

Abstract
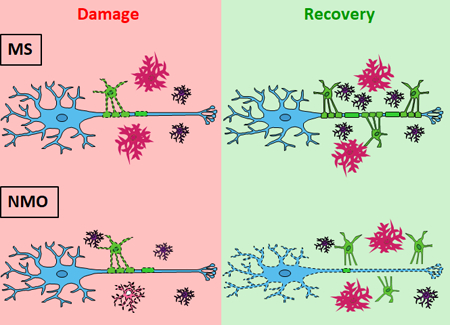
Multiple sclerosis (MS) and neuromyelitis optica (NMO) are inflammatory demyelinating disorders of the central nervous system (CNS) with evidence of antibody-mediated pathology. Using ex vivo organotypic mouse cerebellar slice cultures, we have demonstrated that recombinant antibodies (rAbs) cloned from cerebrospinal fluid plasmablasts of MS and NMO patients target myelin- and astrocyte-specific antigens to induce disease-specific oligodendrocyte loss and myelin degradation. In this study, we examined glial cell responses and myelin integrity during recovery from disease-specific antibody-mediated injury. Following exposure to MS rAb and human complement (HC) in cerebellar explants, myelinating oligodendrocytes repopulated the demyelinated tissue and formed new myelin sheaths along axons. Remyelination was accompanied by pronounced microglial activation. In contrast, following treatment with NMO rAb and HC, there was rapid regeneration of astrocytes and pre-myelinating oligodendrocytes but little formation of myelin sheaths on preserved axons. Deficient remyelination was associated with progressive axonal loss and the return of microglia to a resting state. Our results indicate that antibody-mediated demyelination in MS and NMO show distinct capacities for recovery associated with differential injury to adjacent axons and variable activation of microglia. Remyelination was rapid in MS rAb plus HC-induced demyelination. By contrast, oligodendrocyte maturation and remyelination failed following NMO rAb-mediated injury despite the rapid restoration of astrocytes and preservation of axons in early lesions.
Keywords: glial cell responses, remyelination, multiple sclerosis, neuromyelitis optica, antibody
Main Points
Recovery from MS or NMO damage induces distinct glia responses that result in differential effects on myelin repair in ex vivo models. Astrocyte recovery is insufficient to drive oligodendrocyte maturation in NMO lesions.
Table of Contents Image
TIFF files uploaded with submission.
INTRODUCTION
Myelin provides electrical insulation, ion homeostasis and trophic support (Menichella et al., 2006; Morrison, Lee, & Rothstein, 2013; Nave & Trapp, 2008) to ensheathed axons. Demyelination is critical in the pathogenesis of central nervous system (CNS) disorders such as multiple sclerosis (MS) and neuromyelitis optica (NMO), severely impacting axonal integrity and neuronal function (Lassmann, van Horssen, & Mahad, 2012). Following pathological loss of myelin, remyelination, the formation of new myelin sheaths around axons, occurs (Verden & Macklin, 2016). This process is performed by newly differentiated oligodendrocytes, derived from oligodendrocyte progenitors (OPCs), wrapping and producing myelin around axons. The benefits of remyelination on the restoration of neuronal function have been well documented (Kremer, Gottle, Hartung, & Kury, 2016). While remyelination occurs in MS, it is often inefficient, leading to permanent deficits and dysfunction (Louapre et al., 2015; Lucchinetti et al., 2000). In NMO, remyelination is also limited (Yao, Su, & Verkman, 2016). Currently, there are no therapies that restore myelin in MS and NMO; therefore, understanding the mechanisms that promote and inhibit myelin repair after inflammation injury is vital for developing remyelinative therapies.
Immunoglobulin G (IgG) deposition is a seminal feature of active MS and NMO lesions (Breij et al., 2008; Lucchinetti et al., 2000; Lucchinetti, Bruck, & Lassmann, 2004). Clinical and experimental data have demonstrated that AQP4-IgG is specific (range: 96%–99%) for NMO (Waters et al., 2014) and capable of reproducing disease-specific pathology ex vivo and in vivo. In contrast, the targets of the intrathecal humoral response in MS and their relationship to disease pathogenesis are poorly understood. In surveys of biopsy- and autopsy-derived MS tissues (Breij et al., 2008; Lucchinetti et al., 2000), the most prevalent active lesion subtype (type II) was defined by deposition of Ig and terminal complement at the lesion edge and within macrophages indicating that antibody- and complement-mediated myelin phagocytosis likely play a central role in demyelination in MS patients. A robust and sustained humoral response within the CNS of MS patients is a biochemical hallmark of disease as evidenced by persistent CSF IgG oligoclonal bands and B cell clonal expansion (Hemmer, Cepok, Zhou, & Sommer, 2004; Ritchie et al., 2004).
We have produced IgG1 monoclonal recombinant antibodies (rAbs) from clonally-expanded plasmablasts recovered from NMO (Bennett et al., 2009) and MS (Blauth et al., 2015; Owens et al., 2009) patient CSF. Patient-derived AQP4-specific monoclonal rAbs have been used to direct targeted destruction of primary murine astrocytes and mixed glial cultures in vitro (Liu et al., 2016; Tradtrantip et al., 2012), cerebrum and spinal cord demyelination ex vivo (Liu et al., 2017; Liu et al., 2016; Tradtrantip et al., 2012; Yao et al., 2016; Yao & Verkman, 2017; Zhang, Bennett, & Verkman, 2011), and NMO disease-specific histopathology in vivo (Bennett et al., 2009; Bradl et al., 2009; Ratelade & Verkman, 2014; Saadoun et al., 2010; Tradtrantip et al., 2012). NMO lesions in model systems are dependent on both the AQP4-specificity and effector function of the administered IgG, consistently demonstrating that AQP-IgG is necessary and sufficient for NMO lesion formation. By contrast, CSF-derived rAbs from early active MS patients (Blauth et al., 2015; Liu et al., 2017) have demonstrated 2 patterns of CNS immunoreactivity. Pattern 1 rAbs bind to neurons, neuropil and astrocytes in CNS tissues, while Pattern 2 bind to the surface of oligodendrocyte processes and myelinating axons. When applied to organotypic cerebellar slice cultures ex vivo, myelin-specific MS rAbs cause robust oligodendrocyte loss, demyelination, and microglia activation; however, astrocytes, oligodendrocyte progenitors and neurons remain unaffected (Liu et al., 2017). The injury is distinct from that induced by AQP4-specific NMO rAb which results in astrocyte destruction, oligodendrocyte loss, demyelination, microglia activation, and neuronal death (Liu et al., 2017).
In this study, we evaluated the recovery of cerebellar explants following MS and NMO rAb-mediated injury and observed distinct patterns of glial responses that were coupled with disparate capacities for remyelination. Our results highlight key differences in MS and NMO lesion repair and establish a novel model system for evaluating mechanisms and therapies that may impact recovery in two major human inflammatory demyelinating disorders.
MATERIALS AND METHODS
Animals
The care and euthanasia of animals are in accordance with University of Colorado IUCAC policy for animal use, which is in agreement with the NIH Guide for the Care and Use of Laboratory Animals.
Recombinant Antibodies
Disease-specific and isotype control monoclonal recombinant antibodies (rAbs) MS#30, NMO#53, and ISO were derived from expanded CSF plasma blast clones recovered from patients with relapsing–progressive MS (Owens et al., 2009), AQP4-seropositive NMO (Bennett et al., 2009), and idiopathic chronic meningitis (Blauth et al., 2015), respectively. All rAbs were expressed as full-length bivalent human IgG1 antibodies using a dual vector transient transfection system and purified with protein A-sepharose (Sigma-Aldrich, St. Louis, MO) as previously described (Bennett et al., 2009; Owens et al., 2009). All rAbs were used at 20μg/ml in the slice cultures.
