

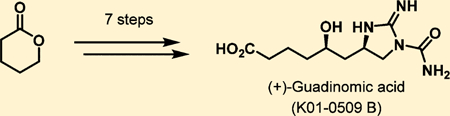
Abstract
Protecting-group-free total synthesis of (+)-guadinomic acid is reported using δ-valerolactone as a readily available starting material. The protocol utilizes the recent hydroxyl-directed guanidylation of unactivated alkenes as an approach for direct stereoselective incorporation of the guanidine unit furnishing the natural product in 7 steps.
Graphical Abstract
The interest in guanidine-containing natural products has been entrenched within the synthetic community because of their rich diversity of biochemical properties on one hand and challenges associated with the preparation of these nitrogen-rich compounds on the other.1 Despite impressive recent achievements in stereoselective synthesis, the synthesis of guanidines is still primarily accomplished by guanidylation of amines with different pyrrazolylcarboxamidines,2 S-alkyliso- thiourea reagents,3 trifluoromethanesulfonyl guanidine reagents (Goodman reagent),4 or cyanamide.5 Implementation of these protocols for the stereoselective preparation of cyclic guanidines is especially challenging and typically requires multistep synthesis of linear chiral amine precursors bearing additional functionality for subsequent guanidine cyclization, resulting in extended synthetic routes and reducing the overall efficiency.
Recently, we developed an alternative method for the stereoselective synthesis of cyclic guanidines by directed vicinal diamination of alkenes. This method enables efficient delivery of guanidine as an entire unit from free guanidine with a high level of stereocontrol.6 Herein, we demonstrate the utility of this method with a short, protecting-group-free synthesis of (+)-guadinomic acid (1).
Guadinomic acid was isolated along with structurally similar guadinomines A—D (Figure 1) from the culture broth of Streptomyces sp. K01–0509 by Omura and co-workers during a search for new anti-infective agents.7 Certain members of the guadinomine family of natural products exhibit high inhibitiory potency toward bacterial type III secretion system (T3SS) used to deliver effector proteins into the host cell during infection process. For instance, guadinomine B is the most potent T3SS inhibitor known to date with IC50 of 14 nM.7 T3SS is used by several virulent strains of medically important Gram-negative pathogens including Escherichia coli, Salmonella spp., Yersinia spp., Chlamydia spp., Vibrio spp., and Pseudomonas spp.8 Therefore, molecules that are able to disrupt this biological mechanism are potentially promising antibiotic agents.
Figure 1.
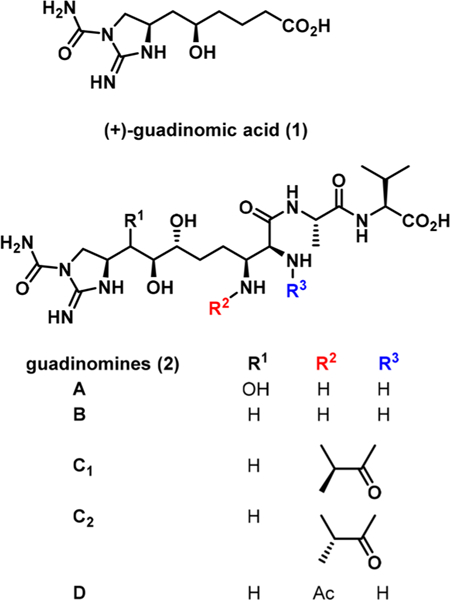
Structures of guadinomic acid (1) and guadinomines A−D(2).
While a few other natural products (enduracidin,9 minosaminomycin,10 and an amino acid derivative from the marine ascidian11) contain a similar five-membered cyclic guanidine unit, the carbomoylated guanidine moiety of guadinomic acid 1 and guadinomines 2 is unique. Omura and co-workers previously accessed guadinomic acid 1 in 15 steps from allyl alcohol 3. The stereochemistry of the molecule was established by a combination of Sharpless epoxidation and an asymmetric Henry reaction.12 A similar protocol was later used by this team to synthesize guadinomines B and C2.13 Later, Tae and co-workers reported a 9-step synthesis of 1 starting from chiral epoxy alkenol 4, not available commercially, as a key building block.14 Both teams utilized guanidylation of a primary amine with Goodman’s reagent followed by Mitsunobu cyclization to access the five- membered guanidine ring of 1. Although these approaches allowed for the expansion of knowledge pertaining to the chemistry and biology of these carbomoylated guanidine alkaloids, in both cases the guanidine was introduced by a laborious multistep protocol that diminished the overall synthetic efficiency. This stimulated our interest in developing a de novo approach to the synthesis of (+)-guadinomic acid 1. We envisioned that the guanidine ring of the molecule can be introduced by a stereoselective diamination of chiral homoallyl alcohol 5, which in turn can be prepared from δ-valerolactone (Scheme 1).
