

Abstract
Carvone is a sustainable and readily available starting material for organic synthesis. Herein, we present the syntheses of various natural product scaffolds that rely on a novel benzannulation involving the α-methyl group (C-10) of carvone to afford a versatile tetralin. The utility of our synthetic approach is highlighted by its application to a short synthesis of the ent-3,4-seco-atisane diterpenoid (−)-crotogoudin. The 13-step enantiospecific synthesis features a regioselective double oxidative dearomatization, a Diels—Alder cycloaddition with ethylene gas (to construct the bicyclo[2.2.2]octane framework), and a final acid-mediated lactonization. The versatility of this benzannulation strategy is demonstrated by its utility in the preparation of the carbon skeleton of ent-3,4-seco-abietane diterpenoids using an intramolecular oxidative dearomatization.
A key aspiration in pursuing total syntheses of complex molecules in the modern era is to maximize sustainable practices.1 Designing highly efficient synthesis strategies, as well as powerful methods to implement them, is paramount to realizing this objective.2 In addition to considerations of strategies and methods, the choice of readily available and sustainable starting materials contributes substantially to achieving the goals of a modern synthesis. In this context, the pool of chiral compounds including amino acids,3 sugars,4 and terpenes5 (the “chiral pool”)6 has served admirably as starting materials for many practical and inspirational total syntheses over the past century. With regard to the total synthesis of terpenoid natural products,7 carvone has been a frequently employed starting material due to its ready availability in both enantiomeric forms, as well as the potential for the orthogonal derivatization of its functional groups.8
Despite the wealth of reactivity that has been established for the α-methyl (C-10), isopropenyl, and enone (i.e., double bond and carbonyl) groups of carvone (Figure 1), we recognized that direct C—C bond formation involving the α-methyl group has been underexplored. Direct C—C bond formation to this methyl substituent holds significant potential in the context of natural product synthesis. Specifically, we envisioned that if benzannulation of the carvone six-membered ring could be achieved by engaging the C-10-methyl and enone carbonyl groups, the stage could be set to access myriad natural product classes. In particular, numerous natural product scaffolds could arise from benzannulation following sequential diastereoselective functionalization α to the enone carbonyl group (i.e., at C-6) of carvone.9 For example, 3,4-seco-atisane natural products10 such as agallochaol C10a (Figure 1A) could be accessed from (S)-carvone whereas 3,4-seco-abietanes including seco-hinokiol11 or callicarpic acid A12 (Figure 1B) could arise from (R)-carvone.
Figure 1.
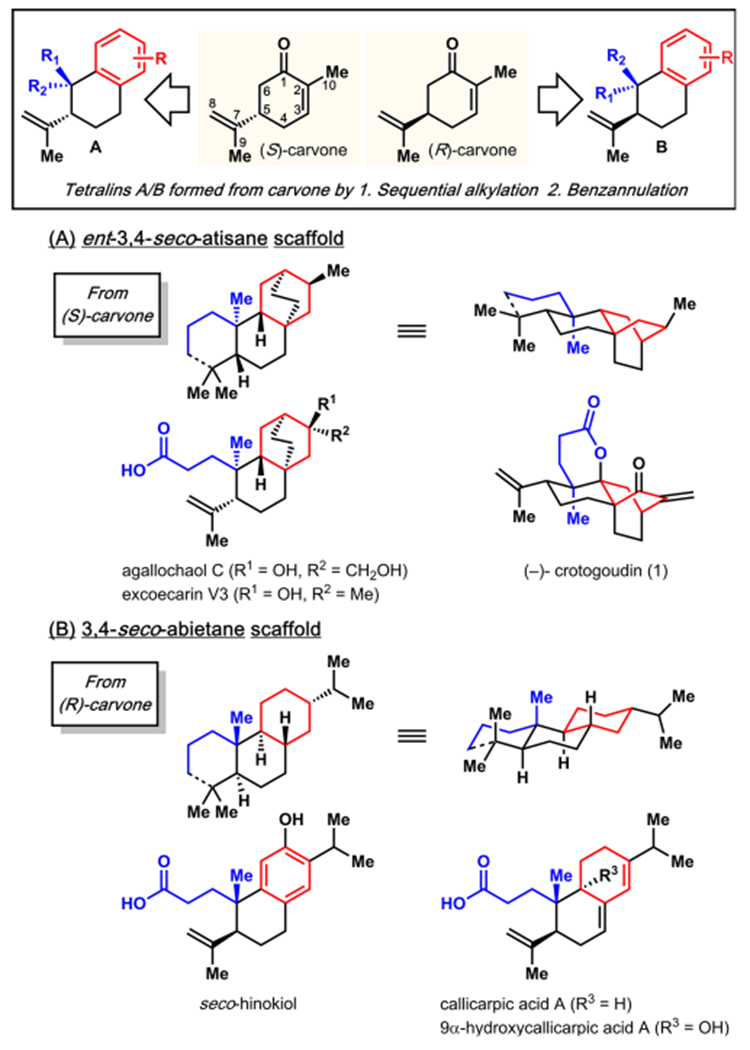
Benzannulation of carvone: a unified approach toward terpenoids.
