

Abstract
Background & Aims:
Metaplastic glands buried under squamous epithelium are frequently detected in patients with Barrett’s esophagus (BE). This sub-squamous intestinal metaplasia (SSIM) might be responsible for cancers that develop despite endoscopic surveillance, and for metaplasia recurrences after endoscopic ablation. To determine whether reflux induces BE cells to undergo epithelial to mesenchymal transition (EMT) that produces SSIM, we assessed EMT in BE cells exposed to acidic bile salts, and in rat and human esophageal tissues.
Methods:
We compared markers of EMT and cell motility in transwell and 3-dimensional organotypic culture systems among dysplastic BE epithelial cell lines, nondysplastic telomerase-immortalized BE cell lines (BAR-T), and BAR-T cells exposed acutely or for 20 weeks (BEC-20W) to acidic bile salts. VEGFA was inhibited with a neutralizing antibody or CRISPR-Cas9n and VEGFR2 was inhibited with SU1498 or shRNA, and cells were analyzed by immunohistochemistry, quantitative PCR, or immunoblotting for markers of VEGF signaling and EMT; cell motility was assessed by transwell. We used immunohistochemistry and quantitative PCR to assess EMT markers in the columnar-lined esophagus of rats with surgically induced reflux esophagitis and in esophagectomy specimens from patients with BE.
Results:
We detected features of EMT (decreased cadherin 1 [CDH1]; increased fibronectin 1, vimentin, and MMP2; and increased motility) in dysplastic BE epithelial cell lines and in BEC-20W cells, but not in unexposed BAR-T cells. Acute acidic bile salt exposure induced expression of the zinc finger E-box binding homeobox 1/2 (ZEB1/2) in BAR-T cells, which reduced their expression of CDH1 and increased motility; inhibitors of VEGF signaling blocked these effects. Columnar-lined esophagus of rats with reflux esophagitis had increased expression of ZEB1/2 and decreased expression of CDH1 compared with controls. Dysplastic BE tissues also had significantly increased levels of ZEB1 and significantly decreased levels of CDH1 compared with non-dysplastic BE tissues.
Conclusions:
In BE cell lines, acidic bile salts induce EMT via VEGF signaling, which increases expression of ZEB1/2, repressors of CDH1. These observations suggest that reflux induces EMT in metaplastic BE tissues, which promotes development of SSIM.
Keywords: gastroesophageal, EAC, carcinogenesis, esophago-jejunostomy
Introduction
Barrett’s esophagus, the condition in which an intestinal-type metaplastic mucosa replaces esophageal squamous mucosa damaged by gastroesophageal reflux of acid and bile, is a major risk factor for esophageal adenocarcinoma.1–4 To prevent this deadly cancer, patients with Barrett’s esophagus are advised to have regular endoscopic surveillance to detect dysplasia, and patients found to have dysplasia are treated with endoscopic eradication therapy, which usually includes radiofrequency ablation (RFA) of the metaplastic mucosa. A randomized, controlled trial has demonstrated that RFA frequently achieves complete eradication of metaplasia and dysplasia in Barrett’s esophagus, and that RFA can prevent dysplasia from progressing to cancer within one year.1 Unfortunately, Barrett’s metaplasia recurs frequently after RFA, with rates approaching 10% per year.2, 3 More worrisome is the observation that invasive adenocarcinoma occurs at the rate of 0.65% per year after apparently successful RFA for dysplasia.2, 3 Therefore, continued endoscopic surveillance is recommended, even after complete eradication of Barrett’s esophagus by RFA.4
The etiology of recurrent Barrett’s metaplasia after RFA is not clear, but sub-squamous intestinal metaplasia (SSIM) is one possibility. SSIM is the condition in which intestinal-type metaplastic glands are present underneath esophageal squamous epithelium in the lamina propria, where they are hidden from endoscopists. Initially, SSIM was thought to be a consequence of incomplete endoscopic ablation procedures that destroyed only the superficial layer of Barrett’s metaplasia, leaving behind a viable, deep glandular layer to be “buried” by neosquamous epithelium that proliferates to cover the ablation wound.5 However, recent reports suggest that the large majority of patients with Barrett’s esophagus who have never been treated with ablation have SSIM underlying esophageal squamous epithelium near its junction with Barrett’s metaplasia.6–8 Conceivably, malignant transformation of this highly prevalent and endoscopically-invisible SSIM might contribute to the frequent failures that have been reported for endoscopic surveillance in preventing cancer in Barrett’s esophagus.9
RFA for Barrett’s esophagus was designed to inflict a thermal injury deep enough to destroy the metaplastic epithelium, but superficial enough to prevent submucosal damage that might result in esophageal stricture formation.10 Thus, the RFA injury might not reach SSIM buried deep in the lamina propria where it is shielded by the overlying stratified squamous epithelium. After this incomplete RFA, the residual metaplastic glands would be covered by neosquamous epithelium, remaining endoscopically undetectable unless they should grow to reach the luminal surface (an event that would be deemed a metaplasia “recurrence”), or they undergo malignant transformation (an event that has been documented).4 Despite the considerable potential importance of SSIM, little is known about its pathogenesis.