Cerebellar Slice Culture
Sagittal cerebellar slices (300µm) were prepared from PLP-eGFP mice (Mallon, Shick, Kidd, & Macklin, 2002) at P10 and cultured on MilliCell 0.4µm membrane inserts (Millipore, Billerica, MA) in slice media (25% Hank’s balanced salt solution (HBSS), 25% heat-inactivated horse serum, 50% minimum essential media (MEM), 125mM HEPES, 28mM D-Glucose, 2mM L-Glutamine, 10U/ml penicillin/streptomycin, all from Life Technologies, Carlsbad, CA) at 37°C (Sheridan & Dev, 2012). Slices were cultured for 7–10 days prior to treatment.
Treatment of Cerebellar Slices
rAbs were applied at 20 µg/ml with 10% (vol/vol) normal human serum (Complement Technology, Tyler, TX) as a source of complement. Cerebellar slices were exposed to media containing treatment reagents both on top (50 µl) and below (240 µl) the membrane inserts for 24 or 48 h, and then were washed and cultured for additional 7–14 days.
Immunostaining
After treatment, cerebellar slices were rinsed twice with PBS and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. For immunohistochemistry, slices were rinsed in PBS and permeabilized in 1.5% or 10% (myelin proteins) Triton X-100 in PBS for 20 min. Slices were rinsed, blocked with 5% normal donkey serum (NDS) in PBS with 0.3% Triton X-100 for 1 h, and incubated with primary antibodies overnight at room temperature. Following 3 washes in PBS, Alexa Fluor-labeled secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were applied (1:800) overnight at room temperature, washed 3 times in PBS and mounted in Fluoromount G (Southern Biotech, Birmingham, AL). The following primary antibodies were used: goat anti-Sox10 (Santa Cruz), rat anti-PLP (Yamamura, Konola, Wekerle, & Lees, 1991), mouse anti-AQP4 (Santa Cruz Biotechnology, Dallas, TX), rabbit anti-S100b (Sigma), rabbit anti-GST-pi (Enzo), mouse anti-Caspr (NeuroMAB), mouse anti-CC1 (Calbiochem), rabbit anti-GFAP (Sigma), rabbit anti-MOG (Abcam, Cambridge, United Kingdom), mouse anti-MBP (Covance, Princeton, NJ), chicken anti- Neurofilament-H (Neuromics, Minneapolis, MN), goat anti-Iba1 (Abcam).
Microscopy
Confocal images were acquired by Leica SP5 laser scanning microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Quantification and Statistical Analysis
Confocal images were quantified with ImageJ (National Institutes of Health open source). To quantify the extent of myelination, we calculated the percentage of total neurofilament-H staining that was co-labeled with MBP using a Matlab algorithm (MathWorks, Natick, MA)(Liu et al., 2017). For each slice, 2–3 images were taken, quantified, and averaged. Slices from 3–4 independent experiments were analyzed. Statistical analyses were performed by unpaired Student’s t test for single comparisons or by two-way ANOVA for grouped comparisons using GraphPad Prism software. Data are expressed as means ± SD of independent experiments (n≥3). Significance is reported for p<0.05.
RESULTS
We have previously demonstrated that patient-derived MS myelin-specific and NMO AQP-specific rAbs bind to the surface of oligodendrocyte processes and astrocytes, respectively (Supporting Information Figure S1; (Liu et al., 2017; Liu et al., 2016)). Due to their distinct glial cell targets, patient-derived MS and NMO rAbs cause distinct patterns of glial and neuronal injury in ex vivo organotypic cerebellar slice cultures (Liu et al., 2017). In this study, we examined the recovery of explanted cerebellar tissue following MS and NMO rAb-mediated damage.
Mature oligodendrocytes repopulate following treatment with MS rAb and human complement
Cerebellar slice cultures prepared from PLP (proteolipid protein)-eGFP mice (Mallon et al., 2002) were treated with myelin-specific MS rAb, MS04–2 #30 (MS#30) or isotype control rAb (ISO) (Blauth et al., 2015) plus 10% human complement (HC) for 24 h. Following treatment, rAb and HC were removed by media exchange, and slices were allowed to recover in culture media (in the absence of rAb+HC) for an additional 14 days (Figure 1a). In agreement with our prior published results (Liu et al., 2017), following 24 h of treatment (T0) with MS#30+HC, eGFP+ oligodendrocyte cell bodies were reduced to 52.8±5.4 % of control slices treated with ISO+HC, and oligodendrocyte processes were fragmented and disrupted. After 3 days of recovery (T3), eGFP+ oligodendrocyte cell bodies and processes began to repopulate the folia. At T7, oligodendrocyte cell bodies had recovered to 99.4±5.1% of control levels (ISO+HC at T0) and oligodendrocyte processes were extensive and well-organized (Figure 1b, d). Similarly, Sox10+ oligodendrocyte lineage cells (Miron, Kuhlmann, & Antel, 2011) were reduced after exposure to MS#30+HC and progressively recovered to normal levels (103.5±2.8 % of control) by T7 (Figure 1c, d). Following treatment with ISO+HC, there was a modest increase in the number of PLP-eGFP+ and SOX10+ cells during the 14 days in culture (Figure 1b, d). Prior to treatment with rAb and HC, most PLP-eGFP+ cells expressed CC1, a differentiated pre-myelinating oligodendrocyte marker (Emery et al., 2009; Liu et al., 2016). Similarly, after recovery from treatment with MS#30+HC, most PLP-eGFP+ oligodendrocytes were CC1+, indicating that regenerated oligodendrocyte cells were well-differentiated (Supporting Information Figure S2). The number of mature myelinating oligodendrocytes was assessed by GST-pi+ (pi form of glutathione-S-transferase) immunostaining (Tamura et al., 2007) (Figure 2a). After exposure to MS#30+HC, the number of GST-pi+ oligodendrocytes was significantly reduced at T0 (57.4±4.8 %); however, by T7 the number had recovered to 102.3±8.1 % of untreated control slices. At T7, approximately 75% of PLP-eGFP+ cells were GST-pi+, similar to that observed in untreated slices (Figure 2b). Together, these results demonstrate that the rapid loss of oligodendrocyte cells induced by treatment with MS rAb plus complement is followed by robust regeneration, differentiation, and maturation into myelinating cells.
FIGURE 1.
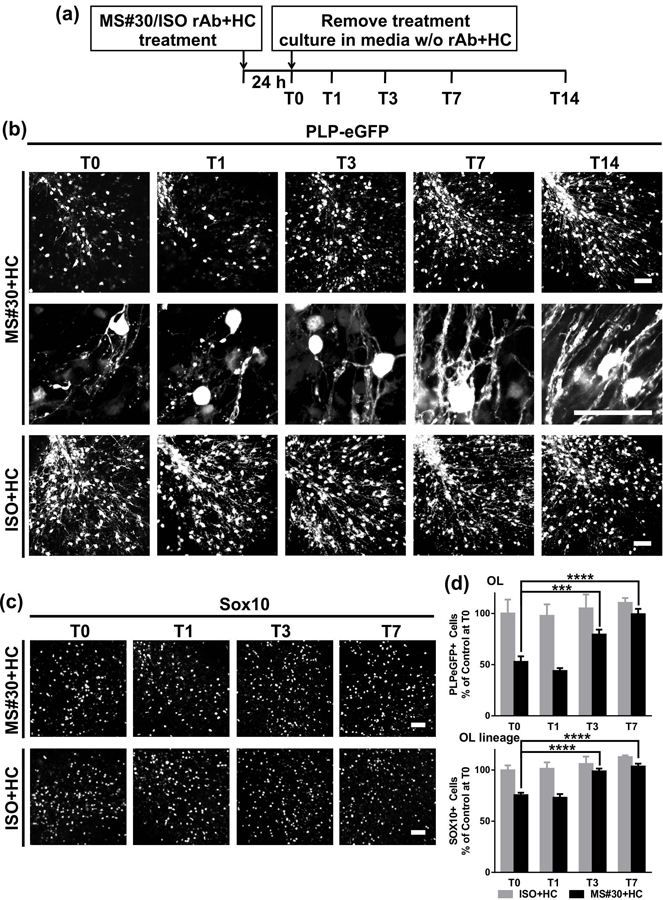
Oligodendrocytes regenerate after treatment with MS#30 plus human complement (HC) was removed. (a) Schematic depicting the treatment and recovery periods. Cerebellar slice cultures prepared from PLP-eGFP mice were treated with 20 μg/ml MS#30 or isotype control rAb (ISO) plus 10% HC for 24 h. Following treatment, rAb and HC were removed by media exchange, and slices were allowed recover in culture media (in the absence of rAb+HC) for an additional 14 days. Slices were fixed, stained, and imaged at T0, T1, T3, T7, and T14. (b) Low and high magnification (upper and lower panels in MS#30+HC) confocal images of PLP-eGFP in slices at T0, T1, T3, T7, T14. (c) Sox10 staining to visualize oligodendrocyte lineage cells. (d) Quantification of PLP-eGFP+ or Sox10+ cell number in slices. Cell count was normalized to control (ISO+HC treated) slice at T0. Statistical analyses were performed by unpaired Student’s t test. ***: p<0.001, ****: p< 0.0001, n=3–4. Scale bars: 50µm.
