencies (Table 1).
Unfortunately, the vast majority of APECED patients will develop several other disease components before reaching a classic diagnostic dyad, which would then raise suspicion for APECED. On average, patients suffer from more than five manifestations and up to 20 conditions have been reported per individual.7,8,31 Given the heterogenous presentation, the disease often goes unrecognized leading to an unfortunate delay in the diagnosis. Within an American cohort that we recently evaluated and reported at the National Institutes of Health, the mean age of achieving a classic diagnostic dyad of APECED was over seven years of age.8 Strikingly, only 20% of those patients developed their first two consecutive disease manifestations among the classic triad of manifestations. Instead, 80% of the patients within our cohort developed a median of three non-triad manifestations before developing a classic diagnostic dyad, which led to prolonged periods of diagnostic uncertainty and development of additional (often irreversible) autoimmune sequelae before a diagnostic dyad was reached and the APECED diagnosis was suspected and eventually established.
Among the early common manifestations that affected children in North and South America before the development of a classic diagnostic dyad, three manifestations were particularly prominent: urticarial eruption, enamel hypoplasia, and intestinal malabsorption. Less often, autoimmune pneumonitis, autoimmune hepatitis or keratoconjuctivitis were noted.8 The urticarial eruption appeared as frequent as CMC as the first manifestation in the American APECED cohort; it is typically a non-pruritic maculopapular rash in the extremities and the torso (Fig. 2) that is not clearly associated with viral illness, vaccination or other inciting event, and is only infrequently accompanied by fever. The eruption is typically self-limited lasting days or weeks, and recurrences are frequent within the first 2–3 years of life. Histologically, the eruption features a characteristic picture of mixed myeloid and lymphoid cell infiltration, occasionally extending to the fat to cause panniculitis, but without vasculitis typically.
Fig. 2. Representative photographs of urticarial eruption in young children with APECED.
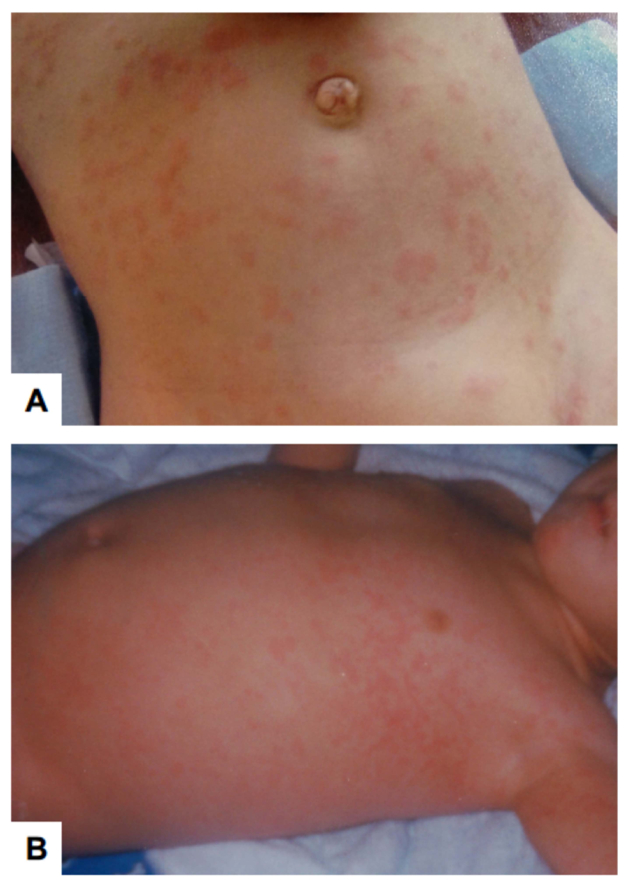
These images are derived from Ferre et al. (ref #8).
Importantly, recognition of these additional manifestations that appear early in the course of the disease may help to reduce diagnostic delay and allow for prompt institution of therapy (see below). Based on the above findings, we proposed expanded diagnostic criteria for American APECED patients by incorporation of the adjunct diagnostic triad of urticarial eruption, enamel hypoplasia and intestinal malabsorption to the classic triad criteria (Fig. 3). Upon such incorporation, a diagnostic dyad within these expanded diagnostic criteria would have been reached with a mean of four years earlier compared to reaching a diagnostic dyad within the classic diagnostic criteria. This earlier diagnosis would have been reached before development of life-threatening Addisonian crisis or hypocalcemic seizures in approximately half of the affected patients.
Fig. 3. Proposed diagnostic algorithm for capturing an earlier diagnosis in children with APECED.
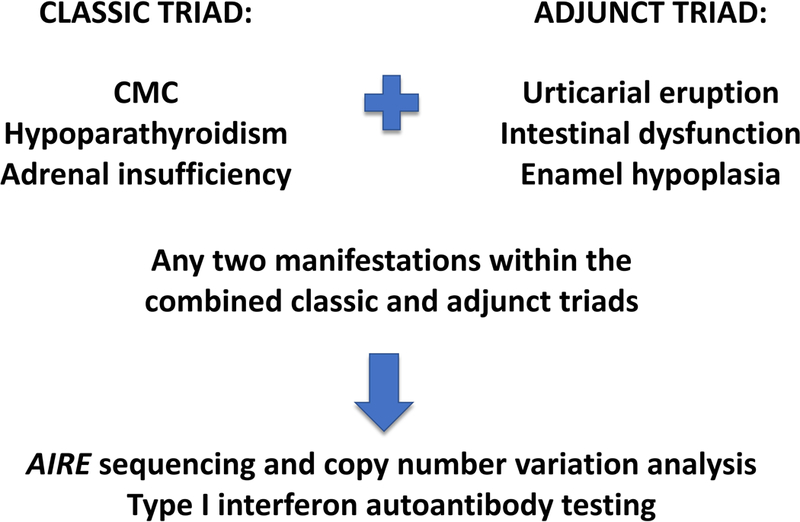
Illustration of the combined classic diagnostic triad and adjunt triad comprised of urticarial eruption, intesntional dysfunction and enamel hypoplasia, which may lead to earlier time to diagnosis in children with APECED via triggering genetic testing for AIRE mutations and deletions/duplications and testing for type I interferon autoantibodies.
We have now validated these expanded diagnostic criteria in other independent patient datasets enrolled consecutively in the National Institutes of Health natural history study from both North/South America and European countries. In these independent datasets, the expanded diagnostic criteria would have also fostered a much earlier diagnosis in both American and European APECED patients and would have assisted in reaching the APECED diagnosis before development of life-threatening endocrine complications in a significant proportion of the patients (Ferre et al., manuscript in preparation). In agreement with the potential clinical utility of these expanded diagnostic criteria, Fierabracci et al.32 recently retrospectively analyzed previously reported APECED cohorts from Finland, Sardinia and Turkey and showed that the application of these expanded diagnostic criteria would have also allowed for earlier recognition and diagnosis in these APECED patients. Collectively, these findings indicate that children with APECED are as likely or even more likely to be seen early in the course of their disease by dermatologists, allergists, dentists, hepatologists, pulmonologists, ophthalmologists and/or gastroenterologists than by endocrinologists. Therefore, the presence of any of these manifestations within the adjunct criteria even in the absence of endocrinopathies in a young child should alert physicians to consider the diagnosis of APECED.
Clinical suspicion for APECED should prompt a) genetic testing for AIRE mutations and/or deletions and b) screening for neutralizing autoantibodies (AAbs) against type I interferons such as interferon-omega (IFN-ω) and interferon-alpha (IFN-α) (Fig. 3). Regarding genetic testing, not all patients with clinical APECED are found to have biallelic AIRE mutations by Sanger sequencing. Some patients have large AIRE deletions, which require copy number variation analysis for establishing the genetic diagnosis, and some other patients have heterozygous AIRE mutations or have wild-type AIRE genotype, indicating that non-coding AIRE variants or non-AIRE coding variation may result in APECED in some patients (see below) (Fig. 3). AAbs against IFN-ω are highly sensitive and specific for APECED (>95%), even as early as the first few months of life before the appearance of the clinical manifestations and the other organ-specific AAbs (see below).33–35 These type I interferon AAbs can also be seen in patients with secondary AIRE deficiency such as those with thymoma36,37 or hypomorphic RAG mutations that present with immune dysregulation, particularly those with late-onset combined immune deficiency and granulomatous/autoimmune manifestations that present with severe viral infections38, and in some patients with late-onset myasthenia gravis.39 The later onset and differential clinical, genetic, laboratory and radiographic features of thymoma, RAG deficiency and myasthenia gravis make them discernible from APECED. Determination of type I interferon AAbs is not commercially available in the US, but the authors of this review can be contacted by physicians to perform the test from serum or plasma in their patients with suspected APECED.
Earlier diagnosis is critical for a number of reasons. First, several of the autoimmune manifestations in APECED are life-threatening including adrenal crisis, hypocalcemic seizures, fulminant hepatitis, autoimmune lung disease and pneumococcal sepsis, which collectively account for the increased mortality in these patients.7,9,18,40 Therefore, earlier diagnosis may allow for earlier screening and recognition of the aforementioned clinical complications. Second, earlier diagnosis may allow for the institution of prophylactic immunomodulation in children with APECED, which may result in prevention of the development of the full-blown syndrome and its irreversible autoimmune sequelae. To that end, a prospective clinical trial aiming at instituting prophylactic immunomodulation in children with APECED very early in the course of their disease is currently under development at the National Institutes of Health.
Clinical Manifestations of APECED Patients
APECED patients present with a characteristic combination of a) a single consistent infectious disease manifestation, CMC, and b) autoimmunity that affects several endocrine and non-endocrine tissues. There is significant patient to patient variability in the phenotypic expression of the syndrome and no clear correlation exists between the presence of a specific AIRE mutation or deletion and the individual patient’s clinical phenotype, even among affected siblings within the same family (Table 1, Fig. 1). This indicates that there are complex interactions between genetic, epigenetic, and environmental factors that likely contribute to the development of organ-specific APECED manifestations in different patients. Most patients with APECED begin having symptoms in early childhood, with CMC and urticarial eruption being the most common initial manifestations of affected patients8, although as noted earlier, the time between the onset of symptoms and the diagnosis of APECED is often long.
