

Abstract
Despite the important advances in chemical and chemoenzymatic synthesis of glycans, access to large quantities of complex natural glycans remains a major impediment to progress in Glycoscience. Here we report a large-scale preparation of N-glycans from a kilogram of commercial soy proteins using oxidative release of natural glycans (ORNG). The high mannose and paucimannose N-glycans were labeled with a fluorescent tag and purified by size exclusion and multidimensional preparative HPLC. Side products are identified and potential mechanisms for the oxidative release of natural N-glycans from glycoproteins are proposed. This study demonstrates the potential for using the ORNG approach as a complementary route to synthetic approaches for the preparation of multi-milligram quantities of biomedically relevant complex glycans.
Keywords: Glycomics, Oxidative release, Natural glycans, Sodium hypochlorite
1. Introduction
Despite the long history of study of carbohydrates as a major class of biomolecules, glycoscience or glycomics has been recognized as an important field in biomedical research only in the last decades, as more and more glycans and glycoconjugates are found to be play important roles in biological pathways and diseases [1–8]. Glycans are composed of multiple monosaccharides that are connected through glycosidic bonds, and each monosaccharide possesses several hydroxyl groups for potential glycosylation, resulting in a variety of different linkages, branching and specific stereochemistry at the anomeric centers. Thus, the analysis of glycan structure is inherently more challenging than the linear polymers of amino acids and nucleotides of proteins/peptides and nucleic acids, respectively. While high-throughput sequencing and automatic synthesis are widely available as routine commercial services for peptides and nucleic acids, similar platforms for glycans are not yet generally available.
Glycans have been routinely released from glycoconjugates by chemical and enzymatic methods [9–13] for glycomics analyses that are based on high performance liquid chromatography (HPLC) and mass spectrometry (MS). While advancement in instrumentation has resulted in more comprehensive glycomics analysis using smaller and smaller amounts of samples, the structural information cannot be directly associated with their biological functions without actually acquiring significant amounts of purified material. It is now being recognized that one of the major impediments in glycoscience is the lack of biomedically relevant complex glycans in significant amounts for their functional study. The lack of these complex glycans as structural standards also seriously hampers the development of high-throughput sequencing technology.
In recent years, glycan microarray analyses, in which many different glycan structures are immobilized onto glass slides for assay with potential glycan binding proteins (GBPs), have become extremely useful in high-throughput study of protein-glycan interactions [14–20]. Screening GBPs on defined glycan microarrays [14] is a powerful initial screen and a method for defining glycan-binding specificity; however, it is a hypothesis-generating platform and detailed study of glycan functions requires data that extend beyond cataloging glycan structures and defining GBP binding specificities. The utility of a glycan microarray is a function of its size, diversity and the biological relevance of the glycan library used for its preparation. Advances in chemoenzymatic synthesis of glycans [21–26] is an approach to increasing diversity, but the synthetic glycans generated still lag far behind the rapid expansion of scientific interest in the biological functions of glycans. Another route to access complex glycans is their isolation from natural sources. Depending on their source, natural glycans are more likely to be biologically relevant for functional study since they were synthesized by a living cell or organism. Several groups reported isolations of gram-level sialylglycopeptide (SGP) [27,28] and milligram-level asparagine-linked Glc1Man9GlcNAc2 [29]. We and others have focused on the use of natural glycans for functional studies using glycan microarray technology, in which hundreds of different, purified glycan structures separated from natural sources can be immobilized onto glass slides for interrogation with glycan binding proteins or microorganisms [30–36]. Through bifunctional fluorescent tagging of released glycans, nmol scale of glycans can be separated, quantified, characterized and printed onto microarrays for functional screening in a process we have termed shotgun glycomics [30,32,36–40]. However, obtaining robust tagged glycan libraries from which to retrieve the relevant glycans for structural analysis requires processing large quantities of cells, tissues or organs [39].
In order to address the difficulty in processing large amounts of natural products to obtain large quantities of biologically relevant glycans for functional studies, we have recently developed a novel chemical method, oxidative release of natural glycans (ORNG) that uses sodium hypochlorite (NaOCl) to release glycans from glycoproteins and glycosphingolipids (materials weight from 500 μg to 220 g) [41]. The procedure is simple, inexpensive and can be applied to kilograms of tissues directly to release multigrams quantities of glycans, which is impractical using traditional chemical or enzymatic approach. To demonstrate the utility of this method, we describe here the application of ORNG at a kilogram scale using commercial soy protein isolates (delipidated soy flour), a common food product containing high mannose and paucimannose N-glycans, for the preparation of large amounts of N-glycans. The procedure involves ORNG treatment of soy proteins, followed by fluorescent tagging using the bifunctional linker, 2-amino-N-(2-aminoethyl)benzamide (AEAB) and preparative 2D-HPLC separation (Fig. 1a). A library of high mannose and paucimannose N-glycans are purified, ranging from low milligram to hundreds of milligrams of purified glycan derivatives. Several side products were observed, which permitted us to propose chemical mechanisms of the oxidative reactions that release N-glycans. This work demonstrates the potential of ORNG in the production of significant quantities of biologically relevant glycans from inexpensive natural materials.
