

Abstract
The Ras-ERK (extracellular signal–regulated kinase) pathway is critical for controlling cell proliferation, and its aberrant activation drives the growth of various cancers. Because many pathogens produce toxins that inhibit Ras activity, efforts to develop effective Ras inhibitors for treating cancer could be informed by studies of Ras inhibition by pathogens. Vibrio vulnificus causes fatal infections in a manner that depends upon multifunctional-autoprocessing repeats-in-toxin (MARTX), a toxin that releases bacterial effector domains into host cells. One of the effector domains delivered by this toxin is the Ras/Rap1-specific endopeptidase (RRSP), which site-specifically cleaves the Switch I domain of the small GTPases Ras and Rap1. We solved the crystal structure of RRSP and found that its backbone shares a structural fold with the EreA/ChaN-like superfamily of enzymes. Unlike other proteases in this family, RRSP is not a metalloprotease. Through nuclear magnetic resonance (NMR) analysis and nucleotide exchange assays, we determined that RRSP processing did not release any fragments of KRAS or cause KRAS to dissociate from its bound nucleotide, but instead only locally impacted the structure. However, this structural alteration of KRAS was sufficient to disable guanine nucleotide exchange factor (GEF)-mediated nucleotide exchange and RAF binding. Thus, RRSP is a bacterial effector that represents a previously unrecognized class of protease that disconnects Ras from its signaling network while inducing limited structural disturbance to its target.
Introduction
Bacterial toxins have evolved to hijack cellular pathways by compromising the innate immune response, thus promoting bacterial invasion, and by modifying epithelial and endothelial cells to promote systemic spread (1). Inhibition or deregulation of signaling through small guanosine triphosphatases (GTPases) is one of the most common strategies for avoiding host innate responses because these proteins serve as central nexuses in many cellular signal transduction pathways. Ras is a small GTPase that directs extracellular signals through mitogen activated protein kinase (MAPK) signaling cascades to promote cell proliferation, differentiation, and survival by specific target gene expression (2). The activity of Ras is finely controlled by guanine-nucleotide-exchange factors (GEFs), which promote formation of the GTP-bound active state, and by GTPase-activating proteins (GAPs), which accelerate the intrinsic GTPase activity to promote formation of the GDP-bound inactive state (3–5). Cycling between GDP and GTP binding causes conformational changes of the Switch I (residues 30–40) and Switch II (residues 60–76) regions of Ras (5). In the GTP-bound active conformation, Ras interacts with downstream effectors to modulate cytoplasmic signaling networks (6). The best characterized downstream effectors of Ras are kinases of the Raf family, which mediate the activation of Ras-MAPK signal transduction pathways (6). Given its prominent role in cell proliferation, Ras signaling is frequently constitutively activated in cancer cells due to mutations in KRAS, NRAS, or HRAS. However, there are currently no drugs targeting Ras in cancer and very few Ras-specific inhibitors useful for studying this protein in cancer cell signaling.
Novel strategies to inhibit Ras could be informed by understanding the natural event of Ras inhibition by bacteria. Several bacteria produce secreted protein toxins that target Ras directly. Multifunctional-autoprocessing repeats-in-toxin (MARTX) toxins are 3500–5300 amino acid protein toxins secreted by Gram-negative bacteria through modified Type I secretion systems (7). These toxins translocate multiple effector domains across the eukaryotic plasma membrane (8), where they are released from the holotoxin into the cytosol by inositol hexakisphosphate–induced autoprocessing (9). The motile, Gram-negative opportunistic human pathogen Vibrio vulnificus causes rapidly progressing, life-threatening sepsis after contaminated seafood is ingested or open wounds are exposed to seawater in a manner that depends upon production of a MARTX toxin (7, 10, 11). Up to seven different variants of the toxin carrying distinct repertoires of effector domains have been identified in different V. vulnificus isolates (7). Remarkably, strains producing the MARTXVv toxin variant that contains the effector domain known as Ras/Rap1-specific endopeptidase (RRSP, also known as DUF5, P. asymbiotica rtxA homology domain, and PMT C1-C2 homology domain) show about 50-fold increased virulence in mice compared to an isogenic strain that produces a toxin without RRSP (12). This potent cytotoxicity of RRSP is due to its direct inhibition of MAPK signaling by proteolytically processing the small GTPases Ras and Rap1. This processing of Ras prevents the subsequent phosphorylation and activation of extracellular signal–regulated kinases (ERKs) (13). RRSP specifically recognizes the Switch I domain of Ras and Rap1 and cleaves it between Tyr32 and Asp33 (13, 14). This activity is also found in toxins of other bacteria, including Photorhabdus spp. insect pathogens and Aeromonas spp. pathogens of fish (13, 14).
Despite the extensive characterization of substrate specificity, the catalytic mechanism by which RRSP cleaves Ras remains unknown because RRSP shows no primary sequence similarity to known protease families. Further, how clipping the Switch I domain leads to inactivation of Ras remains unknown. In this study, we solved the crystal structure of the MARTXVv toxin RRSP effector domain and found it to be similar to that of members of the Ere-like and ChaN superfamily of hydrolytic enzymes (15). Structural alignment identified conserved catalytic residues that were essential for RRSP enzymatic activity and for cytotoxicity. However, RRSP, unlike other known proteases in this family, cleaved its substrate in the absence of metal cofactors. We further show the structural and functional consequences of KRAS cleavage by demonstrating that RRSP processing induced a mainly localized perturbation within and adjacent to the Switch I region without compromising either the overall structural integrity or the nucleotide binding site of KRAS. However, cleaved KRAS could not be stimulated for nucleotide exchange by a GEF and was unable to bind RAF1. Thus, the RRSP toxin effector domain is a unique class of protease that is highly potent for pathogenesis because it completely disconnects the Ras-ERK signaling network, an activity that may prove beneficial to studies of cancer cell signaling.
Results
Crystal structure reveals subdomain architecture of RRSP from the V. vulnificus MARTX toxin
To understand the catalytic mechanism of RRSP, we determined the crystal structure of RRSP from V. vulnificus strain isolate CMCP6 (NCBI locus NC_004460 ((16); residues 3594–4087) at 3.45 Å resolution. The structure was deposited into the Protein Data Bank (PDB) with PDB ID 5W6L (Table 1). There were two molecules in the asymmetric unit, and the structures of the two chains were nearly identical (RMSD 0.488 Å), with chain A selected for further analysis. The structure of RRSP is composed of two subdomains: C1 (residues 3596–3742) and C2 (residues 3743–4087) (Fig. 1A). The C1 subdomain includes seven α-helices (α1–α7), whereas the C2 subdomain is formed by 16 α-helices (α8–α23) and 9 β-strands (β1– β9) (Fig. 1B). The C2 subdomain can be divided into C2A (residues 3743–38430, containing α8– α11 and β1– β3), and C2B (residues 3856–4087, containing α13– α23 and β4– β9), both with α/β folds and connected by the long helix α−12.
Table 1.
X-ray data collection and refinement statistics of V. vulnificus RRSP (PDB ID: 5W6L)
| Crystal | RRSP |
|---|---|
| Data Collection | |
| X-ray wavelength | 0.97872 |
| Space group | I23 |
| a, b, c (Å) | 247.1, 247.1, 247,1 |
| α, β, γ (°) | 90.00, 90.00, 90.00 |
| Resolution (Å) | 30–3.45 (3.51–3.45)a |
| No. unique reflections | 33,078 (1,638) |
| Data redundancy | 6.8 (5.9) |
| Data completeness (%) | 100.0 (99.9) |
| Rsym (%)b | 11.3 (86.6) |
| I/sig | 18.4 (2.0) |
| Refinement | |
| Resolution (Å) | 29.97–3.45 |
| No. unique reflections | 31,061 |
| No. Reflections (Rfree)c | 1,562 |
| Rwork/Rfree (%) | 22.79/24.91 |
| Number of atoms | |
| Protein | 7,662 |
| Ligand/Ion | 17 |
| Solvent | 15 |
| R.m.s. deviations | |
| Bond length (Å) | 0.009 |
| Bond angle (°) | 1.235 |
| Average B factors (Å2) | |
| All proteins atom | 125 |
| Ligand/Solvent | 134 |
| Ramachandran Plot | |
| Outliers (%) | 0.0 |
| Allowed (%) | 6.0 |
| Favored (%) | 94.0 |
| Rotamer outliers (%) | 1.0 |
| No. C-Beta Deviations | 0 |
| All-Atom Clashscore | 2.0 |
Values in parenthesis are for the highest resolution shell.
Rsym = Σ|I-(I)|/ ΣI where I is the observed intensity of a reflection and (I) is the average intensity of all the symmetry related reflections.
For Rfree calculation, 10% of reflections were randomly excluded from the refinement
Fig. 1. Subdomain architecture of RRSP from V. vulnificus strain CMCP6.
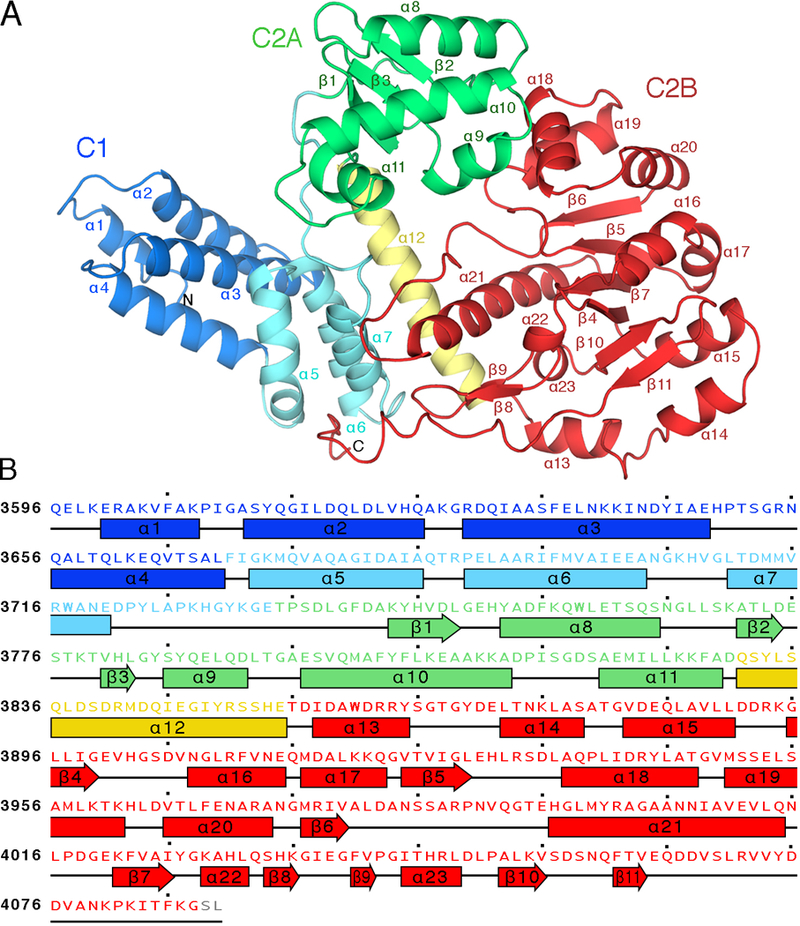
(A) Ribbon representation of RRSP structure. The C1 subdomain includes a conserved membrane localization subdomain (MLD, blue). The C2 subdomain can be divided into C2A (green) and C2B (red), which are connected by a helical linker (yellow). The 23 α helices and 9 β sheets are labelled. (B) Amino acid sequence of RRSP color coded to match the structure in (A) with secondary structure elements (α helices and β sheets) labelled.