Infectious manifestations
Candida is a commensal yeast fungus that colonizes the oral and gastrointestinal mucosal surfaces, the skin and/or the female lower genital tract in up to 60–70% of healthy individuals. When perturbations in host immune responses and/or mucosal microbiome occur, Candida becomes an opportunistic pathogen and may cause mucosal or invasive infections.41 Patients with APECED are at risk for mucosal Candida infections, which typically develop within the first 1–3 years of life, most often in the form of oral and/or esophageal candidiasis. CMC is the “signature” infection in >80% of all reported APECED patients associated with the presence of neutralizing AAbs against Th17 cytokines (see below), with the exception of the Persian Jew APECED population that carries the missense Y85C AIRE mutation and was reported to infrequently (<20%) develop CMC (Fig. 1).15 The reasons for this cohort-specific variation in the prevalence of CMC remain unclear. Future studies are needed to examine whether Th17 AAbs are less prevalent within this patient population. Of note, chronic Candida infections of the oral and esophageal mucosal surfaces can lead to the development of oral or esophageal squamous cell carcinomas in ~5% of adult patients with long-standing mucosal candidiasis.42,43 Carcinoma development in the context of monogenic disorders that manifest with CMC is not restricted to APECED but can also be seen in patients with STAT1 gain-of-function mutations.44,45 The mechanisms for this susceptibility to neoplasm development remain unclear, but may relate to enhanced EGFR signaling triggered by autoreactive CD4+ T cells in the epithelial barrier.46 Candidiasis may also affect the intestinal or vaginal mucosal surfaces or the nails in APECED patients, albeit with much lower frequencies compared to the oral and esophageal mucosa, while involvement of the skin is very uncommon (Lionakis, unpublished observations). As opposed to the susceptibility to mucosal candidiasis, patients with APECED are not at heightened risk of developing invasive Candida infections, which depend on myeloid phagocytes for effective host defense (see below).47–49
Besides CMC, patients with APECED are not consistently susceptible to other infectious complications. A small proportion of patients (~10–15%) develop asplenia, which is not congenital but is most often acquired during childhood (Lionakis, unpublished observations), and as a result of that, they are at risk for development of invasive infections by encapsulated bacteria, such as Streptococcus pneumoniae, Neisseria meningitidis and Capnocytophaga canimorsus, including overwhelming sepsis. Those with asplenia exhibit Howell-Jolly and Pappenheimer bodies in their peripheral blood smear, have an atrophic or absent spleen on abdominal imaging, and require long-term prophylactic antibiotic therapy and vaccinations in order to prevent the development of life-threatening infections by these bacteria (see below).
A small proportion of APECED patients develop recurrent infections by herpetic viruses such as HSV and VZV (~15%, Lionakis, unpublished observations), without an obvious correlation between the titers or neutralizing potential of type I interferon AAbs or the levels of STAT1.50,51 APECED patients have also sporadically been reported to develop Giardia intestinal infections, although the mechanisms for this susceptibility remain elusive.52
Endocrine autoimmune manifestations
Endocrine autoimmune disease is the hallmark of APECED syndrome, as the acronym indicates. Hypoparathyroidism, which typically develops earlier than any other endocrinopathy, and adrenal insufficiency are the most common endocrine manifestations. Additional endocrinopathies that develop with lower frequencies include hypothyroidism, primary ovarian or testicular failure, growth hormone deficiency and hypopituitarism. Some of these endocrine manifestations are associated with the presence of corresponding organ-specific AAbs, although these AAbs do not always predict the development of the corresponding endocrinopathy at the individual patient level (see below; Table 2). Of interest, type I diabetes is an uncommon complication of patients with APECED, which typically occurs later in adolescence or adulthood in ~5–10% of patients.
Table 2.
Autoantigens in APECED patients and their associated disease manifestation.
| Autoantigen | Tissue of origin | Associated clinical manifestation |
|---|---|---|
| IFN-ω | Cytokine | APECED, Thymoma, Myasthenia gravis, RAG deficiency |
| IFN-α | Cytokine | APECED (protection from type 1 diabetes?), Thymoma, Myasthenia gravis, RAG deficiency |
| IFN-β | Cytokine | Unclear |
| IFN-λ | Cytokine | Unclear |
| IL-6 | Cytokine | Unclear |
| IL-17A | Cytokine | Chronic mucocutaneous candidiasis |
| IL-17F | Cytokine | Chronic mucocutaneous candidiasis |
| IL-22 | Cytokine | Chronic mucocutaneous candidiasis |
| NACHT leucine-rich-repeat protein 5 (NALP5) |
Parathyroid, ovary, breast, testis | Hypoparathyroidism |
| Calcium-sensing receptor (CaSR) | Parathyroid, pancreas, kidney | Hypoparathyroidism |
| 21-hydroxylase (CYP21A2) | Adrenal gland | Primary adrenal insufficiency |
| 17-α-hydroxylase (CYP17A1) | Adrenal gland | Primary adrenal insufficiency, ovarian failure |
| Thyroglobulin (TG) | Thyroid | Hypothyroidism |
| Thyroid peroxidase (TPO) | Thyroid | Hypothyroidism |
| Islet antigen-2 (IA-2) | Pancreas | Type I diabetes |
| Potassium channel-regulating protein (KCNRG) |
Lung, cervix | Autoimmune pneumonitis |
| Bactericidal/permeability-increasing fold- containing B1 (BPIFB1) |
Lung, stomach, esophagus, cervix | Autoimmune pneumonitis |
| Aromatic L-amino acid decarboxylase (AADC) |
Kidney, intestine, brain, liver, pancreas |
Autoimmune hepatitis, vitiligo |
| Cytochrome P450 1A2 (CYP1A2) | Liver | Autoimmune hepatitis |
| CYP2A6 | Liver | Autoimmune hepatitis |
| Tryptophan hydroxylase (TPH) | Multiple | Intestinal dysfunction, autoimmune hepatitis, autoimmune gastritis |
| Hydrogen-Potassium ATPase | Stomach | Autoimmune gastritis |
| Intrinsic factor | Stomach | Pernicious anemia |
| Glutamic acid decarboxylase (GAD65) | Pancreas, brain | Intestinal dysfunction, vitiligo |
| Histidine decarboxylase (HDC) | Brain, stomach, lung | Intestinal dysfunction |
| Defensin, alpha 5 (DEFA5) | Paneth cells | Intestinal dysfunction |
| Side chain cleavage enzyme (CYP11A1) | Adrenal, ovary, testis | Primary adrenal insufficiency, ovarian failure, testicular failure |
| Transglutaminase 4 | Prostate | Prostatitis (male infertility) |
| Testis-specific gene 10 protein (TSGA10) | Testis, brain | Unclear |
| Tudor domain containing protein 6 (TDRD6) | Testis, brain | Hypopituitarism (growth hormone deficiency) |
| Melanoma antigen family B 2 (MAGEB2) | Testis, female germ cells | Unclear |
| Protein disulfide isomerase-like testis (PDILT) |
Testis | Unclear |
| Tyrosine hydroxylase (TH) | Brain, adrenal | Alopecia, enamel hypoplasia |
| SOX9/SOX10 | Nervous system, breast | Vitiligo |
| Myelin protein 0 | Nervous system | Neuropathy |
| Interphotoreceptor retinoid‐binding protein | Retina | Retinitis pigmentosa |
Non-endocrine autoimmune manifestations
As mentioned earlier, non-endocrine autoimmune manifestations have recently been recognized as common disease components in certain cohorts of patients with APECED. Urticarial eruption, as mentioned above, is a characteristic early common feature in American APECED patients (Fig. 2). Inflammation in the salivary and lacrimal glands (Sjogren’s-like syndrome) and ocular inflammation in the forms of keratoconjuctivitis or, less often, retinitis, can occur, and the latter can lead to visual loss if left untreated (see below). Among the non-endocrine disease components, the pulmonary and intra-abdominal autoimmune manifestations are worthwhile expanding on further.
Although autoimmune pneumonitis had been reported in only ~2% of all reported APECED patients in the literature, we have recently identified lung autoimmunity in ~40–45% of American APECED patients (Ferre et al., submitted manuscript).8 Pneumonitis in APECED presents clinically with chronic cough and shortness of breath, and is most often misdiagnosed as asthma or recurrent bronchitis. Without immunosuppressive treatment (see below), repeated bouts of inflammation in the airways, the bronchioles and the lung parenchyma result in the development of bronchiectasis leading to recurrent bacterial respiratory infections and eventual hypoxemic respiratory failure. AAbs against BPIFB1 (BPI fold containing family B member 1) and the potassium channel KCNRG appear highly specific for lung autoimmunity in APECED patients (Table 2), but their sensitivity is ~65% and ~30%, respectively. This indicates that other yet-unknown lung autoantigens are the target of autoimmune attack in some patients with APECED pneumonitis. Therefore, because currently known AAbs do not capture all patients with pneumonitis, the best available screening modality for pneumonitis in APECED patients is computed tomography of the chest, which reveals ground-glass opacities and/or bronchiectasis, and also captures the ~5–10% of patients with early lung disease that are as-yet asymptomatic. Immunological analyses in the airways and lung parenchyma of Aire−/− mice and APECED pneumonitis patients have revealed characteristic compartmentalized immunopathology with accumulation of activated neutrophils in the airways and T > B lymphocyte infiltration in intraepithelial, submucosal, interstitial and peribronchiolar/bronchiolar tissue. B cell aggregates are commonly seen deeper in the lung parenchyma (Ferre et al., submitted manuscript). Collectively, autoimmune pneumonitis is a common, early, severe, and often misdiagnosed manifestation of APECED patients, which features a characteristic regional immunopathology that has implications for immunomodulatory treatment (see below).
In addition, there are several intra-abdominal manifestations seen in APECED patients. Autoimmune gastritis features lymphoplasmacytic infiltration in the stomach tissue and can present with abdominal pain, nausea or vomiting. It may be accompanied by pernicious anemia caused by destruction of gastric cells that produce intrinsic factor, which is essential for absorption of vitamin B12 in the gut. Autoimmune gastritis, especially in the presence of intestinal metaplasia, and B12 deficiency both increase the risk for development of gastric adenocarcinoma in APECED patients. The most common intra-abdominal manifestation of APECED patients appears to be intestinal malabsorption of fat that manifests with chronic diarrhea, chronic constipation or an alternating pattern of both. The etiology of fat malabsorption is multifactorial and includes exocrine pancreatic insufficiency, autoimmune enteritis, and/or intestinal dysbiosis (Natarajan et al., manuscript in preparation). Strikingly, autoimmune involvement of the colon is extremely uncommon in APECED patients, with frequency <2–3% (Lionakis, unpublished observations), highlighting the differential role of AIRE in protecting from autoimmune attack the different segments of the gastrointestinal tract.
Autoimmune hepatitis presents with transaminitis, but often is not accompanied by AAbs commonly seen in autoimmune hepatitis in non-APECED patients such as antinuclear antibodies (ANA), anti-mitochondrial antibodies (AMA), anti-smooth muscle antibodies (ASMA), or antibodies against the liver and kidney microsomal antigens (LKM-1) or soluble liver antigens (SLA). Some, but not all, patients with APECED-associated autoimmune hepatitis feature AAbs against cytochrome P450 1A2 (CYP1A2), histidine decarboxylase (HDC) or aromatic L-amino acid decarboxylase (AADC) (Chascsa et al., submitted manuscript).53,54 This indicates that the pathogenesis of APECED-associated autoimmune hepatitis may be distinct from non-APECED autoimmune liver disease, and the full spectrum of liver autoantigens that are targeted by AIRE-deficient lymphocytes in APECED patients remains to be determined. Therefore, transaminitis in APECED patients warrants a liver biopsy and initiation of immunosuppressive treatment (see below).