Fig. 1. Oxidative Release of Natural Glycans from soy proteins.

a) Overview of the procedure for the production of tagged N-glycans released by ORNG method, b) Scheme of oxidative release of N-glycans, tagging the reducing glycans, HPLC separation of the AEAB conjugated glycans, and characterization of AEAB-glycans by MS and NMR. c) Top: mass spectra of N-glycan fraction (Fig. S1a) before conjugation with AEAB (M + Na+); Chemical structures at the reducing end of several side products were shown. Bottom: mass spectra of AEAB conjugated N-glycan fraction (Fig. S1b), d) Normal phase HPLC profile (250 × 4.6 mm amine column) of AEAB conjugated N-glycan fraction (composition of fractions determined by MS), e) C18 reversed phase HPLC profile (250 × 4.6 mm C18 column) of the single peak collected from (d) showing well-separated H8N1-AENB and H8N2-AEAB. (H = hexose, N = N-acetylglucosamine, Fuc = fucose, Xyl = xylose).
2. Results
2.1. ORNG treatment of commercial soy proteins
In previous reports on the process for oxidative release of natural glycans (ORNG) using NaOCl [41], free reducing N-glycans from glycoproteins, such as ovalbumin, bovine IgG and horseradish peroxidase, were obtained. After AEAB conjugation, the N-glycans can be separated and purified by 2D-HPLC for further characterization and studies of biological activity. In order to demonstrate the general utility of this process for the large-scale production of N-glycans, we applied this procedure to commercially soy proteins (Fig. 1). We first evaluated the process using 50 g of soy proteins. After treatment with NaOCl, the reaction solution was acidified with hydrochloric acid and then centrifuged to remove precipitates, presumably partially oxidized non-glycan materials. The supernatant containing glycans released from soy proteins was applied to a size exclusion chromatography (SEC) column of Sephadex G-25 and the N-glycan containing fraction (Fig. S1a) was pooled and lyophilized. The N-glycan fraction was then conjugated with AEAB by reductive amination, and the glycan-AEAB conjugates were separated from excess AEAB on the same SEC column of Sephadex G-25 (Fig. S1b). The N-glycan fraction before (Fig. 1c, top panel) and after (Fig. 1c, bottom panel) AEAB conjugation was analyzed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS). Four major compositions of reducing glycans were observed using positive ionization mode, H8N2, H7N2, H6N2 and H3N2Fuc1Xyl1. In addition, several minor peaks were assigned as ORNG side products, such as H8N1-nitrile, H8N1-erythrose, and H8N1 (Fig. 1c, top panel), with mass difference of Δm = 3, 101, and 203 with H8N2 respectively. Fig. 1c (bottom panel) shows the MS profile of the same 4 major N-glycans after labeling with AEAB. Analytical scale HPLC was used to separate AEAB labeled soy protein N-glycans to evaluate the contents of the N-glycan fraction by MALDI-MS. Separation of the AEAB-labeled mixture of N-glycans suggested that the 4 major N-glycans are well separated by normal phase (NP) HPLC on amino column, due to different size and hydrophilicity (Fig. 1d). When a single peak from NP-HPLC separation was collected and applied to a reverse phase (RP) C18 column, two N-glycans with compositions of H8N2-AEAB and H8N1-AEAB are well separated from each other (Fig. 1e). The different patterns of separation on amine NP and C18 RP-HPLC columns demonstrated the feasibility of using 2D-HPLC systems to separate different high mannose glycans. The minor side products, including the glyconitriles and glycans with erythrose reducing end, provide us with more mechanistic understanding of oxidative degradation of released N-glycans, as proposed in Fig. 2. Presumably, Chlorine substitution of amide proton at the glycan-peptide linkage initiates a six-membered ring pericyclic rearrangement to break C-C bond to generate a glycan-isocyanate, which hydrolyzes into glycosylamine. Further hydrolysis of glycosylamine transforms it to free reducing glycan as the major product. On the other hand, glycosylamine can be further chlorinated and degraded through the six-membered ring pericyclic rearrangement to either glycan with erythrose reducing end (black route) or glyconitrile (red route). Further oxidation of erythrose end glycans may result the complete loss of the reducing end GlcNAc through unknown mechanism, as suggested by compositions with only one GlcNAc.
Fig. 2.

Proposed mechanisms of ORNG method to release N-glycan and the generation of major and minor products.