In this Communication, we report our initial investigations into developing this potentially unifying strategy, which has afforded the frameworks of several terpenoid secondary metabolites via short diastereoselective sequences. The virtues of this approach are borne out in a short, enantiospecific total synthesis of the ent-3,4-seco-atisane diterpenoid (−)-crotogoudin (1)13 in 13 steps from (S)-carvone.
We commenced our studies with the preparation of benzo-fused bicycle 6 (Scheme 1A), bearing allyl and methyl groups at C-6 (carvone numbering). The methyl group is resident in many of the natural products that could arise from this benzannulated intermediate, whereas the choice of the allyl substituent was dictated by its facile introduction as well as its versatility for subsequent derivatizations. Following a well-established sequence, known carvone derivative 2 was easily prepared through a sequential methylation/allylation protocol.9c Conjugate reduction using l-Selectride followed by oxidative workup affords the corresponding ketone,14 which is converted to vinyl triflate 3 upon deprotonation and treatment of the resulting enolate with Comins’ reagent.15–16 Heck reaction of 3 with ethyl acrylate as the cross-coupling partner yields an ethyl enoate (4), which upon saponification provides acid 5, the substrate for benzannulation.
Scheme 1.
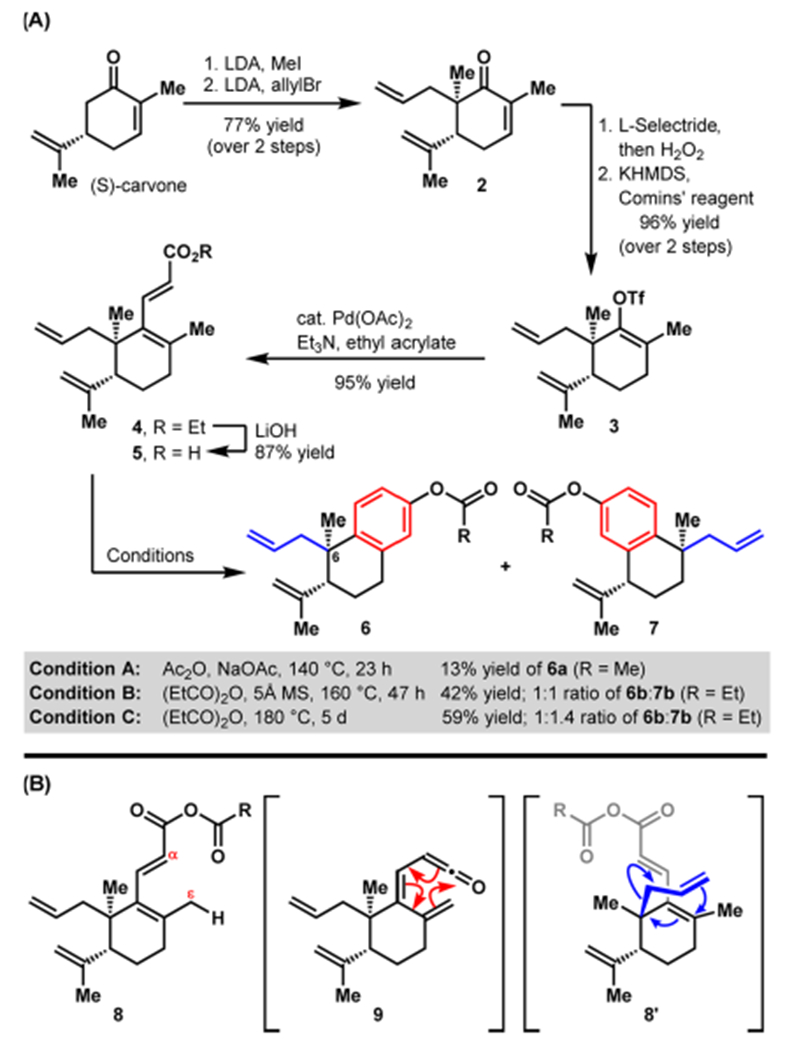
Synthesis of Hexadienoic Acid 5 and Initial Exploration of the Proposed Benzannulation
We anticipated that benzannulation would be achieved by conversion of carboxylic acid 5 to the corresponding ketene17 (9, Scheme 1B) by ε-deprotonation in mixed anhydride intermediate 8.18 In turn, 6π electrocyclization of 9, aromatization, and acylation of the resulting phenol would yield 6, consistent with the precedent of Murali and Krishna Rao.19 Several conditions (A–C), as outlined in Scheme 1A, were explored to effect the benzannulation. Using the conditions reported by Murali and Krishna Rao (Condition A), only a 13% yield of 6a was isolated from a complex reaction mixture.20 A switch