A minority of APECED patients (<10%) develop autoimmunity in the kidneys that manifests with tubulointerstitial nephritis, with sparing of the glomeruli. The presence of autoimmune renal disease is critical to recognize in affected patients and to differentiate from nephrocalcinosis-induced kidney injury, which is more commonly seen and results from chronic oral and intravenous calcium supplementation for hypoparathyroidism, because it can lead to progressive renal failure.55 Indeed, kidney transplantation has been reported in APECED patients with tubulointerstitial nephritis, with recurrence of tubulointerstitial nephritis and renal failure without effective immunomodulatory therapy post-transplantation (see below).56
Interestingly, APECED patients rarely develop certain other autoimmune conditions such as multiple sclerosis, glomerulonephritis, lupus, or rheumatoid arthritis. The underlying protective factors underpinning this finding are not yet fully understood, as some of the peripheral tissue antigens responsible for organ-specific autoimmunity are expressed in mTECs in an Aire-dependent manner. For example, myelin proteolipid protein (PLP), but not myelin oligodendrocyte glycopeptide peptide (MOG), which are autoantigens associated with multiple sclerosis depend differentially on Aire for expression in mTECs and protection from experimental autoimmune encephalomyelitis in mice; yet, APECED patients do not develop multiple sclerosis.57 The immunological shadow of peripheral tissue antigens that Aire projects in the thymus is limited.58 It is likely that other transcription factors expressed in the thymus, such as Fezf2 or others, may guide the expression of a complementary array of tissue specific antigens and may confer tolerance to specific tissues. Indeed, studies using Foxn1-Cre+Fezf2fl/fl mice revealed a different phenotype than Aire−/− models with inflammatory infiltration of the lung, liver, kidney, stomach, small intestine, salivary gland, testis, and brain, some of which (i.e., kidney, brain) are not seen in Aire-deficient mice.59
Ectodermal dystrophy manifestations
Several manifestations of APECED patients have been attributed to ectodermal dystrophy, as the acronym implies. The most common such manifestation is enamel hypoplasia, which manifests early in the course of the disease and its recognition by pediatric dentists should trigger sequencing of the AIRE gene and testing for type I interferon AAbs, especially if accompanied by oral candidiasis or hypoparathyroidism. Other APECED-associated ectodermal dystrophy manifestations include vitiligo, alopecia, and nail dystrophy in the absence of fungal nail disease. The pathogenesis of these manifestations is poorly defined.
AIRE Structure
The AIRE gene is located in the long arm of chromosome 21 at position 22.3 (21q22.3), it contains 14 exons, and it encodes a 545 amino acid protein of 57,727 Da. AIRE is composed of four subunit domains, which are also found in other chromatin-associated proteins including the HSR/CARD (Caspase recruitment) domain in the N-terminal region of the protein, the SAND (Sp100, AIRE, nuclear phosphoprotein 41/75 or NucP41/75 region, and deformed epidermal autoregulatory factor 1 or DEAF1) domain, and two PHD (plant-homeodomain) zinc fingers (PHD-1, PHD-2) in the C-terminal region of the protein (Fig. 4).60,61 The CARD domain is important for the specked nuclear localization of AIRE and the formation of AIRE homo‐dimers and tetramers, which are essential to the functional viability of the AIRE protein.62,63 The SAND domain of several family members binds DNA, but the AIRE SAND domain differs from other SAND domains as it is missing a critical DNA binding motif (KDWK). Indeed, it was recently discovered that AIRE SAND interacts with the ATF7ip (activating transcription factor 7-interacting protein 1)-MBD1 (methyl CpG binding protein 1) complex potentially in concert with PHD-1 to target TSA genes.64 The two PHD zinc fingers suggest that AIRE regulates transcription epigenetically through interaction with unmethylated lysine 4 on histone H3 (H3K4me0).65 Hence, PHD-1 is critical for the transcription-transactivation activity of AIRE and was shown to be essential for preventing autoimmunity in transgenic mouse models.66–68 In addition to these four subunits, there are two NLS (nuclear localization signal) regions implicated in nuclear transport as well as four LXXLL motifs, which mediate interactions with other LXXLL‐containing proteins and may play a role in the homo‐oligomerization of AIRE.61
Fig. 4. Structure of the Autoimmune regulator (AIRE) gene and major mutations.
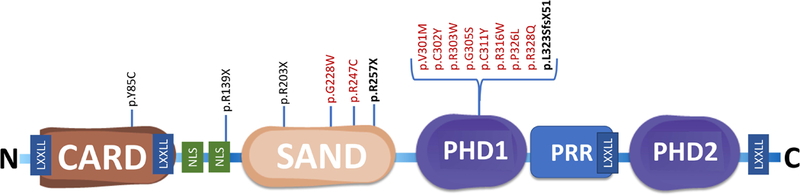
Comprised of four subunits including Caspase recruitment domain (CARD), Sp100, AIRE, nuclear phosphoprotein 41/75 or NucP41/75 region, and deformed epidermal autoregulatory factor 1 or Deaf1 domain (SAND) and two plant-homeodomain (PHD) zinc fingers. A proline rich region (PRR), two nuclear localization signal (NLS) regions and four LXXLL motifs are also displayed. Founder mutations p.Y85C (Persian Jews); p.R139X (Sardinians); p.R257X (Finns); and p.R203X (Sicilians) are displayed. The two most common mutations in our cohort R257X and 13bp mutation, p.L323SfsX51 are shown in bold. Biallelic (homozygous or compound heterozygous) mutations resulting in classical APECED (black). Dominant negative mutations in the SAND or PHD1 domains resulting in APECED-like organ-specific autoimmune manifestations (red). Select icons adapted from Reactome library (ref #148).
AIRE Expression and Function
mTECs and CD4+ T cells
Shortly following the discovery that biallelic mutations in the AIRE gene result in APECED in 199713,14, Anderson and colleagues revealed in 2002 that Aire is predominately expressed within a subset of epithelial cells of the medullary thymus58, although Aire expression has now been shown in extrathymic Aire-expressing cells (eTACs), and B cell and dendritic cell populations (see below).69–72 These specialized cells, referred to as mTECs, facilitate ectopic expression of thousands of self-peptides where they are presented to developing thymocytes (T cells) via major histocompatibility complex (MHC) antigens.61,73,74 T cells that bind to these self-peptide-antigens exceeding an appropriate affinity threshold undergo clonal deletion, thereby preventing their release into circulation.4 In the absence of AIRE this process is defective, self-peptides are not expressed, allowing auto-reactive T cells to escape in the periphery and resulting in lymphocytic tissue infiltration and autoimmunity (Fig. 5).75 This has been illustrated via adoptive transfer studies of Aire-deficient CD4+ T cells into a immunodeficient host, which is sufficient to recapitulate the autoimmune disease seen in Aire-deficient mice. Furthermore, depletion of the CD4+ T cell subset in Aire-deficient mice led to amelioration of the disease process.76 This immunological shadow projected by Aire has been well illustrated through generation of Aire−/− mice in which the Aire-deficient thymic stroma was shown to be critical and sufficient for induction of autoimmunity and development of autoantibodies and lymphocytic infiltration involving specific organs.58,77 Though broadly involved in central tolerance, the immunological shadow that AIRE projects in the thymus is critical to the development of autoimmunity in specific organs as evidenced by disease in patients as well as murine models.58 However, not all TSAs in the thymus require Aire for expression and induction of central immune tolerance. The transcription factor, Fezf2, which is regulated by the LTβR pathway but not the CD40/RANKL-Aire axis, was recently uncovered by Takaba et al.59 to potentially regulate TSAs in mTECs in an Aire-independent manner. Specifically, Fezf2-deficient animals developed organ-specific autoantibodies and lymphocytic infiltration that were distinct from those seen in Aire-deficient mice. Based on these findings, it will be an important direction of future research to determine whether there are patients with loss-of-function FEZF2 mutations who may present with monogenic organ-specific autoimmunity.
Fig. 5. Mechanisms of Aire-dependent induction of central immune tolerance in the thymus.
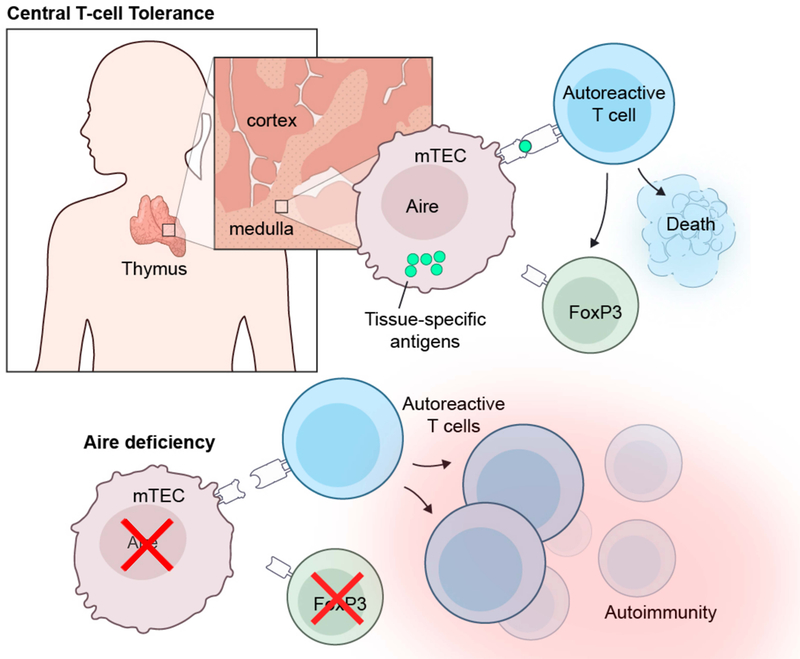
Aire is expressed in a subset of medullary thymic epithelial cells (mTECs) where it is critical for the expression and surface display of peripheral tissue self-antigens (TSAs). Self-reactive T cells with high affinity for TSAs either undergo deletion via apoptosis or become regulatory T cells. In the absence of functional Aire, the expression and surface display of TSAs on mTECs is impaired, which results in the escape of self-reactive T cells in the periphery where they infiltrate tissues and cause the autoimmune manifestations observed in APECED patients.