One plausible mechanism for the development of SSIM involves the migration of Barrett’s epithelial cells into lamina propria of the adjacent squamous epithelium. This process would require the epithelial cells to break their cell-cell connections, pass through the basement membrane, and move into the underlying stroma. Such events are key features of epithelial- mesenchymal transition (EMT), a process that plays a major role in normal embryonic development and wound healing, as well as in the progression of malignancy. During EMT, epithelial cells lose their typical epithelial features (e.g. cell-cell adhesion) by downregulating epithelial genes such as E-cadherin (CDH1), and acquire mesenchymal cell features (e.g. the ability to migrate) by upregulating mesenchymal genes such as fibronectin (FN1), vimentin, and matrix metalloproteases (MMPs).11, 12 Since EMT is a reversible process, Barrett’s cells acquiring mesenchymal cell migratory ability and invading subsquamous lamina propria could then undergo mesenchymal-epithelial transition (MET), reverting back into epithelial cells and thus forming SSIM.13
Non-neoplastic Barrett’s epithelial cells exposed chronically to acid and bile salts in vitro undergo malignant transformation and develop features of EMT,14 and rats with reflux esophagitis induced by esophago-jejunostomy demonstrate EMT of non-neoplastic columnar cells of the adjacent jejunum in the formation of a columnar-lined esophagus (CLE).15 In these models, however, it is not clear if EMT is induced directly by exposure to acid or bile salts or the result of subsequent malignant transformation, or if EMT is the result of the inflammation induced by reflux esophagitis. In experiments described below, we explored our hypothesis that refluxed acid and bile salts directly induce Barrett’s epithelial cells to undergo EMT that results in SSIM. We correlated our in vitro findings in non-neoplastic Barrett’s epithelial cell lines with in vivo data using a rat model of reflux esophagitis and esophagectomy specimens from patients with neoplasia in Barrett’s esophagus.
Materials and methods
Cell lines
We used two non-neoplastic, telomerase-immortalized Barrett’s epithelial cell lines (BAR-T, BAR-10T), and two non-neoplastic, telomerase-immortalized squamous epithelial cell lines from GERD patients with (NES-B10T) and without (NES-G2T) Barrett’s esophagus; all of these cell lines were developed in our laboratory. 16–19 High-grade dysplastic Barrett’s epithelial cell lines (CP-B, CP-C and CP-D) were kindly provided by Dr. Peter Rabinovitch, Fred Hutchinson Cancer Research Center (Seattle, WA). To establish the BEC-20W cell line, BAR-T cells were treated with acidic bile salt medium for 5 minutes a day for 20 weeks as previously described20, 21; short tandem repeat analysis confirmed that the BEC-20W cell line used in this study was indeed derived from BAR-T. We also used the telomerase-immortalized esophageal fibroblast cell line (BEF-T) which was derived from endoscopic biopsies of a patient with long-segment Barrett’s esophagus.22 Culture conditions for the individual cell lines have been previously described. 16, 22, 23 All cell lines were maintained at 37°C in a 5% CO 2 incubator. For individual experiments, BAR-T and BAR-10T cell lines were seeded equally onto collagen IV-coated wells (BD Biosciences, San Jose, CA) in the absence of fibroblast feeder layers, and were maintained in growth medium.