to propionic anhydride as the solvent, which could be heated to 160 °C, led to a substantial increase in yield to 42% and the isolation of desired bicycle 6b and, surprisingly, constitutional isomer 7b in a 1:1 ratio. A Cope rearrangement21 of 8 prior to ketene formation and electrocyclization likely explains the genesis of 7b through conformer 8′. Full conversion of starting material 5 was achieved by heating the reaction mixture to 180 °C for 5 days, resulting in a combined yield of 59% of 6b and 7b (1:1.4 ratio).
In order to obviate the competing Cope rearrangement and with an eye toward application of the benzannulated bicycle to the synthesis of the diterpenoids illustrated in Figure 1, the allyl group of ester 4 was converted to an n-propyl hydroxy group (Scheme 2). This was achieved by chemoselective hydroboration of the allyl group in the presence of the isopropenyl group using Wilkinson’s catalyst (1 mol % loading) and catecholborane followed by oxidation of the resulting alkylborane.22,23 Saponification of the intermediate hydroxyester gave acid 10 in 83% yield over two steps. Benzannulated bispropionate bicycle 11 was formed in 82% yield upon heating 10 in propionic anhydride to 180 °C for 5 days.
Scheme 2.
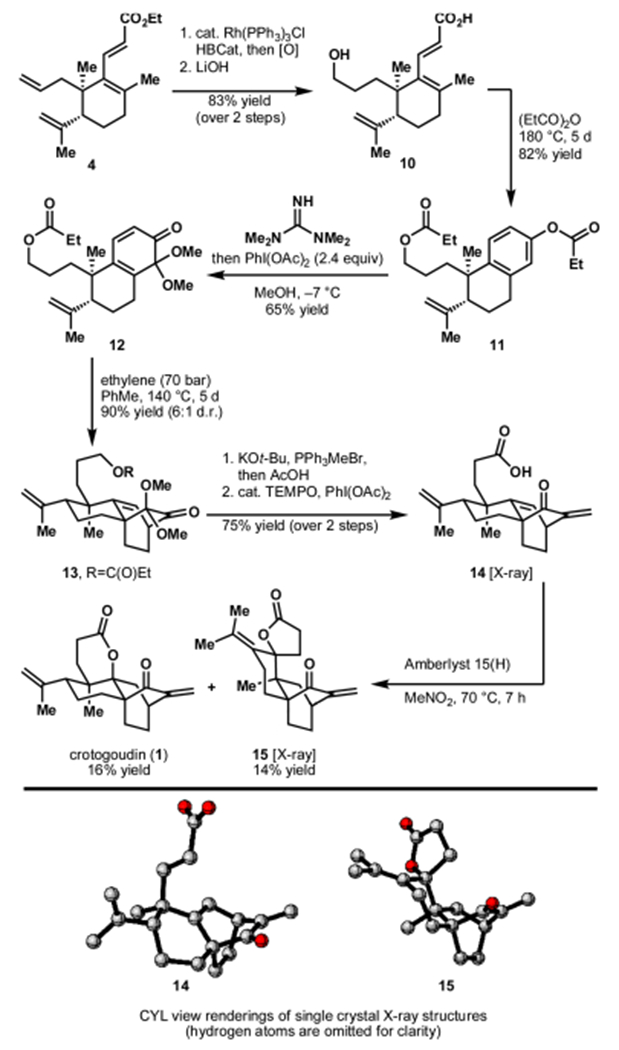
Completion of the Synthesis of Crotogoudin via a Double Oxidative Dearomatization Strategy
Following procedures adapted from Kunesch and Kondo,24 the phenyl propionate in 11 was selectively cleaved using tetramethylguanidine. This set the stage for a position selective oxidative dearomatization to afford dienone 12 (along with the corresponding para-quinol ether and the isomeric masked ortho-benzoquinone as side products in 11–13% yield, respectively).25,23 The observed selectivity in this iodine(III)-mediated oxidative dearomatization is rather unusual and has, to the best of our knowledge, only been reported by Mal and co-workers on simpler substrates.26 Inspired by Fukuyama’s recent synthesis of (−)-lepenine,27 a diastereoselective [4 + 2] cycloaddition of cyclohexadienone 12 with ethylene was