Scheme 1.
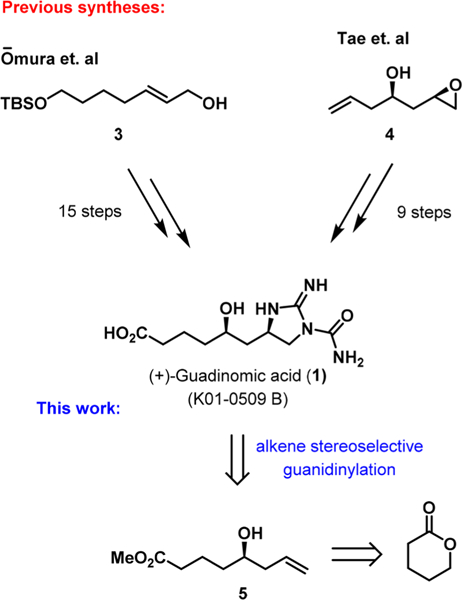
Previous Syntheses of (+)-Guadinomic Acid and Retrosynthetic Analysis
The synthesis commenced with the preparation of aldehyde 6, which was accessed from δ-valerolactone by transesterification with methanol and Swern oxidation. In order to introduce the chiral homoallyllic alcohol, allylation of aldehyde 6 with Leighton’s allylsilane was explored.15 The expected alcohol 5 was obtained in high yield and excellent enantiopurity (89% yield, 95% ee determined after benzoylation of the hydroxy group) when performing the reaction at 0 °C in the presence of 5 mol % of scandium triflate. We then set out to explore stereoselective construction of the five- membered guanidine unit. The reaction between alcohol 5 and 1,1’-carbonyldiimidazole followed by in situ treatment of the intermediate (l-imidazolyl)formate with a solution of guanidine in DMF at ambient temperature furnished carbamate 8 (Scheme 2).
Scheme 2.
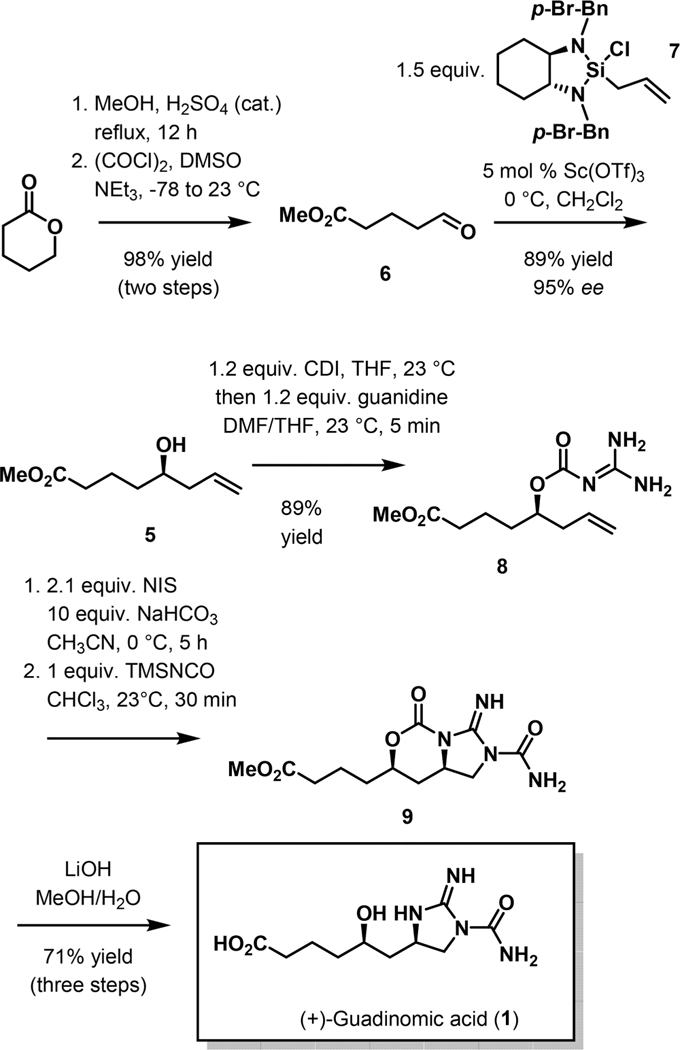
Synthesis of (+)-Guadinomic Acid (1)
With acylguanidine 8 in hand, we focused our efforts on the installation of the 2-iminoimidazolidine unit by intramolecular diamination of the terminal double bond directed by the hydroxy group. Treatment of compound 8 with 2 equiv of N- iodosuccinimide and an excess of sodium bicarbonate in acetonitrile resulted in a clean reaction producing a highly polar cyclic guanidine as a single stereoisomer. We anticipated that treatment of this compound with silyl isocyanate as a precursor of the carbomoyl group would proceed in a regioselective fashion due to the anticipated higher nucleophilicity of the endocyclic nitrogen.16,17 Indeed, treatment of the crude cyclization product with an equimolar amount of (trimethylsilyl)isocyanate resulted in the formation of a single product (9), which, when subjected to hydrolysis under basic conditions, produced (+)-guadinomic acid in 71% yield over 3 steps and following reverse-phase chromatographic purification.