The extreme N-terminus of RRSP (α1–4) is a characterized plasma membrane localization domain (MLD) found in many bacterial toxins (17, 18). It is essential for targeting RRSP to the negatively charged plasma membrane through positively charged lysine and arginine residues and a phenylalanine forming a classic basic-hydrophobic motif (17, 18). Nuclear magnetic resonance (NMR) analysis had previously showed that the RRSP N-terminus is a four-helix bundle in solution with residues essential for membrane targeting found in loops 1 and 3 (19). The RRSP structure here likewise reveals a four-helix bundle within a larger seven-helix C1 subdomain with α5–7 interfacing with the C2 subdomain (Fig. 1A). The resolved structure of the C1 MLD subdomain can be superimposed with similar targeting domains from Pasteurella multocida toxin (PMT), Vibrio cholerae Rho-inactivation domain, and clostridial glucosylating toxins (fig. S1).
The C2 subdomain is sufficient to confer cytotoxicity in mammalian and yeast cells (20). In a Dali server search (21) using the entire RRSP structure (fig. S2), the highest scoring hit (Z-score=31.7) was the structure of the C-terminus of PMT (PDB ID 2EBF) (15). In addition to the C1 MLD of RRSP showing high similarity to the C1 MLD of PMT as noted above, the RRSP-C2 and PMT-C2 domains also showed a high degree of similarity (RMSD 3.3 Å) even though the amino acid sequence identity is only 25% (fig. S3A). This similarity could suggest conservation of function. The PMT C-terminal region consists of three well-defined domains: C1, C2, and C3 (15). The PMT catalytic domain C3 induces the constitutive activation of Gα proteins of the Gq/11, G12/13, and Gi families by deamidating a crucial glutamine in the Switch II domain and blocking GTP hydrolysis activity (22, 23). RRSP does not contain a domain corresponding to the PMT-C3 domain. Further, PMT causes the transactivation of the MAPK cascade by targeting Gq/11 through the p63RhoGEF-RhoA pathway, a process that depends on functional Ras (24), making it unlikely that PMT-C2 would cleave Ras. Indeed, purified recombinant PMT containing only the C1 and C2 domains (rPMT-C1C2) mixed at equimolar concentration with recombinant human KRAS (rKRAS) for 30 min at 37°C did not show Ras processing activity, whereas recombinant (rRRSP) completely cleaved rKRAS in a parallel reaction (fig. S3B). These results demonstrate that although the PMT-C1C2 domains have a similar fold to RRSP, PMT is not able to cleave Ras. These biochemical data reinforce previous cell biological data that PMT-C1C2 in not cytotoxic and does not affect mitogenic signaling (20, 25).
The Dali search also revealed homology of the RRSP-C2B subdomain to the bacterial type III secretion system (T3SS) effector protein HopBA1 of Pseudomonas syringae (PDB ID 5T09 (26), Z-score=11.5), the heme-binding ChaN protein from Campylobacter jejuni (PDB ID 2G5G (27), Z-score=11.0) and two proteins of unknown function from Bacillus cereus, Bcr135 and Bcr136 (PDB ID 3B55 (28) and 2QGM (29), Z-scores=9.2 and 9.1, respectively) (Fig. 2A and fig. S2). All of these proteins have no known function. Thus, alignment of RRSP to other proteins of known structure does not inform about the catalytic mechanism by which RRSP can cleave Ras or is cytotoxic to cells.
Fig. 2. Transitive homology reveals that RRSP is a member of the EreA/ChaN-like family of enzymes.
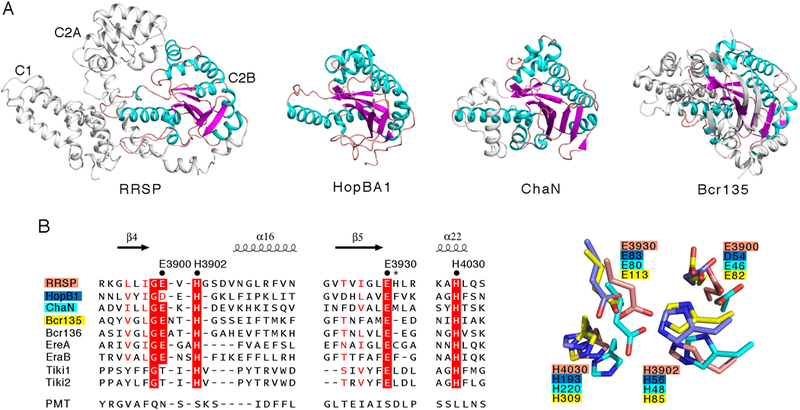
(A) Comparison of the RRSP C2B domain structure (α helices in teal, β sheets in magenta, and flexible linkers in pink) with that of other whole enzymes of the EreA/ChaN-like superfamily: HopBA1, (PDB ID 5T09, (26)), ChaN (PDB ID 2G5G, (72)), and Bcr135 (PDB ID 3B55, (28)). Structural representations were made with PyMOL. (B) Alignment of the primary sequences and structures of the three clusters of amino acids comprising the putative catalytic site of RRSP with those of other known members of the EreA/ChaN-like superfamily enzymes. The conserved residues (black dots) and the non-conserved His3931 (asterisk) that were mutated to alanine in this study are noted. Multiple alignment was performed with ESPript 3.0 (73).
Transitive homology analysis shows unrecognized catalytic residues in RRSP
Transitivity suggests that the function of a protein can be deduced if two proteins with little or no similarity can be clustered with a third protein to which both share similarity (30). The catalytic sites for the erythromycin esterases EreA and EreB (31) and the TIKI metalloproteases (32) were previously modeled based on primary sequence similarity with Bcr136 (31), which revealed structural, but not amino acid sequence, similarity to RRSP in our Dali search (fig. S2). Thus, transitive structure homology suggests that RRSP may belong to a broader family of hydrolytic enzymes, with the RRSP-C2B (residues 3882–4063) defined by the Structural Classification of Proteins (SCOP) database as an α/β fold with a β-strand core of the EreA/ChaN-like superfamily (33). Previous computational analyses predicted RRSP could belong to this family (34, 35).
The structural alignment of RRSP with HopBA1, ChaN, and Bcr135 and identification of the known catalytic residues from EreA, EreB, and Tiki2, all of which were previously modeled using Bcr136, revealed residues Glu3900, His3902, Glu3930, and His4030 as putative catalytic residues for RRSP (Fig. 2B). Notably, all of the identified residues are substituted in PMT (Fig. 2B), despite conservation of structure, which is consistent with PMT lacking Ras protease activity (fig. S3B). The putative catalytic residues are located within a cleft on the protein and are closely oriented, along with two non-conserved residues, Arg4001 and His3931 (Fig. 3A and fig. S4). We generated and purified rRRSPs in which alanine was substituted for each of the six identified residues and tested for their ability to cleave rKRAS in vitro at equimolar concentrations (Fig. 3B and3C). Replacing Glu3900 with alanine (E3900A) has no effect on the ability of rRRSP to cleave rKRAS, whereas replacing His3902 with alanine (H3902A) reduced rKRAS processing nearly 50%. Substitution of the conserved Glu3930 (E3930A) and His4030 (H4030A) residues resulted in proteins that were unable to cleave rKRAS, suggesting a major role for these residues in the catalytic mechanism. Exchange of the non-conserved His3931 with alanine (H3931A) also reduced rKRAS processing activity, whereas exchange of the non-conserved proximal residue Arg4001 to alanine (R4001A) did not affect rKRAS processing.
Fig. 3. RRSP residues that contribute to KRAS processing.
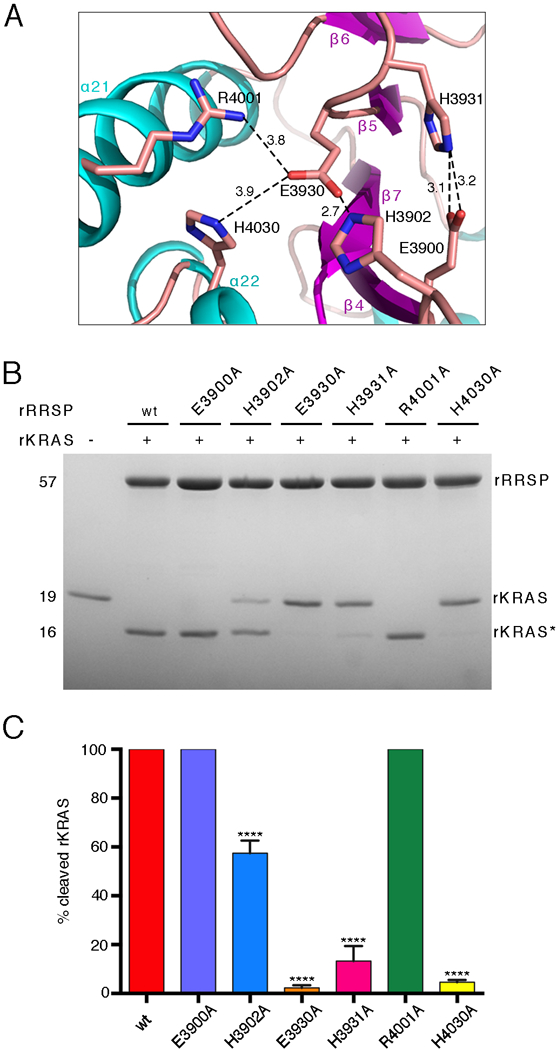
(A) Ribbon representation of the RRSP catalytic site. Sticks indicate the side groups of the residues substituted with alanine in our experiments. Amine groups and carboxyl groups are colored in blue and red, respectively. Polar interactions between residues are depicted by dotted lines with distances indicated in Å. (B) Representative SDS-PAGE analysis of the reaction product(s) of 10 μM rKRAS and 10 μM of each indicated rRRSP mutant following 30 min in vitro incubation. (N=3 independent experiments) (C) Quantification of rKRAS cleavage efficiency of rRRSP mutants shown in (B), bars are color coded by mutant. The bar graph reports means ± SD of three independent biological replicates (ANOVA followed by Dunnett’s multiple comparisons test; ****P≤0.0001).
RRSP is a unique protease in the EreA/ChaN-like family
Some members of the EreA/ChaN-like enzyme family, including the erythromycin esterase EreA and the metalloprotease TIKI2, require metal co-factors for their hydrolytic activity (31, 36), and the crystal structure of Bcr135 included a calcium ion in the putative active site (28). In contrast, EreB does not require a metal co-factor (31), and the structures of ChaN, Bcr136, and HopBA1 lack a divalent ion (26, 27, 29). Given that TIKI2, the only known protease in the EreA/ChaN-like family, is a characterized Co2+/Mn2+-dependent metalloprotease (32), it was surprising that a metal ion was absent from the crystal structure of RRSP. To test whether RRSP activity depended on metal co-factors, we incubated rRRSP with increasing concentrations of the metal ion chelators EDTA or 1,10-phenanthroline and then mixed it with an equimolar concentration of rKRAS. Chelator-treated rRRSP did not show inhibition of activity, even at the highest molar ratio of 1:500 (RRSP:chelator) (Fig. 4A and4B). As a positive control, the nonspecific protease thermolysin was inhibited by both EDTA and 1,10-phenanthroline under the same conditions (Fig. 4C).
Fig. 4. RRSP does not require a metal cofactor.