envisioned. However, in accordance with investigations by Liu and co-workers, compound 12 did not readily undergo the desired Diels—Alder reaction.28 Cycloaddition only proceeded under pressure and at elevated temperature (70 bar, 140 °C, 5 d) to afford tricycle 13 in 90% yield (6:1 d.r.).29 At this stage, Wittig olefination of the ketone group followed by acid treatment removed both the propionyl group and cleaved the dimethyl ketal. The resulting primary hydroxyl was oxidized to the carboxyl group to provide seco-crotogoudin (14) in 75% yield over two steps.30,31 Lactonization of 14 to afford (−)-crotogoudin was fraught with complicating side reactions.23 Ultimately, conditions were identified that provided crotogoudin (1) in 16% yield (2.9% total yield over 13 steps), along with rearranged lactone 15 in 14% yield.32 Current efforts are directed at identifying conditions that provide 1 more selectively and in higher yield.33 Crotogoudin prepared using the strategy outlined here provided spectral and analytical data consistent with those obtained during its previous syntheses by Carreira13c and Liu13b as well as from its isolation by Dumontet and Rasoanaivo from croton goudotii.13a
Notably, our synthesis plan affords opportunities to access other diterpenoid secondary metabolites including the atisane and abietane frameworks outlined in Figure 1. For example, ester cleavage of bispropionate bicycle 11 (Scheme 3) and subsequent intramolecular oxidative dearomatization34 of the intermediate phenol (not shown) provided dienone 16. Selective reduction of the less substituted double bond of the cyclohexadienone moiety of 16 to yield α,β-unsaturated ketone 17 was achieved using a combination of MAD35 and l-Selectride.36 A 1,2-addition of an isopropyl group using Knochel’s method37 readily delivered allylic alcohol 18. The direct treatment of this tertiary alcohol with a proton source results in elimination to key intermediates (19 and 20) for the synthesis of seco-abietane congeners such as 9-hydroxycallicarpic acid A and seco-hinokiol, respectively.
Scheme 3.