FIGURE 2.
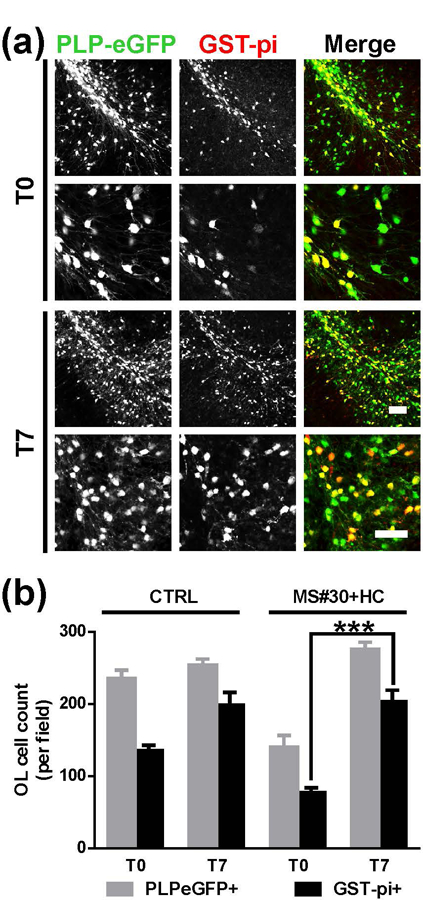
Restoration of mature myelinating oligodendrocytes following treatment with MS#30+HC. (a) Low (upper panels) and high (lower panels) magnification confocal images of PLP-eGFP slices stained with GST-pi (red) to visualize myelinating oligodendrocytes at T0 and T7 after MS#30+HC treatment. (b) Cell count of PLP-eGFP+ or GST-pi+ cells in control slices (no treatment) and slices recovered from MS#30+HC treatment at T0 and T7. Statistical analyses were performed by unpaired Student’s t test. ***: p<0.001, n=3–4. Scale bars: 50µm.
Efficient remyelination after MS#30+HC-mediated demyelination
We next investigated whether the regenerated myelinating oligodendrocytes can re-ensheath axons. The extent of the axon/myelin association was assessed and quantified as a percentage of neurofilament-H (NF-H) positive axons covered by myelin basic protein (MBP). Demyelination was readily apparent 24 h (T0) after exposure to MS#30+HC treatment and recovered over the subsequent 7 days in culture. The percentage of axonal area co-localizing with MBP was 42.8±3.6 % at T0, 69.1±15.0 % at T3, and 89.9±8.5 % at T7. There were no measurable changes of axonal ensheathment in slices exposed to ISO+HC from T0 to T14 (Figure 3a, c). A concomitant increase of myelin proteins, such as PLP and myelin oligodendrocyte glycoprotein (MOG) was observed contemporaneously with remyelination (Figure 2b). In addition, remyelination was associated with restoration of internode structures (Figure 3b). Paranodal contactin-associated protein (Caspr) immunostaining was significantly reduced after treatment with MS#30+HC and gradually reappeared in conjunction with restored myelin coverage (65.8±6.2 % at T0, 94.3±8.4 % at T7, and 97.8±3.9 % at T14) (Figure 3b, d).
FIGURE 3.
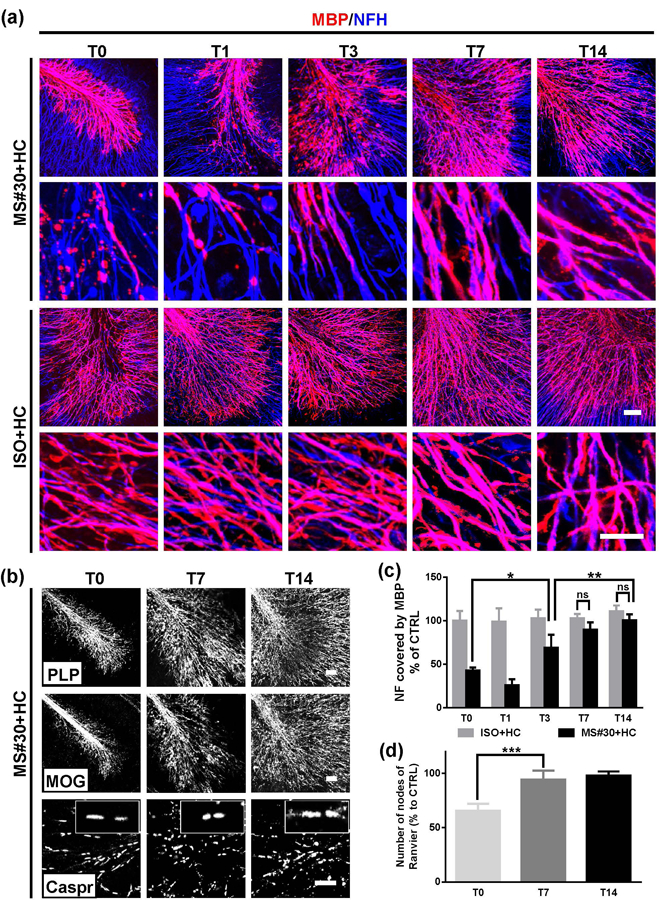
Myelin repair after MS#30+HC demyelination. (a) At the indicated time points after treatment withdrawal, slices were fixed, stained with antibodies against MBP (red) and NF-H (blue), and imaged at low (upper panels) and high (lower panels) magnification using confocal microscopy. (b) Confocal images of slices treated with MS#30+HC and stained with PLP, MOG or Caspr antibodies. (c) The coverage of MBP on NF-H+ axons was calculated at various time points following treatment withdrawal and normalized against control (ISO+HC treated slices) at T0. (d) The number of nodes of Ranvier (paired paranodal Caspar expression) were totaled and then normalized against control (ISO+HC treated slices) at T0. Statistical analyses were performed by unpaired Student’s t test. *: p<0.05, **: p<0.01, ****: p< 0.0001, ns: not significant, n=3–4. Scale bars: 50µm.
The level of myelin repair was not dependent on the extent of MS rAb-mediated demyelination. When slices were exposed to MS#30 +HC for 48h, complete regeneration of oligodendrocytes and extensive axonal ensheathment was still detected (Supporting Information Figure S3) despite severe oligodendrocyte loss (73.1±4.4 %) and demyelination (91.0±8.6 %) (Liu et al., 2017).
Remyelination following MS rAb-induced injury is associated with microglia but not astrocyte activation
Astrocyte morphology and number were evaluated following treatment with MS#30+HC using GFAP (glial fibrillary acidic protein), AQP4, and S100b (S100 calcium-binding protein B) immunostaining. Consistent with our previous report (Liu et al., 2017), demyelination with MS rAb had no immediate effect on astrocyte morphology or network structure, and there were no additional changes during the 7 days of remyelination (Figure 4a). Astrocyte cell number also remained unchanged following injury and during oligodendrocyte recovery (Figure 4b, c).
FIGURE 4.