Three dimensional (3D) organotypic cell culture system and immunohistochemical (IHC) staining
Organotypic cultures were established based on the previously published method for human esophageal epithelial cells. 24 Briefly, acellular collagen matrices using bovine type I collagen (Corning, Bedford, MA) were created on the bottom of 24 mm transwell inserts (Sigma-Aldrich). To prepare the cellular collagen matrices, BEF-T esophageal fibroblasts were suspended in a collagen mixture at a density of 5 × 104 cells/ml. The fibroblast-collagen matrices were cultured for 7 days in fibroblast growth media. On day 2, the fibroblast-collagen matrices were released from the sides of the well to allow for matrix contraction. On day 7, Barrett’s epithelial cells BAR-T and BEC-20W were seeded on the surface of the fibroblast-collagen matrices at a density of 5 × 105 cells/well in epidermalization growth media as per protocol.24 On day 11, the organotypic cultures were raised to the air-liquid interface. On day 14, the cultures were harvested and fixed for 1 hour in 10% buffered formalin phosphate before being paraffin- embedded and sectioned for H&E and immunohistochemical (IHC) staining. Deparaffinized sections were pretreated with 10 mM sodium citrate buffer for antigen unmasking (pH 6.0, boiling temperature, 30 min), blocked in goat serum (Vectastain ABC kit, Vector Laboratories, Inc., Burlingame, CA), incubated with rabbit polyclonal anti-human CDH1 at 4o C overnight (Supplemental Table 1), rinsed, and incubated with anti-rabbit secondary antibody (Vectastain ABC kit). Signals were amplified using Vectastain ABC kit per manufacturer’s instructions. Targeted protein was visualized using diaminobenzidine as substrate.
Acidic bile salt exposure
For individual experiments, cells were cultured in neutral medium (pH 7.2) alone or with periodic exposures to acidic media (pH 5.5 or pH 4.0) containing a mixture of conjugated bile acids (glycocholic acid, glycodeoxycholic acid [both from Calbiochem, San Diego, CA], taurocholic acid, glycochenodeoxycholic acid, taurochenodeoxycholic acid, and taurodeoxycholic acid [Sigma-Aldrich, St. Louis, MO] in a 20:6:3:15:3:1 molar concentration, total concentration 400 μM) as previously described. 19, 25–27 Neutral or acidic bile salt medium was added for times ranging from 5–60 minutes, after which the medium was removed and replaced with neutral growth medium. For long term exposures, cell were treated with acidic bile salt medium (pH 4.0) for 5 minutes, once a day, for 3 days or 1 week.
Rat model of reflux esophagitis and columnar-lined esophagus (CLE)
All earlier animal studies were approved by the animal care committee of the Dallas VA Medical Center. We used previously collected tissues from the CLE of rats for whom reflux was induced by esophago-jejunostomy (EJ); none of these animals developed neoplasia during this study.15 CLE was identified histologically in the portion of esophagus proximal to the EJ anastomosis (identified by the embedded suture) by two gastrointestinal pathologists (AA, RDO). For qRT- PCR, total RNA was isolated using TRIzol (Invitrogen) followed by DNA-free™ DNase Treatment (Ambion, Austin, TX) from CLE at 6, 12, and 16 weeks after the operation (n=3 per time point). For IHC analyses of Cdh1, Zeb1 and Zeb2, formalin-fixed, paraffin-embedded CLE tissues sections were deparaffinized, and HIER was completed with either Bond Epitope Retrieval Solution 1 or Solution 2, followed by incubation with the primary antibody for 30 minutes (Supplemental Table 1) followed by the secondary antibody for 15 minutes. Targeted protein was visualized using diaminobenzidine as substrate.