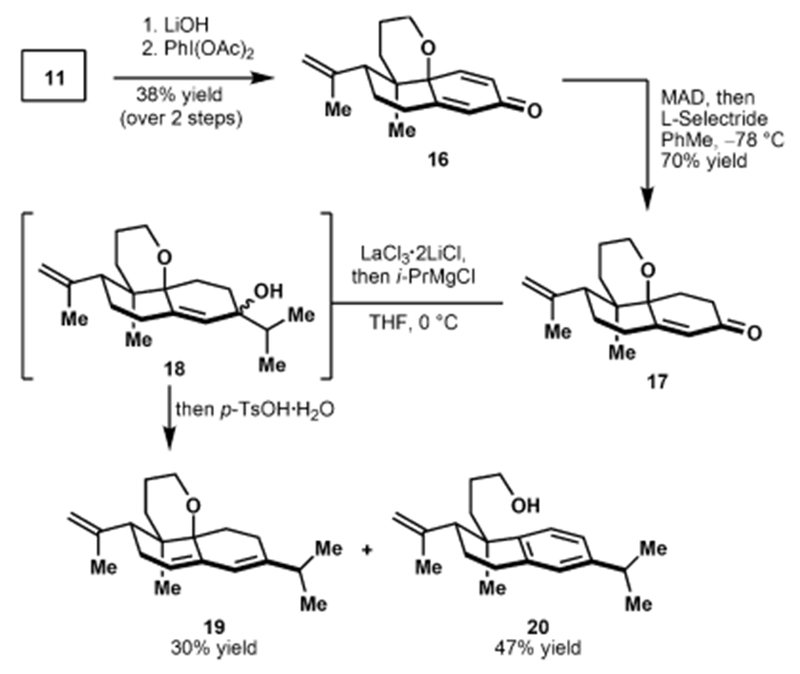
Synthesis of Secondary Metabolite Congeners via an Intramolecular Oxidative Dearomatization Pathway
In conclusion, a novel strategy for the synthesis of diterpenoids using carvone as a starting material has been developed. Several key transformations led to the success of this approach. These include (1) a benzannulation sequence that employs propionic anhydride and (2) a site-selective double oxidative dearomatization reaction that sets the stage for (3) a highly diastereoselective cycloaddition of ethylene to forge the key [2.2.2] bicycle. Our approach has led to an enantiospecific 13-step synthesis of the diterpenoid (−)-crotogoudin and provided a platform for the synthesis of other terpenoids. The application of this plan to the syntheses of other natural products is the subject of ongoing studies in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIGMS RO1 086374). P.F. is thankful for a postdoctoral scholarship from the German Academic Exchange Service (DAAD). K.M. is grateful to the Osaka University Research Abroad Program and the Osaka University Pharmaceutical Sciences Alumni Association (Yakuyukai) for a 1-year leave to undertake these studies at UC Berkeley. We also thank Dr. Antonio DiPasquale (UC Berkeley) and Nicholas S. Settineri (UC Berkeley) for solving the crystal structures of compounds 14 and 15 (X-ray facilities are supported by NIH Shared Instrument Grant S10-RR027172). The authors thank the Catalysis Facility of Lawrence Berkeley National Laboratory, which is supported by the Director, Office of Science, of the US Department of Energy under Contract No. DE-AC02-05CH11231 for generous access to their mass spectrometry instrument. The Bruker AVB-400, DRX-500, AV-500, AV-600 NMR spectrometers are partially funded by NSF grants (CHE-0130862, CHE 82-08992, and CHE 9633007) and NIH grants (1S10RR016634-01, RR02424A-01, and SRR023679A).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06823.