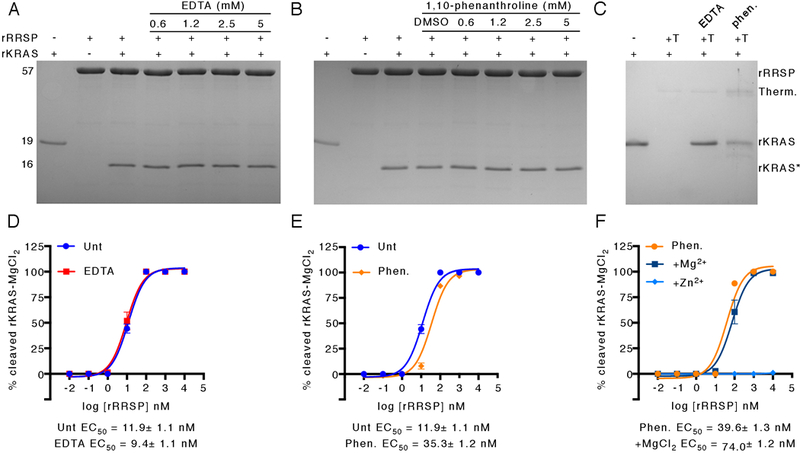
(A, B) SDS-PAGE gels showing cleavage of 10 μM rKRAS that had been buffer-exchanged to remove MgCl2 by 10 μM RRSP that had been pre-incubated with increasing amounts of EDTA (A) or 1,10-phenathroline (B) for 30 min. (C) Cleavage of 10 μM MgCl2-free rKRAS by 10 μM thermolysin (T) that had been pre-incubated with EDTA (A) or 1,10-phenathroline (B) for 30 min. SDS-PAGE gels are representatives of three independent biological replicates. (D-E) Half maximal concentration (EC50) curves for cleavage of 10 μM MgCl2-free rKRAS by increasing concentrations of untreated (Unt) rRRSP compared to rRRSP that had been pre-treated with 5 mM EDTA (D) or 1,10-phenanthroline treated (E). At the highest concentration of rRRSP, KRAS:rRRSP was 1:1 as in panels A and B. (F) EC50 curves for cleavage of 10 μM MgCl2-free rKRAS by rRRSP in the presence of 5 mM MgCl2 or ZnCl2 or by rRRSP pre-treated with 1,10-phenanthroline. All curves are derived from SDS-PAGE analysis (N=3 independent experiments) reported as the means ± SD. EC50 values calculated from non-linear curve fit ± the standard error.
In addition, rRRSP pre-treated with EDTA to chelate divalent cations showed a half maximal inhibition (EC50) value comparable to untreated rRRSP (Fig. 4D). Pre-treatment of rRRSP with 1,10-phenanthroline did reduce the EC50 value about 3-fold, although 100% of rKRAS was still processed within 10 minutes at an rRRSP:rKRAS ratio of 1:10 (Fig. 4E). The activity of chelator-treated rRRSP was not stimulated by MgCl2 carried over with purified rKRAS, because rKRAS that was not subjected to buffer exchange to remove MgCl2 prior to the addition of rRRSP showed identical EC50 values (fig. S5A and S5B). Addition of 5 mM CaCl2, CoCl2, MgCl2, or MnCl2 also did not stimulate chelated rRRSP and, in fact, addition of NiCl2, ZnCl2, or CuCl2 inhibited the reaction (Fig. 4F, fig. S6A, S6B and S6C. These results demonstrate that RRSP is not a metal-dependent protease and does not require divalent ions for its activity. Thus, RRSP uniquely represents a protease within the EreA/ChaN-like family that is not a metal-dependent enzyme. This grouping could eventually also include HopBA1 if this T3SS effector is found to cleave a plant protein as its substrate and does so in the absence of a metal co-factor.
Cleaved KRAS has a disordered Switch I but retains overall structure
The processing catalyzed by RRSP is known to be a specific cleavage in the Switch I domain of Ras, and it is also known that the KRAS N-terminal fragment is not released upon RRSP-mediated processing but remains tethered to the protein (13) and dissociates only upon boiling in SDS-PAGE buffer (Fig. 3B). Thus, the functional and biochemical properties of intact, cleaved KRAS (here called rKRAS*) can be assessed in vitro under non-denaturing conditions. Thermal stability analysis by differential scanning fluorimetry (DSF) showed that the melting temperature (Tm) of cleaved rKRAS* was negatively shifted by about 6.7°C compared to uncleaved rKRAS (fig. S7A), suggesting destabilization of the protein structure. Superimposition of 1H-15N heteronuclear single quantum coherence (HSQC) NMR spectra of rKRAS before and after incubation with rRRSP revealed structural differences concentrated in the Switch I region of rKRAS* (fig. S7B). The disordered Switch I loop region specifically lacks signals for the 1HN protons of Switch I residues Ala29–Ser39, extending to the β-strand 2 residues Tyr40-Lys42 (Fig. 5A). This line broadening surpasses the limit of detection and hence these signals do not appear in the spectra. In addition, residues Asp57-Gly60 of the DXXG motif, which together with Thr35 are essential for interaction with the nucleotide phosphate groups or the Mg2+ ion (37), also showed line broadening. The signal of several residues on β-strand 2 and α-helix 2 were affected by RRSP cleavage as well (Fig. 5A and fig. S7B).
Fig. 5. RRSP-cleaved KRAS maintains its tertiary structure and nucleotide binding.
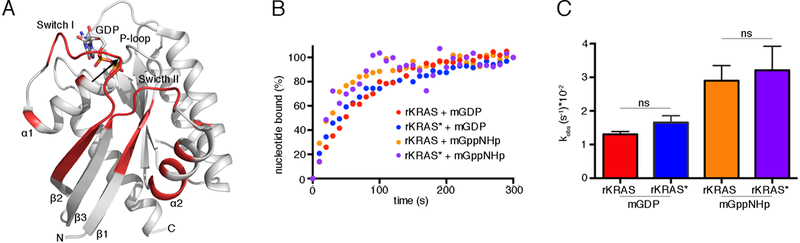
(A) Ribbon representation of the structure of GDP-bound KRAS (PDB ID 4OBE, (74)). Regions structurally affected by RRSP cleavage are colored in red, and the cleavage site is indicated by the black arrow. (B) Representative nucleotide exchange curves for rKRAS and RRSP-cleaved rKRAS (rKRAS*) in the presence of EDTA and either mant-GDP (mGDP) or mant-GppNHp (mGppNHp). (N=3 independent experiments) (C) Bar graph of the observed nucleotide exchange rates (kobs) of rKRAS and rKRAS* in the presence of mGDP or mGppNHp. Data were fit in GraphPad Prism 6.0 to a one-phase exponential association curve to determine kobs values. Data in (C) are means ± SD from three independent biological replicates (ANOVA followed by Dunnett’s multiple comparisons test; ns = non-significant).
These data reveal that RRSP cleavage between the Tyr32 and Asp33 residues of Ras mainly affects the Switch I region and the backbone dynamics downstream of the processing site. However, despite the disappearance of NMR signal around Switch I, rKRAS* is surprisingly not disrupted in its overall tertiary structure, explaining why Ras that is cleaved by RRSP in vivo is easily detected by Western blotting and rKRAS* routinely remains intact through manipulations required for in vitro biochemical assays (13).
Cleaved KRAS retains its bound nucleotide
The observed destabilization of KRAS* Switch I could affect Ras binding to nucleotides (14). To assess whether KRAS* retained the ability to bind nucleotides, we performed an EDTA-catalyzed nucleotide exchange assay. We mixed rKRAS* with a 5-fold molar excess of fluorescent nucleotides, mant-GDP and the non-hydrolyzable GTP analogue mant-GppNHp. The nucleotide exchange assay was initiated by the addition of 20 mM EDTA. KRAS* did not show a significantly higher kobs than rKRAS for mant-GDP or mant-GppNHp (Fig. 5B and5C). This demonstrates that the nucleotide-binding pocket of rKRAS* retains GDP and GTP, confirming that the tertiary structure was not lost after RRSP processing and that bound guanine nucleotides are not expelled. Moreover, the ability of EDTA to catalyze the nucleotide exchange of rKRAS* indicates that, although the processing by RRSP affects the orientation of residues involved in Ras metal binding, Mg2+ must be retained in the KRAS nucleotide-binding pocket. In addition, residues Gly13–Ser17, which bind to the α- and β-phosphates of the nucleotide, and residues Phe28, Asn116, and Asp119, which are critical for binding the guanosine moiety, are not affected by RRSP cleavage, explaining why rKRAS* retains its bound nucleotide (fig. S7B) (37).
Kinetic assays using nucleotide fluorescence detection reveals additional residues that contribute to RRSP activity
Because rKRAS* pre-loaded with mant-GDP retains the nucleotide upon RRSP-mediated processing, we developed a novel kinetic assay for KRAS processing in real time. Upon addition of rRRSP, the cleavage of rKRAS-mantGDP caused a loss in signal fluorescence (Fig. 6A) that was proportional to the amount of cleaved rKRAS (Fig. 6B and6C). Based on the NMR and fluorescence data, this shift is consistent with processing of Switch I, resulting in disordered rearrangement of the Tyr32 and other Switch I residues around the mant fluorophore. As expected, incubation of rRRSP E3930A and rRRSP H4030A with rKRAS-mant-GDP did not show a change in fluorescence, indicating a complete loss of the ability of rRRSP to process rKRAS (Fig. 6D). In this assay, rRRSP H3902A and rRRSP H3931A, both of which showed partial processing activity (Fig. 3B and3C), showed similar slow kobs rates (Fig. 6E). However, rRRSP E3900A and rRRSP R4001A, both of which completely cleaved rKRAS in the endpoint processing assay (Fig. 3B and3C), were significantly slower than wild-type rRRSP (Fig. 6E). Thus, in addition to Glu3930 and His4030 being required for catalysis, and Glu3900 and Arg4001 also contribute substantially to either catalysis or to the structural integrity of the active site.
Fig. 6. Real-time assay of rKRAS processing by RRSP.
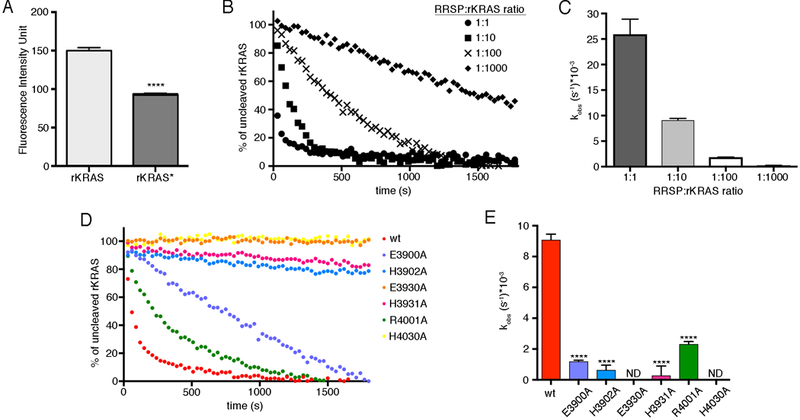
(A) Fluorescence of unprocessed rKRAS bound to mant-GDP and RRSP-processed rKRAS (rKRAS*) that was loaded with mant-GDP prior to incubation with RRSP. Fluorescence was quantified as intensity unit. (B) Representative curves of real-time cleavage assays of mant-GDP–bound rKRAS incubated with wild-type RRSP at the indicated ratios. (N=3 independent experiments) (C) Bar graphs of the observed rKRAS cleavage rates (kobs) of wild-type rRRSP. (D) Representative curves of real-time cleavage assays of mant-GDP–bound rKRAS incubated with wild-type (wt) and the indicated mutant forms of RRSP at 1:10 molar ratio (rRRSPs:rKRAS), (N=3 independent experiments). (E) Bar graphs of the observed rKRAS cleavage rates (kobs) of wild-type (wt) and mutant forms of rRRSP. Lines/bars are color-coded by mutant as in Figure 3. Data were fit in GraphPad Prism 6.0 to a one-phase exponential association curve to determine kobs values. Data in (A), (C) and (E) are means ± SD from three independent biological replicates (ANOVA followed by Dunnett’s multiple comparisons test; ****P≤0.0001).
