

Abstract
In addition to cognitive impairments, neurodevelopmental disorders (NDDs) often result in sensory processing deficits. However, the biological mechanisms that underlie impaired sensory processing associated with NDDs are generally understudied and poorly understood. We found that SYNGAP1 haploinsufficiency in humans, which causes a sporadic neurodevelopmental disorder defined by cognitive impairment, autistic features, and epilepsy, also leads to deficits in tactile-related sensory processing. In vivo neurophysiological analysis in Syngap1 mouse models revealed that upper-lamina neurons in somatosensory cortex (SSC) weakly encode information related to touch. This was caused by reduced synaptic connectivity and impaired intrinsic excitability within upper-lamina SSC neurons. These results were unexpected given that Syngap1 heterozygosity is known to cause circuit hyperexcitability in brain areas more directly linked to cognitive functions. Thus, Syngap1 heterozygosity causes a range of circuit-specific pathologies, including reduced activity within cortical neurons required for touch processing, which may contribute to sensory phenotypes observed in patients.
Keywords: Intellectual Disability, Epilepsy, Autism, Circuitry, Sensory processing, Synapse, Syngap1
INTRODUCTION
Neurodevelopmental disorders (NDD) often result in poor cognitive functioning and impaired behavioral adaptations. While the precise neurobiological and neurophysiological mechanisms that underlie these impairments remain unknown, it is generally understood that they are a consequence of impaired connectivity and processing within neuronal networks1-4. However, it remains unclear what brain areas are disrupted and how neural processing within affected areas contributes to cognitive and behavioral impairments. Therefore, it is critical to identify dysfunctional brain areas that contribute to NDD-relevant phenotypes and to then define the circuit-based mechanisms that contribute to them.
Sensory processing impairments are nearly ubiquitous in NDDs, with a growing appreciation for their central role in behavioral and cognitive deficits associated with these5, and related6, disorders. Mechanistic studies in NDD animal models have traditionally focused on the circuits and synapses located in brain regions associated with cognitive functions4. These studies have demonstrated that NDD risk genes impair synapse properties, such as plasticity and excitatory-inhibitory ratios, which are believed to underlie impaired cognitive functions and common comorbidities, such as epilepsy. However, less attention has been paid to the mechanisms contributing to sensory processing deficits, particularly at the level of primary cortical circuits, which integrate ascending sensory information with top-down modulatory signals from higher cortical areas7. Indeed, higher forms of cognition require information regarding the external environment. Primary sensory cortical areas decode stimulus features8 and facilitate the construction of complex internal representations of the external world9. In this context, disrupted lower-level sensory processing in NDDs could contribute to generalized cognitive and behavioral impairments reported in patients. At present, it remains unclear how impairments in sensory processing contribute to complex cognitive and behavioral phenotypes common to NDD patients. This stems from a relative lack of mechanistic, systems-based studies investigating how highly-penetrant NDD risk factors directly impact the function of circuits that process sensory information.
A tractable entry point for such mechanistic studies is to perform in-depth biological investigations of highly-penetrant rare variants known to cause well-understood childhood NDDs10, 11. The SYNGAP1 gene is frequently mutated in intellectual disability (ID) associated with classically undefined global developmental delay12-15 and is a major risk factor for autism spectrum disorders16, 17. De novo nonsense variants in SYNGAP1 resulting in haploinsufficiency lead to a genetically-defined form of ID (MRD5; OMIM#603384) that may explain up to 1% of these cases18, 19. MRD5 patients suffer from cognitive impairments, such as absent or poor language acquisition, and very low non-verbal IQ19, 20. SYNGAP1 is also a risk factor for epileptic encephalopathies21, 22 and most MRD5 patients have comorbid epilepsy18-20. Currently, the impact of SYNGAP1/Syngap1 pathogenicity on sensory functions is unknown, but is necessary for a deeper understanding of the complex phenotypes observed in this genetically-defined NDD.
We report altered sensory functions in a SYNGAP1 patient population, including behaviors related to abnormal tactile processing. Studies in a series of construct-valid mouse models of SYNGAP1 haploinsufficiency revealed severe impairments in somatosensory cortex (SSC) circuit structure and function. Syngap1 heterozygosity resulted in reduced connectivity and hypoexcitability within upper-lamina SSC glutamatergic neurons, resulting in reduced touch-related activity within these circuits. These results were surprising, as they were distinct from mechanisms described in associational brain areas more directly linked to cognitive function, such as increased synaptic excitability of circuits in the hippocampus23 and prefrontal cortex24. Thus, pathogenicity of Syngap1 causes a range of circuit-specific pathologies. We propose a generalizable scheme where interactions among region-specific circuit pathologies due to causally-linked NDD risk variants drive complex cognitive and behavioral phenotypes observed in patients.
RESULTS
SYNGAP1 heterozygosity in humans leads to touch-related sensory processing defects
To explore how sensory function is impacted by SYNGAP1 pathogenesis, we mined data from a SYNGAP1 patient registry that is a component of an ongoing retrospective MRD5 Natural History Study (NHS). Registries are essential tools to discover common phenotypes in patients with rare genetic disorders25. This registry contains a searchable database with anonymized medical records, including genetic reports and detailed medical histories for SYNGAP1 patients (https://syngap1registry.iamrare.org). Forty-eight unique registry entries contained a comprehensive medical history and completed questionnaire focusing on sensory function. Forty-five of them exhibited features consistent with sensory processing impairments (Supplementary Table 1). Twenty of these entries were supported by detailed narrative accounts describing abnormal responses to tactile stimuli, which included blunted responses to painful touch-related stimuli and/or tactile-seeking or tactile-aversive behaviors (Supplementary Table 2). Genetic reports were available for 17 of the 20. Fourteen of the seventeen reports identified the SYNGAP1 variant as clearly pathogenic (i.e. caused SYNGAP1 heterozygosity) and the cause of their developmental disorder (Supplementary Table 2). The other three reports included variants of undetermined, but potential, clinical significance that require further biological validation, such as predicted splice alterations or missense mutations. Data from these detailed patient entries indicate that disrupting expression or function of SYNGAP1 in humans can lead to tactile-related sensory impairments in addition to cognitive impairment and seizure.
Syngap1 heterozygosity in mice causes touch-related deficits in cortical circuit activation
We next utilized mouse models to understand the biological mechanisms of sensory processing deficits associated with human SYNGAP1 heterozygosity. The barrel field of the SSC processes touch-related sensory information generated by movements and angular deflections of whiskers8. Computations here promote touch-related sensory perception and object localization, and facilitate the creation of spatial maps of the environment8, 9; all processes that facilitate higher-order cognitive functions. First, we mapped cortical receptive fields of whiskers in anesthetized Syngap1 mice by measuring intrinsic optical signals (IOS) generated by whisker deflections (Fig. 1a-b). The amplitude of cortical IOS elicited from C2 and β whisker deflections, as measured by the peak IOS response (Fig. 1c-e) and the area of the absolute value of the thresholded signal (Fig. 1g), was significantly reduced in both whisker-related receptive fields tested in Syngap1 heterozygous mice (Hets). This result was surprising given that Syngap1 pathogenicity has routinely been linked to circuit hyperexcitability 18, 21, 23, 24, 26, 27. However, both the distance between the two fields (Fig. 1f) and their size, as measured by the relative area of the IOS signal (Fig. 1h), did not differ from WT. Reduced cortical activation in Syngap1 mouse barrel SSC was confirmed by widefield, low-resolution imaging of barrel cortex in Thy1-GCaMP6s mice (GP4.3 line28) crossed to Syngap1 mice under anesthesia. In these studies, Syngap1 Het mice also exhibited reduced amplitudes of SSC whisker-evoked responses compared to controls (Supplementary Fig. 1a-l). Moreover, alterations in whisker-evoked signals in SSC were stimulus-dependent, with larger effect sizes occurring in response to stronger stimuli. These findings indicate that receptive field topology is generally unaltered in Syngap1 mice, but that cortical activation driven by whisker input is impaired by Syngap1 heterozygosity.
Figure 1: Reduced sensory-evoked brain activity in Syngap1 SSC.
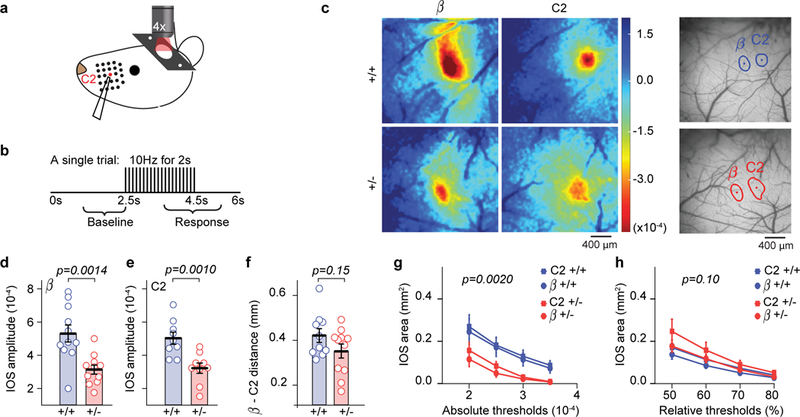
(a-b) Stimulus paradigm used during IOS imaging. (c) Example IOS signals from one animal in each genotype obtained for β and C2 whiskers. (d-f) Scatter plots showing reduced IOS amplitudes in β (d) and C2 (e) whisker fields, but normal inter-barrel distance (f) in adult Syngap1 mutants. Unpaired two-sided t-Tests: t(20)=3.76 p=0.0014 for β amplitude (n=11 WT n=11 Het mice), t(16)=3.70 p=0.001 for C2 amplitude (n=9 WT, n=9 Het mice), t(19)=1.49 p=0.15 for inter-barrel distance(n=10 WT n=11 Het mice). (g, h) Quantification of the area responding to β or C2 whisker stimulation according to absolute (g) or relative thresholding methods (h). 2-way RM-ANOVA for absolute area F(1,36)=11.18 p=0.002 for genotype, F(3,108)=1.74 p=0.163 for genotype*threshold; 2-way RM-ANOVA for relative area F(1,36)=2.8 p=0.1 for genotype, F(3,108)=2.92 p=0.037 for genotype*threshold (n=20 WT n=20 Het IOS imaging sessions from different whiskers). For d-f, open circles represent animal means, black lines indicate population means and error bars indicate SEMs. For g-h, closed circles and squares represent population means and error bars indicate SEMs. Data in this figure were acquired from two independent cohorts of animals that were pooled together.
To better understand altered SSC functional activation in Syngap1 mice, we measured spike-like supra-threshold somatic calcium events in Layer 2/3 SSC neurons. These SSC neurons were chosen because they are readily accessible by two-photon imaging and known to integrate bottom-up sensory signals originating in the periphery with information arriving from higher cortical areas7. Somatic calcium events were measured through in vivo two-photon imaging of GCaMP6 dynamics in awake, head-fixed Syngap1 mice crossed to the GP4.3 line (Fig. 2a-c). These studies were performed in awake animals because anesthesia disrupts neuronal activity and can have complex effects in mouse models of NDD risk genes29. We mapped the cortical receptive field of a single whisker and then loaded this whisker into a small plastic holder. Mice spontaneously whisked during the imaging trials, which resulted in the whisker contacting the sides of the holder (Supplementary Video 1). Thus, recorded activity during these trials was comprised of ongoing, spontaneous activity of unknown origins and activity generated by whisker movements and/or touch. Neurons in L2/3 SSC generally appeared less active in Hets compared to WTs (Fig. 2b). Further analysis revealed that while the size of the responsive population was not affected in Syngap1 Hets (Fig. 2d), neurons that were active during the imaging session had significantly smaller (Fig. 2e) and less numerous (Fig. 2f) events compared to WT neurons. After these imaging sessions, we injected the whisker pad with onabotulinumtoxinA (Botox) to paralyze whisker movements (Supplementary Video 2) and reimaged L2/3 cellular activity in the same animal population one week later. Botox had no effect on the size of the responsive population (Fig. 2d). Botox also had no effect on event amplitudes within genotypes and the difference between genotypes remained even in the presence of whisker paralysis (Fig. 2e). However, Botox had a clear impact on the number of detected events in this experiment. For example, in WT animals, whisker paralysis shifted the cumulative fraction of neuronal activity counts to the left (Fig. 2f). We reasoned that the shift in the “event number” curves after Botox injection reflects the contribution of whisker movements and/or touch to activity within this population. In contrast, Botox did not shift the activity of Het neurons (Fig. 2f). Neurons from Botox(+) Hets appeared to have activity counts that were identical to neurons in Botox(−) Hets, indicating that whisker movements and/or touch results in less activity compared to WTs. Indeed, the Het activity curves were superimposed onto that of Botox-treated WT neurons. While these shifts in spike-like activity may seem subtle, it is known that spiking within a small population of cortical neurons is sufficient to drive a behavioral response30. Thus, it is reasonable that a small, yet highly significant change in the number of detected spike-like events generated in Hets, especially spike-like activity linked to touch, is likely to be behaviorally meaningful to these animals.
Figure 2: Reduced ongoing and whisker-generated activity in SSC L2/3 neurons from awake Syngap1 mice.