Experimental details and complete analytical data for all new compounds (PDF)
Crystallographic data for 14 (CIF)
Crystallographic data for 15 (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Brocksom TJ; Desiderá AL; de Carvalho Alves L; de Oliveira KT Curr. Org. Synth 2015, 12, 496. [Google Scholar]; (b) Chiou W-H; Ojima I In New Methodologies and Techniques for a Sustainable Organic Chemistry; Mordini A, Faigl F, Eds.; Springer: Dordrecht, 2008; Vol. 246, pp 55–83. [Google Scholar]
- (2).(a) Nicolaou KC; Vourloumis D; Winssinger N; Baran PS Angew. Chem., Int. Ed 2000, 39, 44; [PubMed] [Google Scholar]; Angew. Chem 2000, 112, 46. [Google Scholar]; (b) Nicolaou KC; Sorensen EJ Classics in Total Synthesis: Targets, Strategies, Methods; Wiley-VCH: Weinheim, 1996. [Google Scholar]; (c) Nicolaou KC; Snyder SA Classics in Total Synthesis II: More Targets, Strategies, Methods; Wiley-VCH: Weinheim, 2003. [Google Scholar]; (d) Nicolaou KC; Chen JS Classics in Total Synthesis III: Further Targets, Strategies, Methods; Wiley-VCH: Weinheim, 2011. [Google Scholar]; (e) Corey EJ; Cheng X-M The Logic of Chemical Synthesis; John Wiley & Sons: New York, 1995. [Google Scholar]; (f) Hudlicky T; Reed JW The Way of Synthesis: Evolution of Design and Methods for Natural Products; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- (3).Mulzer J In Comprehensive Chirality; Yamamoto H, Carreira EM, Eds.; Elsevier: Amsterdam, 2012; Vol. 2, pp 122–162. [Google Scholar]
- (4).Chida N; Sato T In Comprehensive Chirality; Yamamoto H, Carreira EM, Eds.; Elsevier: Amsterdam, 2012; Vol. 2, pp 207–239. [Google Scholar]
- (5).Gaich T; Mulzer J In Comprehensive Chirality; Yamamoto H, Carreira EM, Eds.; Elsevier: Amsterdam, 2012; Vol. 2, pp 163–206. [Google Scholar]
- (6).(a) Hanessian S Total synthesis of natural products: The “chiron” approach; Pergamon Press: Elmsford, NY, 1983. [Google Scholar]; (b) Blaser HU Chem. Rev 1992, 92, 935. [Google Scholar]
- (7).Ho T-L Enantioselective Synthesis: Natural Products from Chiral Terpenes; Wiley: New York, 1992. [Google Scholar]
- (8).Brill ZG; Condakes ML; Ting CP; Maimone TJ Chem. Rev 2017, DOI: 10.1021/acs.chemrev.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For examples for sequential diastereoselective functionalizations at C-6 of carvone, see:; (a) Abad A; Agulló C; Cuñat AC; de Alfonso Marzal I; Navarro I; Gris A Tetrahedron 2006, 62, 3266. [Google Scholar]; (b) Abad A; Agulló C; Arnó M; Cantín A; Cunãt AC; Meseguer B; Zaragozã RJ. J. Chem. Soc., Perkin Trans. 1 1997, 1837. [Google Scholar]; (c) Gesson J-P; Jacquesy J-C; Renoux B Tetrahedron 1989, 45, 5853. [Google Scholar]; (d) Abad A; Agulló C; Cuñat AC; Navarro I; Ramírez de Arellano MC Synlett 2001, 2001, 349. [Google Scholar]; (e) Miyaoka H; Honda D; Mitome H; Yamada Y Tetrahedron Lett 2002, 43, 7773. [Google Scholar]; (f) Abad A; Agulló C; Cuñat AC; Navarro I Tetrahedron Lett 2001, 42, 8965. [Google Scholar]; (g) Srikrishna A; Pardeshi VH; Satyanarayana G Tetrahedron Lett 2007,48,4087. [Google Scholar]; (h) Shing TKM; Tang Y; Malone JF J. Chem. Soc., Chem. Commun 1989, 1294. [Google Scholar]; (i) Abad A; Agulló C; Arnó M; Cuñat AC; Meseguer B; Zaragozà RJ J. Org. Chem 1998, 63, 5100. [Google Scholar]; (j) Arno M; Gonzalez MA; Zaragoza RJ Tetrahedron 1999, 55, 12419. [Google Scholar]; (k) Shing TKM; Zhu XY; Yeung YY Chem. - Eur. J 2003, 9, 5489. [DOI] [PubMed] [Google Scholar]