Thermal stability analysis suggested that the substitution mutations did not impact the structural stability or RRSP (fig. S8A and S8B). Moreover, surface plasmon resonance (SPR) revealed that rRRSP has an affinity for rKRAS in the high μM range (fig. S9A). This affinity was slightly increased by the E3930A and H4030A mutations (fig. S9B), suggesting that the loss of activity was not due to the absence of protein-protein binding. In agreement, NMR also demonstrated that rRRSP H4030A retains the ability to interact with rKRAS, as evidenced by chemical shift perturbations in Ile46 and Asp54 of rKRAS that depended on the concentration of rRRSP H4030A (fig. S10). Binding to rRRSP H4030A also induced significant signal attenuation in Switch I and Switch II of rKRAS, most likely due to the association with mutated RRSP reducing backbone dynamics in these regions. Overall, these results demonstrate that the substitution mutations did not affect protein stability but rather that RRSP Glu3930 and His4030 are essential for Ras processing and thus likely share a hydrolytic mechanism with erythromycin esterases that similarly use a Glu–His catalytic pair (31).
RRSP cleavage of KRAS Switch I prevents KRAS from interacting with SOS
The son of sevenless (SOS) protein is a Ras GEF that opens the nucleotide-binding site of Ras by displacing Switch I and inserting a helical hairpin. thus allowing GDP-to-GTP exchange (38). The phosphate binding (P)-loop (residues 10–17), Switch I, and Switch II regions of Ras directly interact with SOS (38). NMR analysis revealed a certain degree of disorder within these regions in rKRAS* compared to unprocessed KRAS. Line broadening was also observed in the Switch I region, residues Asp57–Gly60, Met67, Arg73, Thr74, and Gly75, all of which directly interact with SOS (fig. S7B and Fig. 7A) (38). Such structural alteration should compromise SOS function by blocking SOS-catalyzed nucleotide exchange. To test this prediction, we loaded rKRAS with mant-GDP and carried out the SOS-catalyzed nucleotide exchange assay in the presence of GDP (39). Indeed, SOS was not able to significantly accelerate the nucleotide exchange rate of rKRAS* compared to that of uncleaved rKRAS (Fig. 7B–D). The intrinsic nucleotide exchange rate of rKRAS* was 3-fold faster than that of uncleaved rKRAS, suggesting rKRAS* has an open conformation that is favorable for intrinsic nucleotide exchange (Fig. 7D). In addition, the EDTA-catalyzed nucleotide exchange rate of rKRAS* was slightly higher than that of uncleaved rKRAS (Fig. 7D), confirming the presence of Mg2+ in the nucleotide-binding site.
Fig. 7. RRSP processing prevents the interaction of KRAS with GEFs and RAF.
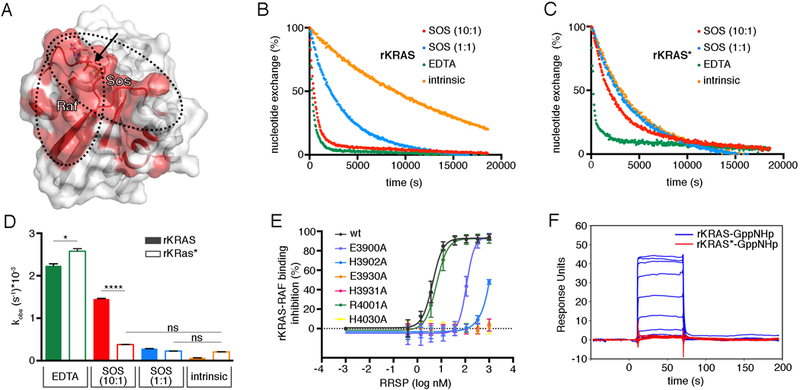
(A) Ribbon and surface representation of GDP-bound KRAS (PDB ID 4OBE, (74)). Regions that are structurally affected by RRSP cleavage are shaded in red, and the cleavage site is indicated by the black arrow. RAF and SOS binding interfaces are indicated with dotted lines. (B-C) Representative curves of intrinsic and SOS-catalyzed nucleotide exchange for rKRAS (B) and rKRAS* (C). (N=3 independent experiments) (D) Bar graph of the intrinsic and SOS-catalyzed nucleotide exchange rates (kobs) of rKRAS (filled bars) and rKRAS* (open bars). Each bar represents the mean ± SD of three independent biological replicates (ANOVA followed by Dunnett’s multiple comparisons test; *P≤0.05 and ****P≤0.0001). Data were fit in GraphPad Prism 6.0 to a one-phase exponential dissociation curve to determine the kobs. (E) Proximity-based AlphaLISA assay between GST-RAF-RBD and avidin-KRAS following pre-incubation of avidin-KRAS with increasing concentrations of each rRRSP mutant. Emission data were normalized to control without addition of RRSP and plotted as 1/x to show percent (%) binding inhibition by rRRSP and mutant variants. Lines are color-coded by mutant as in Fig. 3. Each curve represents the mean ± SD of three independent biological replicates. (F) Binding responses and KD from Biacore SPR analysis of the interaction between immobilized RAF-RBD and injected GppNHp-bound rKRAS or rKRAS*. Representative plot of time (s) vs response unit (RU, defined as binding of 1 pg per mm2) from a single injection is shown (N=3 independent experiments).
The Ras-binding domain of RAF cannot bind to cleaved KRAS
The direct physical interaction of Ras with its downstream effector kinase Raf is essential for the activation of the Ras-MAPK pathway (6). Because the Ras Switch I domain is directly involved in the interaction with Raf (40), RRSP cleavage of Ras between Tyr32 and Asp33 would be predicted to disrupt Raf binding (Fig. fig. S7B and Fig. 7A). To test this hypothesis, we used a proximity-based AlphaLISA binding assay (41). For this assay, biotinylated avidin-tagged KRAS (avi-KRAS) and the Ras binding domain of RAF fused to glutathione-S-transferase (GST-RAF-RBD) are attached to Alpha donor and acceptor beads, respectively, such that binding interaction results in singlet oxygen molecule diffusion detected as light emission from the acceptor bead fluorophore after excitation of the donor bead (42). In this assay, binding of active GppNHp-loaded rKRAS to RAF was proportionally reduced by prior treatment of avi-KRAS with increasing concentrations of rRRSP (Fig. 7E). This inhibition was not observed when avi-KRAS was pre-incubated with rRRSP E3930A, rRRSP H3931A, or rRRSP H4030A mutant proteins (Fig. 7E) that do not cleave rKRAS (Fig. 3B). rRRSP E3900A and rRRSP H3902A were less efficient in inhibiting RAF-RBD–KRAS interaction, and the rRRSP R4001A mutant showed no inhibition (Fig. 7E).
We also used SPR to confirm that RRSP processing of KRAS reduced the interaction between RAF-RBD (residues 55–131) and KRAS. The positive control, uncleaved GppNHp-loaded rKRAS showed strong interaction with RAF-RBD (KD = 335 nM) (Fig. 7F). In contrast, we did not detect an interaction of RAF-RBD with cleaved GppHNp-rKRAS* by SPR, even at the highest concentration of RAF-RBD (20 μM) (Fig. 7F). These data demonstrate that RRSP-mediated processing of the Switch I domain of Ras prevents active KRAS from binding to its downstream effector Raf and that this inhibition depends on the catalytic residues of RRSP.
RRSP processing of Ras in vitro and in cells requires catalytic residues Glu3930 and His4030
To determine the impact of RRSP catalysis on cytotoxicity, we used the anthrax toxin protective antigen (PA) to deliver RRSP fused to the anthrax toxin lethal factor N-terminus (LFN-RRSP) into human epithelial HeLa cells (fig. S11) (13, 20). In lysates from HeLa cells intoxicated with LFN-RRSP+PA for 24 hr, an antibody recognizing the G-domain of all Ras isoforms detected a band of ~15 kDa corresponding to the large fragment of cleaved RAS (Fig. 8A). In addition, phosphorylated ERK1 and ERK2 (ERK1/2) was reduced (Fig. 8B). In contrast, only full-length (21 kDa) RAS and no change in ERK1/2 phosphorylation were detected in HeLa cells intoxicated with catalytically inactive LFN-RRSP E3930A or LFN-RRSP H4030 (Fig 8C). Thus, Glu3930 and His4030 are essential for the Ras-processing activity of RRSP in cells and the downstream failure to activate ERK1/2.
Fig. 8. Glu3930 and His4030 are essential for RRSP cytotoxicity.
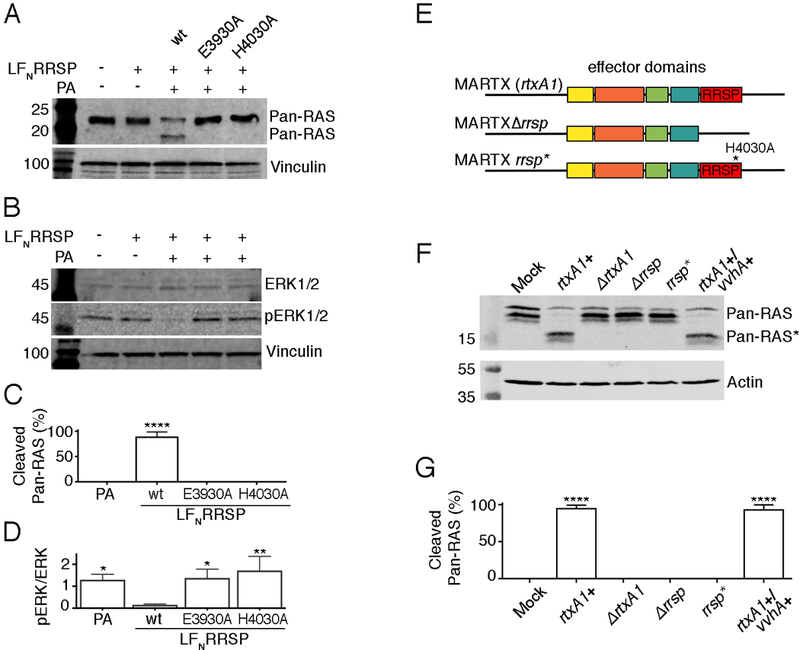
(A, B) Representative immunoblots of lysates from HeLa cells incubated with LFNRRSP, the LFNRRSP E3930A and H4030A mutants, and anthrax toxin protective antigen (PA) as indicated and probed with antibodies that recognize all RAS proteins (Pan-RAS) (A) or total ERK1/2 and phosphorylated ERK1/2 (pERK1/2) (B). (N=3 independent experiments) (C) Percentage of RAS that was cleaved in HeLa cells by wild-type and mutant LFNRRSP as determined by densitometry analysis. (D) The ratio of pERK1/2 to total ERK1/2 in lysates from HeLa cells following treatment with wild-type and mutant LFNRRSP. (E) Schematic representation of MARTX toxin effector domain organization in V. vulnificus CMCP6 strains. (F) Representative immunoblots showing RAS in lysates from T84 cells that has been incubated with V. vulnificus producing the indicated MARTX toxins (N=3). (G) Percentage of Ras that was cleaved in (E) as determined by densitometry analysis. All graphs (C, D, G) show means ± SD from three independent biological replicates (ANOVA followed by Dunnett’s multiple comparisons test; *P≤0.05, **P≤0.01 and ****P≤0.0001). n.d., not detected.