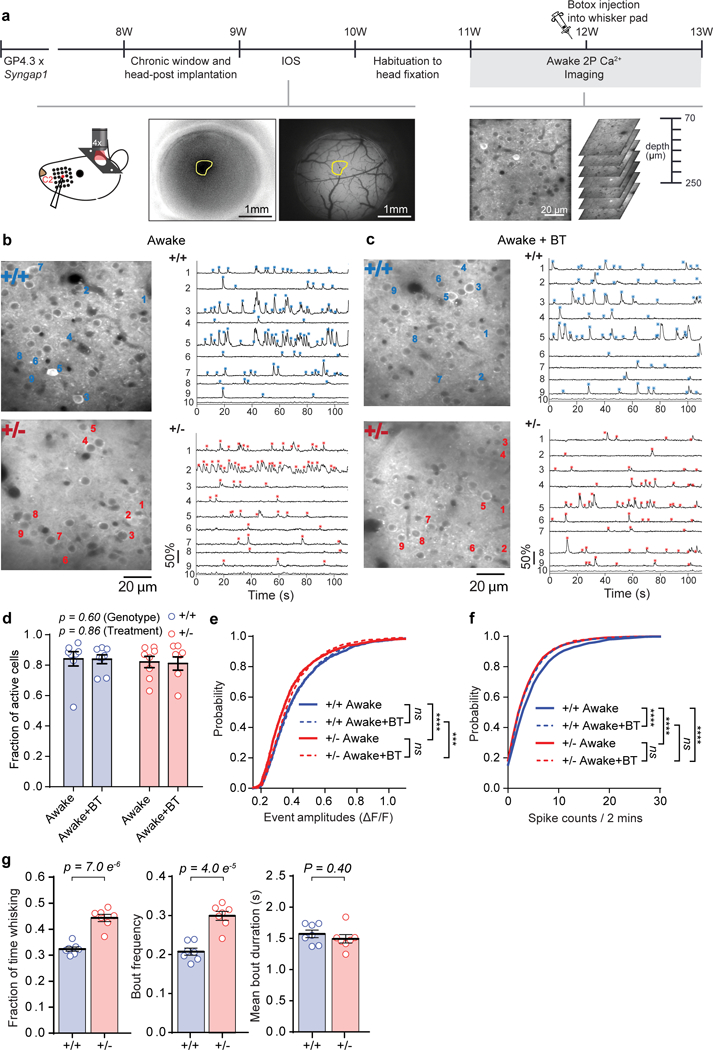
(a) Experimental setup for awake, in vivo two-photon calcium imaging in Syngap1 crossed with Thy-1 Gcamp6s4.3 mice, with and without Botox injection. (b-c) Representative in vivo two-photon microscopy images (left) and ΔF/F traces (right) of spontaneous activity in 9 (1-9) neurons of L2/3 SSC of WT and Het Syngap1 X Thy-1 Gcamp6s4.3 mice, without (b) and with (c) Botox. ROI number 10 is the neuropil signal. Asterisks indicate detected calcium events (blue for WT, red for Het). (d-f) Cellular sensory properties from awake WT and Het mice without and with Botox injection. (d) Scatter plot showing fraction of spontaneously active cells in WT and Het mice (2-way RM-ANOVA, Genotype: F(1, 14) = 0.29, p = 0.60, Treatment: F(1,14) = 0.034, p = 0.86, Interaction: F(1, 14) = 0.012, p = 0.92; WT n=8 mice, Het n=8 mice). (e-f) Cumulative probability plots of ΔF/F amplitudes (e; KS Tests: WT/Botox- vs. Het/Botox-, p= 3.38E-7; WT/Botox+ vs. Het/Botox+, p=0.0002; WT/Botox- vs. WT/Botox+, p= 0.67; Het/Botox- vs. Het/Botox+, p= 0.078; WT: nBotox-=1622 neurons, nBotox+ = 1827 neurons; Het: nBotox- = 1667 neurons, nBotox+ = 1600 neurons) and spike counts (f, KS Tests, WT/Botox- vs. Het/Botox-, p= 1.82E-7; WT/Botox- vs. WT/Botox+, p= 7.99E-6; WT/Botox- vs. Het/Botox+, p= 2.87E-8; Het/Botox- vs. Het/Botox+, p= 0.99; WT/Botox+ vs. Het/Botox+, p=0.24). (g) Whisking behavior in head-fixed Syngap1 mice (Fraction of time whisking: Unpaired t-test t(12)=7.493, p= 7.0E-6; WT n=7 mice, Het n=7 mice. Bout frequency: Unpaired t-test t(12)=6.298, p= 4.0E-5; WT n=7 mice, Het n=7 mice. Mean bout duration: Unpaired t-test t(12)=0.8714, p=0.40; WT n=7 mice, Het n=7 mice). Data were pooled from two independent cohorts of animals and thus obtained from 2169 neurons in 56 imaging planes from 8 WT mice; 1971 neurons in 55 imaging planes from 8 Het mice. In scatter plots, open circles are animal means, black lines indicate population means, and error bars indicate SEMs. ***P<0.001, ****P<0.0001 for cumulative probability plots. All statistical tests were two-sided.
The lack of Botox effects on neuronal activity in Het mice was supported by a cluster analysis of activity counts from neurons imaged in all four conditions. In each condition, active neurons were clustered into low-, medium- and high-activity populations31 based on the number of spikes during the recording session (Supplementary Fig. 2a). From this analysis, there was an overall effect on how the neurons clustered across the four groups (Supplementary Fig. 2b) and there was a difference in each of the three activity clusters when comparing the four experimental groups (Supplementary Fig. 2c). However, post hoc analyses comparing the four groups to each other revealed that all three activity clusters from the Botox(−) WTs were different from the corresponding clusters from all other groups (Supplementary Fig. 2d). The difference arose from more neurons present in the high- and medium-active clusters, and fewer in the low-active cluster, in the Botox(−) WT compared to the other three groups, further supporting the idea that Botox reduced activity within WT, but not Het, SSC circuits. Reduced activity generated from free whisking and/or touch in Syngap1 mice could be caused by impaired whisking behaviors in these animals, such as a decrease in total time whisking. Unexpectedly, Syngap1 Hets spent more time freely whisking compared to WTs (Fig. 2g). Taken together, these findings indicate activity driven by free whisker movement and/or whisker curvature driven by touch is poorly encoded by L2/3 SSC neurons in Syngap1 mice.
To further investigate the possibility that Syngap1 heterozygosity impacts SSC cellular activity generated by whisker curvature, we measured cellular response properties in SSC L2/3 neurons evoked by piezo-driven, passive whisker deflections in awake, head-fixed Syngap1/ GP4.3 mice. We observed an effect of genotype on several measures of stimulus-evoked neuronal activity (Fig. 3a-h), each consistent with reduced activation of L2/3 SSC neurons. A mild stimulus (5 pulses, 5 Hz) resulted in a smaller responsive population in Hets (Fig. 3c), but not the size of individual responses or the probability that a cell responds to the stimulus (Fig. 3d, e). However, a stronger stimulus often used in single whisker-stimulation detection paradigms, such as 60 pulses at 40 Hz, resulted in a smaller active population (Fig. 3f), a reduced amplitude of sensory-evoked events (Fig. 3g), and reduced response probability of responsive cells (Fig. 3h) in Hets. Active whisking could theoretically disrupt the quality of investigator-controlled passive deflections in a genotype-specific manner through degradation of precise whisker control. To control for this possibility, we performed stimulus evoked trials in the same cohort of animals following injection of Botox into the whisker pad. After whisker paralysis, we continued to observe reduced cellular sensory responsiveness arising from passive whisker deflections in Hets (Fig. 3i-n). For both stimuli, we observed a reduction in the response probably of the whisker-responsive population in Hets (Fig. 3k, n). However, other cellular response phenotypes were less pronounced after whisker paralysis, including the absence of change in the event amplitude of the responsive population (Fig. 3j, m) and the size of the responsive population (Fig. 3i, l). Together, these data demonstrate that whisker bending through passive deflections results in reduced activity within SSC of Syngap1 mice.
Figure 3: Reduced whisker responsiveness of SSC neurons in behaving Syngap1 mice.
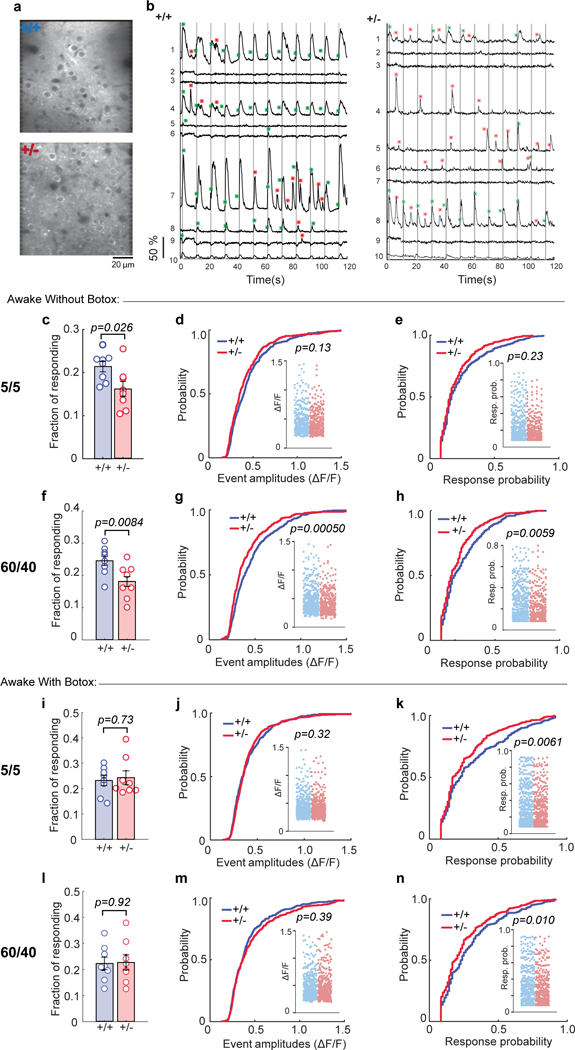
Representative in vivo two-photon microscopy images (a) and representative ΔF/F traces (b) of 9 (1-9) neurons in WT and Het (Botox-) mice in response to 5 passive whisker deflections at 5 Hz. Gray vertical lines indicate the timing of whisker stimuli. Green asterisks indicate calcium events within the response detection window. Red asterisks show spontaneous calcium events. ROI number 10 is the neuropil signal. (c-h) Cellular sensory properties from awake animals in response to 5 pulses at 5 Hz (c-e) and 60 pulses at 40 Hz whisker stimulation (f-h). (c) Scatter plot showing fraction of responding cells in WT and Het mice (Unpaired t-test t(14)=2.48, p=0.026; WT n=8 mice, Het n=8 mice). (d-e) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (d, KS Test, p=0.13; WT: n=406 neurons, Het: n=330 neurons) and response probabilities (e, KS Test, p=0.23) in responding neurons. (f) Scatter plot depicting fraction of responding cells in WT and Het mice (Unpaired t-test t(14)=3.07, p=0.0084; WT n=8 mice, Het n=8 mice). (g, h) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (g, KS Test, p=0.00050; WT: n=467 neurons, Het: n=368 neurons) and response probabilities (h, KS Test, p=0.0059) in responding neurons. (i-n) Cellular sensory properties from awake animals following Botox injection in response to 5 pulses at 5 Hz (i-k) and 60 pulses at 40 Hz whisker stimulation (l-n). (i) Scatter plot depicting fraction of responding cells in WT and Het mice (Unpaired t-test t(14)=0.35, p=0.73; WT n=8 mice, Het n=8 mice). (j, k) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (j, KS test, p=0.32; WT: n=413 neurons, Het: n=416 neurons) and response probabilities (k, KS Test, p=0.0061) in responding neurons. (l) Scatter plot showing fraction of responding cells in WT and Het mice (Unpaired t-test t(14)=0.11, p=0.92; WT n=8 mice, Het n=8 mice). (m, n) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (m, KS Test, p=0.39; WT: n=481 neurons, Het: n=445 neurons) and response probabilities (n, KS Test, p=0.010) in responding neurons. (c-h) Data obtained from 1921 neurons in 54 imaging planes from 8 WT mice; 2044 neurons in 54 imaging planes from 8 Het mice. (c-n) Data was pooled from two independent cohorts of animals and thus obtained from 2169 neurons in 56 imaging planes from 8 WT mice; 1971 neurons in 55 imaging planes from 8 Het mice. In scatter plots, open circles are animal means, closed circles are individual cells, black lines indicate population means, and error bars indicate SEMs. All statistical tests were two-sided.
We next asked if cortex-specific mechanisms contribute to reduced SSC activity observed in Syngap1 Hets by restricting Syngap1 heterozygosity to forebrain glutamatergic neurons. Syngap1 conditional KO mice23 were crossed to a series of Cre drivers that express the recombinase in different sub-classes of forebrain neurons, and then injected these animals with an AAV vector that drives GCaMP6s expression. Syngap1 cKO mice were first crossed to an Emx1-Cre driver32, where Cre expression is restricted to forebrain glutamatergic neurons and glia, and performed in vivo two-photon imaging of calcium dynamics in L2/3 SSC neurons (Fig. 4a-b). We have previously confirmed that this cross results in reduced SynGAP expression within forebrain excitatory neurons24. Two-photon calcium activity measurements were obtained from anesthetized mice to prevent spontaneous whisking, allowing precise control of stimulus conditions. Restricting Syngap1 pathogenicity to this cellular population did not change the size of the active cell population (Fig. 4c), but did disrupt the amplitude and neuronal response probability of sensory-evoked responses (Fig. 4d, e). These data were largely consistent with findings in awake, conventional non-whisking awake Syngap1 animals (i.e. Botox-injected animals shown in Fig. 3i-k), though the effects here were stronger than in this prior experiment. The stronger phenotype in this experiment could be due to the method (virally injected GCaMP6s vs. GP4.3 transgene expression), brain state (anesthesia vs. awake), the cell types expressing Syngap1 heterozygosity (EMX1+-restricted population vs. no restrictions), or some combination of these factors. Never-the-less, reduced sensory responsiveness of L2/3 SSC neurons in Syngap1 mice is a reproducible phenotype and implicates Syngap1-mediated pathology directly within forebrain neurons.