In summary, the carbomoylated guanidine alkaloid (+)-gua- dinomic acid 1 was prepared in 7 steps with an overall yield of 55% from δ-valerolactone. The synthetic design based on directed cycloguanidylation of unactivated alkenes enabled an efficient protecting-group-free synthesis of 1. The brevity and simplicity of the method is expected to improve the synthesis of complex guadinidine natural products.
EXPERIMENTAL SECTION
All reactions were carried out with oven or flame-dried glassware, unless the reaction procedure states otherwise. Tetrahydrofuran (THF) was distilled from sodium-benzophenone in a continuous still under an atmosphere of argon. Dichloromethane (DCM) and acetonitrile were distilled from calcium hydride in a continuous still under and atmosphere of argon. Reaction temperatures were controlled by IKA ETS-D4 fuzzy thermo couples. Analytical thin-layer chromatography (TLC) was performed using precoated TLC plates with Silica Gel 60 F254 (EMD no. 5715–7) and visualized using combinations of UV, anisaldehyde, ceric ammonium molybdate (CAM), potassium permanganate, and iodine staining. Flash column chromatography was performed using 40–63 μm silica gel (EMD, Geduran, no. 1.11567.9026) as the stationary phase. Proton nuclear magnetic resonance spectra were recorded at 500 and 600 MHz on Varian Unity Inova spectrometers. Carbon nuclear magnetic resonance spectra were recorded at 126 and 151 MHz on Varian Unity Inova spectrometers. All chemical shifts were reported in 8 units relative to tetramethylsilane. Optical rotations were measured on a Rudolph Autopol III polarimeter. High-resolution mass spectral data were obtained by the Mass Spectrometry Laboratory at the University of California, Santa Barbara using Waters Micromass QTOF2 tandem ESI mass spectrometer. Low-resolution mass spectral data were obtained using ADVION expression CMS mass spectrometer.
Methyl 5-Oxopentanoate 6.