Patient tissues
This patient study was approved by the Institutional Review Board of the Brigham and Women’s Hospital. Forty-eight cases of formalin-fixed, paraffin-embedded esophagectomy specimens with Barrett’s-associated esophageal adenocarcinoma (N=28) or high-grade dysplasia (N=30) were retrieved from the surgical pathology archives of Brigham and Women’s Hospital (dates of resection between 1994 and 2009); 10 cases had both Barrett’s-associated esophageal adenocarcinoma and high grade dysplasia. None of the cases received neoadjuvant chemotherapy. T stage ranged from Tis to T3, and N stage ranged from N0 to N2.
IHC of Esophagectomy Specimens
All immunohistochemistry was performed on the Leica (Buffalo Grove, IL) Bond III automated stainer, using their Polymer Refine Detection Kit (cat # DS9800). Formalin-fixed, paraffin- embedded esophagectomy specimens were deparaffinized, and heat-induced epitope retrieval (HIER) was completed with either Bond Epitope Retrieval Solution 1 (H1, citrate based, pH 6.0, cat # AR9961) or Bond Epitope Retrieval Solution 2 (H2, EDTA based, pH 9.0, cat # AR9640). This was followed by incubation with the primary antibody for 30 minutes (Supplemental Table 1) followed by the secondary antibody for 15 minutes. Targeted protein was visualized using diaminobenzidine as substrate. Staining intensity of epithelial cells was quantitated using an H- score technique. 28, 29 In brief, staining intensity for ZEB1, CDH1, and FN1 was scored on a 0–3 scale (0=no staining, 1=faint staining, 2=moderate staining, 3=strong staining), and the percentage of cells at each staining intensity was determined. The H-score was calculated using the formula [3 x (% cells with strong staining) + 2 x (% cells with moderate staining) + 1 x (% cells with weak staining)]; possible score range 0–300. For areas of invasive carcinoma, staining was assessed along the invasive front (the leading 1 mm front of invasion) and compared to remaining tissue (i.e. the non-invasive front). Staining also was assessed in areas of high grade dysplasia, and non-dysplastic Barrett’s metaplasia. ZEB1 staining was assessed in the nuclear compartment, FN1 was assessed in the cytoplasmic and membranous areas while CDH1 was assessed in membranous areas only. Tissues were evaluated and scored by two gastrointestinal pathologists (AA, RDO).
Data analyses
All Western blots were repeated in at least two independent experiments. Quantitative data are expressed as the mean ± standard error of the mean (SEM). Statistical analyses were performed using an unpaired Student’s t-test with the Instat for Windows statistical software package (GraphPad Software, San Diego, CA). For quantitative H score data, statistical analyses were performed using a paired Student’s t-test using the GraphPad Prism 7.0 statistical software package (GraphPad Software, San Diego, CA). For multiple comparisons, an ANOVA and the Student-Newman-Keuls multiple-comparisons test was performed using the Instat for Windows statistical software package (GraphPad). P values ≤ 0.05 were considered significant for all analyses.
Results
Dysplastic Barrett’s cells and BEC-20W cells demonstrate features of EMT
We assessed features of EMT in dysplastic Barrett’s cells (CP-B, CP-C, and CP-D) and BEC- 20W cells (BAR-T cells treated with acidic bile salt medium 5 minutes each day for 20 weeks); non-neoplastic BAR-T and BAR-10T served as controls. Compared with BAR-T and BAR-10T cells, dysplastic Barrett’s cells and BEC-20W cells showed decreased expression of CDH1, and increased expression of FN1 and vimentin by IF (Figure 1A); these differences were confirmed by qRT-PCR or Western blot (Supplemental Figure 1A-C). By optic morphology, dysplastic Barrett’s cells and BEC-20W cells appeared spindle-shaped (Figure 1B&C), and showed significantly higher migration rates in transwell assays (without and with a basement membrane- like matrix) than non-dysplastic cells (Figure 1B&C, respectively). Since the BEC-20W cells could degrade a basement membrane-like matrix in order to pass through the pores, we performed Western blot for MMP-2, a matrix metalloprotease (MMP) involved in the breakdown of extracellular matrix. Compared to BAR-T and BAR-10T cells, BEC-20W cells and dysplastic cells exhibited increased expression of MMP-2 (Supplemental Figure 1B&C). To evaluate cell migration in a more tissue-like context, we established three dimensional (3D) organotypic cultures of BAR-T and BEC-20W cells; only the BEC-20W cells migrated deep into the underlying stromal equivalent (Figure 1D).