Figure 4: Reduced sensory responsiveness of L2/3 SSC neurons in Syngap1 mice is cortex-specific.
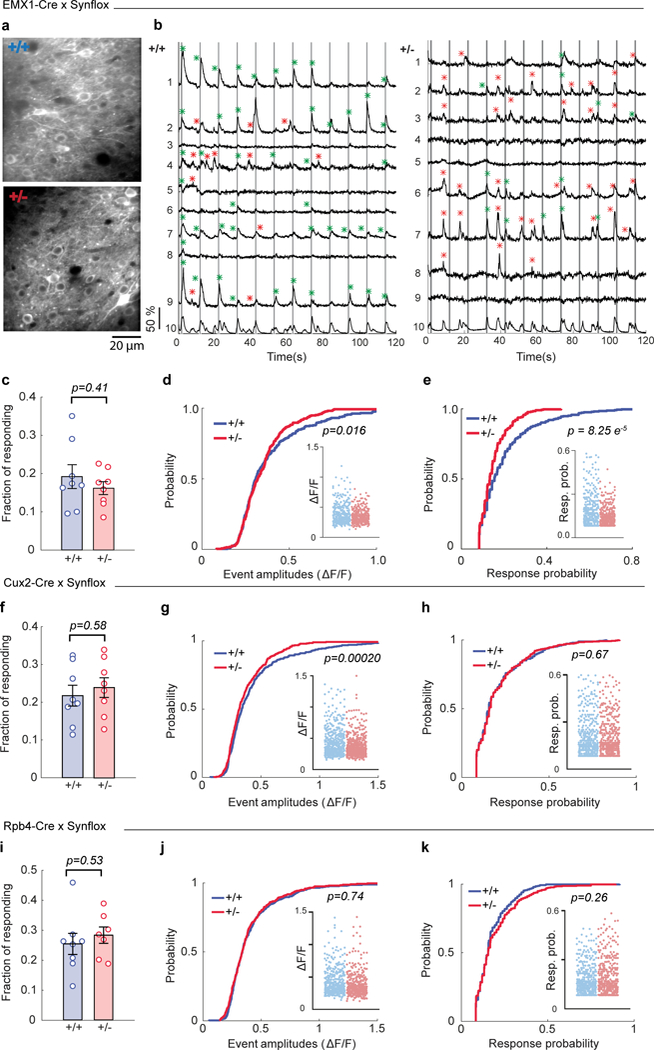
(a) Representative in vivo two-photon microscopy images of L2/3 SSC of WT and Het Emx1-Cre X Syngap1 cKO mice. (b) Representative ΔF/F traces of 9 (1-9) neurons in WT and Het mice in response to 5 passive whisker deflections at 5 Hz (from a). Gray vertical lines indicate the timing of whisker stimulus. Green asterisks indicate calcium events within the response detection window. Red asterisks show spontaneous calcium events. ROI number 10 is the neuropil signal. (c-e) Cellular sensory properties pooled from two independent cohorts of Emx1-Cre X Syngap1 cKO mice in response to 5 pulses at 5 Hz whisker stimulations under anesthesia. (c) Scatter plot showing fraction of responding cells in WT and Het mice (Unpaired t-test t(14)=0.85, p=0.41; WT n=8 mice, Het n=8 mice). (d, e) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (d, KS Test, p=0.016; WT: n=327 neurons, Het: n=306 neurons) and response probabilities (e, KS Test, p= 8.2E-5) in responding neurons. Data obtained from 1671 neurons in 55 imaging planes from 8 WT mice; 1877 neurons in 58 imaging planes from 8 Het mice. (f-h) Cellular sensory properties pooled from two independent cohorts of Cux2-CreERT2 X Syngap1 cKO mice in response to 5 pulses at 5 Hz whisker stimulations under anesthesia. (f) Scatter plot showing fraction of responding cells in WT and Het mice (Unpaired t-test t(14)=0.56, p=0.58; WT n=8 mice, Het n=8 mice). (g, h) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (g, KS Test, p=0.0002; WT: n=435 neurons, Het: n=445 neurons) and response probabilities (h, KS Test, p=0.67) in responding neurons. Data obtained from 2015 neurons in 57 imaging planes from 8 WT mice; 1901 neurons in 56 imaging planes from 8 Het mice. (i-k) Cellular sensory properties pooled from two independent cohorts of Rpb4-Cre X Syngap1 cKO mice in response to 5 pulses at 5 Hz whisker stimulations under anesthesia. (i) Scatter plot showing fraction of responding cells in WT and Het mice (Unpaired t-test t(13)=0.64, p=0.53; WT n=8 mice, Het n=7 mice). Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (j, KS Test, p=0.74; WT: n=340 neurons, Het: n=317 neurons) and response probabilities (k, KS Test, p=0.26) in responding neurons. Data obtained from 1684 neurons in 56 imaging planes from 8 WT mice; 1411 neurons in 46 imaging planes from 7 Het mice. In scatter plots, open circles are animal means, closed circles are individual cells, black lines indicate population means, and error bars indicate SEMs. All statistical tests were two-sided.
Syngap1 cKOs were next crossed to Cre driver lines that induce recombination in non-overlapping EMX1-positive subpopulations and imaged under the same conditions as the EMX1 experiment. Restricting Syngap1 heterozygosity to upper-lamina cortical neurons in Cux2-CreERT2 mice, an extensively validated Cre driver line selective for upper-lamina neurons in neocortex 33 (Supplementary Fig. 3a-b), did not impact the size of the responsive population or neuronal response probability (Fig. 4f, h). However, it caused a weak, but significant, reduction in amplitude of evoked responses (Fig. 4g). Next, we restricted Syngap1 heterozygosity to L5 glutamatergic neurons using the validated Rbp4-Cre diver line33 (Supplementary Fig. 3c-d), and imaged calcium responses in L2/3 barrel cortex neurons. Disrupting Syngap1 only in these neurons did not alter any of the standard measures of neuron responsiveness to sensory stimulation (Fig. 4i-k). While we have measured reduced whisker-evoked activity within Layer 2/3 neurons in both conventional and EMX1-Syngap1 Het mice, the primary cellular origins of how Syngap1 heterozygosity leads to effects on Layer 2/3 neurons is complex and not necessarily autonomous to only these neurons. The inability to fully recapitulate the sensory-responsive phenotypes of EMX1-Cre models in Cux2-Cre or RBP4-Cre models indicates that Syngap1 pathogenicity directly alters the function of multiple EMX1-positive populations that converge to drive the SSC cellular responsiveness phenotypes observed in conventional Syngap1 Hets.
Reduced sensory responsiveness could be caused by decreased excitation or increased inhibition onto, or within, upper-lamina L2/3 SSC neurons. To test the latter possibility, we crossed Syngap1 mice with Gad2-T2A-NLS-MCherry mice7 and injected them with an AAV vector that drives GCaMP6s expression in inhibitory and excitatory neurons (Fig. 5a-b). Under anesthesia, mCherry-negative (excitatory) neurons exhibited a substantial reduction in neuronal activation in response to the passive whisker stimulation (Fig. 5c-e). The reduced activation of neurons was most apparent in the whisker-responsive population (Fig. 5e). Amongst this population of excitatory neurons, Syngap1 disruption appeared to preferentially affect the most active cells. Similarly, there was a reduction in whisker-evoked neuronal activation from mCherry-positive (inhibitory) neurons (Fig. 5f-h). A reduced probability of firing in inhibitory neurons in response to whisker stimulation suggests that Syngap1 heterozygosity does not result in an overactive population of GABA-releasing neurons.
Figure 5: Reduced sensory responsiveness in both excitatory and inhibitory neuronal populations in L2/3 SSC of Syngap1 mutants.
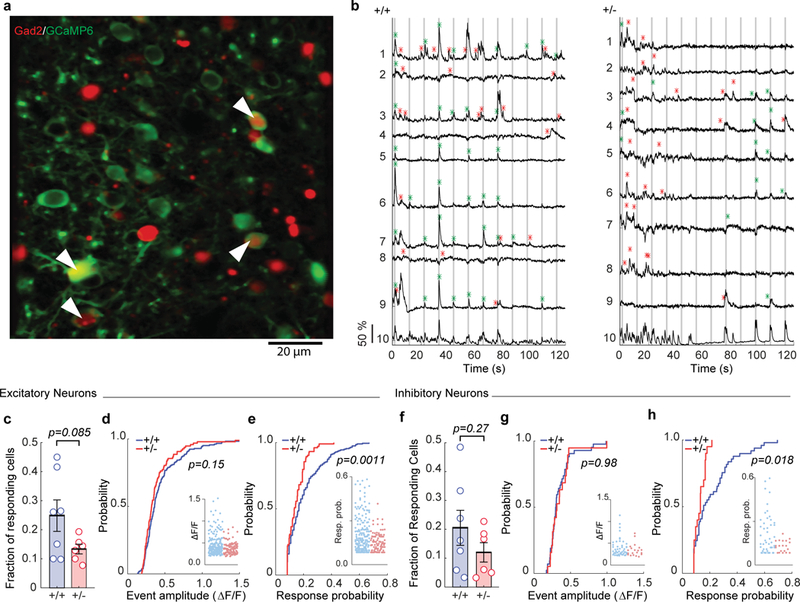
(a) Representative in vivo two-photon microscopy image of L2/3 SSC of a Gad2-T2A-NLS-MCherryXSyngap1 WT mouse expressing GCaMP6s (green) and Mcherry (red). White arrows indicate MCherry positive (inhibitory) neurons expressing GCaMP6s. (b) Representative ΔF/F traces of 9 (1-9) excitatory neurons in WT and Het mice in response to 5 passive whisker deflections at 5 Hz. Gray vertical lines indicate the timing of whisker stimuli. Green asterisks indicate calcium events within the response detection window. Red asterisks show spontaneous calcium events. ROI number 10 is the neuropil signal. (c-h) Cellular sensory properties of excitatory (c-e) and inhibitory (f-h) neurons. (c) Scatter plot showing fraction of excitatory neurons responsive to whisker stimulation (Unpaired t-test t(11)=1.891, p=0.0853; WT n=7 mice, Het n=6 mice). (d, e) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (d, KS Test p=0.1543; WT n=200 neurons, Het n=104 neurons) and response probabilities (e, KS Test, p=0.0011) in responding excitatory neurons. (f) Scatter plot showing fraction of inhibitory neurons responsive to whisker stimulation (Unpaired t-test t(11)=1.164, p=0.2691; WT n=7 mice, Het n=6 mice). (g, h) Cumulative probability and scatter plots (inserts) of ΔF/F amplitudes (g, KS Test, p=0.9848; WT n=49 neurons, Het n=20 neurons) and response probabilities (h, KS Test, p=0.0176) in responding inhibitory neurons. Data in this figure was pooled from two independent cohorts of animals and thus obtained from 850 excitatory and 240 inhibitory cells in 48 imaging planes from 7 WT mice; 825 excitatory and 193 inhibitory cells in 45 imaging planes from 6 Het mice. Open circles are animal means, closed circles are individual cells, black lines indicate population means, and error bars indicate SEMs. All statistical tests were two-sided.
Deficits in touch-related cortical circuit activation in Syngap1 mice are associated with reduced synaptic and intrinsic excitability of upper-lamina SSC neurons
This result prompted us to investigate impaired excitation as a possible cause of reduced sensory-related activity within upper-lamina SSC networks. We performed in vivo whole-cell patch-clamp recordings in L2/3 neurons of barrel cortex to determine how Syngap1 heterozygosity affects whisker-evoked synaptic potentials in anesthetized mice. Synaptic depolarization in response to passive whisker stimulation was reduced in Hets compared to WTs (Fig. 6a-c), a finding consistent with reduced whisker-driven cellular activity within upper-layer SSC circuits. We did not observe any alterations in spontaneous Up/Down state properties or changes in resting membrane potential in neurons from Hets (Supplementary Table 3).
Figure 6: In vivo patch clamp reveals that L2/3 SSC neurons in Syngap1 mutants have reduced sensory-evoked synaptic input.
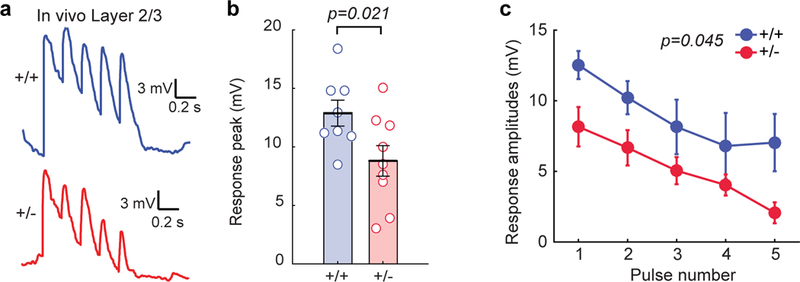
(a) Representative in vivo traces for whole-cell patch clamp experiments in response to passive whisker stimulations. (b) Scatter plot showing the overall response peaks (t-test: t(15)=2.59, p=0.021; n=8 for WT n=9 for Het. (c) Individual response amplitudes (2-way RM-ANOVA, F(1,14)=4.82, p=0.045 for genotype effect, F(4,56)=0.72, p=0.58 for genotype and stimulus interaction) in response to whisker stimulation in L2/3 neurons from in vivo patch clamp recordings. For b, open circles represent animal means, black lines indicate population means and error bars indicate SEMs. For c closed circles represent population means and error bars indicate SEMs. Data obtained from two cohorts of Syngap1 animals. All statistical tests were two-sided.