δ--Valerolactone (2.15 g, 21.6 mmol) and sulfuric acid (0.13 mL, 2.37 mmol) were dissolved in methanol (43.0 mL), and the mixture was heated at reflux for 12 h. The reaction mixture was then cooled to room temperature. Solid sodium bicarbonate (0.50 g, 5.95 mmol) was added, and the mixture was stirred for 10 min. Solids were removed by filtration. After the filtrate was concentrated to half of the original volume, it was diluted with ethyl acetate (50 mL) and washed with water (25 mL). The organic phase was separated, and the aqueous solution was extracted with ethyl acetate (3 X 25 mL). The combined organic phase was washed with water (2 X 30 mL), dried over Na2SO4, and concentrated to dryness to produce methyl 5-hydroxypentanoate as a colorless oil (2.82 g, 21.4 mmol, 99% yield) which was used without further purification. 1H NMR (500 MHz, CDCLl3) δ (ppm): 3.68 (s, 3H), 3.66 (t, J = 6.5 Hz, 2H), 2.36 (t, J = 7.5 Hz, 2H), 1.73 (m, 2H), 1.60 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm): 174.1, 62.2, 51.5, 33.6, 32.0, 21.1. LRMS-APCI (m/z): [M + H]+ calcd for C6H13O3+, 133.1; found, 133.2.
Dimethyl sulfoxide (1.90 mL, 26.0 mmol) was added dropwise to a solution of oxalyl chloride (1.10 mL, 12.8 mmol) in dichloromethane (60 mL) at −78 °C over 30 min. The resulting mixture was stirred for 20 min before a solution of methyl 5-hydroxypentanoate (1.28 g, 9.70 mmol) in dichloromethane (9.0 mL) was added dropwise over 20 min at −78 °C. The mixture was stirred for an additional 20 min, and triethylamine (5.6 mL, 40.0 mmol) was added dropwise over 15 min. After 10 min the reaction mixture was transferred to an ice bath and stirred at 0 °C until complete conversion was observed by TLC. The reaction mixture was then poured into water (60 mL), the organic phase was separated, and the aqueous solution was extracted with dichloromethane (4 X 40 mL). The combined organic phase was washed with water (2 X 80 mL) and brine (2 X 80 mL), dried over Na2SO4, and concentrated to dryness to give a pale-yellow oil (1.25 g, 9.60 mmol, 99% yield) which was used immediately without further purification. 1H NMR (500 MHz, CDCl3) δ (ppm): 9.77 (t, J =1.5 Hz, 1H), 3.67 (s, 3H), 2.53 (dt, J = 1.5, 7.0 Hz, 2H), 2.37 (t, J = 7.0 Hz, 2H), 1.95 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm): 201.4, 173.3, 51.6, 42.9, 32.9, 17.3. HRMS-ESI (m/z): [M + H]+ calcd for C6H11O3, 131.0708; found, 131.0726.
(+)-Methyl (5R)-5-Hydroxyoct-7-enoate 5.
A solution of allylsilane 715 (0.961 g, 1.7 mmol) in dichloromethane (2.4 mL) was added dropwise to a stirring solution of methyl 5-oxopentanoate 6 (128 mg, 0.98 mmol) in dichloromethane (14.4 mL) at 0 °C. Scandium(III) triflate (355 mg, 0.72 mmol) was then added to the stirring reaction mixture in one portion. After stirring for 1 h at ambient temperature, complete disappearance of starting material was observed by TLC, and the reaction was quenched by addition of tetrabutylammonium fluoride (75 mg, 2.88 mmol) solution in tetrahydrofuran (2.88 mL). Then the reaction mixture was stirred for 1 h followed by direct purification by column chromatography (30% EtOAc in hexanes) to produce the product as a colorless oil (150 mg, 0.872 mmol, 89% yield). (c 0.5, CHCl3). 1H NMR (500 MHz, CDO3) δ (ppm): 5.79 (m, 1H), 5.10 (m, 2H), 3.64 (s, 3H), 3.62 (m, 1H), 2.32 (t, J = 7.0 Hz, 2H), 2.26 (m, 1H), 2.13 (m, 1H), 1.90 (d, J = 5.0 Hz, 1H), 1.77 (m, 1H), 1.68 (m, 1H), 1.47 (m, 2H). 13C NMR (151 MHz, CDCl3) δ (ppm): 174.1, 134.6, 118.1, 70.1, 51.5, 41.9, 36.0, 33.8, 21.0. HRMS-ESI (m/z): [M + H]+ calcd for C9H17O3, 173.1178; found, 173.1165.