2.2. Preparative 2D-HPLC purification of AEAB labeled N-glycans
We then carried out the large scale production of soy protein N-glycans from 1 kg soy proteins. For the large-scale separation of AEAB-labeled N-glycans, the N-glycan fraction collected from size exclusion chromatography (Fig. S1b) was applied to a C18 Solid Phase Extraction (SPE) column. Unconjugated glycans are not bound and washed off the column using water while AEAB-conjugated glycans are retained on C18 SPE due to the hydrophobicity of AEAB tag and eluted using 10% acetonitrile. The mixture of glycan-AEAB conjugates was then applied to preparative amino normal phase HPLC system (Fig. 3a). Individual fractions were collected by time (~0.4min/fraction) and pooled based on the HPLC profile into 8 portions (Fig. 3a, F1-F8) from 61.1 min to 109.8 min. Normal phase HPLC fractions from 39.5 min to 53.5 min contain AEAB conjugated glycans of Hex3 and Hex4, which are not N-glycans and probably arise from the degradation of starch; these glycans were not further studied. MALDI-TOF-MS analysis of each of the 8 pooled fractions shown in Fig. 3b indicated decent separation of the N-glycans under this chromatography condition. The 8 fractions were then subjected to preparative C18 reversed phase HPLC for second dimensional purification (Fig. 3c–j). Peaks from each run were collected based on the UV profile and the major components were analyzed by MALDI-MS. Compositions and structures were proposed and shown in Fig. S2. Finally, 17 N-glycans with individual quantities up to 125 mg (Table 1) were purified to apparent homogeneity (> 95%) confirmed by re-profiling on both C18 (Fig. S2) and amine columns (Fig. S3) and analyzed by MALDI-TOF-MS and MS/MS (Fig. 4, Fig. S4). The reducing end structures can be easily discriminated using MS/MS analysis since fragment ions at m/z 407 and m/z 610 correspond to GlcNAc-AEAB and GlcNAc2-AEAB (Fig. 4a, top and middle panels), which can be used to easily confirm the glycan structures with intact reducing end or with loss of one GlcNAc. Fragment Ions of Man6GlcNAc1-erythrose-AEAB at m/z 509 (relatively intense) and 306 (minor) indicate that the terminal GlcNAc was over oxidized to erythrose (Fig. 4a, bottom panel). Every spectrum of paucimannose glycans contained ion at m/z 553 indicating the presence of fucose-GlcNAc at the reducing end (Fig. 4b). In total, over 324.8 mg of pure N-glycans from soy proteins were obtained (Table S1). Based on the yields of H8N2-AEAB (125 mg), H8N1-AEAB (30.4 mg) and H8N1-erythrose-AEAB (2.4 mg) and the small amount of glycan-nitriles shown in Fig. 1c which do not react with AEAB and were not separated, we estimate that > 60% of the glycans are released as intact N-glycans by ORNG.
Fig. 3. Preparative HPLC separation of soy protein N-glycan AEAB conjugates.

a) the elution profile (UV 330 nm) of 0.8 g N-glycan AEAB conjugates on a preparative NP HPLC (50 × 250 mm amino column); 8 pooled fractions: F1-F8 were collected (F1: 61.1 min to 66.2 min; F2: 66.2 min to 71.1 min; F3: 71.1 min to 73.9 min; F4: 73.9 min to 77.9 min; F5: 77.9 min to 85.2min; F6: 85.2 min to 90.4 min; F7: 90.4 min to 97.8 min; F8: 97.8 min to 109.8 min); b) MALDI-TOF-MS of pooled fractions F1-F8 from the preparative NP-HPLC; c-j) Pooled fractions F1-F8 collected from preparative NP HPLC as annotated in (a) were separated on second dimensional preparative reversed phase HPLC using a 21.2 × 250 mm Luna C18(2) column. Further chromatography was used to purify partially purified glycans when needed.
Table 1. Proposed structures of all separated glycan-AEAB conjugates.
Labeling numbers are glycan ID for microarray analysis and a signifies that quantitation was based on fluorescence quantification.
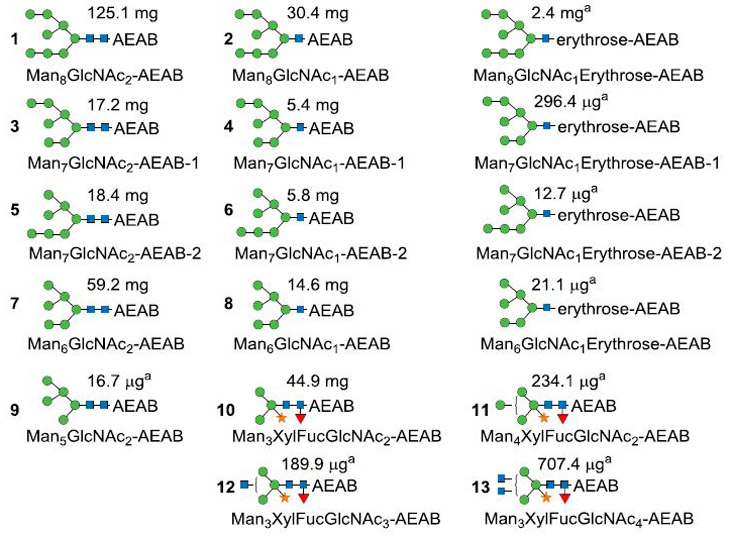 |
Fig. 4. MS/MS of selected high-mannose (M + Na+)(a) and all separated paucimannose glycans (b) showing clear fragmentation patterns.