Alanine substitution at His4030 in the MARTX holotoxin prevents Ras cleavage in cells treated with V. vulnificus
The RRSP in this study was derived from the 5206 a.a. MARTX toxin from V. vulnificus strain CMCP6 (12, 16). To demonstrate the relevance of our biochemical findings in the context of the holotoxin during bacterial exposure, HeLa cells and clinically-relevant T84 intestinal cells, which express the activated mutant KRAS G13D, were treated with live V. vulnificus that had been modified to alter the catalytic residues of RRSP in the holotoxin (Fig. 8D). The modified strains were confirmed to induce MARTX-associated cell rounding upon infection (fig. S12A). To detect RAS processing, cells were treated with V. vulnificus for 1 hr, and whole cell lysates were collected for Western blotting. Lysates from cells treated with V. vulnificus carrying an intact copy of the gene that encodes the MARTXVv toxin (rtxA1+) showed detectable cleavage of RAS (Fig. 8E and fig. S12B). rtxA1+ strains both with and without the secondary virulence factor cytolysin VvhA showed equivalent processing of RAS. Cells treated with rtxA1-null V. vulnificus (∆rtxA1) or a strain with an in-frame deletion in rtxA1 to remove rrsp (∆rrsp) did not cleave RAS, consistent with published observations (13, 43). In contrast to previous studies, the use of a new pan-Ras antibody that recognizes the Ras G-domain (rather than the Switch I domain) facilitated detection of cleaved native Ras, as opposed to ectopically-expressed tagged Ras, indicating that this antibody can be used to detect cleavage of endogenous Ras without overexpression.
To test for contribution of the putative active site residues of RRSP, we used homologous recombination to mutate the His4030 codon to a codon for alanine within the endogenous rtxA1 gene in the V. vulnificus chromosome. This RRSP H4030A strain (rrsp*) retained the ability to induce cell rounding, a phenotype of the multifunctional toxin that is independent of RRSP activity, thereby demonstrating that other portions of the large holotoxin remained intact. This single amino acid change did, however, completely abrogate MARTX-dependent RAS processing, demonstrating that His4030 is an essential residue for MARTX toxin–mediated Ras cleavage in cells (Fig. 8E and Fig. 8F).
Discussion
Bacterial toxins have evolved to hijack cellular pathways to compromise the innate immune response and increase bacterial invasion and systemic spread into the host (1). Among many cellular targets, inhibition or disregulation of small GTPases is one of the most common mechanisms for bacterial disruption of cellular processes (44). RRSP is a toxin effector domain found in V. vulnificus, A. hydrophila, and P. asymbiotica and has been identified by sequence homology in other bacterial pathogens (13). RRSP specifically processes the Switch I domain of Ras and Rap1A (13, 14), but the mechanism of cleavage remained elusive due to the lack of RRSP sequence homology to known protease families. We have revealed the mechanism of action of RRSP to be a proteolytic processing event that dislodges Switch I and alters backbone dynamics downstream of the processing site, without disturbing the overall structure of Ras. We determined that this reaction is a metal-independent catalysis mediated by Glu3930 and His4030, with contributions from His3902 and His3931 and, to a lesser extent, from Glu3900 and Arg4001. This study further demonstrates that RRSP cleavage of KRAS Switch I between residues Tyr32 and Asp33 fully disconnects the Ras-MAPK pathway in human cells. The processing prevents the transition of Ras to an active conformation by blocking the interaction of Ras with GEFs and also impedes the interaction of Ras with the kinase Raf, such that proteins downstream of Raf in the signaling cascade, including ERK1/2, are not activated.
Remarkably, RRSP processing of Switch I does not affect the nucleotide-binding capacity of Ras. Indeed, cleaved KRAS (KRAS*) showed strong affinity for both GDP and GTP, comparable to uncleaved KRAS. This result was surprising because the nucleotide-binding pocket of Ras includes residues within the Switch I domain and the DXXG motif that were found by NMR analysis to undergo conformational changes following RRSP-mediated cleavage Ras. The nucleotide-binding capacity of KRAS* can be explained only by considering that a complex network of interactions engages the nucleotide and its surrounding residues from Ras regions that interact with the guanosine moiety but were not affected by the bisection of Switch I. In particular, the crystal structure of HRAS in the active state shows that Phe28 is positioned perpendicular to the aromatic ring of the nucleotide guanosine moiety, making a tight hydrophobic interaction (37). Amino acid substitution of Phe28 reduces the affinity for GDP and GTP and increases the nucleotide exchange rate (45). Phe28 maintains its orientation after RRSP processing, suggesting that it is one of the crucial residues for the binding of nucleotides to cleaved Ras. However, despite retention of the nucleotide, cleaved Ras can no longer be activated by SOS or activate downstream effector kinases due to structural perturbation of the SOS binding region.
The three-dimensional structure of RRSP shares a protein fold with the C-terminus of the PMT C2 subdomain, as previously predicted by primary sequence alignment (20). However, PMT-C2 does not cleave Ras, revealing that PMT possibly evolved by sequence divergence to have a different yet unknown function, potentially losing the ability to cleave Ras upon combining with the C3 domain that converted PMT from a Ras-inactivating toxin to a Ras-activating toxin.
The structure of RRSP also revealed that the RRSP C2B domain shares an α/β fold with members of the EreA/ChaN-like family, in which Glu3930 and His4030 have been shown to be critical for RRSP processing and cytotoxicity, consistent with the function of the corresponding residues from the erythromycin esterase EreB (31), Wnt metalloprotease TIKI2 (32), and the plant cytotoxin HopBA1 (26). Mutant forms of RRSP bearing substitutions in other modeled active site residues, E3900A and H3902A, showed reduced cleavage of KRAS. Unexpectedly, His3931 was also important for RRSP activity. This histidine is not conserved across the EreA/ChaN-like family but is 100% conserved in RRSP domains from other toxins. The identification of a conserved catalytic site organization among different enzymes with different activities might suggest a common catalytic mechanism that is preserved by the similar structural fold. Morar et al. proposed that EreA and EreB utilize a His-Glu pair to activate a water molecule, which is involved in a nucleophilic attack on the carbonyl carbon group of ester bonds (31). We speculate that RRSP would similarly use activation of water by its catalytic Glu-His pair to initiate the nucleophilic attack on the peptide bond in Ras. Zhang et al. alternatively suggested that the putative His-Glu catalytic residues of TIKI and EreA are crucial for coordinating catalytic divalent ions (Co2+ or Mn2+) to process the peptide bond (32). However, similar to RRSP, EreB and Bcr136 esterase activities are not inhibited by metal chelators. This suggests that the metal dependence of EreA and TIKI proteins might be due to the requirement of a metal ion for structural integrity but not for catalysis (31, 36). Co-crystal structures of EreA/ChaN-like family members with their natural substrates will be critical to further probe the enzymatic activity of the putative catalytic residues to determine if all EreA/ChaN-like members have a shared mechanism of nucleophilic attack for hydrolysis or whether some potentially use water whereas others use a metal ion.
Overall, this study reveals that the catalytic site residues employed by the RRSP effector domain to promote pathogenesis are found within the C2B subdomain. The function of the C2A subdomain remains unknown. A previous study found that ectopic overexpression of C2A, but not C2B, in HeLa cells was sufficient to induce cell rounding and cytotoxicity (20). This finding is consistent with a proposed two-step recognition of the substrate (14). In this model, C2A might be required for specific binding to the backbone of KRAS prior to insertion of Switch I into the active site in C2B. These studies indicate that C2A overexpression may inhibit Ras function by competitive binding, independent of Switch I processing, whereas C2B alone is not sufficient to cleave Ras (20). This may be due to improper folding of C2B when it is expressed alone, to improper membrane targeting of C2B in the absence of the C1 subdomain, or to the absence of putative substrate binding by C2A. The interaction between the C2A and KRAS may account for some of the detected changes in backbone dynamics observed by NMR when KRAS was incubated with increasing concentrations of catalytically inactive RRSP.
During V. vulnificus infection, the loss of signaling through the Ras-ERK pathway would result in reduced innate immune responses essential for host protection, thus enhancing bacterial infection (46–48). Indeed, the presence of RRSP increases the potency of the V. vulnificus MARTX toxin (12). Further, a study found that infection of mice with a V. vulnificus strain that delivers an RRSP+ MARTX toxin induced a less robust immune defense compared to a strain that delivers a MARTX toxin with a different collection of effectors that does not include RRSP (49). Further, toxin modifications to delete RRSP from this MARTX toxin, alone or in combination with another effector, impacted not only ERK, but also AKT signaling (50).
These studies may impact our understanding of tumorigenesis and anti-cancer drug discovery in the future. Indeed, there are currently no direct inhibitors of Ras in clinical use, and there are no chemical inhibitors useful for either in vitro or in vivo studies of Ras. Bacterial toxins like the clostridial toxin TpeL that glucosylates Thr35 in Ras have been used to inhibit Ras to study downstream signaling (24). In this study, rRRSP processing of KRAS in the AlphaLISA proximity assay not only to demonstrate that RRSP blocks Raf recruitment, but also validates this assay as a screening tool for identifying chemical inhibitors that could inhibit Ras-Raf binding. There are no specific inhibitors of the Ras-Raf interaction available, but blocking this interaction would be a useful strategy for targeting Ras activation in cancer. Further, our finding that LFN-RRSP cleaves Ras in vivo creates a tool for inhibiting RAS and pERK in cells in order to probe other aspects of cell biology. Indeed, LFN-RRSP+PA has already been used to demonstrate that desmoplakin activation of ERK1/2 depends on KRAS signaling in cardiomyocytes (51). It has been demonstrated that LFN-RRSP can cleave mutant KRAS G13D from HCT116 colorectal and MDA-MB-231 breast cancer cells (13), and now we report detection of LFN-RRSP –mediated processing of KRAS G13D in T84 colorectal cancer cells. Future studies will address the broad applicability of RRSP as a tool to study the role of KRAS in cell biological processes and cancer and may inform strategies for attenuating Ras signaling in cancer cells.
Materials and Methods
Reagents
E. coli TOP10 cells were obtained from Thermo Life Technologies and E. coli BL21(DE3)Magic and pMCSG7 bacterial expression vector (52) were provided by Andrzej Joachimiak (Argonne National Laboratory, IL). Proteins were expressed in the kanamycin-resistant BL21(DE3)(pMagic) E. coli strain. The pMagic plasmid provides the rare codon tRNA gene argU ((53) and A. Joachimiak, personal communication). Common reagents were obtained from Sigma-Aldrich, Fisher, or VWR. Restriction enzymes StuI, EcoRI, SspI, and BamHI, and Gibson Assembly® Master Mix were obtained from New England Biolabs. Custom DNA oligonucleotide primers and gBlocks™ as listed in table S1 and table S2 were purchased from Integrated DNA Technologies. Plasmids were purified by using Epoch spin columns or Qiagen Midi Prep kit according to the manufacturer’s recommended protocol. Plasmids were introduced into E. coli. BL21(DE3)(pMagic) by electroporation. Recombinant proteins were purified using ÄKTA Xpress Purifier system (GE Healthcare Bio-Sciences). Prepacked columns with Nickel Sepharose resin (Ni-NTA) HisTrap column for affinity chromatography or Superdex 200 (26/60) for size exclusion chromatography (SEC) were purchased from GE Healthcare Bio-Sciences. Anthrax Protective Antigen (PA) was purchased from List Biological Laboratories, Inc.