We further hypothesized that structural impairments in L2/3/4 neurons may contribute to reduced whisker-evoked feed-forward excitation in upper-lamina SSC circuits. L4 stellate cells in SSC receive the bulk of sensory-related information arriving from subcortical areas8, 34. Anatomical assessment of digitally reconstructed L4 neurons showed they were smaller in Syngap1 Hets compared to WTs (Fig. 7a), with dendritic arbors having reduced overall complexity and length. Dendrites from L4 neurons also had reduced spine density (Fig. 7b). Similar anatomical disruptions were found within dendrites and spines of L2/3 pyramidal cells from Hets (Fig. 7c-d), which receive a dense ascending projection from L4 stellate cells that relay sensory-related information to superficial neurons. These findings suggested that there were fewer excitatory synapses in upper-lamina SSC neurons.
Figure 7: Syngap1 heterozygosity degrades synaptic connectivity and reduces intrinsic excitability of upper layer SSC neurons.
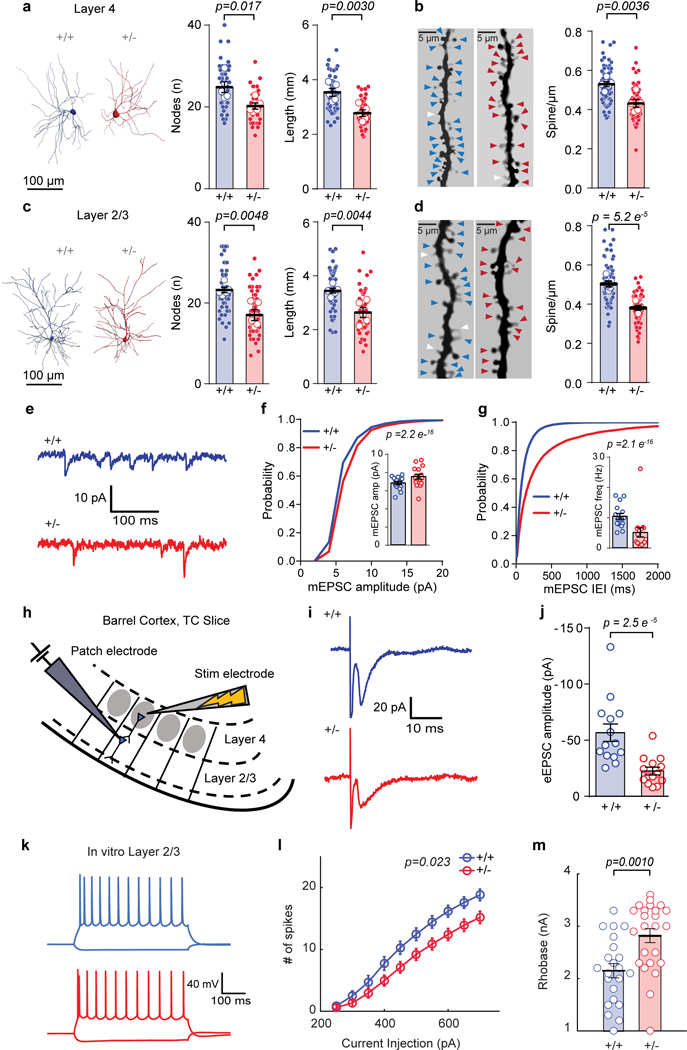
(a, c) Representative 3D reconstruction of L4 (a, left) and L2/3 (c, left) of SSC excitatory neurons depicting dendritic complexity and scatter plot (right) showing the total length and # of nodes by using Sholl analysis. (a) Total length: (WT = 5 mice, Het = 5 mice), unpaired t-Test, t(8)=4.002, p = 0.0030; # of Nodes: unpaired t-Test, t(8)=3.017, p = 0.0166. (c) Total length: (WT = 5 mice, Het = 6 mice), unpaired t-Test, t(9)=3.7713, p = 0.0044; # of Nodes: unpaired t-Test, t(9)=3.7090, p = 0.0048, from a single cohort of animals. (b, d) Examples of L4 (b, left) and L2/3 (d, left) apical dendrites and scatter plots (right) depicting the density of dendritic spines. (b) Layer 4 - spine density: (WT = 5 mice, Het = 5 mice), unpaired t-Test, t(8)=4.059, p = 0.0036. (d) Layer 2/3 - spine density: (WT = 5 mice, Het = 5 mice), unpaired t-Test, t(8)=7.80, p = 5.2E-5, from a single cohort of animals. (e) Representative traces depicting L2/3 excitatory neuron mEPSCs from acute WT and Het TC slices. (f, g) Cumulative probability and scatter plots (inserts) of mEPSC amplitudes (f, Kolmogorov-Smirnov test p= 2.2E-16) and mEPSC IEI (g, Kolmogorov-Smirnov test p= 2.1E-16). (f, g) Data was acquired from a single cohort of animals with n=7986 mEPSC events from16 neurons in 4 WT and n=6765 mEPSC events from 16 neurons in 4 Het mice. (h) Cartoon depicting experimental setup for investigating feed-forward excitation in L2/3 excitatory neurons from L4. (i) Representative traces depicting L2/3 excitatory neuron eEPSCs from acute WT and Het TC slices. (j) Scatter plot of eEPSC amplitudes in L2/3 following stimulation of L4 (Mann-Whitney test, U=14.00, p= 2.5E-5; Data obtained from a single cohort of animals; WT n=14 neurons from 4 mice, Het n=14 neurons from 4 mice). (k) Representative current-clamp traces from L2/3 excitatory neurons from acute WT and Het TC slices and (l) graph depicting a decrease in the number of spikes (2-way RM-ANOVA, F(1,46)=5.51, p=0.023 for genotype effect F(9,414)=5.46, 9.0E-9 for genotype and stimulus interaction, n=22 neurons from 5 WT mice, n=26 neurons from 6 Het mice) in response to current injections. (m) Scatter plot showing increased rheobase (Student’s t-test: t(44)=3.50, p = 0.0010) in the same set of neurons as in l. Data was obtained from a single cohort of animals. For morphology data, open circles are animal means, closed circles are individual cells, black lines indicate population means, error bars indicate SEMs, colored triangles represent spines (blue = WT, red = Het) and white triangles represent filopodia. For f, g, j, m, open circles are individual cells, black lines indicate population means, and error bars indicate SEMs. For l, circles represent population means and error bars indicate SEMs. All statistical tests were two-sided.
Patch-clamp recordings from L2/3 SSC neurons prepared from acute brain slices supported this idea. mEPSC frequency was dramatically reduced in neurons from Hets (Fig. 7e, 7g). Interestingly, mEPSC amplitude was slightly increased in these same neurons (Fig. 7f), possibly reflecting homeostatic compensation arising from too few excitatory synaptic inputs35. To directly measure feed-forward excitation within upper-lamina SSC circuits, we recorded synaptic currents in SSC L2/3 neurons evoked by electrical stimulation of L4 (Fig. 7h). Evoked synaptic currents in L2/3 neurons were reduced in amplitude in Hets compared to WTs (Fig. 7i, j). Finally, it is known that homeostatic compensation of both synaptic and intrinsic neuronal properties contribute to producing stable firing rates. We therefore wondered how intrinsic excitability mechanisms might be engaged to potentially counteract changes in reduced synaptic input onto, and overall activity within, upper-lamina L2/3 SSC neurons. Surprisingly, intrinsic excitability in L2/3 neurons was decreased in Syngap1 Hets compared to controls (Fig. 7k-m), which may also contribute to reduced activity observed in these neurons.
Touch-related behaviors are impaired in Syngap1 mice
Impairments in SSC circuit function in Syngap1 mice may be predictive of whisker-dependent behavioral deficits. To test this, we first explored the ability of constitutive Syngap1 mice to detect novel objects exclusively through touch. Importantly, Syngap1 mice are known to have normal novel object recognition (NOR) memory36. Therefore, we could determine how potential deficits in touch perception may influence novelty detection of similar objects that differ only by subtle change in their surface texture (Fig. 8a). Individual objects with differing textures were equally salient to test subjects (Fig. 8b). During the learning phase, WTs and Hets explored identical objects for a proportionally similar amount of time, though Hets spent more total time exploring objects overall (Fig. 8c-d), which may be related to increased locomotion in this line37. During testing with inclusion of the novel object, WTs distinguished between distinct textures, as evidenced by increased exploration of the novel relative to familiar object (Fig. 8c, e). In contrast, Hets failed to show a bias toward the novel object (Fig. 8d, e). Thus, Hets cannot discriminate between similar objects that differ only by surface texture.
Figure 8: Impaired texture discrimination and whisker-dependent Go/NoGo task performance in Syngap1 mice.
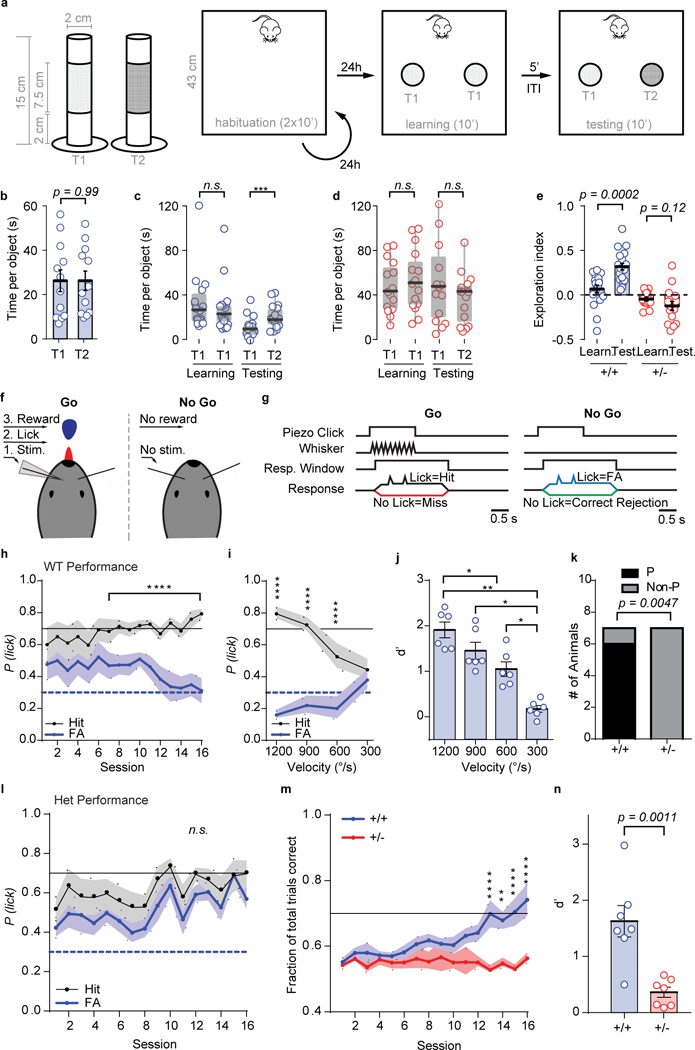
(a) Cartoons depicting different texture roughness of the objects used in Novel Texture Discrimination Task and relative protocol. (b) Scatter plot showing no preference in exploring T1 or T2. (T1 vs T2: n = 12 mice; unpaired t-Test, t(22) = 0.0016, p = 0.9986. (c-d) Box plots (solid line represents median, box represents interquartile range and whiskers represent maximum and minimum values) depicting time spent exploring identical textured objects during the learning phase and time spent exploring the novel (T2) and the old (T1) object for WT (c) and Het groups (d) [(WT mice Friedman test: χ2 (n = 18 mice, df = 3) = 28.87, exact sign. p = 1.4206 E-7; (HET mice, Friedman test: χ2 (n = 15 mice, df = 3) = 15.24, exact sign. p = 0.0016). Pairwise comparisons: (WT(learning) vs WT(testing), Sign test: n = 18 mice, Z = 4.007 p = 0.00006; HET(learning) vs HET(testing), Sign test: n = 15 mice, Z = 2.065, p = 0.04, ns). Statistical significance was accepted at the p < 0.03125. WT(learning) vs HET(learning), Mann-Whitney U test: Z = −2.567 p = 0.01; WT(testing) vs HET(testing), Mann-Whitney U test: Z = −2.821 p = 0.0048). (e) Scatter plot showing Exploration Index for animals in c and d: WT mice, paired t-Test: t(17) = 4.707, p = 0.0002; HET mice paired t-Test: t(14) = 1.641, p = 0.123; One sample test: WT(learning), t(17) = 1.555, p = 0.138; WT(testing), t(17) = 8.579, p = 1.39E-7; HET(learning), t(14) = −2.164, p = 0.048, ns; HET(testing), t(14) = −2.415, p = 0.03, ns. (f) Cartoon representation of Go/NoGo setup. Water-restricted, head-fixed mice were rewarded with water for licking a lick-port in response to a passive whisker (C2) deflection. (g) Detection task trial structure for Step 2 training. Go trials are identical to NoGo trials, except for the passive whisker deflection. Note, NoGo trials include activation of a “dummy piezo” not attached to any whisker to control for noise/vibration associated with piezo activation. (h) Step 2 training learning curve for WT mice showing the probability of licking (P(lick)) on Go (black, hit) or NoGo (blue, FA) trials (n=7 mice; 2-way RM-ANOVA with Bonferroni’s multiple comparison, Trial type: F(1,6)=67.19 p= 0.0002; Session: F(15,90)=0.4827 p=0.9437; Trial type*Session interaction F(15,90)=5.86 p= 2.9E-8). (i, j) Reductions in angular velocity of whisker deflections impairs the ability of “Good Performing” WT mice to discriminate between trial types (i: n=6 mice; 2-way RM-ANOVA with Bonferroni’s multiple comparison, Trial type: F(1,5)=471.1 p= 3.9E-6; Velocity: F(3,15)=1.469 p=0.263; Trial type*Velocity interaction: F(3,15)=30.12 p= 1.4E-6) and results in a reduced discrimination index (j: n=6 mice; RM-ANOVA with Bonferroni’s multiple comparison, F(3,15)=24.52 p= 4.9E-6). (k) Proportion of mice to learn (performers, P vs. non-performers, Non-P) the task (WT n=7, Het n=7: Fisher’s Exact Test: p=0.0047). (l) Step 2 training learning curve for Het mice (n=7 mice; 2-way RM-ANOVA with Bonferroni’s multiple comparison, Trial type: F(1,6)=8.44 p=0.027; Session: F(15,90)=2.416 p=0.0054; Trial type*Session interaction F(15,90)=0.8852 p=0.5825). (m) Learning curves depicting the fraction of total trials correct in Step 2 training (WT n=7, Het n=7; 2-way RM-ANOVA with Bonferroni’s multiple comparison, Genotype: F(1,12)=10.13 p=0.0079; Session: F(15,180)=3.665 p= 1.4E-5; Genotype*Session interaction: F(15,180)=4.398 p= 5.2E-7). (n) Scatter plot showing the discrimination index at the completion of Step 2 training (WT n=7, Het n=7; Unpaired t-test, t(12) = 4.281, p=0.0011). Data for both Novel Texture Discrimination and Go/NoGo Tasks were obtained from two independent cohorts of animals. Open circles are individual animals, closed circles and solid black horizontal lines indicate population means and error bars or shaded area represent the SEMs, except for boxplots in c and d which are described above. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 for post-hoc multiple comparisons. (h, i, l) Solid black and blue dashed lines indicate performance criteria for hit and FAs, respectively. (m) Solid black line indicates performance criteria for total trials correct. All statistical tests were two-sided.