Panel a (from top to bottom): Man6GlcNAc2-AEAB, Man6GlcNAc1-AEAB, Man6GlcNAcl-Erythrose-AEAB; Panel b (from top to bottom): Man3XylFucGlcNAc2-AEAB, Man4XylFucGlcNAc2-AEAB, Man3XylFucGlcNAc3-AEAB, Man3XylFucGlcNAc4-AEAB. Man: mannose; Xyl: xylose; Fuc: fucose; GlcNAc: N-acetyl-glucosamine.
2.3. Glycan characterization by NMR spectroscopy
Six of the purified glycans were characterized by 1H and/or 13C NMR spectroscopy (Fig. 5 and Fig. S5). The availability of these unprecedented quantities (> 10 mg) of N-glycans permits the use of 1H and 13C NMR experiments to analyze the structures of H8N2-AEAB and H8N1-AEAB. The β-configuration of anomeric proton in GlcNAc of Man8GlcNAc2-AEAB can be distinguished clearly by the 3JHH coupling 6.96 Hz at δ 4.58 (Fig. 5b), while there is no related peak for Man8GlcNAc1-AEAB (Fig. 5d) [42]. Comparison of two 13C NMR spectra also confirmed that anomeric position carbon (δ 101.1) in GlcNAc of Man8GlcNAc2-AEAB is absent in Man8GlcNAc1-AEAB (Fig. 5f and h, Table 2). While it is difficult to directly assign chemical shifts to all the protons, Lundborg et al. have developed a useful online computational tool CASPER [43] for accurate prediction of chemical shifts of glycans from structures. To determine the structures of two Man7GlcNAc2-AEAB isomers, we compared experimental NMR data with calculated data from CASPER [43]. Based on the clearly different pattern predicted by CASPER for the two isomers, which match well experimental data, we assigned the structures to the two isomers (Figs. S5 and S6 and Table S2).
Fig. 5. Selected NMR spectra of the N-glycans.

a) Full range 1H spectra and b) zoom in anomeric region of 1H spectra of Man8GlcNAc2-AEAB; c) Full range 1H spectra and d) zoom in anomeric region of 1H spectra of Man8GlcNAc1-AEAB; e) Full range 13C spectra and f) zoom in anomeric region 13C spectra of Man8GlcNAc2-AEAB; g) Full range 13C spectra and h) zoom in anomeric region 13C spectra of Man8GlcNAc1-AEAB. Anomeric region chemical shifts in both 1H and 13C spectra are assigned based on predicted value of methyl-β-glycosides using CASPER [43].
Table 2.
Comparison between experimental NMR data and predicted NMR data of anomeric regions protons and carbons on Man8GlcNAc2-AEAB and Man8GlcNAc1-AEAB.
| NMR Experimental Data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| Man8GlcNAc2-AEAB | 1H | 4.58 (JH-H = 6.96 Hz) | 4.71 | 4.82 | 5.03 | 5.00 | 5.10 | 5.30 | 5.26 | 5.00 |
| 13C | 101.1 | 100.3 | 100 | 97.9 | 102.2 | 102.3 | 100.7 | 100.5 | 102.1 | |
| Man8GlcNAc1-AEAB | 1H | 4.71 | 4.79 | 5.04 | 4.99 | 5.05 | 5.32 | 5.29 | 5.03 | |
| 13C | 99.7 | 99.6 | 97.8 | 102.2 | 102.2 | 100.8 | 100.6 | 102.1 | ||
2.4. Microarray printing and binding assay
To demonstrate the utility of this approach for functional glycomics, we printed an N-glycan microarray from this purified soy protein N-glycan library and analyzed binding of immobilized glycans by two plant lectins (ConA and AAL) (Fig. 6). The glycans with erythrose reducing end were not printed on this microarray because they were minor components and side products of ORNG. The binding affinity results are consistent with the known binding specificities of the two lectins. Con A bound to all high-mannose and paucimannose N-glycans, while AAL only bound fucose-containing glycans (sample ID 10–13, see Table S3). The results also confirmed the biological activities of the purified ORNG N-glycans.
Fig. 6. Plant lectin binding on the glycan microarray of purified soy protein N-glycan-AEAB conjugates.

X-axis represents glycan ID while Y-axis represent relative fluorescence unit of bound proteins (Glycan ID 1–13, Table 1, Glycan ID 14: Biotinylated BSA (negative control)). Error bars represent standard deviation of 6 replicates, a) binding of Concanavalin A (ConA), b) binding of Aleuria aurantia lectin (AAL).