Figure 1.
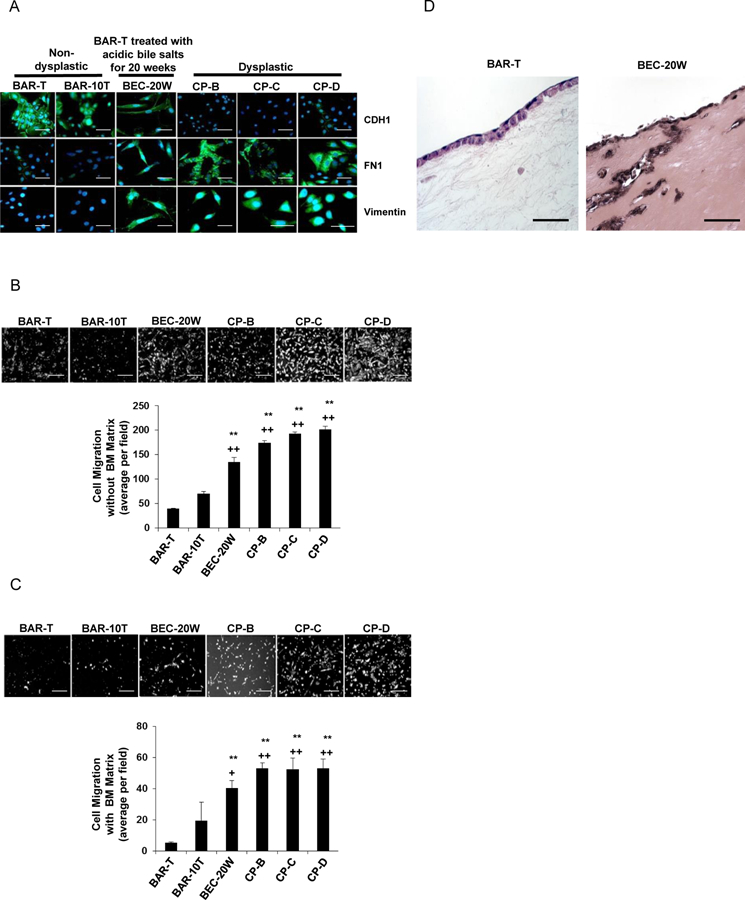
Non-dysplastic Barrett’s cells treated with 20 weeks of acidic bile salt exposure (BEC- 20W) demonstrate features of EMT. (A) Immunofluorescent staining for CDH1, FN1, and vimentin. Cell migration without (B) and with (C) a basement membrane (BM) matrix. (D) Cell migration in 3D organotypic culture. Scale bars = 20 µm. For cell migration assays in B &C, bar graphs depict the mean ± SEM of three independent experiments. **p<0.01 vs. BAR-T; ++p<0.01 vs. BAR-10T.
Acidic bile salts induce EMT features in Barrett’s epithelial cells, but not in esophageal squamous cells
Next, we treated non-neoplastic Barrett’s cells with a single exposure to acidic bile salts for durations ranging from 5–30 minutes. We found that these single exposures decreased CDH1 and increased FN1 and MMP-2 protein expression, and increased vimentin mRNA expression in BAR-T (Figure 2 A&B) and BAR-10T cells (Supplemental Figure 2 A&B). Treatment with a 5- minute acidic bile salt exposure once daily for 3 or 7 days significantly increased BAR-T and BAR-10T migration rates in transwell assays (without and with a basement membrane-like matrix) (Figure 2 C&D for BAR-T and Supplemental Figure 2 C&D for BAR-10T). Treatment of organotypic cultures with acidic bile salts for 7 days resulted in decreased membranous staining for CDH1 in BAR-T cells, which were seen migrating deep into the underlying stromal equivalent (Figure 2E). In contrast, acidic bile salt treatment had no apparent effect on CDH1 levels or cell migration in human esophageal squamous cells (NES-B10T and NES-G4T) (Figure 2F). These data show that acidic bile salts induce EMT features in non-neoplastic Barrett’s epithelial cells, but not in esophageal squamous cells.