Crystallization and structure determination of RRSP
Recombinant RRSP (rRRSP) construct previously designed (13) was expressed in M9 Selenio-Methionine High-yield media according to the vendor instructions (MD045004–50L, Orion Enterprise Inc.). Protein expression was induced by adding 1 mM isopropyl-β-D-thiogalactoside (IPTG) at OD600 0.8 and growth continued at 18°C cells for 18 h. Cells were harvested by centrifugation and rRRSP was purified as previously described (13). The purified protein was concentrated to 15 mg/ml. Sitting drop crystallization plates were set up at room temperature by mixing 1 μl of rRRSP and 1 μl of reservoir solution with 90 μl of reservoir solution. Crystals were grown at 19°C using a reservoir of 0.1 M MOPS pH 6.5, 5% (w/v) PEG 400, 2 M NH4SO4. Harvested crystals were transferred to 4M formate before being frozen in liquid nitrogen. Diffraction data were collected at 100K from a single crystal at experimental station 21ID-F of the Life Science Collaborative Access Team (LS-CAT) at the Advanced Photon Source (APS). Crystals belong to the cubic space group I23 with unit cell parameters 247.1, 247.1, 247.1, 90.0, 90.0, 90.0. Two hundred frames, which correspond to 120° of spindle axes rotation, were collected from a single crystal at the selenium peak (λ=0.97872). Data were indexed, integrated, and scaled using the HKL3000 suite (54). The structure was solved with PHENIX (55), using the Single Anomalous Dispersion (SAD) technique. The initial FOM was 0.40 and after Density Modification (DM)975 out of 1036 residues (two protein chains 494 each and 24 residues of the purification tag) were automatically built into the experimentally obtained electron density maps. The initial model was checked and manually corrected in Coot (56). Another data set was generated for refinement by scaling and merging only the first 100 frames, thus, reducing radiation damage and increasing the resolution of the data. The corrected model was refined in Refmac (57) and solvent molecules were added to the model using ARP/wARP (58) followed by several rounds of refinement in Coot. The TLS group correction to the B-factors (59) was applied at the final steps of refinement. The structure factors and coordinates of the final model were validated using SFCHECK (60) and MolProbity (61) and deposited to the Protein Data Bank with PDB ID 5W6L. The data quality and the quality of the model are summarized in Table 1.
Cloning, expression and purification of RRSP and LFNRRSP proteins
Custom gBlocks™ designed to introduce mutations into C2B domain of RRSP sequence were assembled into the StuI/EcoRI-6xHisTag-RRSP-pMCSG7 (13) digested vector using Gibson Assembly® (New England Biolabs), following the manufacturer’s recommended protocol. For LFNRRSP proteins, DNA sequences corresponding to RRSP wt, RRSP E3930A and RRSP H4030A were amplified from the templates described above, using primers designed for Gibson Assembly®. PCR products were cloned in the pABII expression vector previously digested with BamHI in frame with N-terminal 6xHis tagged LFN domain (62). Plasmids were confirmed to be accurate by DNA sequencing and then transformed into BL21(DE3)Magic cells. All N-terminal 6xHis tagged RRSP and LFNRRSP proteins were purified over a Ni-NTA HisTrap column followed by SEC using the ÄKTA protein purification system, as previously described (13).
Purification of KRAS 1–169
The N-terminal portion of human KRAS (amino acids 1–169) was tagged with 6xHis and purified over a Ni-NTA HisTrap and SEC followed by 6xHis-tag removal by TEV cleavage, using the ÄKTA protein purification system, as previously described (63).
Cloning, expression and purification of PMT C1C2
The codon-optimized DNA sequence corresponding to PMT (residues 561–1088) was amplified from template plasmid previously described (20), using primers designed for ligation-independent cloning. The products were cloned into the pMCSG7 expression vector previously digested with SspI. Plasmid was confirmed to be accurate by DNA sequencing and then transformed into BL21(DE3)Magic cells. Recombinant PMT-C1C2 was purified as described above for rRRSP.
Cloning, expression and purification of SOS
DNA sequence corresponding to SOS1 (residues 566–1049) was amplified from the template plasmid obtained from NCI RAS Initiative RAS Reagents Core fully sequence validated ORFeome clone set (41), using primers designed for Gibson Assembly® (New England Biolabs). PCR products were cloned into the pMCSG7 expression vector previously digested with SspI. The plasmid nucleotide sequence was confirmed by DNA sequencing and then transformed into BL21(DE3)Magic cells. Recombinant SOS was purified as described above for rRRSP.
Expression and purification of 15N KRAS 1–169 for NMR
KRAS 1–169 fused to the C-terminal 6xHis-tagged CPD (63) was expressed in M9 media containing 0.4 M 15NH4Cl (CIL Inc.), 10 mM CaCl2, 10 mM MgCl2 and trace elements. Protein expression was induced by adding 1 mM IPTG at OD600 0.8 and growth continued at 25°C cells for 18 h. Cells were harvested by centrifugation and 15N KRAS 1–169 was purified as previously described (63). Briefly, KRAS 1–169 fused to the C-terminal 6xHis-Tagged CPD was purified over a Ni-NTA HisTrap column and by SEC using the ÄKTA protein purification system, followed by the incubation of 100 μM of InsP6 (phytic acid) to trigger CPD auto-cleavage. Tagless 15N KRAS 1–169 was recovered by removing 6xHis-tagged CPD over a Ni-NTA HisTrap column (63). NMR experiments were performed at 25°C on a Bruker Avance 900 MHz spectrometer equipped with a high sensitivity cryogenic probe. The protein was dissolved in 50 mM Tris citrate pH 6.5, 5 mM MgCl2, 50 mM NaCl, 10 mM β-mercaptoethanol, 20 mM CaCl2 and 10% D2O to achieve the final concentration of 400 μM. . Unlabeled wild type rRRSP was added to KRAS at a 1:1 molar ratio. 15N HSQC experiments were performed and spectra of KRAS before and after the addition of rRRSP were compared. Experiments with rRRSP H4030A were performed similarly to the experiments with wild type rRRSP, except rRRSP H4030A was added to KRAS at 0.01:1, 0.5:1, and 1:1 molar ratios. Sparky (UCSF) (64) was used for KRAS and cleaved KRAS assignments based on HNCA (65), CBCACONH and HNCACB experiments (66). NMRpipe software was used for processing and analyzing the data (67).
In vitro RRSP cleavage assay
rKRAS was incubated with rRRSP or rRRSP mutants or PMT C1C2 at equimolar concentrations (10 μM) in 10 mM Tris pH 7.5, 500 mM NaCl, 10 mM MgCl2 at 37°C for 30 min. Reactions were stopped by adding 6X Laemmli sample buffer and incubating the samples at 90°C for 5 min. Proteins were separated on 18% SDS–polyacrylamide gels and visualized using Coomassie stain.
For assays in the presence of chelators, 10 μM rRRSP was pre-incubated with 0.625, 1.25, 2.5 and 5 mM of EDTA or 1,10-phennthroline at 37°C. After 30 min, 10 μM of rKRAS, previously exchanged in a buffer without MgCl2, was added and the mixture was incubated at 37°C for 30 min. As control, 0.1 μM thermolysin (Sigma) was pre-incubated with 5 mM of EDTA or 1,10-phenanthroline at 37°C. For some assays, rRRSP was first treated in 5 mM EDTA or 5 mM 1,10-phenanthroline for 30 min and then passaged over a PD10 column with exchange to Buffer A (10 mM Tris-HCl, 500mM NaCl, pH 8.0). The treated rRRSP was then added at increasing concentrations to the buffer-exchanged rKRAS. For some assay, rKRAS that was not buffer exchanged was used to show the impact of MgCl2 carryover. For some assays, divalent cations as indicated in the legend were added to a final concentration of 5 mM. These reactions were incubated at 37°C for 10 min and then analyzed by SDS-PAGE as above.
SDS-PAGE analysis and quantification
Cleavage products of rKRAS were quantified from scanned SDS–polyacrylamide gels using NI ImageJ 1.64 (68). The percentage of cleavage product was calculated by the following formula:%Cleavage product=cleaved band/(cleaved rKRAS + uncleaved rKRAS) x100where cleaved rKRAS is the band running at 19 KDa. Each percentage value reported represents the mean ± SD of results from three independent experiments.
Differential Scanning Fluorimetry (DSF)
DSF experiments were performed using a 96-well thin-wall PCR plate (Axigen). Twenty μl reactions consisted of 2 μM of rKRAS, cleaved rKRAS, or rRRSP proteins in a solution of 5X SYPRO orange dye (Thermo Life Technologies), 0.1 mM TRIS, 150 mM NaCl, pH 7.5. Fluorescence intensity was monitored using the StepOnePlusTM Real-Time PCR Systems (Thermo Life Technologies) instrument. Samples were heated from 25 to 95°C at a scan rate of 1°C/min. Data were fit in GraphPad Prism 6.0 (GraphPad Software, Inc.) to Boltzmann equation to extrapolate Tm values that are reported as means ± the standard deviation (SD) from three independent biological replicates.
Nucleotide exchange assay
rKRAS was exchanged in 20 mM Tris pH 7.5, 150 mM NaCl, 5 mM ß-mercaptoethanol using PD10 desalting columns (GE Healthcare). rKRAS was incubated for 1 h at 37°C with rRRSP at a molar ratio of 1:100 (rKRAS:rRSSP). For the EDTA-mediated nucleotide exchange assay, 2 μM rKRAS and 2 μM rKRAS* were mixed with 4 μM mant-GTP (Thermo Life Technologies) or mant-GppNHp (Jena Bioscience) in assay buffer (20 mM Tris pH 7.5, 150 mM NaCl, 5 mM β-mercaptoethanol, 10 mM MgCl2, 20 mM EDTA) and dispensed into a 384-well plate. Fluorescence was measured every 10 s for 5 min at excitation/emission set to 360 nm/440 nm at 25 °C in a Spectramax M3 plate reader (Molecular Devices). Data were fit in GraphPad Prism 6.0 (GraphPad Software, Inc.) to a one-phase exponential association curve to determine the observed nucleotide exchange rate kobs. Data were reported as means and ± SD from three independent biological replicates.
Fluorescence real-time cleavage assay
rKRAS was incubated with 20-fold excess mant-GDP in the presence of 20 mM EDTA. After 1 h at room temperature, MgCl2 was added to a final concentration of 40 mM and the mix was incubated for 30 min at 25°C. The protein was exchanged in 20 mM Tris pH 7.5, 150 mM NaCl, 10 mM MgCl2, 5 mM β-mercaptoethanol using PD10 desalting columns (GE Healthcare); 1 μM of mant-GDP loaded rKRAS was dispensed in a 384-well plate and the cleavage reaction was started by adding each rRRSP at equimolar concentration with rKRAS. Fluorescence was measured every 25 s for 30 min at excitation/emission set to 360 nm/440 nm at 25°C in a Spectramax M3 plate reader (Molecular Devices). Data were fit in GraphPad Prism 6.0 (GraphPad Software, Inc.) to a single-exponential decay curve to determine the observed rKRAS processing kobs. Data were reported as means ± SD from three independent biological replicates.
SOS-mediated nucleotide exchange assay
rKRAS was loaded with mant-GDP, as described above. For the assay, 1 μM rKRAS and 1 μM rKRAS* were in the assay buffer (20 mM Tris pH 7.5, 150 mM NaCl, 5 mM β-mercaptoethanol, 10 mM MgCl2, 5 mM GDP) and dispensed into a 384-well plate. The SOS-mediated nucleotide exchange assay was initiated by the addition of 1 μM and 10 μM SOS to the reaction mix. Fluorescence was measured every minute for 5 h at excitation/emission set to 360 nm/440 nm at 25°C in a Spectramax M3 plate reader (Molecular Devices). Data were fit in GraphPad Prism 6.0 (GraphPad Software, Inc.) to a single-exponential decay curve to determine the observed rKRAS processing kobs. Data were reported as means ± SD from three independent biological replicates.