In order to determine the enantiopurity of (+)-methyl (5R)-5- hydroxyoct-7-enoate 5 by HPLC/UV, benzoyl chloride (30 μL, 0.26 mmol) was added dropwise to a solution of alcohol 5 (30 mg, 0.18 mmol) and pyridine (30 μL, 0.37 mmol) in dichloromethane (0.6 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h and an additional hour at room temperature. After TLC showed complete consumption of the starting material the reaction mixture was diluted with dichloromethane (10 mL) and washed with 1 M HCl (10 mL). The organic layer was separated, and the aqueous solution was extracted with dichloromethane (3 X 7 mL). The combined organic phase was washed with water (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated to dryness. The crude product was purified by column chromatography (10% EtOAc in hexanes) to give corresponding benzoate of alcohol 5 as a pale-yellow oil (30.0 mg, 60% yield, 0.11 mmol). (c 0.20, CHCl3). 1H NMR (600 MHz, CDCl3) δ (ppm): 8.03 (d, J = 6.0 Hz, 2H), 7.55 (t, J = 6.0 Hz, 1H), 7.43 (t, J = 6.0 Hz, 2H), 5.81 (dddd, J = 7.0, 7.0, 10.0, 17.0 Hz, 1H), 5.18 (m, 1H), 5.11 (m, 1H), 5.07 (m, 1H), 3.65 (m, 3H), 2.46 (t, J = 6.5 Hz, 2H), 2.35 (dd, J = 5.0, 7.5 Hz, 2h), 7.73 (m, 4H). 13C NMR (126 MHz, CDCl3) δ (ppm): 173.6, 166.1, 133.3, 132.8, 130.5, 129.5,128.3, 118.0, 73.4, 51.5, 38.5, 33.7, 32.9, 20.7. HRMS-ESI (m/ z): [M + Na]+ calcd for C16H20NaO4 299.1259; found, 299.1271. The enantiopurity of the material was determined by HPLC analysis. [Chiralcel AD-H; 1% i-PrOH-Hexanes; flow rate = 1 mL/min; detection at 215 nm; t1 = 23.36 min. (major), t2 = 21.81 min. (minor)].
(+)-Methyl (5R)-5-(((Diaminomethylene)carbamoyl)oxy)oct-7-enoate) 8.
To a solution of methyl (5R)-5-hydroxyoct-7-enoate 5 (200 mg, 1.16 mmol) in tetrahydrofuran (4.0 mL), 1,1’-carbon- yldiimidazole (227 mg, 1.40 mmol) was added at ambient temperature, and the reaction was stirred overnight to prepare corresponding N-alkoxycarbonyl imidazole derivative.
Guanidine hydrochloride (0.46 g, 4.8 mmol) was added to a freshly prepared solution of sodium methoxide obtained by dissolving 110 mg (4.8 mmol) of sodium in dry methanol (3.6 mL). The reaction mixture was stirred for 10 min and concentrated to dryness under reduced pressure. The material was dried under vacuum for 10 min and dissolved in 8.2 mL of DMF. A crude solution of N- alkoxycarbonyl imidazole obtained in the first step was added dropwise over 10 min to the solution of guanidine in DMF. The reaction was stirred for an additional 10 min and poured into 60 mL of water. The product was extracted with EtOAc (4 X 30 mL). The combined organic phase was sequentially washed with water (2 X 60 mL), saturated aqueous ammonium chloride (2 X 60 mL) and brine (60 mL), then dried over Na2SO4, and the organic solvent was removed under reduced pressure. The crude product was subjected to column chromatography (5% MeOH in dichloromethane -→10% MeOH in dichloromethane) to deliver the product (267 mg, 1.04 mmol, 89% yield) as a colorless oil. (c 0.43, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 5.74 (ddt, J = 17.2, 10.1, 7.1 Hz, 1H), 5.06 (m, 1H), 5.03 (m, 1H), 4.73 (m, 1H), 3.65 (s, 3 H), 2.32 (m, 4 H), 1.69 (m, 1H), 1.60 (m, 1H), 1.55 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm): 174.3, 161.8, 161.7, 133.6, 117.7, 73.3, 51.6,38.7,33.5, 33.0, 20.6. HRMS-ESI (m/z): [M + H]+ calcd for C11H20N3O4, 258.1454; found, 258.1473.