In addition to AIRE’s role in negative selection of self-reactive effector T cells, AIRE has also been implicated in the positive selection of thymic FoxP3+ regulatory T cells (Tregs) and a recent study demonstrated that a defect in the neonatal output of thymic Tregs (tTregs) in Aire−/− mice drives organ-specific autoimmunity.78–82 In the presence of functional AIRE, T cell clones that recognize self-antigens with an intermediate affinity are positively selected and differentiate into the T regulatory subset. These specialized cells demonstrate immunosuppressive effects to halt autoreactive effector T cells in the periphery and have been shown to be important in maintenance of self-tolerance.80 Which of these Aire-dependent functions is the predominant mechanism of tolerance responsible for disease development and/or whether these mechanisms operate in concert remains to be fully elucidated. Current evidence would suggest that the negative selection of effector T cells may play a more crucial role in pathogenesis over the positive selection of Tregs. For example, through performing double Aire-sufficient/Aire-deficient thymus transplant experiments in nude mice, Tregs derived from a functional Aire thymus were not sufficient to prevent autoimmunity.3 In fact, these mouse data would suggest that transplantation of allogeneic cultured thymus alone without the concurrent surgical removal of endogenous AIRE−/− thymic tissue and the elimination of peripheral AIRE−/− lymphocytes is unlikely to be beneficial in APECED patients. In addition, genetic crossing of Aire-deficient and Foxp3-deficient mice to generate Aire-Foxp3 double-deficient animals results in much more severe autoimmune disease relative to each deficient mouse strain alone83. Moreover, in contrast to APECED patients, patients with FOXP3 mutations who lack Tregs develop Immune Dysregulation, Polyendocrinopathy, Enteropathy X-linked (IPEX) syndrome, which is characterized by absent or defective Tregs and results in severe widespread autoimmunity early in life with organ specificity that is distinct from APECED patients such as the high prevalence of type I diabetes or glomerulonephritis.84 The dichotomy in clinical presentation and disease onset and severity of these two monogenic diseases illustrates that although AIRE regulates thymic Treg generation through positive selection, the development of autoreactive CD4+ effector T cells may play a primary role in the pathogenesis in APECED patients. Beyond the important role of these AIRE-dependent functions in induction of tolerance and prevention of autoimmunity, there is emerging literature on how therapeutic manipulation of the Aire-dependent thymic development of tumor-associated Tregs and of the Aire-dependent negative selection of tumor-specific effector T cells may have implications in designing novel anti-tumor therapeutic strategies.85–87
CD8+ T cells and γδ T cells
In addition to AIRE−/− CD4+ effector and/or regulatory T cells that drive the autoimmune disease, other lymphoid cell types likely play a role in modulating the disease development, as shown in the Aire−/− mouse model. For example, adoptive transfer of Aire−/− CD8+ T cells into SCID (severe combined immunodeficiency) recipient mice was sufficient to drive autoimmune disease in peripheral nerve tissue, although to a lesser extent compared to adoptive transfer of Aire−/− CD4+ T cells.88 How Aire may affect CD8+ T cell development in the thymus is less defined relative to its role in CD4+ T cell selection. In addition, recent evidence implicates γδ T cells in the development of organ-specific autoimmune disease in the retina and lung of Aire-deficient mice. Specifically, Aire, via its regulation of IL-7 expression in mTECs, was shown to modulate the accumulation of thymic IL-17A+ Vγ6+ Vδ1+ T cells.89 Aire-deficient mice have expanded populations of thymic and peripheral perinatally generated IL-17A+ Vγ6+ Vδ1+ T cells, and deletion of γδ T cells in Aire-deficient mice ameliorates retinal and pulmonary autoimmune disease. A putative corresponding population of γδ T cells in humans, IL-17A+ Vγ9+ Vδ2+ T cells, are expanded in the peripheral blood of APECED patients, indicating that the role of these cells in mediating organ-specific autoimmune disease in humans deserves further investigation.
B cells and AAbs
Aire−/− auto-reactive B cells have also been suggested to be drivers of organ-specific autoimmunity in mice, predominantly in the lungs, although another study revealed a broader organ distribution for the role of Aire-deficient B cells in autoimmunity.76,90 In fact, APECED patients exhibit a significant expansion in the autoreactive CD21lo CD38+ B cell subset from an early age, and Aire−/− mice have increased peripheral accumulation of CD11b+ CD11c+ B cells, which are thought to functionally and phenotypically correspond to human CD21lo CD38+ B cells.8 These findings may have translational implications with regard to the potential utility of anti-CD20-targeted immunotherapy in affected patients (see below). The mechanisms by which AIRE promotes central and/or peripheral tolerance of B cells, and whether these tolerance defects relate to B cell-intrinsic or B cell-extrinsic mechanisms remain unclear.72
In addition, the relative contribution of B cell-dependent production of AAbs over B cell-dependent T cell priming and expansion via antigen presentation in promoting autoimmunity in APECED patients is not fully understood (see below).90 In fact, the extent to which AAbs play a direct role in pathogenesis is unclear, as transfer experiments of Aire-deficient serum have not resulted in development of autoimmune disease in mice. At the clinical level, the sensitivity and specificity of organ-specific AAbs is not 100%, and although positivity for certain AAbs may predict the development of certain disease components in certain cases, this is not a universal rule for all patients (Lionakis, unpublished observations).
However, the identification and characterization of a wide array of tissue-specific AAbs through the years has provided important information regarding organ-specific autoantigens that are targeted by autoimmunity in humans. Several AAbs have been identified in APECED and their role in autoimmunity has, in some instances, been extended beyond APECED. These include 21-OH (21-hydroxylase) for primary adrenal insufficiency91,92, NALP5 (NACHT, leucine-rich repeat, pyrin domain–containing protein 5) for hypoparathyroidism93, TPH (tryptophan hydroxylase) for intestinal malabsorption94, TGM4 (transglutaminase 4) for prostate-associated male infertility95, TDRD6 (tudor domain containing protein 6) for hypopituitarism 96, BPIFB1 and KCNRG for autoimmune lung disease26,97, and others (Table 2).8,98–110 Future work will be needed to uncover other autoantigens that may provide explanation for the pathogenesis of the autoimmune attack in the spleen, salivary and lacrimal glands, nails and skin of APECED patients.
In addition to tissue-specific autoantibodies, as mentioned earlier, nearly all patients with APECED produce autoantibodies directed against type I interferons, which have early diagnostic utility.33,35 Of note, the occurrence of AAbs against GAD65 (glutamic acid decarboxylase) in the majority of APECED patients8 without the accompanying development of type I diabetes has led to investigations for the identification of potential protective factors that may prevent the development of type I diabetes in this patient population. To that end, Meyer and colleagues recently reported on the inverse correlation between the presence of high-affinity, neutralizing AAbs against type I interferons with type I diabetes in APECED patients, suggesting that these AAbs may harbor diabetes-ameliorating potential and, thus, may have therapeutic utility.111 Studies in additional APECED cohorts will be needed to verify these interesting translational research findings.
The identification of neutralizing AAbs against IL-17A, IL-17F and IL-22, which are critical cytokines involved in the mucosal defense against Candida has been associated with the susceptibility to CMC in APECED patients.112,113 How deficiency in a thymic transcription regulator leads to universal and isolated susceptibility to a single infectious disease, CMC, is indeed an intriguing question, and the Th17 cytokine AAbs suggest a potential autoimmune origin for CMC. However, in most reported studies, the correlation between the presence of Th17 cytokine AAbs and CMC is ~70%, indicating that other factors may also contribute to development of mucosal fungal disease in APECED.8,112,113 Alterations in C-type lectin receptor signaling in macrophages and impaired production of antimicrobial factors in mucosal secretions have also been proposed as potential contributing factors.114–116
Thus far, the antifungal immune studies in APECED patients have focused on their systemic, but not their mucosal, immune responses. Because the immune response requirements for effective mucosal versus systemic host defense against Candida are strictly segregated, it is likely that yet-unknown defects at the mucosal level in these patients may also contribute to CMC susceptibility. To that end, we have established a mouse model of mucosal candidiasis in Aire−/− deficient mice and have performed parallel studies of mucosal immunity in mucosal biopsy samples obtained from APECED patients. We have found that pathogenic T cells, not autoantibodies, drive fungal susceptibility at the mucosa via impairing the integrity of the mucosal barrier (Break et al, manuscript in preparation).
eTACs
Besides Aire-expressing mTECs, the identification of eTACs within secondary lymphoid organs of mice and humans has resulted in great interest in the potential extrathymic role of AIRE expression in induction of tolerance. The Anderson lab has shown that mouse eTACs are a MHCIIhi, EpCAMhi, CD45lo, CD80lo, CD86lo bone marrow-derived antigen presenting cell population that expresses distinct self-antigens and is able to interact with and delete naïve autoreactive T cells through Treg-independent mechanisms that are still not fully characterized.69,71
AIRE-expressing cells that exhibit a dendritic cell-like morphology and express HLA-DR, CD11c, CD40, CD83, CD208 and CCR7 have also been reported in human peripheral lymphoid tissues such as lymph nodes (predominantly abdominal), tonsils and gut-associated lymphoid tissue (GALT), but not in spleen.70 These cells, which appear to correspond to a subset of activated interdigitating dendritic cells, express TSAs such as insulin and “tolerogenic” molecules such as IL-10 or indoleamine 2,3-dioxygenase, raising the possibility of serving a potential tolerogenic function. Studies employing bone marrow replacement of eTACs or gene therapy within bone marrow-derived eTACs into Aire deficient and other studies using conditional Aire-knockout mice will be critical to directly examine the potential role of eTACs in organ-specific autoimmune disease and their ability to ameliorate disease. In fact, a preliminary report using conditional Aire-knockout mice revealed that Aire protects from infertility derived from autoimmune disease within the reproductive organs via expression in cells outside the thymic stromal or hematopoietic compartments, presumably within the gonads.117 Future research will be required to determine whether different eTAC subsets exist in humans in order to examine their functional and regulatory roles, and compare whether their numbers and/or effector functions are defective in patients with APECED.
AIRE Partners and Regulation
AIRE appears to be tightly regulated with recent identification of important enhancer elements activated by nuclear factor-κB (NF-κB) signaling that are necessary for AIRE’s expression. For example, the conserved non‐coding sequence 1 (CNS1) proximal to the Aire promoter is a cis-regulatory element critically important for AIRE expression in mTECs.118,119 Multiple additional regulators have also been recently reported to control AIRE expression and/or function. These include FBXO3 (F-box protein 3 E3), which ubiquitinates AIRE and regulates its transcriptional activity, SIRT1 (Sirtuin 1), which is abundantly found in mTECs where it acetylates Aire and regulates the expression of Aire-dependent TSA-encoding genes, and HIPK2 (homeodomain-interacting protein kinase 2), which phosphorylates AIRE and suppresses its coactivator activity, although its impact on the expression of Aire-dependent TSA-encoding genes in mTECs is modest.120–122 HIPK2 was recently shown to also modulate the development of thymic tuft cells, which represent a subset of thymic epithelial cells that rely on the taste chemosensory molecule TRPM5 for their thymic function and pass through an Aire-dependent phase for their thymic development.123 Moreover, the lysyl‐hydroxylase Jmjd6 (Jumonji domain-containing protein 6) affects splicing of intron 2 of the Aire gene and is required for expression of mature Aire in mTECs, and Dgcr8 (DiGeorge syndrome critical region gene 8) is important for accumulation of Aire-expressive mTECs in the thymus.124,125 Recent work from Herzig et al.126 further demonstrated a very complex method of Aire expression regulation resulting from positive and negative mechanisms and the coordinated effort of multiple transcription factors, Irf4, Irf8, Tbx21, Tcf7 and Ctcfl. Whether patients with APECED-like clinical disease with wild-type AIRE genotype have mutations in the aforementioned AIRE partners and regulators remains to be determined in future studies.