We next used a discrimination task that requires the perception of a stimulus similar to what was used for evoking neuronal activity in our functional studies (Figs. 1-6). We selected a Go/NoGo paradigm, where water-deprived mice learn to perceptually report passive deflections of their whiskers by licking a water-dispensing port during a short “answer” period38, 39 (Fig. 8f). This type of learning paradigm is attractive for probing touch-related behaviors because it requires an intact ability to perceive a whisker stimulation39 and activation of whisker-responsive cortical circuits to sufficiently drive learning40. Task performance is quantified by the probability of correct choices during two trial types, “Go” (presence of whisker stimulus cue) and “NoGo” (absence of whisker cue) (Fig. 8g). WTs learned to discriminate between trial types after sufficient training (Fig. 8h; Supplementary Fig. 4a-b) with trial discrimination dependent on the strength of whisker stimulation (Fig. 8i-j), confirming task performance is driven by a stimulus precept computed by the function of whisker-sensitive circuits. Age-matched cohorts of Syngap1 WTs and Hets were then trained, with 86% of WT mice and 0% of Het mice reaching the training goals (Fig. 8k). WTs learned to lick during “Go” trials and withhold licks during “NoGo” trials (Fig. 8h; Supplementary Fig. 4a-b). In contrast, Hets did not improve accuracy in either trial type, likely explaining their static task performance over time (Fig. 8l; Supplementary Fig. 4c-d). Importantly, we observed a significant difference between genotypes in the fraction of correct answers over the entire training interval (Fig. 8m) and in the trial discrimination index (Fig. 8n), an objective measure of overall task performance at the completion of training. There was no difference between genotypes in relative weight gain/loss over the training interval (Supplementary Fig. 4e) and Hets exhibited normal licking, indicating familiarity with use of the port (Supplementary Fig. 4f-g). There was also no difference between genotypes in the response times for either trial type (Supplementary Fig. 4h-i) or total trials during training (Supplementary Fig. 4j-l). We also found no evidence of impulsive responding in Hets (Supplementary Fig. 4m-o). Together, these data indicate that poor task performance in Hets was not related to an obvious lack of motivation, training participation, or generalized impairment in the instrumental response.
DISCUSSION
The principle finding of this study is that disease-linked SYNGAP1/Syngap1 variants lead to impaired sensory processing. Sensory abnormalities are common in NDD patient populations41, 42. Moreover, there is a correlation between the severity of sensory disruptions and behavioral phenotypes in ASD populations43. Human imaging studies from NDD5 and schizophrenia patients6 have identified altered function of primary sensory areas, leading to the idea that altered sensory processing contributes directly to the complex phenotypes observed in patients. However, the neurophysiological mechanisms that lead to NDD-associated sensory impairments are poorly understood. By mining a SYNGAP1 patient registry within a retrospective Natural History Study, we confirmed that clinically-significant sensory alterations exist in patients with SYNGAP1 haploinsufficiency, including abnormal responses to painful tactile stimuli and unusual touch-related behaviors (Supplementary Table 2). Moreover, by utilizing Syngap1 mouse models, we uncovered unexpected circuit-level mechanisms consistent with impaired touch-related cortical sensory processing. Thus, reverse translation of NDDs caused by single rare variants, such as SYNGAP1, hold promise for better understanding these pathobiological mechanisms.
Our findings demonstrate that Syngap1 heterozygosity reduces activity within upper-lamina SSC circuits of awake Syngap1 mice. Reduced measures of sensory-evoked activity in vivo persisted in awake animals with paralyzed whiskers and in animals under anesthesia. These results were unexpected because many prior reports describe increased excitability and synaptic function in neurons from Syngap1 heterozygous mutant animals23, 24, 27, 44-47. Indeed, Syngap1 heterozygosity is believed to generally enhance neuronal excitation through shifting the synaptic E-I ratio in several types of neurons in areas of the brain linked directly to cognitive processing23, 44, 48, 49. Syngap1 is a risk factor for severe epilepsy21, and prior studies in this same Syngap1 mouse line used in this study have described seizure and circuit hyperexcitability in the hippocampus and prefrontal cortex23, 24. Syngap1 pathogenicity also accelerates the maturation of excitatory synapses during development, including several inputs within the hippocampus23 and the thalamo-cortical synapse in L4 somatosensory cortex27. Moreover, Syngap1 pathogenicity reduces the level of GABAergic connectivity onto SSC pyramidal neurons26. This loss of inhibitory control contributes to changes in oscillatory cortical rhythms in Syngap1 mice and is also consistent with increased excitability of neural circuits. The most parsimonious explanation for the differences between these prior reports and the current study is that Syngap1 heterozygosity has cell- and region-specific effects. For instance, L2/3/4 SSC neurons in adult constitutive Syngap1 mice had smaller dendritic fields with reduced spine density, which contributed to reduced feed-forward excitation during sensory stimulation. However, deep-layer SSC neurons from adult Syngap1 mutants were previously shown to have normally-sized arbors and spine densities50. Therefore, the overall impact of Syngap1 heterozygosity on brain function is defined by a range of circuit-specific impairments that disrupt neuronal excitability and function in complex ways. Our findings, therefore, give insight into the complexity of how a single gene causally-linked to severe ID and epilepsy can disrupt the structure and function of neurons and circuits linked to cognitive processes. We propose a generalizable scheme where interactions among region-specific circuit pathologies caused by causally-linked NDD risk variants drive complex cognitive and behavioral phenotypes observed in patients. The challenge will be to understand how disparate circuit pathologies interact to disrupt behavior, impair cognition, and promote seizure. Furthermore, it will be important to understand to what extent other highly-penetrant ID risk genes cause similarly complex region- and/or circuit-specific pathologies.
Our results support the conclusion that measures of reduced neuronal activity within upper-lamina of SSC circuits in Syngap1 mice were caused by a combination of lower intrinsic excitability and reduced synaptic connectivity onto L2/3 neurons in this area. This interpretation is supported by several key pieces of data. First, measures of reduced activity of SSC L2/3 neurons can be attributed to effects of Syngap1 within this cellular population. Restricting Syngap1 heterozygosity to forebrain glutamatergic neurons and some glia [i.e. EMX1(+) populations] was sufficient to impair SSC neuronal activity evoked from passive whisker deflections. This indicates that a primary cause of impaired sensory-evoked activity within Syngap1 mutants arises, at least in part, from altered function of cortical circuits, rather than from disruptions in sensory coding occurring in sub-cortical areas, such as in the mechanoreceptor neurons or thalamic relays. Second, evoked L2/3 SSC neuronal activation was depressed in both GABAergic and glutamatergic cellular populations, indicating that reduced activity was not a consequence of increased inhibition. Third, in vivo patch-clamp recording demonstrated reduced whisker-evoked membrane depolarization in L2/3 neurons from Het mice, which is consistent with our primary observation of reduced cellular activity of neurons in this area. In measurements from acute slices, these neurons also had reduced intrinsic excitability, which may contribute to reduced membrane depolarization observed in vivo. Fourth, we observed that both L4 and L2/3 neurons from Het mice had smaller dendritic fields. We also observed that these shorted dendritic fields contained fewer dendric spines. This anatomical defect translated into reduced functional synaptic connectivity, as we observed reduced mEPSC frequency in Het neurons even though there was a slight increase in mEPSC amplitude. Increased amplitude mEPSC in L2/3 neurons from Hets is consistent with our prior observation of larger dendritic spines in these cells50, which may be a homeostatic adaptation to neurons that have reduced activity35. We also observed direct evidence of reduced feed-forward excitation within upper-lamina of SSC. There was a substantial decrease in synaptic input from L4-to-L2/3 in thalamocortical slices prepared form Syngap1 mice. A strength of this study is that large impairments in synaptic function within whisker-responsive circuits translated into a significant reduction in touch-related proxy measures of somatic spiking (i.e. super-threshold somatic GCAMP6 events) within these same circuits. Taken together, these data are consistent with a model where L2/3 neurons in Syngap1 mice poorly encode incoming sensory information due to reduced synaptic excitation within upper-lamina of SSC. Given that L2/3 neurons integrate bottom up sensory codes with top-down modulatory information 7, it is possible that the cortex-specific circuit pathologies uncovered in this study disrupt sensory processing related to learning and/or behavioral adaptations. Future studies will be necessary to causally-link activity deficits within cortical circuits that respond to sensory input in Syngap1 mice to relevant behavioral phenotypes, such as poor learning.
ONLINE METHODS
Collection and analysis of data from the Retrospective SYNGAP1 Natural History Study Registry
The SYNGAP1 Patient Registry (https://syngap1registry.iamrare.org) is funded through the National Organization of Rare Disorders. This study was approved through the Hummingbird Institutional Review Board and meets all relevant ethical regulations for protections for human subjects. It is actively managed by a board of trustees comprised of a team of seven stakeholders, including parents with affected children, clinician-scientists that care for MRD5 patients, and neurobiologists that study the gene. The SYNGAP1 (MRD5) Natural History Study Registry is a retrospective longitudinal web-based observational natural history study. Parents or guardians provided informed consent prior to depositing medical history data into the registry. Participants with SYNGAP1 (MRD5) will be followed throughout the course of their lives with either the participant or authorized respondents contributing data at varying intervals throughout the course of the study. Initially, when a new patient is registered, data is collected on demographics, quality of life, medical history including genetic reports, disease phenotypes, event episodic data, retrospective data, participant review of systems, medication and diagnostic data. Each registrant is given a unique identifier to facilitate anonymization of patient data. Initial data collection is done through a series of questionnaires, including a survey of sensory and sensory-related issues. The structure of the database and all questionnaires were reviewed and approved by the members of the Board of Trustees.
To acquire information of possible sensory alterations in the SYNGAP1 patient population, the registry database was queried for all entries that answered the sensory questionnaire. The questionnaires for each anonymized entry were then exported to a spreadsheet for analysis (Supplementary Table 1). Entries that noted obvious impairments in tactile alterations, typically though narrative descriptions in columns E, F and/or K of Supplementary Table 1, were placed in Supplementary Table 2. The Registry was then revisited to determine if genetic reports were available for these entries. Genetic reports were available for 17 of these 20 entries. The presence/absence of an anonymized genetic report for each entry was noted in Column A and for entries with a report, the type of variant was listed in Column B and pathogenicity in Column C. Importantly, decisions on which entries to include in Supplementary Table 2 were made with no prior knowledge of each patient’s genotype. Entries containing narratives were unedited, including spelling errors, except when necessary to protect patient information (i.e. redact a name).