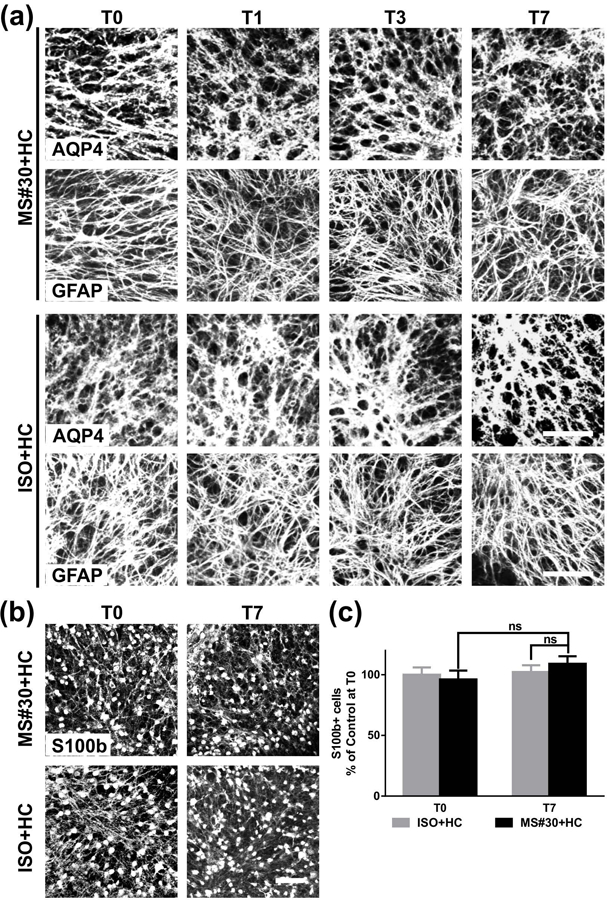
Number and morphology of astrocytes following MS#30+HC treatment. (a) Confocal images of cerebellar slices treated with either MS#30 or ISO plus HC and stained for (a) AQP4 and GFAP or (b) S100b. (c) quantification of S100b staining in slices. No significant difference of S100b+ cells at T0 and T7 in MS#30+HC treatment or between MS#30+HC and ISO+HC. Statistical analyses were performed by multiple unpaired Student’s t test. ns: not significant, n=3–4. Scale bars: 50µm.
Microglia cell number and activation was assayed by Iba1 (ionized calcium binding adaptor molecule 1) immunostaining (Figure 5). Increased numbers of Iba1+ microglia were observed in MS#30+HC-treated slices relative to ISO+HC sections at T0 (56.5±8.3 %). As recovery ensued, the number of microglia peaked at T3 (39.5±8.1 % increase from T0, more than double the number of microglia compared to ISO+HC) followed by a decline to IC+HC levels by T7 (Figure 5a, b). Microglia in MS#30+HC sections altered their appearance from a ramified morphology to an ameoboid-like shape consistent with activation (Salter & Stevens, 2017). From T0 to T3, microglia typically exhibited a reactive morphology with an enlarged cell body containing limited short and thickened processes (Figure 5a, b). By T3 after MS#30+HC treatment, both total number and fraction of reactive microglia (63.4±5.3 % and 17.6±4.4 %, respectively) were increased relative to T0 (Figure 5b, c). Overall, 71.7±2.7 % of total microglia at T3 displayed a reactive morphology compared to 38.5±2.2 % in the ISO+HC control (Figure 5c). Following successful remyelination by T7, most microglial morphology returned to the ramified shape with long, branching processes and small cell bodies characteristic of resting cells. In the control ISO+HC treated sections, resting ramified-shaped microglia were predominant from T1 to T7, though some microglia at T0 (~50%) displayed reactive morphology due to the pre-exposure of sections to HC at T0 (Liu et al., 2017) (Figure 5a, single cell panels). Overall, recovery from MS rAb-induced demyelination in this model is coincident with extensive microglia but not astrocytic activation.
FIGURE 5.
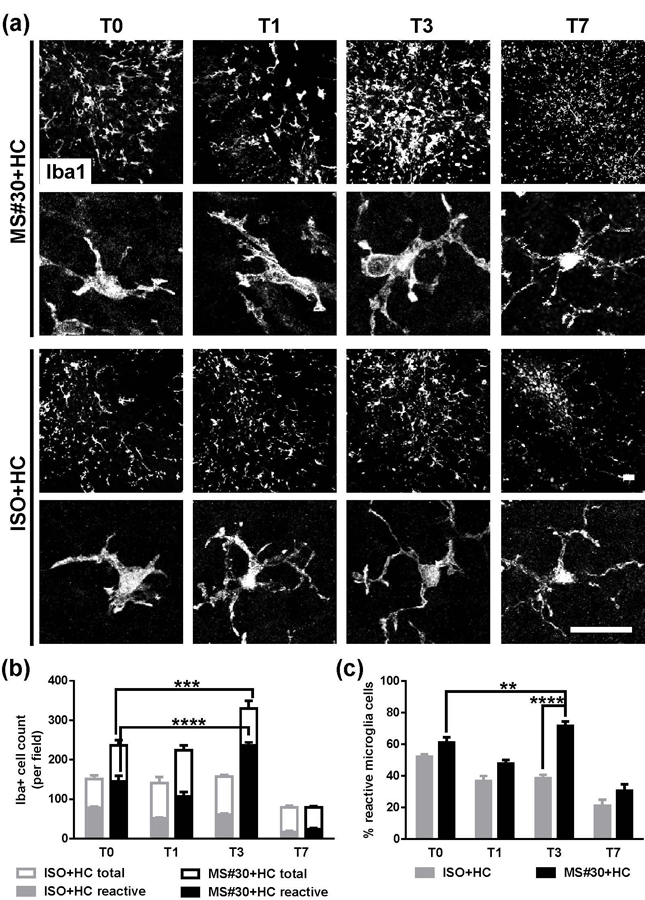
Increased microglia activation in slices recovering from MS#30+HC treatment. (a) Confocal images of slices stained with Iba1 after MS#30+HC or ISO+HC treatment withdrawal. Representative single cell images (lower panels) indicate the morphological change of microglia during the recovery time. (b and c) Quantification of total and reactive microglia in slices at indicated time points after withdrawal of MS#30+HC or ISO+HC treatment. Total (Iba1+) microglia were identified as reactive based on shortened and thick processes and enlarged cell bodies. Statistical analyses were performed by multiple unpaired Student’s t test. **: p<0.01, ***: p<0.001, ****: p< 0.0001, n=3–4. Scale bars: 25µm.
A second myelin-specific rAb (MS rAb 07–7#49, MS#49) derived from CSF plasmablasts of an isolated optic neuritis patient, who progressed to clinically-definite MS, displayed the similar glial damage and demyelination pattern as MS#30 (Liu et al., 2017). Slices treated with MS#49+HC demonstrated identical glial recovery and myelin repair pattern to that observed with MS#30+HC. In the presence of HC, MS#49 caused oligodendrocyte loss, demyelination and microglia activation (Supporting Information Figure S4, T0). Slices recovering from MS#49+HC demonstrated oligodendrocyte repopulation and efficient myelin repair (Supporting Information Figure S4a, b). Recovery from MS#49-induced demyelination was also coincident with increased microglial activation at T3 (Supporting Information Figure S4c).
Oligodendrocytes repopulate but fail to remyelinate following NMO rAb-induced damage
AQP4-specific NMO rAb treatment initiates complement-dependent astrocyte destruction followed by loss of oligodendrocytes and demyelination (Liu et al., 2017). Cerebellar slice cultures were treated with NMO rAb#53 (NMO#53)+HC for 24h, washed, and then recovered in culture media for 7 days (Figure 6a). NMO#53+HC caused massive disruption of the astrocyte network and loss of astrocyte cell number as visualized by AQP4, GFAP and S100b immunostaining (see T0; Figure 6b, c). By T3, astrocyte morphology was restored to baseline (Figure 6b; AQP4 and GFAP panels) and astrocyte cell number gradually increased from 49.9±8.6 % of control at T0, to 73.9±3.5% at T3, and 95.2±8.3 % at T7 (Figure 6b, c; S100b panels).
FIGURE 6.