AIRE mutations in APECED patients
APECED follows an autosomal recessive pattern of inheritance with more than 100 AIRE mutations having been reported throughout the entire gene.127 Depending on the patient cohort, homozygous or compound heterozygous AIRE mutations are seen more often. As mentioned earlier, homozygosity for the p.Y85C, p.R139X or p.R257X mutations is observed in the vast majority of Persian Jew, Sardinian or Finnish APECED patients, respectively. The 13-bp deletion 1094–1106del13 is enriched among North American, Norwegian, British and Irish APECED patients, and in North American patients, compound heterozygous AIRE mutations are most often seen, consonant with the greater genetic and ethnic diversity of the population.
A few important points are worthwhile highlighting about the genetics of APECED patients: first, as mentioned above, there is no clear genotype-phenotype correlation with organ-specific autoimmune manifestations and significant clinical variability is seen amongst patients (even siblings) with identical AIRE genetic variants.127 These findings imply the presence of other disease-modifying genetic variants in APECED patients.128 Indeed, this hypothesis was recently supported in mice with mutations in both Aire and the inhibitory protein tyrosine kinase Lyn, which developed autoimmune uveitis, underscoring the cooperation between central and peripheral tolerance checkpoints in the organ-specific phenotypic expression of autoimmune disease.129 Similarly, mice with deficiency in both Aire and the T cell peripheral tolerance checkpoint Cbl-b (Casitas B-lineage lymphoma), which modulates CD28 co-stimulation, developed lethal exocrine pancreatic autoimmune disease.130 Therefore, despite APECED being a monogenic disorder, these findings underscore the importance of other to be discovered genetic modifiers that may promote or ameliorate susceptibility of organ-specific manifestations of the disease. Investigation of such variants is an important direction of future research.
Second, patients with isolated hypoparathyroidism have been discovered with pathogenic biallelic AIRE mutations131,132, raising the possibility of the existence of several unrecognized APECED patients with biallelic AIRE mutations who may develop isolated organ-specific autoimmune manifestations that are typical of APECED syndrome but have not developed a classic diagnostic dyad to raise awareness for AIRE genetic testing. Third, in the North American APECED population, we have witnessed a significant proportion (~15%) that does not have biallelic AIRE mutations or deletions and yet, the clinical presentation of the patients is indistinguishable from patients with biallelic AIRE mutations.8 Whole exome and whole genome sequencing in these patients are a first step toward identifying potential digenic models of disease or non-AIRE coding variants that may phenocopy AIRE deficiency or non-coding variants that may affect AIRE expression and/or function.
Recently, dominant-negative AIRE mutations involving the SAND or PHD-1 domains have also been identified, some of which have a relatively high frequency in the general population (up to 1 in 1000). These patients present with much less severe APECED-like disease, often with later onset, manifested by limited organ involvement such as pernicious anemia or vitiligo or an isolated endocrinopathy; AAbs are not always positive in these patients.133–136 These findings indicate that such dominant-negative AIRE variants may modulate the phenotypic expression of common organ-specific autoimmune diseases. In that light, AIRE variants along with other risk loci were recently reported to be associated with Addison’s disease.137 Last, an intriguing recent report by the Matsumoto lab indicated that overexpression of human AIRE in mice resulted in the development of a paradoxical muscle-specific autoimmune disease, associated with impaired maturation of mTECs and defective expression of Aire-dependent TSAs.138 These data both illustrate the importance for tight control of AIRE expression and function and raise the possibility that gain-of-function AIRE genetic variants may exist in humans, perhaps having a potential association with organ-specific autoimmune diseases such as polymyositis or dermatomyositis.
Management of APECED Patients
As the onset of symptoms, disease severity and progression, and number of disease manifestations are highly variable amongst patients the majority of patients are best served by a multidisciplinary team of providers at a tertiary care-level facility. At the initial visit, healthcare providers should perform a comprehensive evaluation with special attention to involvement of tissue sites reported.8 The integration of specialists from immunology, infectious disease, endocrinology, pulmonology, hepatology, gastroenterology, dermatology, nephrology and dentistry provides the best possible evaluation for the individual patient. Subsequent visits over time should continue surveillance for the development of other disease manifestations and involvement of additional organ systems. The principles of evaluation and management of the APECED patients include treatment of CMC and other infectious complications, hormone and/or electrolyte replacement for irreversible endocrinopathies, and immunomodulation for non-endocrine end-organ disease.
CMC and other infectious complications
Management of CMC is challenging in APECED patients because a substantial proportion of them develops azole-resistant Candida albicans and Candida glabrata strains, thus limiting long-term oral treatment options.139–141 In our experience, the use of oral swiss/swallow suspension of amphotericin B provides the best long-term preventive strategy in these patients with azole-resistance Candida strains. This topical therapeutic approach works best when it follows an induction treatment phase with a fungicidal parenteral drug such as an echinocandin in order to prevent recurrence of candidiasis rather than being effective as treatment of acute severe oral or esophageal candidiasis. Novel antifungal agents with remarkable in vitro and in vivo activity against azole-susceptible and -resistant Candida strains require future investigation in APECED patients with severe azole-resistant CMC140,141, as does the Als3-based fungal vaccine that boosts IL-17-mediated anti-Candida immunity and is the first fungal vaccine shown to display efficacy in humans.142,143 Routine screening for gastrointestinal cancers is a recommended practice for all patients. Patients should undergo routine oral examination in partnership with dental colleagues, and patients with intestinal metaplasia or dysplastic features on gastric biopsies should be evaluated by gastric mapping.
Screening for asplenia should be performed in all patients via peripheral blood smear and abdominal ultrasound. In the subset of patients identified with asplenia, we initiate prophylactic oral penicillin VK or amoxicillin (or azithromycin in penicillin-allergic individuals). These individuals should be provided with a prescription of extended-spectrum fluoroquinolones (e.g., levofloxacin) to be available for initiation at the onset of fever. They should receive vaccination against pneumococcus (both 13- and 23-valent vaccines), meningococcus, and Haemophilus influenzae type b as well as annual vaccination for influenza.
Management of endocrinopathies
Close monitoring of hormone and electrolyte replacement is paramount to avoid acute Addisonian or hypocalcemic crises. Patients with adrenal insufficiency should carry an emergency card or medical alert bracelet and stress dose corticosteroids should be made available. Long-term calcium replacement results in nephrocalcinosis, which can lead to kidney stones and renal failure. The advent of recombinant PTH may help some APECED patients with hypoparathyroidism, especially in the setting of renal insufficiency and/or severe intestinal malabsorption.144,145
Management of non-endocrine end-organ disease
Immunosuppressive treatment is critical for the control of autoimmunity in the lungs, liver, small intestine, eye or kidney with a goal to prevent irreversible end-organ damage. The use of cyclosporine ophthalmic solution is efficacious in patients with autoimmune keratoconjunctivitis. Azathioprine has a long track record of success in the treatment of autoimmune hepatitis in patients with APECED with a favorable tolerability profile without hematological, hepatic or infectious complications (Chascsa et al, submitted manuscript). In APECED patients with TPMT (thiopurine S-methyltransferase) genotypes that predict poor metabolism of azathioprine, mycophenolate or mTOR inhibition are acceptable alternative options in order to avoid azathioprine-induced toxicity. We avoid cyclosporine or tacrolimus due to prevalent renal-associated toxicity in APECED patients. mTOR inhibitors appear particularly effective for biopsy-proven autoimmune enteritis. Less experience exists regarding the optimal treatment of tubulointerstitial nephritis, but azathioprine or mycophenolate appear to be effective in halting the progression of the disease, when started early. Monotherapy with rituximab appears ineffective in the small number of treated APECED patients with pneumonitis, nephritis or enteritis. Rituximab appears to be effective against APECED-associated autoimmune hepatitis, typically in combination with a T cell immunomodulator in the rare occasions where T cell-targeted monotherapy is ineffective (Lionakis, unpublished observations). In agreement with Aire−/− mouse studies of T and B cell depletion, autoimmune pneumonitis responds dramatically at the clinical, radiographic, and pulmonary function levels with the combination of azathioprine and rituximab. The majority of treated patients remain in remission on azathioprine monotherapy without the need for re-challenging with rituximab (Ferre et al., submitted manuscript).
Currently, there is no consensus with regard to initiation of prophylactic immunomodulation in patients with APECED as an approach to prevent development of subsequent autoimmune manifestations. Future studies will be critical to examine the role of such a preventive immunosuppressive approach, especially in patients who are diagnosed very early in the course of their disease.
Conclusion and Future Prospects
The past 20 years have provided significant advances in our understanding of the clinical, genetic, molecular and immunological mechanisms that underpin the development of APECED. Description of the functional and regulatory mechanisms of AIRE has offered key information regarding mechanisms of self-tolerance and autoimmunity. The implications of dominant-negative heterozygous AIRE mutations and the collaboration of AIRE with other at-risk loci and peripheral tolerance checkpoints may shed light on the pathogenesis of other autoimmune disease states. Identification of patients early in the course of the disease is crucial and may allow for instituting disease-modifying interventions prior to the development of irreversible end-organ complications. The challenge moving forward will be to translate our improved understanding of the clinical, genetic, molecular and cellular features of AIRE deficiency into the development of novel strategies that will improve the diagnosis, risk stratification, immunotherapy, prognosis and quality of life of APECED patients.
Acknowledgments:
This work was supported by the Division of Intramural Research, National Institutes of Health, National Institute of Allergy and Infectious Diseases. We thank Ryan Kissinger for his assistance with Figures 1 and 4.