Mice
All animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all protocols were approved by The Scripps Institutional Animal Care and Use Committee. The design and maintenance of the conventional and conditional Syngap1 lines have been described previously23, 51. Thy1-GCaMP6s4.3 (#024275), Gad2-NLS-mCherry (#023140), and the TdTomato Ai9 (#007905) reporter lines were purchased from Jackson Laboratories. Rbp4-Cre (037128-UCD) and Cux2-CreERT2 (032779-MU) were purchased from MMRC. Both males and females were used in all experiments indiscriminately, except for the Go/NoGo task, where only males were used. All animals were older than 6 weeks of age at the beginning of experiments. Data collection occurred from mice >8 weeks of age. Mice were housed 4-5 per cage on a 12-hour normal light-dark cycle, except for Go/NoGo experiments, where mice were housed on a reverse light-dark cycle. For experiments requiring chronic cranial window and head-post implantation, mice were singly housed following surgery, with added environmental enrichment consisting of cardboard huts or plastic running wheels (Bio-Serv, Flemington, NJ) for the remainder of the study. Animals expressing the Cux2-CreERT2 allele were injected with tamoxifen (once) at PND2 as previously described50. Data collection was semi-randomized. Experimentalists were blind to genotype at the time of data acquisition and analysis. Generation of multiple transgenic mouse lines was labor, time and resource intensive. Additionally, most experiments required 1-3 months to complete, even with small sample sizes. This prevented us from picking WT and Het animals completely at random. Therefore, to obtain comparable sample sizes between genotypes, animal cohorts were generated by allocating equal (if possible) number of age-matched Syngap1 WT and Het littermates from separate litters, usually more than two. Then, animals were assigned a number to hide identity of genotype and/or group assignment. For imaging and behavior tasks, animals were recorded once per day in a randomized order while blinded to genotype. This process enabled balanced populations across experimental groups while minimizing potential biases. Only animals (or equivalent biological specimens) that died (or became non-responsive) during the course of the study or data collection procedures were excluded from analysis. The Life Sciences Reporting Summary contains additional details on data exclusions for specific experiments.
Intrinsic Optical Signal (IOS) Imaging
Animals were anesthetized with 1.6 g/kg urethane (Sigma-Aldrich, St. Louis, MO), followed by implantation of a custom head plate. The skull was scraped gently with a scalpel but otherwise left intact. The skull was thinned removing most of the spongy bone. Following gluing of the head plate, the skull was sealed with 1.5 % low melting point agarose dissolved in lactate ringer’s solution under a glass coverslip. Imaging was performed under a 4X objective on an upright microscope frame (BW51X; Olympus, Tokyo, Japan). The skull was illuminated with a 630 nm LED light mounted on the 4X objective52. The images were acquired with a Zeiss Axiocam camera (Carl Zeiss Microscopy Inc, Thornwood, NY) controlled by μManager software (Open Imaging, Inc.). Acquisition rate was approximately 10Hz. Whiskers were deflected using a piezoelectric bending actuator controlled by a linear voltage amplifier (Piezo Systems Inc, Woburn, MA). A single sinusoidal wave with a 5 ms rise and a 5 ms decay times were generated using Clampex software (Molecular Devices, Sunnyville, CA). Bending of the piezo was calibrated using a laser based displacement device (LD1610-0.5 Micro - Epsilon, Raleigh, NC). A single whisker deflection was approximately 200 μm at 2 mm away from the whisker pad (~6 0 or 1200 0/s). Each IOS imaging trial consisted of a 2 s baseline imaging period followed by 20 deflections at 10 Hz. 50-70 trials were performed for each whisker (now called an imaging session) and averaged using IO and VSD Signal Processor plugin in ImageJ 52. Images taken between 1 s to 3 s after the start of the stimulus, were averaged, and defined as the response. IOS images were obtained by calculating (response-baseline)/baseline value for each pixel using custom scripts written in MATLAB (MathWorks, Natick, MA) according to established procedures 53, 54. The animals that died during an intrinsic imaging experiment (due to anesthesia) or animals on which no visually reliable IOS map could be obtained (due to blood vessel contamination etc.) were excluded from the analysis (less than 5% of the cases). Image analysis was performed with the investigator blind to animal genotype. Briefly, images were first filtered with a Gaussian filter. Afterwards a baseline and a response region was manually selected in the final IOS image to minimize contamination by blood vessels53, 55. Response size was determined as the minimum value of the response region subtracted by median of the baseline region. Image thresholding was performed in the response region in order to determine the area of activation. Thresholds based on the absolute response size were specified in the figures. Relative thresholding values were set at 50-80 % of the response size for each image.
GCaMP Widefield Imaging (GWI):
Thy-1 Gcamp6s4.3 mice were implanted with a chronic cranial window according to established procedures56, 57. Briefly animals were anesthetized with avertin (Tribromoethanol, Sigma-Aldrich, St. Louis, MO) and intra-peritoneally injected with dexamethasone (4 mg/kg), rimadyl (carprofen 10mg/kg) and enroflox (enrofloxacin 5mg/kg). A 3 mm cranial window was made over the barrel cortex (A/P −2.0 mm, D/V +3.5 mm). The cranial window was sealed using two 3 mm glass coverslips glued onto a 5 mm glass coverslip. Animals were supplied with rimadyl for one week in drinking water for pain management. Following 2-3 weeks recovery from the surgery, animals were anesthetized with 1.6 g/kg urethane. Widefield fluorescent imaging of GCaMP6 was performed through the cranial window using 490 nm illumination under a standard 4X objective. Signals were acquired using a standard eGFP epifluorescence filter set. Whisker deflection and acquisition parameters were the same as with the IOS imaging, although we varied frequency and the number of deflections as specified in the figures. ΔF/F images were obtained by calculating (response-baseline)/baseline for each pixel similar to intrinsic imaging. A fixed rectangle of 150 μm centered at the functionally defined barrel center (90 % relative thresholding) was used for creating individual and averaged ΔF/F traces. Bias was minimized in these analyses. Image analyses were performed blind to the animal genotypes and performed by automated MATLAB scripts (minimal investigator-driven selection artifacts).
In vivo GCaMP imaging in barrel cortex:
For awake Thy-1 Gcamp6s4.3 experiments, both male and female mice at least 6 weeks of age were fitted with a chronic cranial window and implanted with a titanium head-post according to established procedures with minor alterations39. Briefly, animals were anesthetized with isoflurane (5 % induction, 1.5-2 % maintenance) and IP injected with a cocktail of dexamethasone (4 mg/kg), rimadyl (carprofen 10 mg/kg) and enroflox (enrofloxacin 5 mg/kg). Animals were mounted on a stereotaxic frame (David Kopf Instruments, Tujunga, CA) and body temperature was maintained with a thermal regulator (Harvard Apparatus, Holliston, MA). The scalp was shaved and sterilized with alternating swabs of betadine and 70 % alcohol. A small skin flap was removed, periosteum gently cleared, and the skull was scraped with a scalpel. A small, circular craniotomy was made over the left barrel cortex (3 mm diameter; center relative to bregma: lateral 3.5 mm; posterior 1.8 mm) using a dental drill, and the dura was left intact. The cranial window was sealed by gluing, two 3 mm glass coverslips glued onto a 5 mm glass coverslip, directly to the bone (VetBond, 3M). The titanium head-post was implanted by adhering it directly to the bone using VetBond, and then dental cement (Metabond, Parkell, Edgewood, NY). Animals recovered on a warm blanket before being placed back in their home cage. Rimadyl was injected (5 mg/kg) for three consecutive days after surgery for pain management. Following 1 week recovery from surgery, IOS imaging was performed through the cranial window, as described above, using light (0.5-1 %) isoflurane anesthesia to locate principle whisker areas (typically C2 whisker). The following week, animals were slowly habituated to head-fixation with increasing time spent under head-fixation, up to one hour, which was continuously monitored via IR videography (Basler, acA640-120um). Mice were head-fixed in a custom-built stainless-steel body tube and mounting brackets. Noise associated with the resonant scanner was recorded and continuously played through speakers (Avisoft-UltraSoundGate, Avisoft Bioacoustics) within the microscope enclosure during habituation and imaging. Once mice were comfortable with head-fixation (typically four days), awake in vivo GCaMP imaging was performed. Mice were then injected with 0.5 MU of onabotulinumtoxinA (Botox, Allergan; prepared in PBS) into the right whisker pad under isoflurane anesthesia using a Hamilton syringe (2.5 μL 62 RN model, beveled tip), and allowed to recover for 2 days before repeating awake in vivo GCaMP imaging. Whisker paralysis, as demonstrated in Supplemental Movie 2, lasted ~5 days.
For Gad2-T2A-NLS-MCherry X Syngap1 (i.e. Fig. 5) and Cre-driver experiments (i.e. Fig. 4), both male and female mice, at least 6 weeks of age were fitted with a chronic cranial window and implanted with a titanium head-post according to procedures detailed above. GCaMP6s was expressed following transduction with a rAAV (AAV1.Syn.GCaMP6s.WPRE.SV40, University of Pennsylvania Vector Core). During cranial window surgeries, iontophoresis via pulled-glass capillary micropipettes (Harvard Apparatus, Holliston, MA, 1.2 mm O.D., 0.69 mm I.D., inner tip diameter 20 um) was used for infection at 4-6 sites within the craniotomy. Iontophoresis was performed with 5 uA at 7s ‘on’ and 7 s ‘off’ cycles for 5 min total per injection site at a depth of ~200 μm. Following 2-3 weeks recovery from surgery, IOS imaging was performed as described above, using light (0.5-1 %) isoflurane anesthesia, to locate principle whisker areas that overlapped with GCaMP6s expression (typically C2 whisker). In vivo GCaMP imaging was performed ~4 weeks following surgery under light (1-1.5 % isoflurane) anesthesia.
Imaging was performed with a VivoScope two-photon microscope equipped with a resonant scanner (Scientifica, UK). The light source was a Mai Tai HP 100 femtosecond-pulse laser (Spectra-Physics) running at 940 nm for GCaMP and 1040 nm for Mcherry imaging. The objective was a 16× water immersion lens with 0.8 NA (Nikon). Images were acquired using ScanImage 5 (vidriotechnologies.com). Functional images (512×512 pixels, 4X zoom, 150×150 μm) of L2/3 cells (70-250 μm under the pia) were collected at 10 Hz. 100% laser power was 165 mW at the front aperture of the objective. For all GCaMP imaging experiments, we used 30-60% power depending on the imaging depth. A similar number of imaging sessions at similar depths (~ 7 sessions starting > 70 μm under the pia in 30 μm intervals) were acquired for each animal.
Analysis of GCaMP activity in barrel cortex:
Calcium images were corrected for motion artifacts using the moco plugin in ImageJ58. All subsequent analyses were performed in MATLAB R2015b using FluoroSNNAP15.04.08 plugin59 with the following parameter choices. Regions of interest (ROIs) corresponding to identifiable cell bodies were selected manually. The fluorescence time course was measured by averaging all pixels within the ROI, then corrected for neuropil contamination. The neuropil ROIs were also manually drawn where there are no visible cell bodies and were the same for all cells within an imaging frame. After neuropil correction, the ΔF/F of each ROI was calculated as (F−F0)/F0, where F0 was the mean of the lower 50% of the proceeding 10 s period. For the first 10 s period, minimum value of F0 was used (Patel et al., 2015). A template search-based algorithm was used in order to detect calcium events using built-in templates in FluoroSNNAP15.04.08. A correlation coefficient of 0.8 and ΔF/F amplitude threshold of 15 % was used during template search. Spontaneous activity was recorded over 2 mins. Neurons with at least one detected calcium event during a 2 mins period were classified as spontaneously active neurons. Whisker stimulation induced activity was also recoded over a 2 mins period from the same ROIs. Whisker stimulation consisted of 5 whisker stimulations at 5 Hz or 60 whisker stimulations at 40 Hz, with an intertrain interval of 10 s with the same whisker stimulation parameters used in IOS imaging (~6°). Therefore, each single stimulus train lasted for 1 or 1.5 s. A total of 12 trains were given during a 2 mins period. Raw response probability was calculated as the total number of calcium events during the 1 or 1.5 s stimulation window divided by the total number of trains (12). In order to adjust for the probability of obtaining calcium events non-specifically during the stimulation detection window, we subtracted expected number of spontaneous calcium events from the raw calcium event count before dividing by 12. Cells with response probabilities greater than 0.083 (responding 1 out 12 stimulation) are considered responders. For Gad2-T2A-NLS-MCherry X Syngap1 experiments, mCherry fluorescence was recorded for 1 minute. Images were flattened and merged onto GCaMP6s images to delineate inhibitory neurons from excitatory neurons. ROIs for excitatory and inhibitory neurons were analyzed in a similar fashion described above.
In vivo whole cell recordings:
A custom in vivo whole cell patch clamp system was built as described 60. Mice were anesthetized with 1.6 g/kg urethane (Sigma-Aldrich, St. Louis, MO), followed by implantation of a custom head plate and a 1 mm craniotomy was made over the barrel cortex. Recordings were performed the same day in current clamp mode with the following internal solution in the electrode (mM): 130 potassium gluconate, 5 KCl, 10 HEPES, 10 sodium phosphocreatine, 0.4 EGTA, 1 Na-GTP and 4 Mg-ATP (pH 7.3, 285-290 mOsm). Electrophysiological signals were amplified with Multiclamp 700B (Molecular Devices, Sunnyville, CA), filtered at 2 KHz, digitized (10 KHz) with NI USB-6363 (National Instruments, Austin, TX) and recorded using NI acquisition system in MATLAB61. UP/DOWN states were identified as described29. Briefly, the signal was first filtered with a median filter (100 ms window) in order to eliminate action potentials. Voltage segments lasting at least 100 ms and higher or lower than a third of the standard deviation away from the signal mean were classified as either up or down states respectively. In order to obtain synaptic currents, whiskers were deflected 5 times at 5 Hz with intensity parameters used during imaging experiments. In order to obtain synaptic amplitudes, 50 trials were averaged, excluding the trials with the action potentials. Action potentials were defined as samples where the speed of depolarization exceeded 6 mV/ms and whose values were more than two standard deviations away from the mean.