3. Discussion
Cataloging the predicted structures of glycans that comprise profiles of various glycomes including cultured cell lines and animal tissues and organs represents useful comparative information. However, these data alone are insufficient in deciphering the biological function of glycans. We developed the concept of shotgun glycomics as an initial functional approach to glycomic analysis [32,44] where the glycans from cells, tissues or organs are obtained as tagged glycan libraries in quantities sufficient for printing microarrays. After interrogation of microarrays with biologically relevant GBPs, the bound and functionally relevant glycans can be retrieved from the library for structural analysis. For example, using this approach, we were able to identify a number of human milk glycans that could be considered as “candidate” glycans to function as receptors for human rotaviruses [37]. However, further analysis and functional study of candidate glycans generally requires larger quantities of material. Since glycans, unlike nucleic acids and protein, cannot be amplified, we developed ORNG, which allows us to process large quantities of natural products to generate large amounts of natural glycans that are very challenging to obtain in significant quantities using traditional chemical or enzymatic approaches. Due to the large quantities of glycans obtained by ORNG, they are more easily purified and analyzed and extremely valuable for functional studies. Once purified and characterized, these glycans can also serve as structure standards to facilitate our understanding of their behavior in analyses by MS and HPLC.
Large quantities of glycans have been generated from natural sources such as mammalian milk [45] and in a specialized case from hen's eggs which, contain large quantities (8 mg/egg yolk) of a free glycopeptide containing a complex bi-antennary glycan [46]. In the case of milk glycans and the egg yolk glycopeptide, the products are available without the necessity to release them from larger conjugates. Nevertheless, significant amount of labor is required to extract these glycans. In this study, we demonstrate the capability of ORNG to process natural materials at a kilogram-scale by starting from 1 kg of commercial soy proteins.
From 1 kg of soy proteins, we extracted an N-glycan fraction using ORNG and were able to purify 17 N-glycans totaling 320 mg of AEAB-conjugated glycan. The process to extract N-glycans is rapid and simple, using inexpensive reagents, household bleach and HCl. While the final yields are not high, it is straightforward to reproduce or scale up this simple process. The scale of the ORNG process is only limited by the size of the reaction vessel and the ability to process the large volumes of the process stream. The availability of the large quantity of the crude, labeled glycan permitted a relatively rapid purification process that could be monitored using the UV absorbance or fluorescence of the AEAB-labeled material. To our knowledge, this represents the first approach to generating such quantities of N-glycans released from glycoprotein.
The crude mixture of AEAB-labeled glycans are rapidly separated from excess AEAB and any unlabeled glycans using Sephadex G25 column followed by a C18 Solid Phase Extraction (SPE) column. Since AEAB conjugated glycans are retained on C18 SPE but unlabeled glycans are not, this provides a quick way to purify the glycan-AEAB conjugates from unconjugated glycans before 2D preparative HPLC separation. The first dimension HPLC on amino column provides separations based on size and hydrophilicity of glycans, while the second dimension HPLC on C18 column separates glycan-AEAB conjugates based on subtle hydrophobicity differences among isomeric glycans. It appears that HexNAc provides more retention power than hexose on C18 column as Man8HexNAc1-AEAB and Man8HexNAc2-AEAB are well separated. The fact that even the isomers of Man7HexNAc2 and Man7HexNAc1 are resolved by this separation scheme strongly support the ability of common preparative separation systems to resolve complex glycan mixtures. Since significant amounts of each component as well as over-oxidized side products are available, structural analysis is relatively straightforward. For example, we were able to distinguish the two Man7 isomers using their clearly different NMR spectra with assistance of CASPER [43] prediction.
A summary of the glycans that we prepared from soy proteins are shown in Table 1, which includes the predicted structure(s) and the amounts purified. The structures observed are largely consistent with previous findings on the major soybean storage protein β-conglycinin [47]. Since we can process large amounts of starting material, we were able to purify and characterize not only 44.9 mg Man3GlcNAc2Xyl1Fuc1, but also 3 paucimannose glycans commonly found in rice, but rare in soybeans [48]. Man8GlcNAc2 was found to be the most abundant glycan, while the more processed high mannose N-glycans were less abundant. Interestingly, we did not isolate Man9GlcNAc2, which is known to be the major N-glycan on soybean agglutinin [49]. It is possible that soybean agglutinin is lost during the commercial preparation of soy proteins.
Currently there are only a limited number of vendors that provide complex glycans as standards (20–100 μg) for HPLC and MS analysis and mg quantities of glycans of limited diversity. During our decade-long work with these commercial complex glycans for the development of microarrays, we have lower than expected purity and in some cases even wrong structures, in sharp contrast with other commercial biochemical such as peptides and oligonucleotides, which are much easier to characterize and quantify. Nevertheless, given the current status of glycoscience and the small quantities of glycans provided, this is merely surprising. In this study, we have purified and characterized a significant number of complex glycans in large quantities. With extensive HPLC purification, all the glycans are obtained with > 95% purity, exceeding the purity generally available commercially. The ORNG method for the production of large amount of natural complex glycans holds great promise for obtaining unprecedented quantities of glycans with larger scaled processes.