(+)-Guadinomic Acid 1.
A solution of compound 8 (48 mg, 0.187 mmol) in 3.7 mL of acetonitrile was added to a 10 mL round- bottom flask equipped with a magnetic stirring bar and an argon inlet adapter. Sodium bicarbonate (157 mg, 1.90 mmol) was added, and the mixture was cooled to 0 °C before freshly recrystallized N- iodosuccinimide (88 mg, 0.39 mmol) was added in one portion. The heterogeneous mixture was vigorously stirred for 5 h followed by quench with 10% aqueous sodium sulfite (300.0 mL). The crude product was extracted with EtOAc (30 mL). The organic layer was separated and concentrated to dryness under reduced pressure. The residue was dissolved in ethyl acetate (30 mL) and washed with the same sodium sulfite solution used previously. The organic layer was separated, and the aqueous phase was additionally extracted with ethyl acetate (3 X 15 mL). The combined organic phase was dried over Na2SO4 and concentrated to dryness under reduced pressure to give the crude product as a yellow solid that was used in the next step without further purification.
The material obtained in the previous step was dissolved in dry CDCl3 (2.0 mL), and trimethylsilyl isocyanate (24.0 μL, 0.19 mmol) was titrated into the solution in 6 μL increments and monitored by 1H NMR. This process typically took 3 h and required a total of 1 equiv of isocyanate. When complete disappearance of starting material was observed by 1H NMR, a saturated solution of brine (2 mL) was added, and the mixture was stirred for 15 min. The product was extracted with dichloromethane (2 X 10 mL). The combined organic phase was washed with brine (10 mL), dried over Na2SO4, and concentrated to dryness under reduced pressure to give a crude product 9 as a white solid which was submitted to hydrolysis without purification.
A solution of lithium hydroxide monohydrate (12.0 mg, 0.29 mmol) was added to the solution of compound 9 in a mixture of methanol/water (0.5 mL, 1:1, v/v). The reaction mixture was stirred vigorously for 2 h at room temperature. The basic solution was then neutralized with trifluoroacetic acid, and the product was directly isolated by column chromatography on C18 reverse-phase silica gel (100% H2O) to provide (+)-guadinomic acid 1 as a white solid (35.0 mg, 71% yield, 0.14 mmol). LH and 13C NMR data for obtained (+)-guadinomic acid 1 are in full accordance with those reported in the literature.12 (c 0.2, CH3OH). 1H NMR (500 MHz, D2O) 5 (ppm): 4.25 (m, 2H), 3.78 (m, 2H), 2.26 (t, J = 7.0 Hz, 2H), 1.83 (m, 2H), 1.68 (m, 1H), 1.58 (m, 1H), 1.49 (m, 2H). 13C NMR (126 MHz, D2O) δ (ppm): 177.5, 171.9, 149.5, 135.8, 129.4, 128.9,128.1, 124.4, 123.1, 122.4, 121.0, 119.3, 115.2, 83.6, 56.1, 51.4, 35.3, 28.2. HRMS-ESI (m/z): [M + H] + calcd for C10H19O4 259.1406; found, 259.1428.
ACKNOWLEDGMENTS
This work was supported by NIH (NIGMS R01–077379 and NIEHS R03 ES025345–01). Dr. Hongjun Zhou is acknowledged for assistance with NMR spectroscopy. Dr. Dmitriy Uchenik and the UCSB mass spectroscopy facility are thanked for assistance with mass spectral analysis.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS_Publications_website at DOI: 10.1021/acs.joc.8b01214.