- (10).(a) Wang J-D; Li Z-Y; Guo Y-W Helv. Chim. Acta 2005, 88, 979. [Google Scholar]; (b) Wang J-D; Li Z-Y; Xiang W-S; Guo Y-W Helv. Chim. Acta 2006, 89, 1367. [Google Scholar]; (c) Li Y; Liu J; Yu S; Proksch P; Gu J; Lin W Phytochemistry 2010, 71, 2124. [DOI] [PubMed] [Google Scholar]; (d) Konishi T; Yamazoe K; Kanzato M; Konoshima T; Fujiwara Y Chem. Pharm. Bull. 2003, 51, 1142. [DOI] [PubMed] [Google Scholar]
- (11).Cantrell CL; Richheimer SL; Nicholas GM; Schmidt BK; Bailey DT J. Nat. Prod . 2005, 68, 98. [DOI] [PubMed] [Google Scholar]
- (12).Chen J-J; Wu H-M; Peng C-F; Chen I-S; Chu S-DJ Nat. Prod 2009, 72, 223. [DOI] [PubMed] [Google Scholar]
- (13).For isolation of crotogoudin, see:; (a) Rakotonandrasana OL; Raharinjato FH; Rajaonarivelo M; Dumontet V; Martin M-T; Bignon J; Rasoanaivo P J. Nat. Prod 2010, 73, 1730. [DOI] [PubMed] [Google Scholar]; For previous total syntheses, see:; (b) Song L; Zhu G; Liu Y; Liu B; Qin S J. Am. Chem. Soc 2015,137, 13706. [DOI] [PubMed] [Google Scholar]; (c) Breitler S; Carreira EM Angew. Chem., Int. Ed 2013, 52, 11168. [DOI] [PubMed] [Google Scholar]; For synthetic studies, see:; (d) Guo Y; Liu Q; Jia Y Chem. Commun 2015, 51, 889. [DOI] [PubMed] [Google Scholar]; (e) Behera TK; Singh V Tetrahedron 2014, 70, 7983. [Google Scholar]; (f) Ushakov DB; Maier ME Synlett 2013, 24, 705. [Google Scholar]; For recent reviews on the synthesis of atisane-type diterpenoids, see:; (g) Zhu G; Wadavrao SB; Liu B Chem. Rec 2017, 17, 584. [DOI] [PubMed] [Google Scholar]; (h) Zhu G; Liu R; Liu B Synthesis 2015, 47, 2691. [Google Scholar]
- (14).Fortunato JM; Ganem BJ Org. Chem 1976, 41, 2194. [Google Scholar]
- (15).For N-(5-chloro-2-pyridyl)triflimide, see:; Comins DL; Dehghani A Tetrahedron Lett 1992, 33, 6299. [Google Scholar]
- (16).A potential one-pot conversion of 2 to 3 was investigated; for carvone, see:; Crisp GT; Scott WJ Synthesis 1985,1985, 335. [Google Scholar]; Steric bulk conferred by the substituents at C-6 of enone 2 presumably hamper the reactivity of the intermediate enolate and result in only a poor yield of triflate 3.
- (17).(a) Dehmlow EV; Slopianka M Angew. Chem., Int. Ed. Engl 1979, 18, 170. [Google Scholar]; (b) Moore HW; Decker OHW Chem. Rev 1986,86, 821. [Google Scholar]; (c) Serra S; Fuganti C; Brenna E Chem. -Eur.J 2007,13, 6782. [DOI] [PubMed] [Google Scholar]; (d) Tidwell TT Ketenes II; John Wiley & Sons: Hoboken, NJ, 2006. [Google Scholar]; (e) Fu N; Tidwell TT In Organic Reactions; Denmark SE et al. , Eds.; John Wiley & Sons, Inc.: 2004; Vol. 87, pp 257–505. [Google Scholar]; (f) Danheiser RL; Dudley GB; Austin WF In Science of Synthesis (Houben-Weyl); Danheiser RL, Ed.; Georg Thieme Verlag: Stuttgart, 2006; Vol. 23, pp 493–568. [Google Scholar]
- (18).Deprotonation of the vinyl methyl ε position presumably occurs faster than at the endocyclic ε position. However, deprotonation of the latter position followed by equilibration and benzannulation cannot be ruled out.
- (19).Murali D; Krishna Rao GS Synthesis 1987, 1987, 254. [Google Scholar]
- (20).An extensive investigation of various reaction conditions was performed. Addition of acids (2-nitrobenzoic acid, camphorsulfonic acid, pyridinium p-toluene-sulfonate), bases (collidine, K2CO3, diisopropylethylamine) or cosolvents to compound 5 in acetic anhydride did not lead to a significant improvement in the reaction outcome. Attempts to form dienylketene 9 from mixed anhydrides with either TFAA or tosyl chloride were also not successful.