Figure 2.
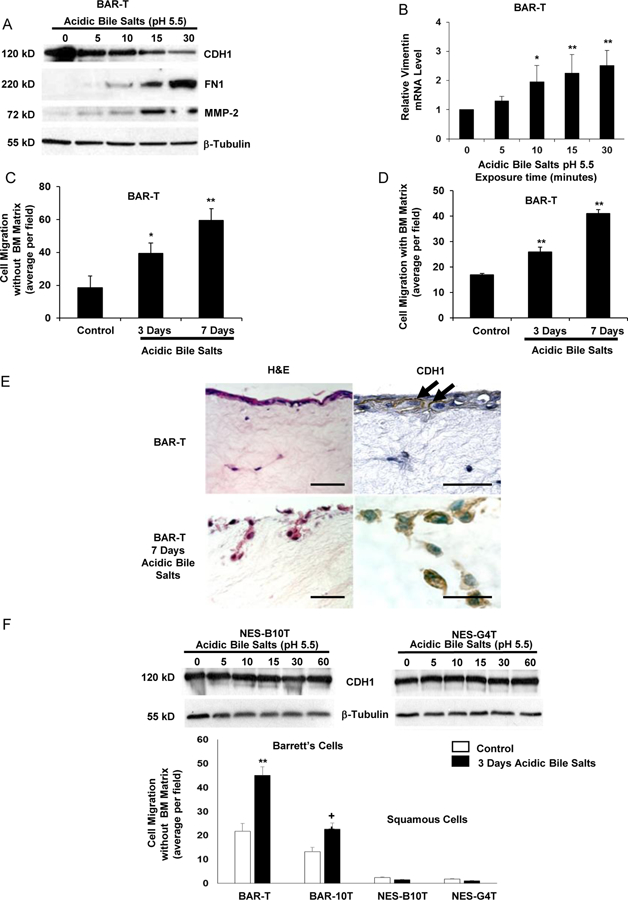
Acidic bile salts induce features of EMT in non-neoplastic Barrett’s (BAR-T) cells, but not in non-neoplastic esophageal squamous (NES) cells. (A) Western blot for CDH1, FN1, and MMP-2; β-tubulin served as a loading control. (B) Representative qPCR for vimentin mRNA level after acidic bile salt treatment relative to untreated (0 minutes) cells; qPCR assays were performed in triplicate in at least two independent experiments. Cell migration after 3 and 7 days of acidic bile salt exposure without (C) and with (D) a basement membrane (BM) matrix. (E) Cell migration and CDH1 immunostaining of BAR-T cells grown in 3D organotypic culture after 7 days of acidic bile salts. Arrows depict membranous CDH1 staining. Scale bars = 20 µm. (F) Western blot for CDH1 and cell migration in NES cells. Bar graphs depict the mean ± SEM, *p<0.05 and **p<0.01 compared with 0 minutes or control; +p<0.05 compared with BAR-10T control. For cell migration assays in C, D, & F, bar graphs depict the mean ± SEM of three independent experiments.
Zinc finger E-box-binding (ZEB) proteins regulate CDH1 expression and cell migration in non-neoplastic Barrett’s epithelial cells
The master regulators that drive EMT by repressing epithelial genes and activating mesenchymal genes include the zinc finger family of transcription factors (ZEB1, ZEB2, SNAIL, SLUG) and the basic helix-loop-helix transcription factor twist related protein 1 (TWIST).11 By Western blot, dysplastic Barrett’s cells and BEC-20W cells showed markedly greater nuclear levels of ZEB1, ZEB2, SNAIL, and SLUG than BAR-T and BAR-10T cells; increased expression of TWIST in the dysplastic cells was not observed consistently (Supplemental Figure 3A). To determine which transcription factors mediate loss of CDH1 expression in BAR-T cells, we knocked down or overexpressed ZEB1, ZEB2, SNAIL, and SLUG using siRNAs or cDNAs, respectively. Efficiency of the siRNAs and cDNAs was determined by Western blotting (Supplemental Figure 3B&C). We found that knockdown of ZEB1 and ZEB2 markedly increased CDH1 protein, whereas overexpression of ZEB1 and ZEB2 markedly decreased CDH1 protein in BAR-T cells (Figure 3A). Furthermore, knockdown of ZEB1 or ZEB2 reduced cell migration (Figure 3B), whereas stable overexpression of ZEB1 or ZEB2 (Supplemental Figure 3D) increased cell migration (Figure 3B). We found that a single exposure to acidic bile salts increased nuclear ZEB1 and ZEB2 proteins and reduced CDH1 mRNA expression in BAR-T cells (Figures 3C&D). Knockdown of ZEB1 or ZEB2 eliminated the acidic bile salt-induced reduction in CDH1 mRNA in BAR-T cells (Figure 3E).