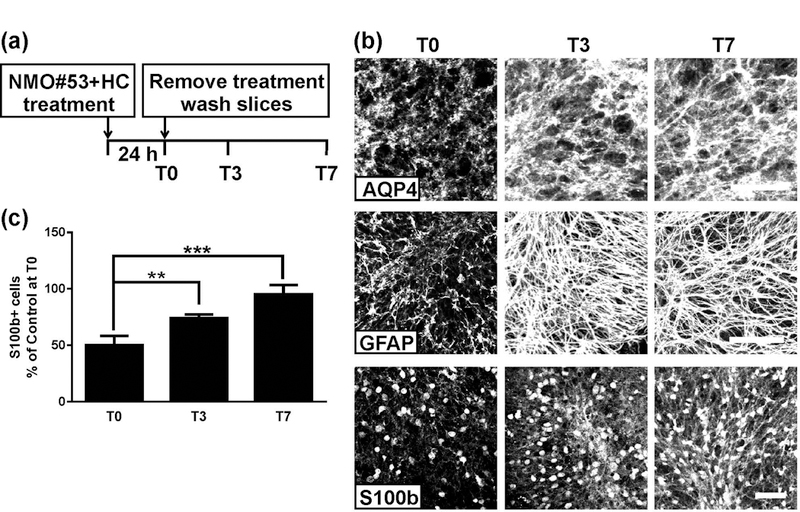
Astrocyte repair after treatment with NMO#53+HC. (a) Schematic depicting the treatment and recovery periods. Cerebellar slice cultures prepared from PLP-eGFP mice were treated with 20 μg/ml NMO#53 rAb plus 10% HC for 24 h. Following treatment, rAb and HC were removed by media exchange, and slices were allowed recover in culture media (in the absence of rAb+HC). Slices were fixed, stained, and imaged at T0, T3, and T7. (b) Confocal images of slices stained with AQP4, GFAP or S100b at the indicated time points. (c) Quantification of S100b+ cell number in slices. Cell count was normalized to control (ISO+HC treated) slice at T0. Statistical analyses were performed by unpaired Student’s t test. **: p<0.01, ***: p<0.001, n=3–4. Scale bars: 50µm.
We next examined whether astrocyte repair coincided with oligodendrocyte repopulation and myelin regeneration. The number of PLP-eGFP+ oligodendrocytes and Sox10+ oligodendrocyte lineage cells was significantly greater in the slices at T7 than T0 (Figure 7a). At T7, the numbers of PLP-eGFP+ and Sox10+ cells were restored to 100.1±7.5 % and 93.8±9.6 % of ISO+HC control slices, respectively (Figure 7c). All the PLP-eGFP+ oligodendrocyte cells recovering from NMO#53+HC treatment were CC1+, indicating that regenerated oligodendrocyte cells had differentiated to the early myelinating stage (Supporting Information Figure S2). Despite restoration of oligodendrocyte numbers, the extension of eGFP+ processes from oligodendrocyte cell bodies was limited and fragmented in appearance (Figure 7a, high magnification PLP-eGFP panels). In contrast, oligodendrocyte processes were extensive and well-organized at T7 in slices recovering from treatment with MS#30+HC (Figure 1b, high magnification PLP-eGFP panels). Moreover, the number of GST-pi+ oligodendrocytes remained significantly reduced (48.7±3.4 % to untreated controls) when compared to MS#30+HC slices at T7 (Figure 7b, c). Hence, in NMO rAb+HC treated slices, differentiated early myelinating oligodendrocytes (CC1+) repopulate but do not mature fully (GST-pi+).
FIGURE 7.
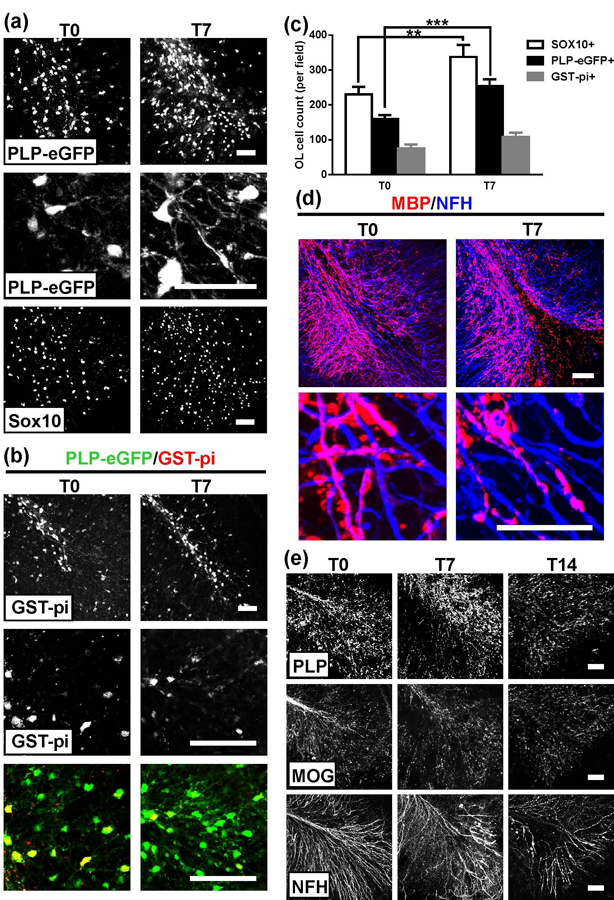
Oligodendrocyte repopulation without myelin repair following treatment with NMO#53+HC. Low (upper panel) and high (lower panel) magnification confocal images of (a) PLP-eGFP and Sox10 and (b) GST-pi in treated slices at T0 and T7 (high magnification in lower panels in PLP-eGFP and GST-pi images). (c) Quantification of Sox10+, PLP-eGFP+ and GST-pi+ cell number in slices. Statistical analyses were performed by unpaired Student’s t test. **: p<0.01, ***: p<0.001, n=3–4. (d) Low (upper panel) and high (lower panel) magnification confocal images of slices stained with MBP (red) and NF-H (blue) antibodies at T0 and T7. (e) Confocal images of slices stained with PLP, MOG or NF-H antibodies at T0, 7 and 14. Scale bars: 50µm.
Consistent with the lack of oligodendrocyte maturation, slices treated with NMO#53+HC showed no restoration of axonal ensheathment or paranode immunostaining at T7 (Figure 7d and data not shown). Even with extended recovery time (T14), myelin protein expression on axons had not returned to baseline and axonal NFH staining showed increased deterioration and fragmentation (Figure 7e). At T14, the number of PLP-eGFP+ oligodendrocyte cells was comparable to that at T7 (data not shown). Together, these results indicate that astrocyte repair in NMO is insufficient for complete oligodendrocyte maturation and remyelination.
Reduced microglia activation following NMO#53+HC-induced injury
Similar to that observed after treatment with MS#30+HC, Iba1+ microglia were significantly increased at T0 following exposure to NMO#53+HC (Figure 8a). However, in contrast to MS rAb-treated slices, both total Iba1+ cell number and the reactive-shaped Iba1+ cells gradually reduced during recovery from NMO rAb-induced demyelination (Figure 8a, b, c). Because the extent of demyelination did not differ significantly between MS rAb- and NMO rAb-treated slices at T0 (Figures 2b, 7c) (Liu et al., 2017), increased microglia activation during remyelination from MS#30-mediated injury may reflect more than debris clearance.
FIGURE 8.
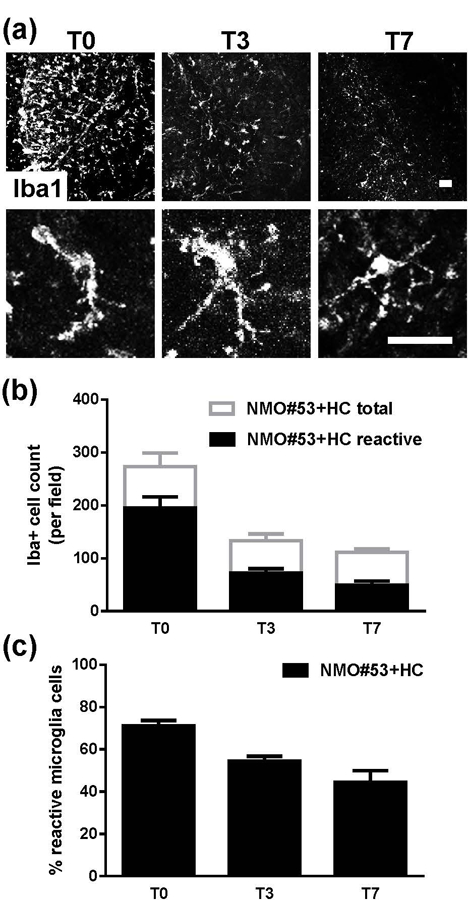
Microglia activation in slices recovered from NMO#53+HC treatment. (a) Confocal images of slices stained with Iba1 at T0, T3 and T7 following NMO#53+HC treatment withdrawal. Representative single cell images (lower panels) depict the morphological change of microglia during recovery. (b and c) Quantification of total (Iba1+) and reactive (short, thick processes with enlarged cell bodies) microglia in slices at the indicated time points following NMO#53+HC treatment withdrawal.