- (21).Cope AC; Hardy EM J. Am. Chem. Soc 1940, 62, 441. [Google Scholar]
- (22).(a) Evans DA; Fu GC; Hoveyda AH J. Am. Chem. Soc 1988, 110, 6917. [Google Scholar]; (b) Yoshinari T; Ohmori K; Schrems MG; Pfaltz A; Suzuki K Angew. Chem., Int. Ed 2010, 49, 881. [DOI] [PubMed] [Google Scholar]
- (23).See the Supporting Information for details.
- (24).(a) Oyama K.-i.; Kondo T Org. Lett 2003, 5, 209. [DOI] [PubMed] [Google Scholar]; (b) Kunesch N; Miet C; Poisson J Tetrahedron Lett 1987, 28, 3569. [Google Scholar]
- (25).For related observations regarding selectivity of bicylic phenols in aminomethylation, bromination, or oxidative dearomatization with IBX, see:; (a) Lange J; Hoogeveen S; Veerman W; Wals H Heterocycles 2000, 53, 197. [Google Scholar]; (b) Nilsson JLG; Selander H; Sievertsson H; Skanberg I; Svensson K-G Acta Chem. Scand 1971, 25, 94. [Google Scholar]; (c) Huang Y; Zhang J; Pettus TRR Org. Lett 2005, 7, 5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Roy HN; Sarkar MS; Mal D Synth. Commun 2005, 35, 2183. [Google Scholar]; (b) Mal D; Roy HN J. Chem. Soc., Perkin Trans. 1 1999, 3167. [Google Scholar]
- (27).Nishiyama Y; Han-ya Y; Yokoshima S; Fukuyama T J. Am. Chem. Soc 2014, 136, 6598. [DOI] [PubMed] [Google Scholar]
-
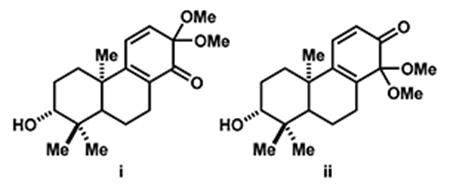
(28).In Liu’s synthesis of (±)-crotogoudin [ref 13b] i and ii were reduced or hydrolyzed by reacting with ethylene (7 MPa) in CH2Cl2 at 70 °C for 48 h. Dienone 12 did not react under these conditions.
- (29).Attempted Mukaiyama hydration of cycloadduct 13 under the conditions described by Liu and co-workers (see ref 13b) resulted in highly complex product mixtures.
- (30).Epp JB; Widlanski TS J. Org. Chem 1999, 64, 293. [DOI] [PubMed] [Google Scholar]
- (31).The absolute stereoconfiguration of compound 14 was unambiguously determined by single crystal X-ray analysis, and assignment of the stereocenters in cycloadduct 13 was thus inferred.
- (32).Compound 15 may arise from an initial isomerization of the isopropenyl double bond to the corresponding tetrasubstituted alkene. Protonation of the endocyclic double bond in the bicyclo[2.2.2]octane and a subsequent 1,2-methyl migration will then yield an allylic cation which may be engaged by the carboxyl group to form the γ-lactone.
- (33).An iodolactonization using 14 only engaged the isopropenyl group to form a seven-membered lactone. See the Supporting Information for details.
- 34.(a) Tamura Y; Yakura T; Haruta J; Kita Y J. Org. Chem 1987, 52, 3927. [Google Scholar]; (b) Bauer RA; Wenderski TA; Tan DS Nat. Chem. Biol 2012, 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) For a review, see:; Pouysegu L; Deffieux D; Quideau S Tetrahedron 2010, 66, 2235. [Google Scholar]
- (35).MAD = Methylaluminium bis(2,6-di-tert-butyl-4-alkylphen-oxide):; Maruoka K; Nonoshita K; Yamamoto H Tetrahedron Lett 1987, 28, 5723. [Google Scholar]
- (36).Doty BJ; Morrow GW Tetrahedron Lett 1990, 31, 6125. [Google Scholar]
- (37).Krasovskiy A; Kopp F; Knochel P Angew. Chem., Int. Ed 2006, 45, 497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.