1H and 13C NMR spectra of all synthesized compounds and HPLC trace analysis of chiral intermediates (PDF)
REFERENCES
- (1).For a review see: (a) Berlinck R G. S; Romminger S The Chemistry and Biology of Guanidine Natural Products. Nat. Prod. Rep. 2016, 33, 456–490. [DOI] [PubMed] [Google Scholar]; (b) Ma Y; De S; Chen C Syntheses of Cyclic Guanidine-Containing Natural Products. Tetrahedron 2015, 71, 1145–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mailyan AK; Eickhoff JA; Minakova AS; Gu Z; Lu P; Zakarian A Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C—N Bonds in Total Synthesis. Chem. Rev. 2016, 116, 4441—4557. [DOI] [PubMed] [Google Scholar]
- (2).(a) Bernatowicz MS; Wu Y; Matsueda GR Urethane Protected Derivatives of 1-Guanylpyrazole for the Mild and Efficient Preparation of Guanidines. Tetrahedron Lett. 1993, 34, 3389–3392. [Google Scholar]; (b) Drake B; Patek M; Lebl MA Convenient Preparation of Monosubstituted N,N’-di(Boc)-Protected Guanidines. Synthesis 1994, 1994, 579–582. [Google Scholar]; (c) Katritzky AR; Parris RL; Allin SM; Steel PS Benzotriazole-1-carboxamidinium Tosylate: An Alternative Method for the Conversion of Amines to Guanidines. Synth. Commun. 1995, 25, 1173–1186. [Google Scholar]
- (3).(a) Poss MA; Iwanowicz E; Reid JA; Lin J; Gu Z Mild and Efficient Method for the Preparation of Guanidines. Tetrahedron Lett. 1992, 33, 5933–5936. [Google Scholar]; (b) Yong YF; Kowalski JA; Lipton MA Facile and Efficient Guanylation of Amines Using Thioureas and Mukaiyama’s Reagent. J. Org. Chem. 1997, 62, 1540–1542. [Google Scholar]; (c) Levallet C; Lerpiniere J; Ko SY The HgCl2-Promoted Guanylation Reaction: The Scope and Limitations. Tetrahedron 1997, 53, 5291–5304. [Google Scholar]
- (4).(a) Feichtinger K; Zapf C; Sings HL; Goodman M Diprotected Triflylguanidines: A New Class of Guanidinylation Reagents. J. Org. Chem. 1998, 63, 3804–3805. [Google Scholar]; (b) Feichtinger K; Sings HL; Baker TJ; Matthews K; Goodman MJ Triurethane- Protected Guanidines and Triflyldiurethane-Protected Guanidines: New Reagents for Guanidinylation Reactions. J. Org. Chem. 1998, 63, 8432–8439. [Google Scholar]
- (5).(a) Davis TL Guanidine Nitrate. Org. Synth. 1927, 7, 46. [Google Scholar]; (b) Dukat M; AbdelRahman A; Ismaiel A; Ingher S; Teitler M; Gyermek L; Glennon RA Structure–Activity Relationships for the Binding of Arylpiperazines and Arylbiguanides at 5-HT3 Serotonin Receptors. J. Med. Chem. 1996, 39, 4017–4026. [DOI] [PubMed] [Google Scholar]; (c) Tsubokura K; Iwata T; Taichi M; Kurbangalieva A; Fukase K; Nakao Y; Tanaka K Direct Guanylation of Amino Groups by Cyanamide in Water: Catalytic Generation and Activation of Unsubstituted Carbodiimide by Scandium(III) Triflate. Synlett 2014, 25, 1302– 1306. [Google Scholar]; (d) Li J; Neuville L Copper-Catalyzed Oxidative Three- Component Synthesis of N, N’,N”-Trisubstituted Guanidines. Org. Lett. 2013, 15, 6124–6127. [DOI] [PubMed] [Google Scholar]; (e) Looper RE; Haussener TJ; Mack JBC Chlorotrimethylsilane Activation of Acylcyanamides for the Synthesis of Mono-N-acylguanidines. J. Org. Chem. 2011, 76, 6967– 6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mailyan AK; Young K; Chen JL; Reid BT; Zakarian A Stereoselective Synthesis of Cyclic Guanidines by Directed Diamina- tion of Unactivated Alkenes. Org. Lett. 2016, 18, 5532–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Iwatsuki M; Uchida R; Yoshijima H; Ui H; Shiomi K; Matsumoto A; Takahashi Y; Abe A; Tomoda H; Omura SJ Guadinomines, Type III Secretion System Inhibitors, Produced by Streptomyces sp. K01–0509. J. Antibiot. 