DISCUSSION
We have previously demonstrated that patient-derived MS and NMO rAbs target disease-specific epitopes resulting in distinct patterns of glial and neuronal injury (Liu et al., 2017). In the current study, we demonstrate that these disease-specific injuries establish distinct environments that alter glial cell responses and remyelination (Table 1). Oligodendrocytes repopulate after both MS- and NMO-mediated injury; however, while oligodendrocytes mature fully into functional myelinating cells and form new myelin sheaths after exposure to MS myelin-specific rAb and HC, regenerated oligodendrocytes fail to reach a functional myelinating stage following NMO AQP4 rAb-mediated injury.
Table 1.
Summary of MS and NMO rAb induced glial responses during damage and repair in cerebellar slices.
| Complement dependent damage |
Recovery after treatment withdrawal |
||
|---|---|---|---|
| MS#30 | OL | lossa | mature myelinating OL regeneration |
| AST | no change | no change | |
| Axon | preserved | preserved | |
| Myelin | demyelination | remyelination | |
| MG | activation | increased activation | |
| NMO#53 | OL | loss | differentiated early myelinating OL regeneration |
| AST | lossa | regeneration | |
| Axon | preserved at early timeb | damage | |
| Myelin | demyelination | no remyelination | |
| MG | activation | reduced activation | |
primary damage.
axon preserved at 24 h, neuron cell death observed from 48 h of NMO#53+HC treatment ((Liu et al., 2017) and Figure 7).
Abbreviations:
OL oligodendrocyte
AST astrocyte
MG microglia.
Remyelination has been studied in ex vivo and in vivo animal models and shown to be quite robust (Verden & Macklin, 2016). In ex vivo slice culture systems using rodent brain or spinal cord, efficient remyelination from lysolecithin-induced demyelination occurs 7 to 14 days after treatment removal (Birgbauer, Rao, & Webb, 2004; Miron et al., 2010; Zhang, Jarjour, Boyd, & Williams, 2011). We observed a similar time frame for remyelination of cerebellar slices after inflammatory demyelination induced by MS rAb and complement. Remyelination is often inefficient and limited in MS patients, leading to permanent deficits and dysfunction (Louapre et al., 2015; Lucchinetti et al., 2000), and there are no therapies proven to repair myelin damage in MS (Harlow, Honce, & Miravalle, 2015). Understanding why remyelination often fails in MS after inflammation injury is essential to the development of effective remyelination and repair strategies. To a certain extent, this failure results from the inability of OPCs to successfully generate new mature myelinating oligodendrocytes, although the underlying reasons for this are not yet fully understood (Kremer et al., 2016). Dissecting the processes mediating recovery in the MS antibody-driven cerebellar explant model may offer novel insights into mechanisms that govern post-inflammatory remyelination.
There is limited histopathologic description of remyelination in human NMO lesions (Ikota, Iwasaki, Kawarai, & Nakazato, 2010). This is due in part to our limited understanding of the dynamics of NMO lesion formation and repair in vivo. NMO lesions demonstrate combined astrocyte, oligodendroglial, and neuronal pathology that likely limit remyelination through diverse mechanisms: (i) inhibition of oligodendroglial differentiation; (ii) inhibition of oligodendrocyte progenitor migration through blood-brain barrier injury; and (iii) impaired myelin wrapping secondary to irreversible axonal destruction (reviewed in (Weber, Derfuss, Metz, & Bruck, 2018; Yao et al., 2016). It is unclear how primary astrocyte destruction results in downstream oligodendrocyte loss, myelin destruction, and neuronal injury in NMO lesions. Potential pathogenic mechanisms include excitotoxicity, altered ion homeostasis, increased oxidative stress, and reduced axonal metabolic support from astrocyte (Brown & Ransom, 2007; Ransom & Fern, 1997) (reviewed in (Bennett & Owens, 2017)). Identifying the link between astrocyte damage and demyelination may ultimately help to understand the relative remyelination deficiency in NMO. The impaired microglial activation observed in our ex vivo NMO injury model may be a factor in one or more of these processes.
In our ex vivo cerebellar model, CC1+ differentiated oligodendrocytes were regenerated but failed to mature to the myelinating stage despite the restoration of astrocyte cell numbers and morphology. Yao et al. (2016) reported a similar lack of remyelination in cerebellar slices treated with AQP4-specific antibody and human complement. Using low concentrations of AQP4 rAb and human complement, they observed reduced CC1+ oligodendrocyte differentiation. The difference in oligodendrocyte differentiation observed herein may be due to different genetic backgrounds (CD1 vs C57) and/or sources of human complement. Differential oligodendrocyte progenitor damage might also account for the reduced oligodendrocyte differentiation into CC1+ cells in that study (Yao et al., 2016). In this study, recovery of slices from 24 h treatment of NMO#53+HC myelin involved increased deterioration and fragmentation of axons at T7 and T14 (Figure 7). It is possible that lack of remyelination leads to neuronal injury as indicated by fragmented axons. Alternatively, injured neurons and reduced neuronal activity (Liu et al., 2017) may have reduced signals that enhance oligodendrocyte ensheathment or produced inhibitory signals that block oligodendrocyte maturation and myelination (Mount & Monje, 2017).
Although we and others have observed a rapid repopulation of astrocytes in cerebellar slices recovering from NMO rAb-mediated damage, the regenerated cells may not function equivalently to nascent astrocytes during recovery (Figure 7). There is abundant and growing evidence for astrocytes to actively participate in both MS lesion development and repair (Brosnan & Raine, 2013). It has been shown that reactive astrocytes are key players in driving CNS inflammation and are directly implicated in suppressing remyelination in an EAE model (Brambilla et al., 2014). Further characterization of the sub-type and function of repaired astrocytes in lesions may reveal the role of astrocyte in remyelination suppression.
We previously reported that approximately 50% of OPCs are lost in slices treated with rAb NMO#53+HC, whereas slices treated with MS#30+HC show no reduction in OPCs (Liu et al., 2017; Liu et al., 2016). The equivalent repopulation of oligodendrocytes after both MS- and NMO-mediated injury in the current study demonstrates that initial OPC differentiation was not impaired by NMO rAb-mediated injury despite a lower number of surviving cells. However, the failure of oligodendrocytes to reach a functional myelinating stage following NMO#53-mediated injury indicates that the milieu produced by NMO-specific rAbs is not conducive to full recovery, while that produced by myelin-specific MS rAbs is distinctly conducive to complete remyelination.