Dendrite reconstructions: AAV systemic Injections, Tissue Clearing, Sholl Analysis
Dendrite reconstructions were performed in mice (Syngap1+/− Hets crossed to Ai9+/+) that were injected with an rAAV9-packaged Cre-expressing virus via the superficial temporal vein (STV) of PND1 mouse pups as described previously62. Briefly, pups were sedated by covering them with ice for 3 min. STV is visualized using a handheld transilluminator (WeeSight; Respironics, Murrysville, PA, USA), and a pair of standard reading glasses. Virus solution was prepared by 1:50 dilution of stock solution in Dulbecco’s phosphate-buffered saline (PBS), also supplemented with 0.001% pluronic-F68. Virus solution (50 nL) was injected using a 100 nL Nanofil syringe attached with a 34-gauge Nanofil beveled needle (World Precision Instruments, Sarasota, FL). A correct injection was verified by noting blanching of the vein. After the injection, pups were returned to the incubator until active and then returned to their dam.
For dendritic tracing, PND 60 animals were deeply anesthetized with pentobarbital (Nembutal) and transcardially perfused with 4 % PFA/PBS (wt/vol). Extracted brains were post-fixed in 4 % PFA/PBS at 4°C for 10 h and cryoprotected in 20 % sucrose/PBS (wt/vol) at 4°C for 24 h. Brains were cut on a vibratome (500 μm thickness) collecting the somatosensory cortices. Slices were immediately submerged in Scale A2 solution in order to clear the tissue. When the tissue was transparent (at least two weeks), brain slices were mounted in Petri dishes, covered in agarose, and imaged using standard confocal microscopy. Three-dimensional image stacks were collected (x: 2048, y: 2048 pxl; step size 1 μm) using confocal microscopy equipped with water immersion objective lens (ULTRA 25x, numerical aperture 1.05, Olympus). A computer-based tracing system (Neurolucida360; MicroBrightField) was used to generate three-dimensional neuron tracings that were subsequently visualized and analyzed with NeuroExplorer (MicroBrightField). In order to select a neuron, the following criteria were strictly followed: 1) neuron was selected starting toward the middle of the stack (~150 μm ± 30 μm) to ensure the accurate reconstruction of an entire dendritic arbor; 2) neuron was distinct from other neurons to allow for identification of branches; 3) neuron was not truncated in some obvious way. For every reconstructed neuron, an estimate of dendritic complexity within layer 2/3 and 4 neurons was obtained with Sholl analysis. A 3D Sholl analysis was then performed in which concentric spheres of increasing radii (20 μm increments) were layered around the cell body until branches were completely enveloped. The total length of branches, the number of dendritic intersections at each sphere, and the dendritic orders were measured 50. Neurons were traced by an experimenter blind to genotype.
Spine Density Analysis
Because the clearing tissue method enhanced the quality of our images, spine density was determined using the same set of images previously acquired for the tracing experiment. As previously described 50, ten to fifteen dendritic segments of somatosensory cortex L2/3 and L4 (20-120 μm in length) at PND 60 mice were collected and considered for analysis. All measurements were performed by an experimenter blind to the experimental conditions. Pictures were visualized and elaborated with Neurolucida 360 software (MicroBrightField).
Acute slice preparation and in vitro electrophysiology
Acute thalamocortical (TC) slices (350 μm) from 8 week old Syngap1 WT and Het mice were cut using standard methods as previously described63. Ice-cold cutting solution contained (in mM): 110 choline-Cl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.5 CaCl2, 5 MgCl2, 25 Glucose, 10 Ascorbic acid, 5 Pyruvic acid (pH 7.4, ~300 mOsm). The slices were then warmed to 35˚C for 40 minutes in standard artificial cerebrospinal fluid (aCSF), composed of (mM): 125 NaCl, 2.5 KCl, 24 NaHCO3, 2 CaCl2, 1.25 NaH2PO4, 2 MgSO4, and 10 D-Glucose, and equilibrated with 95 % O2 and 5 % CO2 (pH 7.4, ~300 mOsm). Following this, slices were maintained in bubbled aCSF at room temperature until transferred to a submerged-type recording chamber (Warner Instruments, Hamden, CT). All experiments were performed at 32˚C±2 (2-3 mL/min).
Whole-cell patch clamp experiments were conducted from visually identified L2/3 neurons using infrared DIC optics and regular spiking was confirmed in current clamp. Recordings were made using borosilicate glass pipettes (3-6 MΩ; 0.6 mm inner diameter; 1.2 mm outer diameter; Harvard Apparatus). All signals were amplified using Multiclamp 700B (Molecular Devices, Sunnyville, CA), filtered at 2 KHz, digitized (10 KHz), and stored on a personal computer for off-line analysis. Analog to digital conversion was performed using the Digidata 1440A system (Molecular Devices). Data acquisitions and analyses were performed using pClamp 10.2 software packsge (Clampex and Clampfit programs; Molecular Devices).
For current clamp and evoked excitatory postsynaptic current (eEPSC) recordings, the following internal solution was used (in mM): 130 potassium gluconate, 5 KCl, 10 HEPES, 0.25 EGTA, 10 phosphocreatine disodium, 0.5 Na-GTP and 4 Mg-ATP (pH 7.3, 285-290 mOsm). For miniature excitatory postsynaptic current (mEPSC) recordings, the following internal solution was used (in mM): 120 CsCl, 5 NaCl, 1 MgCl2, 10 HEPES, 10 EGTA, 3 Mg-ATP and 0.3 Na-GTP (pH 7.3, 285-290 mOsm). Cells with access resistance >30 MΩ or were unstable (>20 % change) were discarded from further analysis.
In eEPSC experiments, slices were incubated with 100 μM picrotoxin. L4 was stimulated by placing a concentric bipolar stimulating electrode (FHC, 25 μm inner diameter; 125 μm outer diameter, ME, USA) in the center of a barrel. L2/3 neurons were recorded directly above the stimulus site. The stimulation intensity (0.2 ms, constant-current pulses) was regulated by a stimulus isolation unit (ISO-Flex, A.M.P.I). A minimum stimulus intensity protocol was employed (6 sweeps with 15 s ISI) in current clamp, as eEPSPs appeared to be more stable than eEPSCs recorded in voltage clamp. The stimulus intensity was gradually increased until the emergence of an eEPSP with a 30-50 % failure rate. This stimulus intensity was used in voltage-clamp mode to elicit eEPSCs from 15 sweeps (15 s ISI). eEPSCs were quantified by averaging the peak amplitudes, within a 30 ms post-stimulus window, excluding failures. Only one neuron was recorded from a single barrel column and a maximum of 2 neurons per slice. mEPSCs were recorded in TTX (1 μM), picrotoxin (100 μM) and APV (100 μM) at a holding potential of −75 mV. For each recording, the first 500 events were analyzed using Mini Analysis software (Synaptosoft Inc, NJ). Intrinsic properties of neurons were recorded at the resting membrane potential and measured as described24.
Novel Texture Discrimination Task (NoTeDt)
Before testing for NoTeDt, mice were first habituated to an Open Field Arena. Cohorts of mice were handled for several minutes on three separate days prior to commencement of behavioral testing. On the first day of behavioral assay, mice were subjected to the open field habitation phase, during which an animal was allowed to explore a custom-made clear acrylic arena (43 cm x 43 cm x 32h cm) for 10 minutes, two daily sessions, for two days. Opaque white acrylic dividers surrounded each arena to prevent distractions from activities in adjacent boxes. Activity was monitored with two CCTV cameras (Panasonic WV-BP334) feeding into a computer equipped with Ethovision XT 11.5 for data acquisition and analyses. A white noise generator (2325-0144, San Diego Instruments) was set at 70 dB to mask external noises and provide a constant noise level. Fluorescent linear strip lights placed on each of the four walls of the behavioral room adjacent to the ceiling provided a lower lighting (100 lux) environment. Distance travelled was analyzed as an indicator of familiarization to the experimental environment.
NoTeDt was performed in the same arenas used for open field habituation. The cylindrical-shaped objects were conceived to promote whiskers-like explorations. Objects were designed by optimizing criteria from previously described and validated texture-based object discrimination tasks64, 65. Briefly, white polyoxymethylene objects were 15 cm tall, fixed to a 5 cm diameter base and the rough portion of the object covered 7.5 cm of the surface starting 2 cm from its base. Two different grades of roughness (T1 and T2) were chosen and tested to determine whether the objects shared similar degree of attractiveness.
After the habituation phase (see open field section for details), mice were engaged in exploring two identical objects (T1) for 10 minutes (learning phase). After 5 minutes, necessary for rearranging the arena (cleaning and replacing one of the objects T1 with the novel object T2), mice were exposed to the testing phase, engaging in the exploration of both old and new objects. The location of the novel object was pseudo-randomized and counterbalanced between groups. Time spent in exploring textures was assessed by manually scoring each trial (offline), during both learning and testing phases. Mice that did not explore the objects during the learning phase, explored only one of the two objects during the testing phase, or had a total investigation time of less than 20 s during the learning phase, were excluded from the study for lack of adequate exploratory activity. Behavioral scoring was performed by an experimenter blind to the experimental conditions.
Whisker-dependent Go/NoGo task
Syngap1 WT and Het littermates (male, housed in a reverse light-dark room) were trained one session per day (~5 days/week) to perform a head-fixed whisker-dependent Go/NoGo task39. The behavioral apparatus was controlled by open-source BControl software (C. Brody, Princeton University) on a RT Linux machine66. A custom titanium head-post was implanted onto the skull at six to ten weeks after birth. Briefly, animals were anesthetized with isoflurane (5% induction, 1.5-2% maintenance) and injected IP with rimadyl (carprofen 10 mg/kg) and enroflox (endrofloxacin 5 mg/kg). A small flap of skin was removed over the midline, and the skull scraped with a scalpel before a thin layer of cyanoacrylate glue (Vetbond, 3M) was applied. The head-post was implanted onto this layer of glue and secured with dental cement (Metabond). Animals were then singly housed and monitored for 1 week following surgery, with added environmental enrichment (running wheel). All behavioral sessions occurred during the dark phase. Weekly and throughout the experiment, all whiskers were trimmed to the base except for the C2 whisker on either side. Following recovery from head-post surgery, mice were placed on water restriction (1 ml/day) for 10 days prior to and then throughout training (food ad libitum). Water-restricted mice were monitored for health issues and weighed daily. On day six of water restriction, habituation to head-fixation commenced with increasing time spent under head-fixation, up to one hour. Mice were head-fixed in a custom-built stainless-steel body tube, inside a fully enclosed light and soundproof box, which was continuously monitored via IR videography. On the last day of habituation, video recordings (30 FPS, Basler, acA640-120um) were obtained (45 mins duration) under IR illumination to determine basic whisking properties (bouts and bout durations). Whisking was scored manually, offline (Solomon Coder) by an experimenter blinded to mouse genotypes.
Mice moved through a series of training steps. First, lick-port training (1 session lasting 10 mins) allowed mice to associate water availability via licking from a lick-port, with the C2 whisker inserted into a pipette (with ~ 2 mm at the base exposed) attached to a piezo actuator (Physik Instrumente), however, no passive whisker deflections were introduced. The metal lick-port was positioned directly in front and slightly below the mouth, within reach of the tongue. Detection of licks was performed electronically67 and precise water delivery (8 μl/reward) was controlled with a solenoid valve (The Lee Company, CT). Next, Step 1 training lasted for 2 sessions and represented the first-time mice were exposed to passive whisker deflections. Here, sessions consisted of 90% Go and 10% NoGo trials. For Go trials, the whisker was deflected by the piezo actuator, controlled by a linear voltage amplifier (Physik Instrumente) and a waveform generator (BK Precision), for 1.5 s with a 40 Hz sinusoidal wave (rostral to caudal, 1200 °/s). Bending of the piezo was calibrated using a laser-based displacement device (LD1610-0.5 Micro - Epsilon). For NoGo trials, a “dummy” piezo, placed just above the whisker deflecting piezo was driven with the same stimulation, but was not attached to a whisker. The response window was defined from 0.1-4 s after the start of piezo stimulation. For Go trials, hits consisted of trials where mice licked for a water reward within the response window, and misses were scored as lack of licking during this same period. For NoGo trials, a false alarm (FA) resulted when the animal licked during the response window and a correct rejection occurred when the animal withheld licking. No punishments were given for FAs and no auditory cue was presented. Inter-trial interval remained constant at 4 s, however, mice were required to withhold licking for 1.5 s before the piezo was stimulated for trials to proceed. Mice performed the task until satiated. Step 2 training was similar to Step 1, except sessions consisted of 50% Go and 50% NoGo trials (200-350 trials/session) with no more than 3 consecutive trials of the same type. The response window was shortened to 2 s. Training persisted until mice reached several performance criteria for at least 2 consecutive days. 1) Overall performance reached > 70% correct for all trials, 2) Hits > 70%, 3) FAs < 30%, and 4) discrimination index (d’) > 1.1. D’ was calculated in Excel (Microsoft) as d’ = z(hit) - z(FA), with z scores computed using the function NORMSINV. If mice did not reach criteria by 21 days, training was halted. “Good performers” graduated to Step 3, whereby the stimulus duration was reduced to 0.5 s. Mice continued this training until the same criteria was met as in Step 2. These mice graduated to a reduced stimulation protocol. Here, sessions consisted of the same experimental setup as Step 3, except angular velocity of passive whisker deflections were reduced from 1200 °/s to a range between 300-900 °/s.