2008, 61, 222–229. [DOI] [PubMed] [Google Scholar]; (b) Iwatsuki M; Uchida R; Yoshijima H; Ui H; Shiomi K; Kim Y-P; Hirose T; Sunazuka T; Abe A; Tomoda H; Omura S Guadinomines, Type III Secretion System Inhibitors, Produced by Streptomyces sp. K01–0509. J. Antibiot. 2008, 61, 230–236. [DOI] [PubMed] [Google Scholar]; (c) Holmes TC; May AE; Zaleta-Rivera K; Ruby JG; Skewes-Cox P; Fischbach MA; DeRisi JL; Iwatsuki M; Omura S; Khosla C Molecular Insights into the Biosynthesis of Guadinomine: A Type III Secretion System Inhibitor. J. Am. Chem. Soc. 2012, 134, 17797–17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ghosh P Microbiol. Process of Protein Transport by the Type III Secretion System. Mol Biol. Rev. 2004, 68, 771–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Horii S; Kameda YJ Enduracidin, A New Antibiotic. V. Structures of New Basic Amino Acids, Enduracidine and Alloendur- acidine. J. Antibiot. 1968, 21, 665–667. [PubMed] [Google Scholar]; (b) Higashide E; Hatano K; Shibata M; Nakazawa K Enduracidin, A New Antibiotic. I. Streptomyces Fungicidicus No. 5477, An Enduracidin Producing Organism. J. Antibiot. 1968, 21, 126–137. [PubMed] [Google Scholar]; (c) Asai M; Muroi M; Sugita N; Kawashima H; Mizuno K; Miyake A Enduracidin, ANew Antibiotic. II. Isolation and Characterization. J. Antibiot. 1968, 21, 138–146. [DOI] [PubMed] [Google Scholar]
- (10).(a) Hamada M; Kondo J; Yokoyama T; Miura K; Iinuma K; Yamamoto H; Maeda K; Takeuchi T; Umezawa H Minosaminomycin, a New Antibiotic Containing Myo-inosamine. J. Antibiot. 1974, 27, 81–83. [DOI] [PubMed] [Google Scholar]; (b) Iinuma K; Kondo S; Maeda K; Umezawa H Structure of Minosaminomycin. J. Antibiot. 1975, 28, 613–615. [DOI] [PubMed] [Google Scholar]
- (11).Garcia A; Vazquez MJ; Qumoa E; Riguera R; Debitus C New Amino Acid Derivatives from the Marine Ascidian Leptoclinides dubius. J. Nat. Prod. 1996, 59, 782–785. [Google Scholar]
- (12).Tsuchiya S; Sunazuka T; Hirose T; Mori R; Tanaka T; Iwatsuki M; Omura S Asymmetric Total Synthesis of (+)-K01– 0509 B: Determination of Absolute Configuration. Org. Lett. 2006, 8, 5577–5580. [DOI] [PubMed] [Google Scholar]
- (13).Hirose T; Sunazuka T; Tsuchiya S; Tanaka T; Kojima Y; Mori R; Iwatsuki M; O mura S Total Synthesis and Determination of the Absolute Configuration of Guadinomines B and C2. Chem. - Eur. J. 2008, 14, 8220–8238. [DOI] [PubMed] [Google Scholar]
- (14).Kim H; Kim M-Y; Tae J Concise Asymmetric Total Synthesis of ent-Guadinomic Acid from an Epoxy Alkenol. Synlett 2009, 2009, 2949–2952. [Google Scholar]
- (15).Kim H; Ho S; Leighton JL A More Comprehensive and Highly Practical Solution to Enantioselective Aldehyde Crotylation. J. Am. Chem. Soc. 2011, 133, 6517–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ishikawa F; Kosasayama A; Nakamura S; Konno T Cyclic Guanidines. I. Synthesis of Hypoglycemic 1-Substituted 2-Imino-1, 3- diazacycloalkanes. Chem. Pharm. Bull. 1978, 26, 3658–3665. [DOI] [PubMed] [Google Scholar]
- (17).Mailyan AK; Chen JL; Li W; Keller AA; Sternisha SM; Miller BG; Zakarian A Short Total Synthesis of [15N5]-Cylindrospermopsins from 15NH4Cl Enables Precise Quantification of Freshwater Cyanobacterial Contamination. J. Am. Chem. Soc. 2018, 140, 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]