We observed a significant difference in microglia activation following MS and NMO rAb-mediated injury (Table 1). Microglial numbers and reactivity increased in slices recovering from MS rAb+HC-induced damage (Figure 5), whereas microglial activation continually declined during recovery from NMO rAb demyelination (Figure 8). Microglia are the principal resident immune cells in the CNS and play an essential and versatile role in CNS development, maintenance, repair (Sierra et al., 2016) and in pathological context (Salter & Stevens, 2017; Wieghofer, Knobeloch, & Prinz, 2015). Although microglia-induced neuroinflammation is implicated in the occurrence and progression of many neurological disease models (Clark et al., 2016; Colonna & Butovsky, 2017), they also exhibit protective and regenerative properties (Miron et al., 2013; Rice et al., 2017; Spangenberg et al., 2016; Szalay et al., 2016) . In demyelination models, increasing evidence has suggested that microglia play an important role in remyelination. Minocycline-induced inhibition of microglia activation significantly impairs remyelination following ethidium bromide injection in rat cerebellum (Li, Setzu, Zhao, & Franklin, 2005), and activation of microglia or adoptive transfer of cytokine-induced adult microglia in the EAE model is associated with milder disease and improved recovery (Mikita et al., 2011; Zhang, Lund, Mia, Parsa, & Harris, 2014). In the cuprizone model of demyelination, genetic depletion of microglia function in CX3CR1 knockout mice results in insufficient myelin debris clearance and impaired remyelination (Lampron et al., 2015), and remyelination following focal lysolecithin-induced demyelination in the mouse corpus callosum is impaired following microglia depletion using clodronate liposomes. Conversely, in a traumatic spinal cord injury mouse model, microglial activation by CNS IL-4 injection increases oligodendrogenesis (Fenn, Hall, Gensel, Popovich, & Godbout, 2014), and oligodendrocyte differentiation is enhanced in vitro in the presence of microglia cells and conditioned media (Miron et al., 2013).
Microglia function in remyelination likely involves phagocytosis of myelin debris and the secretion of factors to recruit oligodendrocyte progenitors and promote oligodendrocyte regeneration (Lloyd & Miron, 2016; McMurran, Jones, Fitzgerald, & Franklin, 2016; Olah et al., 2012). However, without any characterization of the regenerated oligodendrocytes in these studies, it is not clear whether microglia promote oligodendrocyte progenitor differentiation or oligodendrocyte maturation from the early myelinating to mature myelinating stage. Following NMO rAb-mediated demyelination, there is limited microglial activation and remyelination despite normal oligodendrocyte differentiation. Hence, microglia activation may play an important role in the development of oligodendrocytes to a mature myelinating stage. Understanding the role of microglial activation in the later stages of functional remyelination will complement ongoing efforts to identify factors promoting oligodendrocyte differentiation (Cole et al., 2017).
Recently, autoantibodies against myelin oligodendrocyte glycoprotein (MOG-IgG) have been associated with various CNS inflammatory demyelinating diseases, such as acute disseminated encephalomyelitis (ADEM), monophasic or recurrent optic neuritis (ON) or transverse myelitis (TM), and AQP4-seronegative neuromyelitis optica spectrum disorders (NMOSD) (Ramanathan, Dale, & Brilot, 2016). MOG-IgG, however, is rarely identified in MS patients (Peschl, Schanda, et al., 2017). The MS patient-derived, myelin-specific rAbs used in this study (MS#30 and MS#49) are not directed against MOG (Blauth et al., 2015; Owens et al., 2009) and behave quite differently from patient-derived anti-MOG IgG. While the murine anti-MOG monoclonal antibody 8–18-C5 causes complement-mediated demyelination in in vitro, ex vivo, and in vivo assays (Kerlero de Rosbo, Honegger, Lassmann, & Matthieu, 1990; Linington et al., 1989; Saadoun et al., 2014), serum IgG from high-titer MOG-seropositive patients induces only mild complement-dependent tissue injury in ex vivo organotypic slice cultures and produces no injury in the experimental autoimmune encephalitis (EAE) Lewis rat model (Peschl, Bradl, Hoftberger, Berger, & Reindl, 2017). Additionally, when tested alongside anti-AQP4 NMO rAb, intracerebral micro-injection of MOG-positive serum IgG failed to induce CNS demyelination (Saadoun et al., 2014). Therefore, the complement-mediated effects of MS patient-derived myelin-specific rAbs on organotypic cerebellar slices are not merely representative of any myelin-specific autoantibody but reflect MS-specific injury.
In summary, identifying distinct glia responses and effects on myelin repair in MS and NMO will lead to a better understanding of the mechanisms governing remyelination in these distinct disorders. This information will be crucial for the successful development of distinct therapeutic strategies to facilitate recovery in affected patients. Our results suggest that microglia activation may play a crucial role in promoting oligodendrocyte maturation and effective remyelination. Importantly, astrocyte recovery is insufficient to drive oligodendrocyte maturation in NMO lesions. Our novel ex vivo models of rAb-mediated demyelination/remyelination will serve as new experimental models to identity potential therapies that promote effective remyelination in MS or NMO.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Collaborative Research Grant from National Multiple Sclerosis Society (W.B.M.), NEI EY022936 (J.L.B.), UM1 AI110498 (J.L.B.) NINDS NS072141 and NMSS Research Grant (G.P.O.), NS25304 (W.B.M.) and the Guthy-Jackson Charitable Foundation (J.L.B.).
We are indebted to Adeline Matschulat, Kimberley Clawson-Stone and Hannah Schumann for the preparation of recombinant antibodies. We thank Kristin Schaller for assistance with mouse breeding.
Footnotes
CONFLICTS OF INTEREST STATMENT
The authors declare that they have no competing interests.
REFERENCES
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, … Hemmer B (2009). Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol, 66(5), 617–629. 10.1002/ana.21802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JL, & Owens GP (2017). Neuromyelitis Optica: Deciphering a Complex Immune-Mediated Astrocytopathy. J Neuroophthalmol, 37(3), 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgbauer E, Rao TS, & Webb M (2004). Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J Neurosci Res, 78(2), 157–166. [DOI] [PubMed] [Google Scholar]
- Blauth K, Soltys J, Matschulat A, Reiter CR, Ritchie A, Baird NL, … Owens GP (2015). Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid cause demyelination of spinal cord explants. Acta Neuropathol, 130(6), 765–781. 10.1007/s00401-015-1500-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, … Lassmann H (2009). Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol, 66(5), 630–643. 10.1002/ana.21837 [DOI] [PubMed] [Google Scholar]
- Brambilla R, Morton PD, Ashbaugh JJ, Karmally S, Lambertsen KL, & Bethea JR (2014). Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia, 62(3), 452–467. [DOI] [PubMed] [Google Scholar]
- Breij EC, Brink BP, Veerhuis R, van den Berg C, Vloet R, Yan R, … Bo L (2008). Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol, 63(1), 16–25. 10.1002/ana.21311 [DOI] [PubMed] [Google Scholar]
- Brosnan CF, & Raine CS (2013). The astrocyte in multiple sclerosis revisited. Glia, 61(4), 453–465. [DOI] [PubMed] [Google Scholar]
- Brown AM, & Ransom BR (2007). Astrocyte glycogen and brain energy metabolism. Glia, 55(12), 1263–1271. 10.1002/glia.20557 [DOI] [PubMed] [Google Scholar]
- Clark KC, Josephson A, Benusa SD, Hartley RK, Baer M, Thummala S, … Dupree JL (2016). Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia, 64(7), 1190–1209. 10.1002/glia.22991 [DOI] [PubMed] [Google Scholar]
- Colonna M, & Butovsky O (2017). Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol, 35, 441–468. 10.1146/annurev-immunol-051116-052358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, … Barres BA (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell, 138(1), 172–185. 10.1016/j.cell.2009.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Hall JC, Gensel JC, Popovich PG, & Godbout JP (2014). IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci, 34(26), 8904–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow DE, Honce JM, & Miravalle AA (2015). Remyelination Therapy in Multiple Sclerosis. Front Neurol, 6, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer B, Cepok S, Zhou D, & Sommer N (2004). Multiple sclerosis -- a coordinated immune attack across the blood brain barrier. Curr Neurovasc Res, 1(2), 141–150. [DOI] [PubMed] [Google Scholar]
- Ikota H, Iwasaki A, Kawarai M, & Nakazato Y (2010). Neuromyelitis optica with intraspinal expansion of Schwann cell remyelination. Neuropathology, 30(4), 427–433. 10.1111/j.1440-1789.2009.01071.x [DOI] [PubMed] [Google Scholar]
- Kerlero de Rosbo N, Honegger P, Lassmann H, & Matthieu JM (1990). Demyelination induced in aggregating brain cell cultures by a monoclonal antibody against myelin/oligodendrocyte glycoprotein. J Neurochem, 55(2), 583–587. [DOI] [PubMed] [Google Scholar]