SPR measurements
SPR binding experiments were performed on a Biacore T200 instrument (GE). Human RAF1-RBD (RAF1 residues 51–131) and biotinylated avi-KRAS (KRAS with a C-terminal 15 amino acid AviTag™ (GLNDIFEAQKIEWHE)) proteins for this assay are described by Esposito et al. (41). Avi-KRAS was loaded with GppNHp and incubated with rRRSP (1:1 molar ratio) for 30 minutes at 37°C. Binding measurements of RAF1-RBD on avi-KRAS-GppNHp and cleaved avi-KRAS-GppNHp were carried out as follows. Neutravidin (Pierce) was amine coupled to the carboxymethylated dextran surface of a CM5 sensor chip (GE) using standard amine coupling chemistry. The surface was activated with 0.1 M N-hydroxysuccinimide and 0.4 M N-ethyl-N’-(3-dimethylaminopropyl) carbodiimide at a flow rate of 20 μl/min. Neutravidin was diluted to 20 μg/ml in 10 mM sodium acetate (pH 4.5) and injected on all 4 flow cells until a density of approximately 5000 RU was attached. Activated amine groups were quenched with an injection of 1 M ethanolamine (pH 8.0). Avi-KRAS-GppNHp and the cleaved avi-KRAS-GppNHp were captured on flow cells 2 and 4, flow cell 1 and 3 were used for referencing purposes. After the protein capture a series of buffer injections were performed in the running buffer (20 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 5 μM GppNHp) to establish a stable baseline. RBD was diluted in running buffer from 20 μM – 4nM and injected over the captured Avi-KRAS protein surface for 1 min at 30 μl/min at 25°C. At the end of the injection, bound RAF1-RBD was removed by a 30 sec injection of 1M NaCl. The data were fit using the Biacore T200 evaluation software. Binding of rKRAS to rRRSP was performed by amine coupling rRRSP wild type, E3930A, or H4030A protein on a CM5 sensor chip as outlined above. Serial dilutions of rKRAS-GDP (500 – 3.9 μM) were injected over the rRRSP proteins in 20 mM Hepes, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM TCEP, 5 μM GDP. The RRSP surfaces were regenerated with an injection of 1M NaCl. The data were analyzed using Biacore T200 evaluation software and steady state binding was fit using Origin software.
GST-RAF-RBD/rKRAS AlphaLISA binding assay
The effect of KRAS cleavage by rRRSP on effector binding was studied by in vitro protein-protein interaction using PerkinElmer’s AlphaLISA Technology as previously described (41, 42). RAF1-RBD (RAF residues 51–131) and biotinylated avi-KRAS proteins for this assay are described by Esposito et al. (41). Briefly, avi-KRAS binds the donor bead and GST-RAF-RBD binds the acceptor bead. Upon laser excitation of the donor bead, donor bead generated singlet state oxygen molecules react with the acceptor bead to generate chemiluminescence that then activates a fluorophore in the same bead for detectable emission at 520–620 nm (42). To determine the impact of rRRSP processing on KRAS interaction with RAF, avi-KRAS-GppNHp was incubated with rRRSP proteins (titration: 1 μM-0.4 nM) for 1 h at 37°C in a shaker (300 rpm), in the assay buffer (50 mM Tris pH 7.5, 5 mM NaCl, and 0.1 mM DTT). Followed by the addition of GST-RAF-RBD, and the binding reactions were carried out for 1 h at room temperature with shaking (700 rpm). In the final reaction avi-KRAS-GppNHp and GST- GST-RAF-RBD were 300 nM. After incubation, 30 μl mix of Streptavidin Alpha Donor beads (PerkinElmer) and Glutathione Acceptor beads (PerkinElmer) in Bead Buffer (50 mM Tris pH 7.5, 0.01 % Tween), were added to each reaction and the mix was incubated for 1 h at room temperature and protected from light. The reactions were read on a PerkinElmer EnVision® Multilabel Reader. Emission data were normalized to control sample without addition of RRSP and then plotted vs concentration using GraphPad Prism 6.0 (GraphPad Software, Inc.) as log(inhibitor) vs normalized response curve to determine IC50 values. Data were reported as means ± SD from three independent biological replicates.
HeLa cells intoxication
Human HeLa female epithelial cells (American Type Culture Collection CCL-2) were seeded into 6 well cell culture-treated plate and grown to 70%, at which time the cells were intoxicated with 3 nM of LFNRRSP proteins in combination with 7 nM of PA for 24 h. Cells were collected in the lysis buffer and total protein content was determined by BCA kit. Cell lysates were separated on 18% SDS–polyacrylamide gels followed by immunoblotting using an antibody that recognizes all forms of Ras (CPTC-KRAS4b-2, Developmental Studies Hybridoma Bank at Iowa University), and antibodies that recognize ERK1/2 (4696S Cell Signaling Technology), phosphorylated ERK/2 (4377S Cell Signaling Technology), and vinculin (ABCAM). Band quantification was performed by NIH ImageJ 1.64 (68) and band intensities values were normalized with vinculin.
Generation of RRSP H4030A (rrsp*) in V. vulnificus strains
Custom gBlock™ corresponding to regions upstream and downstream of RRSP H4030A was designed and assembled into digested pDS132 (69) using Gibson Assembly® according to the manufacturer’s protocols. The resulting plasmid was transformed to DH5α and it was purified by mini prep. It was then transformed to SM10λpir (70). The RRSP H4030A plasmid was transferred to V. vulnificus ΔvvhA CMCP6 strain by conjugation followed by selection for double homologous recombination using sucrose counterselection to isolate recombinants as previously described (71). Strain was validated to acquire the mutation by sequencing of amplified DNA.
Bacterial challenge of T84 and HeLa cells
V. vulnificus rifampicin-resistant isolates of strains CMCP6, CMCP6 ΔvvhA, CMCP6 ΔvvhAΔrtxA1 (12), CMCP6 ΔvvhAΔrrsp (43) and CMCP6 ΔvvhArrsp* were grown at 30°C in Luria–Bertani medium with 50 μg/ml rifampicin. Overnight cultures were diluted 1:1000 and grown at 30°C with shaking until the OD600 reached 0.5–0.6. Bacteria from 1–2mL were pelleted by centrifugation and resuspended in PBS to equal bacterial concentrations. Media were exchanged over 1 × 106 human T84 male intestinal epithelial cells (American Type Culture Collection CCL-248) or 2 × 105 HeLa cells previously seeded into 6 well cell culture-treated plate overnight for antibiotic-free media. V. vulnificus strains in PBS (multiplicity of infection =100) or an equal volume of buffer was added to media over cells and plates. After 60 min, both HeLa cells and T84 cells were collected for immunoblotting analysis, as described above. HeLa cells were also photographed before collection to validate that introduced mutations did not affect other biological activities of the toxin including cell rounding.
Supplementary Material
Acknowledgments:
The authors thank Zdzislaw Wawrzak, Matthew Lam, Hannah Gavin, Sara Fernandez Dunn, and Chi-Hao Luan for assistance in technical development of the project. X-ray data collection was done at the LS-CAT beamlines 21ID-F at the Advanced Photon Source Science User Facility operated for the U.S. Department of Energy. Services were provided by the Northwestern Genomics Core, Structural Biology Core, and High Throughput Analytical Laboratory and by the Nuclear Magnetic Resonance Laboratory of the University of Illinois-Chicago.
Funding: This work has been funded in whole or in part with Federal funds under NCI/NIH contract number HHSN261200800001E for the FNLCR and NIAID/NIH contract numbers HHSN272201200026C (to WFA) and HHSN272201700060C (to KJFS). for the Center for Structural Genomics of Infectious Diseases. Additional funding from PanCan/FNCLR KRAS and Chicago Biomedical Consortium Fellowships (to MB), DOD CDMRP Grant CA160954 (to AB), NIAID/NIH Fellowship T32AI007476 (to PJW), NIH grants R01AI092825, R01AI098369 (to KJFS) and R01CA188427 (to VG), the NCI-Ras Initiative at FNLCR (to AGS and MH), Robert H. Lurie Cancer Research Center, Northwestern Medicine Catalyst Fund, and a pilot grant from the Northwestern University Skin Diseases Research Center funded by NIH/NIAMS P30 AR057216. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Competing interests:M.B. and K.J.F.S. have a patent pending related to use of RRSP in study and treatment of cancer and other Ras-related syndromes.
Data and materials availability:The structure of RRSP has been reviewed and deposited into PDB (www.rcsg.org) with the PDB ID 5W6L. Recombinant overexpression plasmids for proteins described herein and newly created bacterial strains are available from Northwestern University with a Material Transfer Agreement. Reagents related to the AlphaLISA and SPR are available from the NCI-Ras Initiative with a Materials Transfer Agreement. All other data needed to evaluate the conclusions in the study are present in the paper or the Supplementary Materials.