A detailed reaction mechanism was proposed for the oxidative reactions in Fig. 2. Pericyclic rearrangement through six-membered ring transition states is likely responsible for irreversible C-C bond cleavage and further degradation. Further understanding of the mechanism will contribute to continued optimization of the ORNG protocol. For example, it is possible to reduce the glyconitrile using catalytic hydrogenation to regenerate free reducing glycans. Stronger oxidation might enrich more N-glycans losing terminal GlcNAc, which are useful substrates for endoglycosidase catalyzed N-glycopeptide synthesis [50]. The byproducts observed contribute to the complexity of the glycan mixture, but as demonstrated here they can be well separated by 2D-HPLC and easily identified using MS and MS/MS. Thus, the oxidative byproducts are not considered as a major obstacle to applications involving the ORNG procedure. Due to the easily scalable nature of ORNG, we anticipate that a 10-fold increase in the starting material will allow us to generate gram quantities of the major N-glycans. We anticipate that increased quantities of released glycan will permit the separation, detection and characterization of previously unidentified, minor, biologically relevant glycans.
3.1. Experimental procedures
All chemicals and HPLC solvents were purchased from Sigma-Aldrich, Acros, Oakwood Chemicals, and Fisher Scientific. Milli-Q water was used to prepare all aqueous solutions. C18 Sep-pak was from Waters, Inc. Sodium hypochlorite solutions are from Pure Bright (6% NaOCl), and prepared freshly by addition of water. Bleach stored for more than 6 months under room temperature as 6% NaOCl has been used successfully.
3.2. Mass spectrometry (MS)
A Bruker Daltonics Ultraflex-II MALDI-TOF/TOF system and an anchorchip target plate were used for MS analysis. Reflective positive mode was used for glycans. 2,5-dihydroxybenzoic acid (DHB) (10 mg/mL in 50% acetonitrile with 0.1% trifluoroacetic acid) was used as matrix.
3.3. High-performance liquid chromatography analyses
A Shimadzu HPLC CBM-20A system with UV detector SPD-20A and fluorescence detector RF-10Axl was used for analytical HPLC profiles. A Shimadzu HPLC LC-8a preparative system with UV detector SPD-10Av was used for preparative HPLC separation. UV absorption at 330 nm or fluorescence at 330 nm excitation and 420 nm emission was used for detection of 2-amino-N-(2-aminoethyl) benzamide (AEAB) tag. Phenomenex amino columns were used for normal phase HPLC. Phenomenex C18(2) columns were used for reverse-phase HPLC separation. For normal phase HPLC, the mobile phases were acetonitrile, water, and aqueous ammonium acetate buffer at pH 4.5. A linear gradient from 20 mM ammonium acetate in 80% acetonitrile to 200 mM ammonium acetate in 10% acetonitrile for either 50 min (analytical run) or 150 min (preparative run) was used. For reversed phase HPLC, the mobile phases were acetonitrile, water, and trifluoroacetic acid and linear gradients from 2% to 10% acetonitrile/0.1% trifluoroacetic acid over 25–80 min were applied.
3.4. NMR spectroscopy
N-glycans of Man8GlcNAc2-AEAB, Man8GlcNAc1-AEAB, Man8GlcNAc1erythrose-AEAB, Man7GlcNAc2-AEAB-1, Man7GlcNAc2-AEAB-2, and Man3GlcNAc2Xyl1Fuc1-AEAB were analyzed by NMR spectroscopy for structural elucidation. 1H and 13C spectra were acquired in D2O at 25 °C, D2O chemical shift is referenced at 4.79 ppm [51]. NMR spectra were recorded on a Bruker AVANCE III HD 600 MHz spectrometer equipped with a Prodigy cryoprobe.
3.5. Sodium hypochlorite release of N-glycans from soy proteins
1.0 kg soy proteins (Cat.# 6636) purchased from a local Sprouts Farmers Market (Phoenix, AZ) was separated into 250 g portions, and 6 L of H2O and 3 L of 6% NaOCl were added subsequently under open air to each and stirred for about 30 min. NaOCl is consumed within several minutes and extension of reaction time up to 1 h does not vary results. The reaction time was not critical for the oxidation. The reaction was quenched with concentrated HCl so that the final pH is 4.8. A total of 36 L of reaction solution was centrifuged to remove precipitated material. The supernatant was concentrated under reduced pressure to ~ 500 mL, and 500 mL absolute ethanol was added. The mixture was stirred for 10 min. The mixture was centrifuged to remove precipitates. Another 2000 mL absolute ethanol was added and the precipitate containing the bulk of the glycans was collected after centrifugation. The pellet was dissolved into 100 mL H2O and subjected to Sephadex G-25 size exclusion chromatography. The hexose-containing eluate from 800 mL to 1000 mL were collected. After lyophilizing the collected solution, solid materials were reacted with AEAB•2HCl and NaCNBH3 in dimethyl sulfoxide and acetic acid mixture solution (7:3, v/v) at 65 °C stirring for 2h [52]. The reaction mixture is precipitated with 10 vol of acetonitrile and centrifuged to collect the pellet. The pellet containing glycan-AEAB conjugates was dissolved in 100 mL water and applied to Sephadex G-25 size exclusive chromatography to collect eluate from 850 mL to 1250 mL. This 400 mL fraction was then lyophilized to 10.3 g solid material. The solid material was dissolved in 100 mL H2O and passed through a Sep-Pak C18 cartridge (120 g) and washed with 3 × 100 mL 0.1 M acetic acid solution. Conjugated glycans were eluted with 300 mL 10% acetonitrile and 0.1 M acetic acid. The elution was lyophilized to generate 0.8 g white solid powder.