- Kremer D, Gottle P, Hartung HP, & Kury P (2016). Pushing Forward: Remyelination as the New Frontier in CNS Diseases. Trends Neurosci, 39(4), 246–263. [DOI] [PubMed] [Google Scholar]
- Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, … Rivest S (2015). Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med, 212(4), 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J, & Mahad D (2012). Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol, 8(11), 647–656. [DOI] [PubMed] [Google Scholar]
- Li WW, Setzu A, Zhao C, & Franklin RJ (2005). Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J Neuroimmunol, 158(1–2), 58–66. [DOI] [PubMed] [Google Scholar]
- Linington C, Morgan BP, Scolding NJ, Wilkins P, Piddlesden S, & Compston DA (1989). The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain, 112 ( Pt 4), 895–911. [DOI] [PubMed] [Google Scholar]
- Liu Y, Given KS, Harlow DE, Matschulat AM, Macklin WB, Bennett JL, & Owens GP (2017). Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun, 5(1), 25 10.1186/s40478-017-0428-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Harlow DE, Given KS, Owens GP, Macklin WB, & Bennett JL (2016). Variable sensitivity to complement-dependent cytotoxicity in murine models of neuromyelitis optica. J Neuroinflammation, 13(1), 301 10.1186/s12974-016-0767-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd AF, & Miron VE (2016). Cellular and Molecular Mechanisms Underpinning Macrophage Activation during Remyelination. Front Cell Dev Biol, 4, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louapre C, Govindarajan ST, Gianni C, Langkammer C, Sloane JA, Kinkel RP, & Mainero C (2015). Beyond focal cortical lesions in MS: An in vivo quantitative and spatial imaging study at 7T. Neurology, 85(19), 1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, & Lassmann H (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol, 47(6), 707–717. [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Bruck W, & Lassmann H (2004). Evidence for pathogenic heterogeneity in multiple sclerosis. Ann Neurol, 56(2), 308 10.1002/ana.20182 [DOI] [PubMed] [Google Scholar]
- Mallon BS, Shick HE, Kidd GJ, & Macklin WB (2002). Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J Neurosci, 22(3), 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurran CE, Jones CA, Fitzgerald DC, & Franklin RJ (2016). CNS Remyelination and the Innate Immune System. Front Cell Dev Biol, 4, 38 10.3389/fcell.2016.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, & Paul DL (2006). Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci, 26(43), 10984–10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, … Petry KG (2011). Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler, 17(1), 2–15. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, … ffrench-Constant C (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci, 16(9), 1211–1218. 10.1038/nn.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Kuhlmann T, & Antel JP (2011). Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim Biophys Acta, 1812(2), 184–193. [DOI] [PubMed] [Google Scholar]
- Miron VE, Ludwin SK, Darlington PJ, Jarjour AA, Soliven B, Kennedy TE, & Antel JP (2010). Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am J Pathol, 176(6), 2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, & Rothstein JD (2013). Oligodendroglia: metabolic supporters of axons. Trends Cell Biol, 23(12), 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount CW, & Monje M (2017). Wrapped to Adapt: Experience-Dependent Myelination. Neuron, 95(4), 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, & Trapp BD (2008). Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci, 31, 535–561. [DOI] [PubMed] [Google Scholar]
- Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, & Boddeke HW (2012). Identification of a microglia phenotype supportive of remyelination. Glia, 60(2), 306–321. [DOI] [PubMed] [Google Scholar]
- Owens GP, Bennett JL, Lassmann H, O’Connor KC, Ritchie AM, Shearer A, … Gilden D (2009). Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann Neurol, 65(6), 639–649. 10.1002/ana.21641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschl P, Bradl M, Hoftberger R, Berger T, & Reindl M (2017). Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front Immunol, 8, 529 10.3389/fimmu.2017.00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschl P, Schanda K, Zeka B, Given K, Bohm D, Ruprecht K, … Reindl M (2017). Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflammation, 14(1), 208 10.1186/s12974-017-0984-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan S, Dale RC, & Brilot F (2016). Anti-MOG antibody: The history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev, 15(4), 307–324. [DOI] [PubMed] [Google Scholar]
- Ransom BR, & Fern R (1997). Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia, 21(1), 134–141. [PubMed] [Google Scholar]
- Ratelade J, & Verkman AS (2014). Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol Immunol, 62(1), 104–113. 10.1016/j.molimm.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RA, Pham J, Lee RJ, Najafi AR, West BL, & Green KN (2017). Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia, 65(6), 931–944. 10.1002/glia.23135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie AM, Gilden DH, Williamson RA, Burgoon MP, Yu X, Helm K, … Owens GP (2004). Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol, 173(1), 649–656. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, & Papadopoulos MC (2010). Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain, 133(Pt 2), 349–361. 10.1093/brain/awp309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, Owens GP, Bennett JL, Vincent A, & Papadopoulos MC (2014). Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain. Acta Neuropathol Commun, 2, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, & Stevens B (2017). Microglia emerge as central players in brain disease. Nat Med, 23(9), 1018–1027. 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- Sheridan GK, & Dev KK (2012). S1P1 receptor subtype inhibits demyelination and regulates chemokine release in cerebellar slice cultures. Glia, 60(3), 382–392. 10.1002/glia.22272 [DOI] [PubMed] [Google Scholar]
- Sierra A, de Castro F, Del Rio-Hortega J, Rafael Iglesias-Rozas J, Garrosa M, & Kettenmann H (2016). The “Big-Bang” for modern glial biology: Translation and comments on Pio del Rio-Hortega 1919 series of papers on microglia. Glia, 64(11), 1801–1840. 10.1002/glia.23046 [DOI] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, … Green KN (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain, 139(Pt 4), 1265–1281. 10.1093/brain/aww016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, … Denes A (2016). Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun, 7, 11499 10.1038/ncomms11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Kataoka Y, Cui Y, Takamori Y, Watanabe Y, & Yamada H (2007). Intracellular translocation of glutathione S-transferase pi during oligodendrocyte differentiation in adult rat cerebral cortex in vivo. Neuroscience, 148(2), 535–540. [DOI] [PubMed] [Google Scholar]
- Tradtrantip Lukmanee, Zhang Hua, Saadoun Samira, Phuan Puay-Wah, Lam Chiwah, Papadopoulos Marios C., … Verkman AS (2012). Anti–Aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Annals of Neurology, 71(3), 314–322. 10.1002/ana.22657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verden D, & Macklin WB (2016). Neuroprotection by central nervous system remyelination: Molecular, cellular, and functional considerations. J Neurosci Res, 94(12), 1411–1420. 10.1002/jnr.23923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, & Wingerchuk DM (2014). Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol, 5(3), 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MS, Derfuss T, Metz I, & Bruck W (2018). Defining distinct features of anti-MOG antibody associated central nervous system demyelination. Ther Adv Neurol Disord, 11, 1756286418762083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieghofer P, Knobeloch KP, & Prinz M (2015). Genetic targeting of microglia. Glia, 63(1), 1–22. 10.1002/glia.22727 [DOI] [PubMed] [Google Scholar]
- Yamamura T, Konola JT, Wekerle H, & Lees MB (1991). Monoclonal antibodies against myelin proteolipid protein: identification and characterization of two major determinants. J Neurochem, 57(5), 1671–1680. [DOI] [PubMed] [Google Scholar]
- Yao X, Su T, & Verkman AS (2016). Clobetasol promotes remyelination in a mouse model of neuromyelitis optica. Acta Neuropathol Commun, 4(1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, & Verkman AS (2017). Complement regulator CD59 prevents peripheral organ injury in rats made seropositive for neuromyelitis optica immunoglobulin G. Acta Neuropathol Commun, 5(1), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Jarjour AA, Boyd A, & Williams A (2011). Central nervous system remyelination in culture--a tool for multiple sclerosis research. Exp Neurol, 230(1), 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bennett JL, & Verkman AS (2011). Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Annals of Neurology, 70(6), 943–954. 10.1002/ana.22551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Lund H, Mia S, Parsa R, & Harris RA (2014). Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia, 62(5), 804–817. 10.1002/glia.22643 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.