References and Notes
- 1.Diacovich L, Gorvel JP, Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol 8, 117–128 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M, RAS oncogenes: the first 30 years. Nat Rev Cancer 3, 459–465 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, Kim SH, Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247, 939–945 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Wittinghofer A, Pai EF, The structure of Ras protein: a model for a universal molecular switch. Trends Biochem Sci 16, 382–387 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Wennerberg K, Rossman KL, Der CJ, The Ras superfamily at a glance. J Cell Sci 118, 843–846 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Roskoski R Jr., RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun 399, 313–317 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Satchell KJ, Multifunctional-autoprocessing repeats-in-toxin (MARTX) toxins of Vibrios. Microbiol Spectr 3, VE-0002–2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim BS, Gavin HE, Satchell KJ, Distinct roles of the repeat-containing regions and effector domains of the Vibrio vulnificus multifunctional-autoprocessing repeats-in-toxin (MARTX) toxin. mBio 6, e00324 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egerer M, Satchell KJ, Inositol hexakisphosphate-induced autoprocessing of large bacterial protein toxins. PLoS Pathog 6, e1000942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daniels NA, Vibrio vulnificus oysters: pearls and perils. Clin Infect Dis 52, 788–792 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Jones MK, Oliver JD, Vibrio vulnificus: disease and pathogenesis. Infect Immun 77, 1723–1733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwak JS, Jeong HG, Satchell KJ, Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proc Natl Acad Sci U S A 108, 1645–1650 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antic I, Biancucci M, Zhu Y, Gius DR, Satchell KJ, Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat Commun 6, 7396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biancucci M, Rabideau AE, Lu Z, Loftis AR, Pentelute BL, Satchell KJF, Substrate Recognition of MARTX Ras/Rap1-Specific Endopeptidase. Biochemistry 56, 2747–2757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitadokoro K, Kamitani S, Miyazawa M, Hanajima-Ozawa M, Fukui A, Miyake M, Horiguchi Y, Crystal structures reveal a thiol protease-like catalytic triad in the C-terminal region of Pasteurella multocida toxin. Proc Natl Acad Sci U S A 104, 5139–5144 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim YR, Lee SE, Kim CM, Kim SY, Shin EK, Shin DH, Chung SS, Choy HE, Progulske-Fox A, Hillman JD, Handfield M, Rhee JH, Characterization and pathogenic significance of Vibrio vulnificus antigens preferentially expressed in septicemic patients. Infect Immun 71, 5461–5471 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geissler B, Tungekar R, Satchell KJ, Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Proc Natl Acad Sci U S A 107, 5581–5586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geissler B, Ahrens S, Satchell KJ, Plasma membrane association of three classes of bacterial toxins is mediated by a basic-hydrophobic motif. Cell Microbiol 14, 286–298 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hisao GS, Brothers MC, Ho M, Wilson BA, Rienstra CM, The membrane localization domains of two distinct bacterial toxins form a 4-helix-bundle in solution. Protein Sci 26, 497–504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antic I, Biancucci M, Satchell KJ, Cytotoxicity of the Vibrio vulnificus MARTX toxin effector DUF5 is linked to the C2A subdomain. Proteins 82, 2643–2656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holm L, Rosenstrom P, Dali server: conservation mapping in 3D. Nucleic Acids Res 38, W545–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orth JH, Fester I, Siegert P, Weise M, Lanner U, Kamitani S, Tachibana T, Wilson BA, Schlosser A, Horiguchi Y, Aktories K, Substrate specificity of Pasteurella multocida toxin for alpha subunits of heterotrimeric G proteins. FASEB J 27, 832–842 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orth JH, Preuss I, Fester I, Schlosser A, Wilson BA, Aktories K, Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc Natl Acad Sci U S A 106, 7179–7184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegert P, Schmidt G, Papatheodorou P, Wieland T, Aktories K, Orth JH, Pasteurella multocida toxin prevents osteoblast differentiation by transactivation of the MAP-kinase cascade via the Galpha(q/11)--p63RhoGEF--RhoA axis. PLoS Pathog 9, e1003385 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aminova LR, Luo S, Bannai Y, Ho M, Wilson BA, The C3 domain of Pasteurella multocida toxin is the minimal domain responsible for activation of Gq-dependent calcium and mitogenic signaling. Protein Sci 17, 945–949 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishimura MT, Anderson RG, Cherkis KA, Law TF, Liu QL, Machius M, Nimchuk ZL, Yang L, Chung EH, El Kasmi F, Hyunh M, Osborne Nishimura E, Sondek JE, Dangl JL, TIR-only protein RBA1 recognizes a pathogen effector to regulate cell death in Arabidopsis. Proc Natl Acad Sci U S A 114, E2053–E2062 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan ACK, Lelj-Garolla B, Rosell FI, Pedersen KA, Mauk AG, Murphy MEP, Cofacial heme binding is linked to dimerization by a bacterial heme transport protein. Journal of Molecular Biology 362, 1108–1119 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Vorobiev SM, Chen Y, Kuzin AP, Seetharaman J, Wang H, Cunningham K, Owens L, Maglaqui M, Xiao R, Acton TB, Montelione GT, Tong L, Hunt JF, Crystal structure of the Q81BN2_BACCR protein from Bacillus cereus. Northeast Structural Genomics Consortium target BcR135 RCSB Protein Data Bank. 2007. (doi:10.2210/pdb3b55/pdb). [Google Scholar]
- 29.Kuzin AP, Abashidze M, Jayaraman S, Wang H, Fang Y, Maglaqui M, Ma L-C, Xiao R, Liu J, Baran MC, Acton TB, Rost B, Montelione GT, Hunt JF, Tong L, Crystal structure of succinoglycan biosynthesis protein at the resolution 1.7 A. Northeast Structural Genomics Consortium target BcR136 RCSB Protein Data Bank 2007. (doi: 10.2210/pdb2qgm/pdb). [Google Scholar]
- 30.Bolten E, Schliep A, Schneckener S, Schomburg D, Schrader R, Clustering protein sequences--structure prediction by transitive homology. Bioinformatics 17, 935–941 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Morar M, Pengelly K, Koteva K, Wright GD, Mechanism and diversity of the erythromycin esterase family of enzymes. Biochemistry 51, 1740–1751 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, MacDonald BT, Gao H, Shamashkin M, Coyle AJ, Martinez RV, He X, Characterization of Tiki, a new family of WNT-specific metalloproteases. J Biol Chem 291, 2435–2443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murzin AG, Brenner SE, Hubbard T, Chothia C, SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247, 536–540 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Bazan JF, Macdonald BT, He X, The TIKI/TraB/PrgY family: a common protease fold for cell signaling from bacteria to metazoa? Dev Cell 25, 225–227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Pulido L, Ponting CP, Tiki, at the head of a new superfamily of enzymes. Bioinformatics 29, 2371–2374 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Abreu JG, Yokota C, MacDonald BT, Singh S, Coburn KL, Cheong SM, Zhang MM, Ye QZ, Hang HC, Steen H, He X, Tiki1 is required for head formation via Wnt cleavage-oxidation and inactivation. Cell 149, 1565–1577 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knihtila R, Holzapfel G, Weiss K, Meilleur F, Mattos C, Neutron Crystal Structure of RAS GTPase Puts in Question the Protonation State of the GTP gamma-Phosphate. J Biol Chem 290, 31025–31036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J, The structural basis of the activation of Ras by Sos. Nature 394, 337–343 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM, K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thapar R, Williams JG, Campbell SL, NMR characterization of full-length farnesylated and non-farnesylated H-Ras and its implications for Raf activation. J Mol Biol 343, 1391–1408 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Esposito D, Stephen AG, Turbyville TJ, Holderfield M, New weapons to penetrate the armor: Novel reagents and assays developed at the NCI RAS Initiative to enable discovery of RAS therapeutics. Semin Cancer Biol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bosse R, Illy C, Chelsky D, “Principles of AlphaScreen: Amplified Luminescent proximity homogenous assay,” AlphaScreen Application Note (2002). [Google Scholar]
- 43.Gavin HE, Beubier NT, Satchell KJ, The Effector Domain Region of the Vibrio vulnificus MARTX Toxin Confers Biphasic Epithelial Barrier Disruption and Is Essential for Systemic Spread from the Intestine. PLoS Pathog 13, e1006119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aktories K, Bacterial protein toxins that modify host regulatory GTPases. Nat Rev Microbiol 9, 487–498 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Reinstein J, Schlichting I, Frech M, Goody RS, Wittinghofer A, p21 with a phenylalanine 28----leucine mutation reacts normally with the GTPase activating protein GAP but nevertheless has transforming properties. J Biol Chem 266, 17700–17706 (1991). [PubMed] [Google Scholar]
- 46.Abraham SJ, Muhamed I, Nolet R, Yeung F, Gaponenko V, Expression, purification, and characterization of soluble K-Ras4B for structural analysis. Protein Expr Purif 73, 125–131 (2010). [DOI] [PubMed] [Google Scholar]
- 47.David MD, Cochrane CL, Duncan SK, Schrader JW, Pure lipopolysaccharide or synthetic lipid a induces activation of p21Ras in primary macrophages through a pathway dependent on Src family kinases and PI3K. J Immunol 175, 8236–8241 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Scheele JS, Marks RE, Boss GR, Signaling by small GTPases in the immune system. Immunol Rev 218, 92–101 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Murciano C, Lee CT, Fernandez-Bravo A, Hsieh TH, Fouz B, Hor LI, Amaro C, MARTX Toxin in the Zoonotic Serovar of Vibrio vulnificus Triggers an Early Cytokine Storm in Mice. Front Cell Infect Microbiol 7, 332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen CL, Chien SC, Leu TH, Harn HI, Tang MJ, Hor LI, Vibrio vulnificus MARTX cytotoxin causes inactivation of phagocytosis-related signaling molecules in macrophages. J Biomed Sci 24, 58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kam CY, Dubash AD, Magistrati E, Polo S, Satchell KJF, Sheikh F, Lampe PD, Green KJ, Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. J Cell Biol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI, A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif 25, 8–15 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Wu N, Christendat D, Dharamsi A, Pai EF, Purification, crystallization and preliminary X-ray study of orotidine 5’-monophosphate decarboxylase. Acta Crystallogr D Biol Crystallogr 56, 912–914 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M, HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murshudov GN, Vagin AA, Dodson EJ, Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Perrakis A, Morris R, Lamzin VS, Automated protein model building combined with iterative structure refinement. Nature Structural Biology 6, 458–463 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Painter J, Merritt EA, Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62, 439–450 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Vaguine AA, Richelle J, Wodak SJ, SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr 55, 191–205 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spyres LM, Qa’Dan M, Meader A, Tomasek JJ, Howard EW, Ballard JD, Cytosolic delivery and characterization of the TcdB glucosylating domain by using a heterologous protein fusion. Infect Immun 69, 599–601 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biancucci M, Dolores JS, Wong J, Grimshaw S, Anderson WF, Satchell KJ, Kwon K, New ligation independent cloning vectors for expression of recombinant proteins with a self-cleaving CPD/6xHis-tag. BMC Biotechnol 17, 1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Monleon D, Colson K, Moseley HN, Anklin C, Oswald R, Szyperski T, Montelione GT, Rapid analysis of protein backbone resonance assignments using cryogenic probes, a distributed Linux-based computing architecture, and an integrated set of spectral analysis tools. J Struct Funct Genomics 2, 93–101 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Clore GM, Bax A, Driscoll PC, Wingfield PT, Gronenborn AM, Assignment of the side-chain 1H and 13C resonances of interleukin-1 beta using double- and triple-resonance heteronuclear three-dimensional NMR spectroscopy. Biochemistry 29, 8172–8184 (1990). [DOI] [PubMed] [Google Scholar]
- 66.Grzeslek S, Bax A, Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc 114, 6291–6293 (1992). [Google Scholar]
- 67.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Philippe N, Alcaraz JP, Coursange E, Geiselmann J, Schneider D, Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51, 246–255 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Miller VL, Mekalanos JJ, A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170, 2575–2583 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fullner KJ, Mekalanos JJ, Genetic characterization of a new type IV pilus gene cluster found in both classical and El Tor biotypes of Vibrio cholerae. Infect. Immun. 67, 1393–1404 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chan AC, Lelj-Garolla B, I. R. F, Pedersen KA, Mauk AG, Murphy ME, Cofacial heme binding is linked to dimerization by a bacterial heme transport protein. J Mol Biol 362, 1108–1119 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Robert X, Gouet P, Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD, In situ selectivity profiling and crystal structure of SML-8–73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A 111, 8895–8900 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Y, Huang C, Yin L, Wan M, Wang X, Li L, Liu Y, Wang Z, Fu P, Zhang N, Chen S, Liu X, Shao F, Zhu Y, Nepsilon-Fatty acylation of Rho GTPases by a MARTX toxin effector. Science 358, 528–531 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Pruitt RN, Chagot B, Cover M, Chazin WJ, Spiller B, Lacy DB, Structure-Function Analysis of Inositol Hexakisphosphate-induced Autoprocessing in Clostridium difficile Toxin A. J Biol Chem 284, 21934–21940 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reinert DJ, Jank T, Aktories K, Schulz GE, Structural basis for the function of Clostridium difficile toxin B. J Mol Biol 351, 973–981 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Jank T, Ziegler MO, Schulz GE, Aktories K, Inhibition of the glucosyltransferase activity of clostridial Rho/Ras-glucosylating toxins by castanospermine. FEBS Lett 582, 2277–2282 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Craven R, Lacy DB, Clostridium sordellii Lethal-Toxin Autoprocessing and Membrane Localization Activities Drive GTPase Glucosylation Profiles in Endothelial Cells. mSphere 1, e00012–00015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kamitani S, Kitadokoro K, Miyazawa M, Toshima H, Fukui A, Abe H, Miyake M, Horiguchi Y, Characterization of the membrane-targeting C1 domain in Pasteurella multocida toxin. J Biol Chem 285, 25467–25475 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Varela Chavez C, Hoos S, Haustant GM, Chenal A, England P, Blondel A, Pauillac S, Lacy DB, Popoff MR, The catalytic domains of Clostridium sordellii lethal toxin and related large clostridial glucosylating toxins specifically recognize the negatively charged phospholipids phosphatidylserine and phosphatidic acid. Cell Microbiol 17, 1477–1493 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.