Footnotes
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Bibliography
- 1.Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat. Rev. Immunol 2016;16(4):247–258. doi: 10.1038/nri.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metzger TC, Anderson MS. Control of central and peripheral tolerance by Aire. Immunol. Rev 2011;241(1):89–103. doi: 10.1111/j.1600-065X.2011.01008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity 2005;23(2):227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol 2014;14(6):377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeVoss J, Hou Y, Johannes K, et al. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exp. Med 2006;203(12):2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Husebye ES, Perheentupa J, Rautemaa R, Kämpe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J. Intern. Med 2009;265(5):514–529. doi: 10.1111/j.1365-2796.2009.02090.x. [DOI] [PubMed] [Google Scholar]
- 7.Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med 1990;322(26):1829–1836. doi: 10.1056/NEJM199006283222601. [DOI] [PubMed] [Google Scholar]
- 8.Ferre EMN, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J. Clin. Endocrinol. Metab 2017;102(9):3546–3556. doi: 10.1210/jc.2017-00139. [DOI] [PubMed] [Google Scholar]
- 10.Thorpe ES, Handley HE. Chronic tetany and chronic mycelial stomatitis in a child aged 4 and half years. The American Journal of Diseased Children 1929;(38):328– 338. [Google Scholar]
- 11.Talbot NB, Butler AM, Maclachlan EA. The effect of testosterone and allied compounds on the mineral, nitrogen, and carbohydrate metabolism of a girl with addison’s disease. J. Clin. Invest 1943;22(4):583–593. doi: 10.1172/JCI101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutphin A, Albright F, McCune DJ. Five Cases (Three in Siblings) of Idiopathic Hypoparathyroidism Associated with Moniliasis1. The Journal of Clinical Endocrinology & Metabolism 1943;3(12):625–634. doi: 10.1210/jcem-3-12-625. [DOI] [Google Scholar]
- 13.Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet 1997;17(4):399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 14.Nagamine K, Peterson P, Scott HS, et al. Positional cloning of the APECED gene. Nat. Genet 1997;17(4):393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 15.Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J. Med. Genet 1992;29(11):824–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosatelli MC, Meloni A, Meloni A, et al. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum. Genet 1998;103(4):428–434. doi: 10.1007/s004390050846. [DOI] [PubMed] [Google Scholar]
- 17.Wolff ASB, Erichsen MM, Meager A, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J. Clin. Endocrinol. Metab 2007;92(2):595–603. doi: 10.1210/jc.2006-1873. [DOI] [PubMed] [Google Scholar]
- 18.Bruserud Ø, Oftedal BE, Landegren N, et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J. Clin. Endocrinol. Metab 2016;101(8):2975–2983. doi: 10.1210/jc.2016-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myhre AG, Halonen M, Eskelin P, et al. Autoimmune polyendocrine syndrome type 1 (APS I) in Norway. Clin. Endocrinol. (Oxf.) 2001;54(2):211–217. doi: 10.1046/j.1365-2265.2001.01201.x. [DOI] [PubMed] [Google Scholar]
- 20.Stolarski B, Pronicka E, Korniszewski L, et al. Molecular background of polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome in a Polish population: novel AIRE mutations and an estimate of disease prevalence. Clin. Genet 2006;70(4):348–354. doi: 10.1111/j.1399-0004.2006.00690.x. [DOI] [PubMed] [Google Scholar]
- 21.Podkrajsek KT, Bratanic N, Krzisnik C, Battelino T. Autoimmune regulator-1 messenger ribonucleic acid analysis in a novel intronic mutation and two additional novel AIRE gene mutations in a cohort of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. J. Clin. Endocrinol. Metab 2005;90(8):4930–4935. doi: 10.1210/jc.2005-0418. [DOI] [PubMed] [Google Scholar]
- 22.Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med 2018;378(12):1132–1141. doi: 10.1056/NEJMra1713301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahaly GJ. Polyglandular autoimmune syndromes. Eur. J. Endocrinol 2009;161(1):11–20. doi: 10.1530/EJE-09-0044. [DOI] [PubMed] [Google Scholar]
- 24.Betterle C, Greggio NA, Volpato M. Clinical review 93: Autoimmune polyglandular syndrome type 1. J. Clin. Endocrinol. Metab 1998;83(4):1049–1055. doi: 10.1210/jcem.83.4.4682. [DOI] [PubMed] [Google Scholar]
- 25.Pollak U, Bar-Sever Z, Hoffer V, Marcus N, Scheuerman O, Garty BZ. Asplenia and functional hyposplenism in autoimmune polyglandular syndrome type 1. Eur. J. Pediatr 2009;168(2):233–235. doi: 10.1007/s00431-008-0735-9. [DOI] [PubMed] [Google Scholar]
- 26.Alimohammadi M, Dubois N, Sköldberg F, et al. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen. Proc. Natl. Acad. Sci. USA 2009;106(11):4396–4401. doi: 10.1073/pnas.0809986106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominguez M, Crushell E, Ilmarinen T, et al. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) in the Irish Population. Journal of Pediatric Endocrinology and Metabolism 2006;19(11). doi: 10.1515/JPEM.2006.19.11.1343. [DOI] [PubMed] [Google Scholar]
- 28.Meloni A, Willcox N, Meager A, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J. Clin. Endocrinol. Metab 2012;97(4):1114–1124. doi: 10.1210/jc.2011-2461. [DOI] [PubMed] [Google Scholar]
- 29.Valenzise M, Fierabracci A, Cappa M, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy: report of seven additional sicilian patients and overview of the overall series from sicily. Horm Res Paediatr 2014;82(2):127–132. doi: 10.1159/000363537. [DOI] [PubMed] [Google Scholar]
- 30.F Magitta N, Pura M, S Bøe Wolff A, et al. Autoimmune polyendocrine syndrome type I in Slovakia: relevance of screening patients with autoimmune Addison’s disease. Eur. J. Endocrinol 2008;158(5):705–709. doi: 10.1530/EJE-07-0843. [DOI] [PubMed] [Google Scholar]
- 31.Perheentupa J Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab 2006;91(8):2843–2850. doi: 10.1210/jc.2005-2611. [DOI] [PubMed] [Google Scholar]
- 32.Fierabracci A, Pellegrino M, Frasca F, Kilic SS, Betterle C. APECED in Turkey: A case report and insights on genetic and phenotypic variability. Clin. Immunol 2018;194:60–66. doi: 10.1016/j.clim.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 33.Meager A, Visvalingam K, Peterson P, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med 2006;3(7):e289. doi: 10.1371/journal.pmed.0030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolff ASB, Sarkadi AK, Maródi L, et al. Anti-cytokine autoantibodies preceding onset of autoimmune polyendocrine syndrome type I features in early childhood. J. Clin. Immunol 2013;33(8):1341–1348. doi: 10.1007/s10875-013-9938-6. [DOI] [PubMed] [Google Scholar]
- 35.Meloni A, Furcas M, Cetani F, et al. Autoantibodies against type I interferons as an additional diagnostic criterion for autoimmune polyendocrine syndrome type I. J. Clin. Endocrinol. Metab 2008;93(11):4389–4397. doi: 10.1210/jc.2008-0935. [DOI] [PubMed] [Google Scholar]
- 36.Wolff ASB, Kärner J, Owe JF, et al. Clinical and serologic parallels to APS-I in patients with thymomas and autoantigen transcripts in their tumors. J. Immunol 2014;193(8):3880–3890. doi: 10.4049/jimmunol.1401068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng MH, Fan U, Grewal N, et al. Acquired autoimmune polyglandular syndrome, thymoma, and an AIRE defect. N. Engl. J. Med 2010;362(8):764–766. doi: 10.1056/NEJMc0909510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walter JE, Rosen LB, Csomos K, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J. Clin. Invest 2015;125(11):4135–4148. doi: 10.1172/JCI80477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meager A, Wadhwa M, Dilger P, et al. Anti-cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clin. Exp. Immunol 2003;132(1):128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaidi G, Bhatia V, Sahoo SK, et al. Autoimmune polyendocrine syndrome type 1 in an Indian cohort: a longitudinal study. Endocr. Connect 2017;6(5):289–296. doi: 10.1530/EC-17-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lionakis MS, Levitz SM. Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu. Rev. Immunol 2018;36:157–191. doi: 10.1146/annurev-immunol-042617-053318. [DOI] [PubMed] [Google Scholar]
- 42.Rautemaa R, Hietanen J, Niissalo S, Pirinen S, Perheentupa J. Oral and oesophageal squamous cell carcinoma--a complication or component of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, APS-I). Oral Oncol 2007;43(6):607–613. doi: 10.1016/j.oraloncology.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 43.Bensing S, Brandt L, Tabaroj F, et al. Increased death risk and altered cancer incidence pattern in patients with isolated or combined autoimmune primary adrenocortical insufficiency. Clin. Endocrinol. (Oxf.) 2008;69(5):697–704. doi: 10.1111/j.1365-2265.2008.03340.x. [DOI] [PubMed] [Google Scholar]
- 44.Sampaio EP, Ding L, Rose SR, et al. Novel signal transducer and activator of transcription 1 mutation disrupts small ubiquitin-related modifier conjugation causing gain of function. J. Allergy Clin. Immunol 2018;141(5):1844–1853.e2. doi: 10.1016/j.jaci.2017.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016;127(25):3154–3164. doi: 10.1182/blood-2015-11-679902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu F, Willette-Brown J, Song N-Y, et al. Autoreactive T cells and chronic fungal infection drive esophageal carcinogenesis. Cell Host Microbe 2017;21(4):478–493.e7. doi: 10.1016/j.chom.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Netea MG, Joosten LAB, van der Meer JWM, Kullberg B-J, van de Veerdonk FL. Immune defence against Candida fungal infections. Nat. Rev. Immunol 2015;15(10):630–642. doi: 10.1038/nri3897. [DOI] [PubMed] [Google Scholar]
- 48.Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, Kullberg BJ. Invasive candidiasis. Nat. Rev. Dis. Primers 2018;4:18026. doi: 10.1038/nrdp.2018.26. [DOI] [PubMed] [Google Scholar]
- 49.Desai JV, van de Veerdonk FL, Lionakis MS. Understanding the role of host immune responses in invasive candidiasis. Intensive Care Med 2018;44(8):1310–1314. doi: 10.1007/s00134-017-4988-5. [DOI] [PubMed] [Google Scholar]
- 50.Nagafuchi S, Umene K, Yamanaka F, et al. Recurrent herpes simplex virus infection in a patient with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy associated with L29P and IVS9–1G>C compound heterozygous autoimmune regulator gene mutations. J. Intern. Med 2007;261(6):605–610. doi: 10.1111/j.1365-2796.2007.01786.x. [DOI] [PubMed] [Google Scholar]
- 51.Zimmerman O, Rosen LB, Swamydas M, et al. Autoimmune regulator deficiency results in a decrease in STAT1 levels in human monocytes. Front. Immunol 2017;8:820. doi: 10.3389/fimmu.2017.00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kluger N, Jokinen M, Krohn K, Ranki A. Gastrointestinal manifestations in APECED syndrome. J. Clin. Gastroenterol 2013;47(2):112–120. doi: 10.1097/MCG.0b013e31827356e1. [DOI] [PubMed] [Google Scholar]