3.6. 2D-HPLC separation
Amino normal phase HPLC separation: 0.8 g white solid powder containing AEAB conjugated glycans was dissolved into 10 mL 30% acetonitrile in H2O (v/v). Then the 10-mL solution was injected into preparative amino normal phase HPLC column (column size: 250 × 50 mm). Fractions (0.4 min) were collected and selected fractions were analyzed for glycans using MALDI. C18 reversed phase HPLC separation: 8 combined fractions obtained from amino normal phase HPLC separation (Fig. 3a) were individually lyophilized, redissolved in water and then applied to preparative C18 reversed phase HPLC (column size: 250 × 50 mm). Peaks were collected and characterized.
3.7. Microarray printing, binding assay and scanning
Contact printing on NHS-activated slides was carried out using an Aushon's 2470 microarrayer. N-glycans were printed at 100 μM in 100 mM sodium phosphate (pH 8.5) in replicates of 6. Biotinylated-Aleuria aurantia lectin (AAL, Vector Labs) was assayed at 1 μg/mL. Biotinylated -Concanavalin A was assayed at 0.1 μg/mL. Cyanine 5-streptavidin (Invitrogen) 5 μg/mL was used for the detection of binding using a fluorescent scanner (Innopsys).
Supplementary Material
Acknowledgements
This work was supported by NIH Common Fund Glycoscience (U01GM116254 to X.S.) and partially by a STTR grant (R41GM122139 to X.S.). This study was supported in part by the Emory Comprehensive Glycomics Core (ECGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities.
Footnotes
Conflicts of interest
David F. Smith and Xuezheng Song are co-founders of NatGlycan LLC, which is commercializing the ORNG technology.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.carres.2018.05.002.
References
- [1].Hart GW, Copeland RJ, Cell 143 (2010) 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jaeken J, Matthijs G, Annu. Rev. Genom. Hum. Genet 2 (2001) 129–151. [DOI] [PubMed] [Google Scholar]
- [3].Mrksich M, Chem. Biol 11 (2004) 739–740. [DOI] [PubMed] [Google Scholar]
- [4].Shriver Z, Raguram S, Sasisekharan R, Nat. Rev. Drug Discov 3 (2004) 863–873. [DOI] [PubMed] [Google Scholar]
- [5].Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Essentials of Glycobiology, second ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y, 2009. [PubMed] [Google Scholar]
- [6].Zhao YY, Takahashi M, Gu JG, Miyoshi E, Matsumoto A, Kitazume S, Taniguchi N, Cane. Sci 99 (2008) 1304–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zoldos V, Horvat T, Lauc G, Curr. Opin. Chem. Biol 17 (2013) 34–40. [DOI] [PubMed] [Google Scholar]
- [8].Ohtsubo K, Marth JD, Cell 126 (2006) 855–867. [DOI] [PubMed] [Google Scholar]
- [9].Wiegandt H, Baschang G, Naturforsch Z. B Chem. Sci 20 (1965) 164–166. [PubMed] [Google Scholar]
- [10].Yosizawa Z, Sato T, Schmid K, Biochim. Biophys. Acta 121 (1966) 417–420. [DOI] [PubMed] [Google Scholar]
- [11].Elder JH, Alexander S, Proc. Natl. Acad. Sci. U.S.A 79 (1982) 4540–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ito M, Yamagata T, J. Biol. Chem 264 (1989) 9510–9519. [PubMed] [Google Scholar]
- [13].Plummer TH Jr., Tarentino AL, Glycobiology 1 (1991) 257–263. [DOI] [PubMed] [Google Scholar]
- [14].Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC, Proc. Natl. Acad. Sci. U.S.A 101 (2004) 17033–17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].De PJL, Seeberger PH, Mol. Biosyst 4 (2008) 707–711. [DOI] [PubMed] [Google Scholar]
- [16].Feizi T, Fazio F, Chai W, Wong CH, Curr. Opin. Struct. Biol 13 (2003) 637–645. [DOI] [PubMed] [Google Scholar]
- [17].Oyelaran O, Gildersleeve JC, Curr. Opin. Chem. Biol 13 (2009) 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Paulson JC, Blixt O, Collins BE, Nat. Chem. Biol 2 (2006) 238–248. [DOI] [PubMed] [Google Scholar]
- [19].Rillahan CD, Paulson JC, Annu. Rev. Biochem 80 (2011) 797–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stevens J, Blixt O, Paulson JC, Wilson IA, Nat. Rev. Microbiol 4 (2006) 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons G-J, Science 341 (2013) 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa SA, Linhardt RJ, Liu J, Science 334 (2011) 498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hsu C-H, Hung S-C, Wu C-Y, Wong C-H, Angew. Chem. Int. Ed 50 (2011) 11872–11923. [DOI] [PubMed] [Google Scholar]