Statistics:
Data analyses were conducted in SPSS (IBM Corp, version 20), MATLAB (Mathworks, version 2015b, Natick, MA) and GraphPad Prism 7 (GraphPad Software, CA). D’Agostino-Pearson omnibus normality test was applied to determine data distributions and the appropriate parametric or non-parametric statistical test was performed accordingly. However, for experiments with small sample sizes (N<8 subjects per group), statistical tests tend to lack power to detect deviations from normality, and therefore a more subjective approach was used. For these cases where data appeared to approximate a normal distribution, we assumed normality and used parametric statistical tests (i.e. Fig. 5c, Fig. 8n, but not limited to these panels). For analysis of imaging data, the following tests were used: two-sided student’s t-test was used to compare IOS amplitudes, IOS inter-barrel distance, GWI integrated ΔF/F, GWI ratio of 4th/1st pulse to first pulse and the fraction of responding cells (except for Fig. 2d, where 2-way RM-ANOVA was used) between genotypes; 2-way RM-ANOVA was used to compare area measures for both relative and absolute thresholding methods for IOS and for peak responses and integrated ΔF/F in GWI experiments; Kolmogorov-Smirnov test was used to compare event amplitudes, event counts/2 minutes, and response probabilities, with Bonferroni-adjusted P values to control for type I errors when multiple pairwise comparisons were made. For cluster analyses of spike counts, we pooled all spikes for each genotype and treatment, then performed a two-step cluster analysis. We specified a fixed number of cluster to three, for low-, medium- and high-responsive neurons and used the Euclidean distance for computing the similarity between clusters. The number of neurons in each cluster were then separated into groups based on genotypes and treatment for further analysis. Differences in the clustered data were analyzed using Chi-square analysis. Pairwise post-hoc comparisons between clusters and groups were performed using Chi-square analysis with Bonferroni-adjusted P values to control for type I errors. For analysis of the neuronal morphology of layer 2/3, and 4 neurons, two-sided student’s t-test, assessing the contribution of total length, # of nodes and the spine density were performed. For electrophysiology data, two-sided student’s t-test or Mann-Whitney U test when applicable or Kolmogorov-Smirnov test was used to compare measurements between WT and Het mice. 2-way RM-ANOVA was used to compare response amplitudes of responses to each whisker deflection and the number of spikes in response to current injections. NoTeDt behavioral data are expressed as the median ± interquartile except data showed in fig 1b and fig.1e which are expressed as the mean ± SEM. Non-parametric Friedman and Mann-Whitney U-tests were used to analyze WT and Het performance in both learning and testing phases. Post-hoc comparisons were performed by using sign test and the false discovery rate (FDR)-controlling procedure Benjamini/Hochberg, was used to compute the adjusted level of significance. One sample t-tests were performed against a chance value of 50% to determine if the mice were able to discriminate between the novel and the old textures. For Go/NoGo behavioral data, the following tests were used: 2-way RM-ANOVA with Bonferroni’s multiple comparisons post-hoc test was used to compare performance (P(lick)) of WT and Het mice, performance with reduced angular velocity of whisker stimulation, normalized weights, number of trials performed, response times, and number of total licks; one-way repeated measures ANOVA with Bonferroni’s multiple comparisons post-hoc test was used to compare d’ with reduced angular velocity of whisker stimulation; two-sided student’s t-test or Mann-Whitney U-tests were used to compare d’ between WT and Hets, mean water licks, total reward licks, mean trials/session, total trials performed, mean licks/trial (total), number of total licks and basic whisking parameters; fisher’s exact test was used to compare proportion of performers vs. non-performers in WT and Het mice. Unless otherwise stated, data represents mean ± SEM. No statistical test was used to predetermine sample sizes, however, our sample sizes are similar to those previously reported in the field68.
Supplementary Material
ACKNOWLEDGEMENTS:
This work was supported in part by NIH grants from the National Institute of Mental Health [MH096847 and MH108408 (GR), MH105400 (CAM)], the National Institute for Neurological Disorders and Stroke [NS064079 (GR); NS083894 (JMC)], and the National Institute for Drug Abuse [DA034116, DA036376 (CAM)]. JLH is supported by a National Institute for Neurological Disorders and Stroke Mentored Clinical Scientist Research Career Development Award [NS091381]. The SYNGAP1 Natural History Study and Patient Registry is supported by a grant to MW, JLH and GR from The National Organization of Rare Disorders (NORD).
Footnotes
COMPETING FINANCIAL INTEREST STATEMENTS: The authors declare no competing financial interests. MW is a paid employee of Bridge-the-GAP Educational Research Foundation. GR and JLH are unpaid scientific advisors to Bridge-the-GAP Educational Research Foundation.
Accession Codes: None
DATA AVAILABILITY STATEMENT: The data that support the findings of this study are available from the corresponding author upon request.
Code Availability:
Custom code was written in MATLAB to analyze intrinsic imaging experiments. Minor code modifications to the FluoroSNNAP15.04.08 plugin 59 were used to analyze somatic GCaMP6 signals. These scripts are available upon reasonable request.
REFERENCES
- 1.Sahin M & Sur M Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hull JV, et al. Resting-State Functional Connectivity in Autism Spectrum Disorders: A Review. Front Psychiatry 7, 205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Just MA, Cherkassky VL, Keller TA & Minshew NJ Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain 127, 1811–1821 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Zoghbi HY & Bear MF Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harbor perspectives in biology 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson CE & Baron-Cohen S Sensory perception in autism. Nat Rev Neurosci 18, 671–684 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Javitt DC & Sweet RA Auditory dysfunction in schizophrenia: integrating clinical and basic features. Nat Rev Neurosci 16, 535–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peron SP, Freeman J, Iyer V, Guo C & Svoboda K A Cellular Resolution Map of Barrel Cortex Activity during Tactile Behavior. Neuron 86, 783–799 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Feldmeyer D, et al. Barrel cortex function. Prog Neurobiol 103, 3–27 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Pluta SR, Lyall EH, Telian GI, Ryapolova-Webb E & Adesnik H Surround Integration Organizes a Spatial Map during Active Sensation. Neuron 94, 1220–1233 e1225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogden KK, Ozkan ED & Rumbaugh G Prioritizing the development of mouse models for childhood brain disorders. Neuropharmacology 100, 2–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoischen A, Krumm N & Eichler EE Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci 17, 764–772 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deciphering Developmental Disorders, S. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamdan FF, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 360, 599–605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Deciphering Developmental Disorders, S. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Roak BJ, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun 5, 5595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamdan FF, et al. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry 69, 898–901 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Berryer MH, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Human mutation 34, 385–394 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Parker MJ, et al. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Med Genet A 167A, 2231–2237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mignot C, et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet 53, 511–522 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Carvill GL, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45, 825–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Stulpnagel C, et al. SYNGAP1 Mutation in Focal and Generalized Epilepsy: A Literature Overview and A Case Report with Special Aspects of the EEG. Neuropediatrics 46, 287–291 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Clement JP, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozkan ED, et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 82, 1317–1333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gliklich RE, D.N., Leavy MB, editors. Registries for Evaluating Patient Outcomes: A User’s Guide [Internet]: Rare Disease Registries (Chapter 20). Agency for Healthcare Research and Quality (US) (2014). [PubMed] [Google Scholar]
- 26.Berryer MH, et al. Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nat Commun 7, 13340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clement JP, Ozkan ED, Aceti M, Miller CA & Rumbaugh G SYNGAP1 links the maturation rate of excitatory synapses to the duration of critical-period synaptic plasticity. J Neurosci 33, 10447–10452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dana H, et al. Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS One 9, e108697 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goncalves JT, Anstey JE, Golshani P & Portera-Cailliau C Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci 16, 903–909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfe J, Houweling AR & Brecht M Sparse and powerful cortical spikes. Curr Opin Neurobiol 20, 306–312 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Margolis DJ, et al. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat Neurosci 15, 1539–1546 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Gorski JA, et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22, 6309–6314 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray LT, et al. Layer-specific chromatin accessibility landscapes reveal regulatory networks in adult mouse visual cortex. Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diamond ME & Arabzadeh E Whisker sensory system - from receptor to decision. Prog Neurobiol 103, 28–40 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Nelson SB & Valakh V Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 87, 684–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muhia M, Yee BK, Feldon J, Markopoulos F & Knuesel I Disruption of hippocampus-regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. Eur J Neurosci 31, 529–543 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Guo X, et al. Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology 34, 1659–1672 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachidhanandam S, Sermet BS & Petersen CC Parvalbumin-Expressing GABAergic Neurons in Mouse Barrel Cortex Contribute to Gating a Goal-Directed Sensorimotor Transformation. Cell Rep (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang H, Kwon SE, Severson KS & O’Connor DH Origins of choice-related activity in mouse somatosensory cortex. Nat Neurosci 19, 127–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sachidhanandam S, Sreenivasan V, Kyriakatos A, Kremer Y & Petersen CC Membrane potential correlates of sensory perception in mouse barrel cortex. Nat Neurosci 16, 1671–1677 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Ben-Sasson A, et al. A Meta-Analysis of Sensory Modulation Symptoms in Individuals with Autism Spectrum Disorders. Journal of Autism and Developmental Disorders 39, 1–11 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Marco EJ, Hinkley LB, Hill SS & Nagarajan SS Sensory processing in autism: a review of neurophysiologic findings. Pediatr Res 69, 48R–54R (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robertson AE & Simmons DR The relationship between sensory sensitivity and autistic traits in the general population. J Autism Dev Disord 43, 775–784 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Araki Y, Zeng M, Zhang M & Huganir RL Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85, 173–189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kopanitsa MV, et al. Chronic treatment with a MEK inhibitor reverses enhanced excitatory field potentials in Syngap1(+/−) mice. Pharmacol Rep 70, 777–783 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Rumbaugh G, Adams JP, Kim JH & Huganir RL SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A 103, 4344–4351 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vazquez LE, Chen HJ, Sokolova I, Knuesel I & Kennedy MB SynGAP regulates spine formation. J Neurosci 24, 8862–8872 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walkup WG, et al. A model for regulation by SynGAP-alpha1 of binding of synaptic proteins to PDZ-domain ‘Slots’ in the postsynaptic density. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng M, Bai G & Zhang M Anchoring high concentrations of SynGAP at postsynaptic densities via liquid-liquid phase separation. Small GTPases, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aceti M, et al. Syngap1 haploinsufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol Psychiatry 77, 805–815 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 51.Kim JH, Lee HK, Takamiya K & Huganir RL The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci 23, 1119–1124 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison TC, Sigler A & Murphy TH Simple and cost-effective hardware and software for functional brain mapping using intrinsic optical signal imaging. Journal of neuroscience methods 182, 211–218 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Chen-Bee CH, Kwon MC, Masino SA & Frostig RD Areal extent quantification of functional representations using intrinsic signal optical imaging. Journal of neuroscience methods 68, 27–37 (1996). [PubMed] [Google Scholar]
- 54.Chen-Bee CH, et al. Visualizing and quantifying evoked cortical activity assessed with intrinsic signal imaging. Journal of neuroscience methods 97, 157–173 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Chen-Bee CH & Frostig RD Variability and interhemispheric asymmetry of single-whisker functional representations in rat barrel cortex. Journal of neurophysiology 76, 884–894 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Goldey GJ, et al. Removable cranial windows for long-term imaging in awake mice. Nature protocols 9, 2515–2538 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holtmaat A, et al. Imaging neocortical neurons through a chronic cranial window. Cold Spring Harb Protoc 2012, 694–701 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dubbs A, Guevara J & Yuste R moco: Fast Motion Correction for Calcium Imaging. Front Neuroinform 10, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel TP, Man K, Firestein BL & Meaney DF Automated quantification of neuronal networks and single-cell calcium dynamics using calcium imaging. J Neurosci Methods 243, 26–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Desai NS, Siegel JJ, Taylor W, Chitwood RA & Johnston D MATLAB-based automated patch-clamp system for awake behaving mice. Journal of neurophysiology 114, 1331–1345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desai NS & Walcott EC Synaptic bombardment modulates muscarinic effects in forelimb motor cortex. J Neurosci 26, 2215–2226 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ozkan ED, et al. Input-specific regulation of hippocampal circuit maturation by non-muscle myosin IIB. J Neurochem 134, 429–444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agmon A & Connors BW Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379 (1991). [DOI] [PubMed] [Google Scholar]
- 64.Heyser CJ & Chemero A Novel object exploration in mice: not all objects are created equal. Behav Processes 89, 232–238 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Wu HP, Ioffe JC, Iverson MM, Boon JM & Dyck RH Novel, whisker-dependent texture discrimination task for mice. Behavioural brain research 237, 238–242 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Gaffield MA, Amat SB, Bito H & Christie JM Chronic imaging of movement-related Purkinje cell calcium activity in awake behaving mice. J Neurophysiol 115, 413–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slotnick B A simple 2-transistor touch or lick detector circuit. J Exp Anal Behav 91, 253–255 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He CX, et al. Tactile Defensiveness and Impaired Adaptation of Neuronal Activity in the Fmr1 Knock-Out Mouse Model of Autism. J Neurosci 37, 6475–6487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.