- 53.Husebye ES, Gebre-Medhin G, Tuomi T, et al. Autoantibodies against aromatic L-amino acid decarboxylase in autoimmune polyendocrine syndrome type I. J. Clin. Endocrinol. Metab 1997;82(1):147–150. doi: 10.1210/jcem.82.1.3647. [DOI] [PubMed] [Google Scholar]
- 54.Gebre-Medhin G, Husebye ES, Gustafsson J, et al. Cytochrome P450IA2 and aromatic L-amino acid decarboxylase are hepatic autoantigens in autoimmune polyendocrine syndrome type I. FEBS Lett 1997;412(3):439–445. [DOI] [PubMed] [Google Scholar]
- 55.Kluger N, Kataja J, Aho H, Rönn A-M, Krohn K, Ranki A. Kidney involvement in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy in a Finnish cohort. Nephrol. Dial. Transplant 2014;29(9):1750–1757. doi: 10.1093/ndt/gfu064. [DOI] [PubMed] [Google Scholar]
- 56.Ulinski T, Perrin L, Morris M, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome with renal failure: impact of posttransplant immunosuppression on disease activity. J. Clin. Endocrinol. Metab 2006;91(1):192–195. doi: 10.1210/jc.2005-1538. [DOI] [PubMed] [Google Scholar]
- 57.Tagawa A, Aranami T, Matsumoto M, Yamamura T. Autoimmune regulator gene,Aire , is involved in central tolerance to the DM20 isoform of proteolipid protein and the prevention of autoimmune inflammation. Clin. Exp. Neuroimmunol 2014;5(3):304–314. doi: 10.1111/cen3.12127. [DOI] [Google Scholar]
- 58.Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002;298(5597):1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 59.Takaba H, Morishita Y, Tomofuji Y, et al. Fezf2 Orchestrates a Thymic Program of Self-Antigen Expression for Immune Tolerance. Cell 2015;163(4):975–987. doi: 10.1016/j.cell.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 60.Mathis D, Benoist C. Aire. Annu. Rev. Immunol 2009;27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532. [DOI] [PubMed] [Google Scholar]
- 61.Abramson J, Husebye ES. Autoimmune regulator and self-tolerance - molecular and clinical aspects. Immunol. Rev 2016;271(1):127–140. doi: 10.1111/imr.12419. [DOI] [PubMed] [Google Scholar]
- 62.Kumar PG, Laloraya M, Wang CY, et al. The autoimmune regulator (AIRE) is a DNA-binding protein. J. Biol. Chem 2001;276(44):41357–41364. doi: 10.1074/jbc.M104898200. [DOI] [PubMed] [Google Scholar]
- 63.Björses P, Pelto-Huikko M, Kaukonen J, Aaltonen J, Peltonen L, Ulmanen I. Localization of the APECED protein in distinct nuclear structures. Hum. Mol. Genet 1999;8(2):259–266. [DOI] [PubMed] [Google Scholar]
- 64.Waterfield M, Khan IS, Cortez JT, et al. The transcriptional regulator Aire coopts the repressive ATF7ip-MBD1 complex for the induction of immunotolerance. Nat. Immunol 2014;15(3):258–265. doi: 10.1038/ni.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Org T, Chignola F, Hetényi C, et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep 2008;9(4):370–376. doi: 10.1038/sj.embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koh AS, Kingston RE, Benoist C, Mathis D. Global relevance of Aire binding to hypomethylated lysine-4 of histone-3. Proc. Natl. Acad. Sci. USA 2010;107(29):13016–13021. doi: 10.1073/pnas.1004436107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koh AS, Kuo AJ, Park SY, et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc. Natl. Acad. Sci. USA 2008;105(41):15878–15883. doi: 10.1073/pnas.0808470105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Björses P, Halonen M, Palvimo JJ, et al. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am. J. Hum. Genet 2000;66(2):378–392. doi: 10.1086/302765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gardner JM, Metzger TC, McMahon EJ, et al. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4+ T cells. Immunity 2013;39(3):560–572. doi: 10.1016/j.immuni.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Poliani PL, Kisand K, Marrella V, et al. Human peripheral lymphoid tissues contain autoimmune regulator-expressing dendritic cells. Am. J. Pathol 2010;176(3):1104–1112. doi: 10.2353/ajpath.2010.090956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardner JM, Devoss JJ, Friedman RS, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 2008;321(5890):843–847. doi: 10.1126/science.1159407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamano T, Nedjic J, Hinterberger M, et al. Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity 2015;42(6):1048–1061. doi: 10.1016/j.immuni.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 73.Abramson J, Giraud M, Benoist C, Mathis D. Aire’s partners in the molecular control of immunological tolerance. Cell 2010;140(1):123–135. doi: 10.1016/j.cell.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 74.Meredith M, Zemmour D, Mathis D, Benoist C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat. Immunol 2015;16(9):942–949. doi: 10.1038/ni.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mathis D, Benoist C. Back to central tolerance. Immunity 2004;20(5):509–516. doi: 10.1016/S1074-7613(04)00111-6. [DOI] [PubMed] [Google Scholar]
- 76.Devoss JJ, Shum AK, Johannes KPA, et al. Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J. Immunol 2008;181(6):4072–4079. doi: 10.4049/jimmunol.181.6.4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramsey C, Winqvist O, Puhakka L, et al. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum. Mol. Genet 2002. [DOI] [PubMed] [Google Scholar]
- 78.Savage PA, Leventhal DS, Malchow S. Shaping the repertoire of tumor-infiltrating effector and regulatory T cells. Immunol. Rev 2014;259(1):245–258. doi: 10.1111/imr.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. Aire Enforces Immune Tolerance by Directing Autoreactive T Cells into the Regulatory T Cell Lineage. Immunity 2016;44(5):1102–1113. doi: 10.1016/j.immuni.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015;348(6234):589–594. doi: 10.1126/science.aaa7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kekäläinen E, Tuovinen H, Joensuu J, et al. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Immunol 2007;178(2):1208–1215. [DOI] [PubMed] [Google Scholar]
- 82.Laakso SM, Laurinolli T-T, Rossi LH, et al. Regulatory T cell defect in APECED patients is associated with loss of naive FOXP3(+) precursors and impaired activated population. J. Autoimmun 2010;35(4):351–357. doi: 10.1016/j.jaut.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 83.Chen Z, Benoist C, Mathis D. How defects in central tolerance impinge on a deficiency in regulatory T cells. Proc. Natl. Acad. Sci. USA 2005;102(41):14735–14740. doi: 10.1073/pnas.0507014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hannibal MC, Torgerson T. IPEX Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al. , eds. GeneReviews(®) Seattle (WA): University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 85.Bakhru P, Zhu M-L, Wang H-H, et al. Combination central tolerance and peripheral checkpoint blockade unleashes antimelanoma immunity. JCI Insight 2017;2(18). doi: 10.1172/jci.insight.93265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khan IS, Mouchess ML, Zhu M-L, et al. Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J. Exp. Med 2014;211(5):761–768. doi: 10.1084/jem.20131889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malchow S, Leventhal DS, Nishi S, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science 2013;339(6124):1219–1224. doi: 10.1126/science.1233913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Su MA, Davini D, Cheng P, et al. Defective autoimmune regulator-dependent central tolerance to myelin protein zero is linked to autoimmune peripheral neuropathy. J. Immunol 2012;188(10):4906–4912. doi: 10.4049/jimmunol.1200493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fujikado N, Mann AO, Bansal K, et al. Aire Inhibits the Generation of a Perinatal Population of Interleukin-17A-Producing γδ T Cells to Promote Immunologic Tolerance. Immunity 2016;45(5):999–1012. doi: 10.1016/j.immuni.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gavanescu I, Benoist C, Mathis D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc. Natl. Acad. Sci. USA 2008;105(35):13009–13014. doi: 10.1073/pnas.0806874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr. Rev 2002;23(3):327–364. doi: 10.1210/edrv.23.3.0466. [DOI] [PubMed] [Google Scholar]
- 92.Winqvist O, Karlsson FA, Kämpe O. 21-Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet 1992;339(8809):1559–1562. doi: 10.1016/0140-6736(92)91829-W. [DOI] [PubMed] [Google Scholar]
- 93.Alimohammadi M, Björklund P, Hallgren A, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N. Engl. J. Med 2008;358(10):1018–1028. doi: 10.1056/NEJMoa0706487. [DOI] [PubMed] [Google Scholar]
- 94.Ekwall O, Hedstrand H, Grimelius L, et al. Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet 1998;352(9124):279–283. doi: 10.1016/S0140-6736(97)11050-9. [DOI] [PubMed] [Google Scholar]
- 95.Landegren N, Sharon D, Shum AK, et al. Transglutaminase 4 as a prostate autoantigen in male subfertility. Sci. Transl. Med 2015;7(292):292ra101. doi: 10.1126/scitranslmed.aaa9186. [DOI] [PubMed] [Google Scholar]
- 96.Bensing S, Fetissov SO, Mulder J, et al. Pituitary autoantibodies in autoimmune polyendocrine syndrome type 1. Proc. Natl. Acad. Sci. USA 2007;104(3):949–954. doi: 10.1073/pnas.0610070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shum AK, Alimohammadi M, Tan CL, et al. BPIFB1 is a lung-specific autoantigen associated with interstitial lung disease. Sci. Transl. Med 2013;5(206):206ra139. doi: 10.1126/scitranslmed.3006998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Winqvist O, Gustafsson J, Rorsman F, Karlsson FA, Kämpe O. Two different cytochrome P450 enzymes are the adrenal antigens in autoimmune polyendocrine syndrome type I and Addison’s disease. J. Clin. Invest 1993;92(5):2377–2385. doi: 10.1172/JCI116843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Landegren N, Sharon D, Freyhult E, et al. Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1. Sci. Rep 2016;6:20104. doi: 10.1038/srep20104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Söderbergh A, Myhre AG, Ekwall O, et al. Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I. J. Clin. Endocrinol. Metab 2004;89(2):557–562. doi: 10.1210/jc.2003-030279. [DOI] [PubMed] [Google Scholar]
- 101.Kisand K, Peterson P. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J. Clin. Immunol 2015;35(5):463–478. doi: 10.1007/s10875-015-0176-y. [DOI] [PubMed] [Google Scholar]
- 102.Hedstrand H, Ekwall O, Olsson MJ, et al. The transcription factors SOX9 and SOX10 are vitiligo autoantigens in autoimmune polyendocrine syndrome type I. J. Biol. Chem 2001;276(38):35390–35395. doi: 10.1074/jbc.M102391200. [DOI] [PubMed] [Google Scholar]
- 103.Hedstrand H, Ekwall O, Haavik J, et al. Identification of tyrosine hydroxylase as an autoantigen in autoimmune polyendocrine syndrome type I. Biochem. Biophys. Res. Commun 2000;267(1):456–461. doi: 10.1006/bbrc.1999.1945. [DOI] [PubMed] [Google Scholar]
- 104.Sköldberg F, Portela-Gomes GM, Grimelius L, et al. Histidine decarboxylase, a pyridoxal phosphate-dependent enzyme, is an autoantigen of gastric enterochromaffin-like cells. J. Clin. Endocrinol. Metab 2003;88(4):1445–1452. doi: 10.1210/jc.2002-021761. [DOI] [PubMed] [Google Scholar]