- [24].Seeberger PH, Chem. Soc. Rev 37 (2008) 19–28. [DOI] [PubMed] [Google Scholar]
- [25].Sears P, Wong C-H, Science 291 (2001) 2344–2350. [DOI] [PubMed] [Google Scholar]
- [26].Plante OJ, Palmacci ER, Seeberger PH, Science 291 (2001) 1523–1527. [DOI] [PubMed] [Google Scholar]
- [27].Sun B, Bao W, Tian X, Li M, Liu H, Dong J, Huang W, Carbohydr. Res 396 (2014) 62–69. [DOI] [PubMed] [Google Scholar]
- [28].Liu L, Prudden AR, Bosman GP, Boons G-J, Carbohydr. Res 452 (2017) 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang N, Seko A, Takeda Y, Ito Y, Carbohydr. Res 411 (2015) 37–41. [DOI] [PubMed] [Google Scholar]
- [30].de Boer AR, Hokke CH, Deelder AM, Wuhrer M, Anal. Chem 79 (2007) 8107–8113. [DOI] [PubMed] [Google Scholar]
- [31].Song X, Xia B, Stowell SR, Lasanajak Y, Smith DF, Cummings RD, Chem. Biol 16 (2009) 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song X, Lasanajak Y, Xia B, Heimburg-Molinaro J, Rhea JM, Ju H, Zhao C, Molinaro RJ, Cummings RD, Smith DF, Br. J. Pharmacol 8 (2011) 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Song X, Lasanajak Y, Xia B, Smith DF, Cummings RD, ACS Chem. Biol 4 (2009) 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Song X, Smith DF, Cummings RD, Anal. Biochem 429 (2012) 82–87. [DOI] [PubMed] [Google Scholar]
- [35].Song X, Xia B, Lasanajak Y, Smith DF, Cummings RD, Glycoconj. J 25 (2008) 15–25. [DOI] [PubMed] [Google Scholar]
- [36].Song X, Xia B, Stowell SR, Lasanajak Y, Smith DF, Cummings RD, Chem. Biol 16 (2009) 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yu Y, Lasanajak Y, Song X, Hu L, Ramani S, Mickum ML, Ashline DJ, Venkataram Prasad BV, Estes MK, Reinhold VN, Cummings RD, Smith DF, Mol. Cell. Proteomics 13 (2014) 2944–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu Y, Mishra S, Song X, Lasanajak Y, Bradley KC, Tappert MM, Air GM, Steinhauer DA, Haider S, Cotmore S, Tattersall P, Agbandje-McKenna M, Cummings RD, Smith DF, J. Biol. Chem 287 (2012) 44784–44799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Byrd-Leotis L, Liu R, Bradley KC, Lasanajak Y, Cummings SF, Song X, Heimburg-Molinaro J, Galloway SE, Culhane MR, Smith DF, Steinhauer DA, Cummings RD, Proc. Natl. Acad. Sci. U.S.A. 111 (2014) E2241–E2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].van Diepen A, van der Plas A-J, Kozak RP, Royle L, Dunne DW, Hokke CH, Int. J. Parasitol 45 (2015) 465–475. [DOI] [PubMed] [Google Scholar]
- [41].Song X, Ju H, Lasanajak Y, Kudelka MR, Smith DF, Cummings RD, Nat. Meth 13 (2016) 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Govindaraju V, Young K, Maudsley AA, NMR Biomed 13 (2000) 129–153. [DOI] [PubMed] [Google Scholar]
- [43].Lundborg M, Widmalm G, Anal. Chem 83 (2011) 1514–1517. [DOI] [PubMed] [Google Scholar]
- [44].Zaia J, Br. J. Pharmacol. 8 (2011) 55–57. [Google Scholar]
- [45].Kobata A, Yamashita K, Tachibana Y, Methods Enzymol. 50 (1978) 216–220. [DOI] [PubMed] [Google Scholar]
- [46].Seko A, Koketsu M, Nishizono M, Enoki Y, Ibrahim HR, Juneja LR, Kim M, Yamamoto T, Biochim. Biophys. Acta 1335 (1997) 23–32. [DOI] [PubMed] [Google Scholar]
- [47].Picariello G, Amigo-Benavent M, del Castillo MD, Ferranti P, Chromatogr J. A 1313 (2013) 96–102. [DOI] [PubMed] [Google Scholar]
- [48].Horiuchi R, Hirotsu N, Miyanishi N, Carbohydr. Res 418 (2015) 1–8. [DOI] [PubMed] [Google Scholar]
- [49].Evers DL, Hung RL, Thomas VH, Rice KG, Anal. Biochem 265 (1998) 313–316. [DOI] [PubMed] [Google Scholar]
- [50].Amin MN, McLellan JS, Huang W, Orwenyo J, Burton DR, Koff WC, Kwong PD, Wang L-X, Nat. Chem. Biol 9 (2013) 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI, Organometallics 29 (2010) 2176–2179. [Google Scholar]
- [52].Song X, Xia B, Stowell SR, Lasanajak Y, Smith DF, Cummings RD, Chem. Biol 16 (2009) 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.