- 105.Obermayer-Straub P, Perheentupa J, Braun S, et al. Hepatic autoantigens in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Gastroenterology 2001;121(3):668–677. [DOI] [PubMed] [Google Scholar]
- 106.Bratland E, Magitta NF, Bøe Wolff AS, et al. Autoantibodies against aromatic amino acid hydroxylases in patients with autoimmune polyendocrine syndrome type 1 target multiple antigenic determinants and reveal regulatory regions crucial for enzymatic activity. Immunobiology 2013;218(6):899–909. doi: 10.1016/j.imbio.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 107.Reimand K, Perheentupa J, Link M, Krohn K, Peterson P, Uibo R. Testis-expressed protein TSGA10 an auto-antigen in autoimmune polyendocrine syndrome type I. Int. Immunol 2008;20(1):39–44. doi: 10.1093/intimm/dxm118. [DOI] [PubMed] [Google Scholar]
- 108.Perniola R, Falorni A, Clemente MG, Forini F, Accogli E, Lobreglio G. Organ-specific and non-organ-specific autoantibodies in children and young adults with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Eur. J. Endocrinol 2000;143(4):497–503. [DOI] [PubMed] [Google Scholar]
- 109.Gavalas NG, Kemp EH, Krohn KJE, Brown EM, Watson PF, Weetman AP. The calcium-sensing receptor is a target of autoantibodies in patients with autoimmune polyendocrine syndrome type 1. J. Clin. Endocrinol. Metab 2007;92(6):2107–2114. doi: 10.1210/jc.2006-2466. [DOI] [PubMed] [Google Scholar]
- 110.Kärner J, Pihlap M, Ranki A, et al. IL-6-specific autoantibodies among APECED and thymoma patients. Immun. Inflamm. Dis 2016;4(2):235–243. doi: 10.1002/iid3.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Meyer S, Woodward M, Hertel C, et al. AIRE-Deficient Patients Harbor Unique High-Affinity Disease-Ameliorating Autoantibodies. Cell 2016;166(3):582–595. doi: 10.1016/j.cell.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Puel A, Döffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med 2010;207(2):291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kisand K, Bøe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med 2010;207(2):299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.de Albuquerque JAT, Banerjee PP, Castoldi A, et al. The role of AIRE in the immunity against candida albicans in a model of human macrophages. Front. Immunol 2018;9:567. doi: 10.3389/fimmu.2018.00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindh E, Brännström J, Jones P, et al. Autoimmunity and cystatin SA1 deficiency behind chronic mucocutaneous candidiasis in autoimmune polyendocrine syndrome type 1. J. Autoimmun 2013;42:1–6. doi: 10.1016/j.jaut.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 116.Pedroza LA, Kumar V, Sanborn KB, et al. Autoimmune regulator (AIRE) contributes to Dectin-1-induced TNF-α production and complexes with caspase recruitment domain-containing protein 9 (CARD9), spleen tyrosine kinase (Syk), and Dectin-1. J. Allergy Clin. Immunol 2012;129(2):464–72, 472.e1. doi: 10.1016/j.jaci.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 117.Dobeš J, Edenhofer F, Vobořil M, et al. A novel conditional Aire allele enables cell-specific ablation of the immune tolerance regulator Aire. Eur. J. Immunol 2018;48(3):546–548. doi: 10.1002/eji.201747267. [DOI] [PubMed] [Google Scholar]
- 118.LaFlam TN, Seumois G, Miller CN, et al. Identification of a novel cis-regulatory element essential for immune tolerance. J. Exp. Med 2015;212(12):1993–2002. doi: 10.1084/jem.20151069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Haljasorg U, Bichele R, Saare M, et al. A highly conserved NF-κB-responsive enhancer is critical for thymic expression of Aire in mice. Eur. J. Immunol 2015;45(12):3246–3256. doi: 10.1002/eji.201545928. [DOI] [PubMed] [Google Scholar]
- 120.Shao W, Zumer K, Fujinaga K, Peterlin BM. FBXO3 protein promotes ubiquitylation and transcriptional activity of AIRE (autoimmune regulator). J. Biol. Chem 2016;291(34):17953–17963. doi: 10.1074/jbc.M116.724401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rattay K, Claude J, Rezavandy E, et al. Homeodomain-interacting protein kinase 2, a novel autoimmune regulator interaction partner, modulates promiscuous gene expression in medullary thymic epithelial cells. J. Immunol 2015;194(3):921–928. doi: 10.4049/jimmunol.1402694. [DOI] [PubMed] [Google Scholar]
- 122.Chuprin A, Avin A, Goldfarb Y, et al. The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat. Immunol 2015;16(7):737–745. doi: 10.1038/ni.3194. [DOI] [PubMed] [Google Scholar]
- 123.Miller CN, Proekt I, von Moltke J, et al. Thymic tuft cells promote an IL-4-enriched medulla and shape thymocyte development. Nature 2018;559(7715):627–631. doi: 10.1038/s41586-018-0345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yanagihara T, Sanematsu F, Sato T, et al. Intronic regulation of Aire expression by Jmjd6 for self-tolerance induction in the thymus. Nat. Commun 2015;6:8820. doi: 10.1038/ncomms9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Khan IS, Taniguchi RT, Fasano KJ, Anderson MS, Jeker LT. Canonical microRNAs in thymic epithelial cells promote central tolerance. Eur. J. Immunol 2014;44(5):1313–1319. doi: 10.1002/eji.201344079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herzig Y, Nevo S, Bornstein C, et al. Transcriptional programs that control expression of the autoimmune regulator gene Aire. Nat. Immunol 2017;18(2):161–172. doi: 10.1038/ni.3638. [DOI] [PubMed] [Google Scholar]
- 127.Bruserud Ø, Oftedal BE, Wolff AB, Husebye ES. AIRE-mutations and autoimmune disease. Curr. Opin. Immunol 2016;43:8–15. doi: 10.1016/j.coi.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 128.Proekt I, Miller CN, Lionakis MS, Anderson MS. Insights into immune tolerance from AIRE deficiency. Curr. Opin. Immunol 2017;49:71–78. doi: 10.1016/j.coi.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Proekt I, Miller CN, Jeanne M, et al. LYN- and AIRE-mediated tolerance checkpoint defects synergize to trigger organ-specific autoimmunity. J. Clin. Invest 2016;126(10):3758–3771. doi: 10.1172/JCI84440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Teh CE, Daley SR, Enders A, Goodnow CC. T-cell regulation by casitas B-lineage lymphoma (Cblb) is a critical failsafe against autoimmune disease due to autoimmune regulator (Aire) deficiency. Proc. Natl. Acad. Sci. USA 2010;107(33):14709–14714. doi: 10.1073/pnas.1009209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sahoo SK, Zaidi G, Srivastava R, et al. Identification of autoimmune polyendocrine syndrome type 1 in patients with isolated hypoparathyroidism. Clin. Endocrinol. (Oxf.) 2016;85(4):544–550. doi: 10.1111/cen.13111. [DOI] [PubMed] [Google Scholar]
- 132.Li D, Streeten EA, Chan A, et al. Exome sequencing reveals mutations in AIRE as a cause of isolated hypoparathyroidism. J. Clin. Endocrinol. Metab 2017;102(5):1726–1733. doi: 10.1210/jc.2016-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oftedal BE, Hellesen A, Erichsen MM, et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity 2015;42(6):1185–1196. doi: 10.1016/j.immuni.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 134.Cetani F, Barbesino G, Borsari S, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J. Clin. Endocrinol. Metab 2001;86(10):4747–4752. doi: 10.1210/jcem.86.10.7884. [DOI] [PubMed] [Google Scholar]
- 135.Su MA, Giang K, Zumer K, et al. Mechanisms of an autoimmunity syndrome in mice caused by a dominant mutation in Aire. J. Clin. Invest 2008;118(5):1712–1726. doi: 10.1172/JCI34523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abbott JK, Huoh Y-S, Reynolds PR, et al. Dominant-negative loss of function arises from a second, more frequent variant within the SAND domain of autoimmune regulator (AIRE). J. Autoimmun 2018;88:114–120. doi: 10.1016/j.jaut.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eriksson D, Bianchi M, Landegren N, et al. Common genetic variation in the autoimmune regulator (AIRE) locus is associated with autoimmune Addison’s disease in Sweden. Sci. Rep 2018;8(1):8395. doi: 10.1038/s41598-018-26842-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nishijima H, Kajimoto T, Matsuoka Y, et al. Paradoxical development of polymyositis-like autoimmunity through augmented expression of autoimmune regulator (AIRE). J. Autoimmun 2018;86:75–92. doi: 10.1016/j.jaut.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 139.Rautemaa R, Richardson M, Pfaller M, Koukila-Kähkölä P, Perheentupa J, Saxén H. Decreased susceptibility of Candida albicans to azole antifungals: a complication of long-term treatment in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) patients. J. Antimicrob. Chemother 2007;60(4):889–892. doi: 10.1093/jac/dkm299. [DOI] [PubMed] [Google Scholar]
- 140.Break TJ, Desai JV, Healey KR, et al. VT-1598 inhibits the in vitro growth of mucosal Candida strains and protects against fluconazole-susceptible and -resistant oral candidiasis in IL-17 signalling-deficient mice. J. Antimicrob. Chemother 2018;73(8):2089–2094. doi: 10.1093/jac/dky170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Break TJ, Desai JV, Natarajan M, et al. VT-1161 protects mice against oropharyngeal candidiasis caused by fluconazole-susceptible and -resistant Candida albicans. J. Antimicrob. Chemother 2018;73(1):151–155. doi: 10.1093/jac/dkx352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ibrahim AS, Luo G, Gebremariam T, et al. NDV-3 protects mice from vulvovaginal candidiasis through T- and B-cell immune response. Vaccine 2013;31(47):5549–5556. doi: 10.1016/j.vaccine.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Edwards JE, Schwartz MM, Schmidt CS, et al. A Fungal Immunotherapeutic Vaccine (NDV-3A) for Treatment of Recurrent Vulvovaginal Candidiasis-A Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Infect. Dis 2018;66(12):1928–1936. doi: 10.1093/cid/ciy185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mannstadt M, Clarke BL, Vokes T, et al. Efficacy and safety of recombinant human parathyroid hormone (1–84) in hypoparathyroidism (REPLACE): a double-blind, placebo-controlled, randomised, phase 3 study. Lancet Diabetes Endocrinol 2013;1(4):275–283. doi: 10.1016/S2213-8587(13)70106-2. [DOI] [PubMed] [Google Scholar]
- 145.Winer KK, Yanovski JA, Cutler GB. Synthetic human parathyroid hormone 1–34 vs calcitriol and calcium in the treatment of hypoparathyroidism. JAMA 1996;276(8):631–636. [PubMed] [Google Scholar]
- 146.Pearce SH, Cheetham T, Imrie H, et al. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am. J. Hum. Genet 1998;63(6):1675–1684. doi: 10.1086/302145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Proust-Lemoine E, Saugier-Véber P, Lefranc D, et al. Autoimmune polyendocrine syndrome type 1 in north-western France: AIRE gene mutation specificities and severe forms needing immunosuppressive therapies. Horm Res Paediatr 2010;74(4):275–284. doi: 10.1159/000297714. [DOI] [PubMed] [Google Scholar]
- 148.Sidiropoulos K, Viteri G, Sevilla C, et al. Reactome enhanced pathway visualization. Bioinformatics 2017;33(21):3461–3467. doi: 10.1093/bioinformatics/btx441. [DOI] [PMC free article] [PubMed] [Google Scholar]