Figure 3.
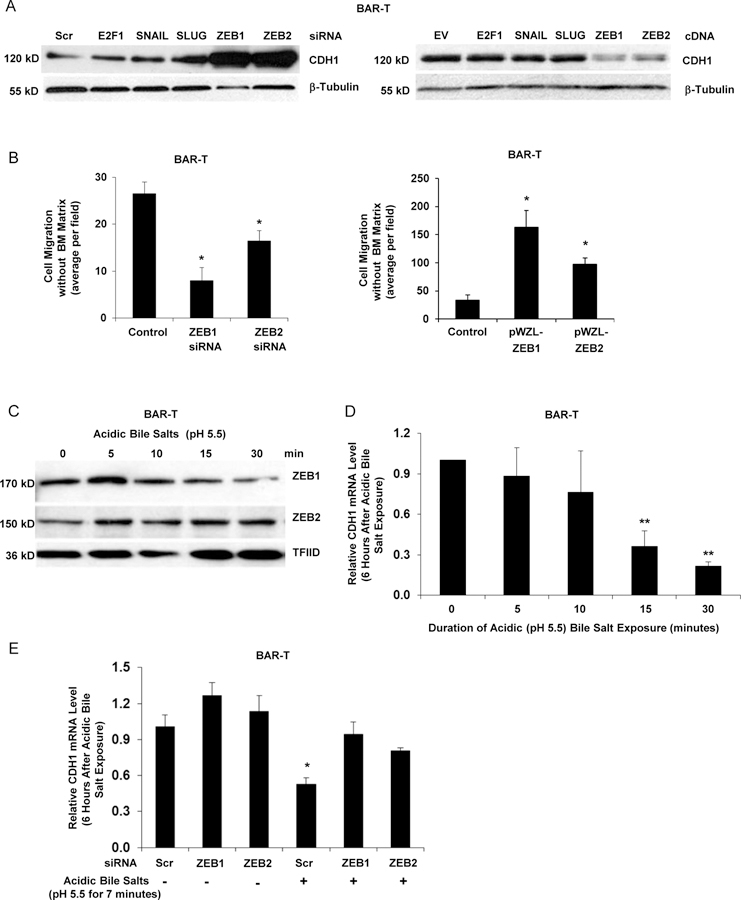
ZEB1 and ZEB2 regulate features of EMT in response to acidic bile salt exposure in non-neoplastic Barrett’s cells. (A) Western blot for CDH1 at 72 hours following siRNA knockdown or cDNA expression of SNAIL, SLUG, ZEB1 or ZEB2; β-tubulin served as a loading control. Scrambled (Scr) siRNA, empty vector (EV) and non-related E2F1 served as controls. (B) Cell migration with knockdown (left panel) or stable overexpression (right panel) of ZEB1 or ZEB2. (C) Western blot for nuclear levels of ZEB1 and ZEB2 immediately following exposure to acidic bile salts for times ranging from 5–30 minutes; TFIID served as a loading control. Representative qPCR for CDH1 mRNA level 6 hours after exposure to acidic bile salts for times ranging from 5–30 minutes relative to untreated (0 minutes) cells without (D) and with (E) knockdown of ZEB1 or ZEB2; qPCR assays were performed in triplicate in at least two independent experiments. Bar graphs depict the mean ± SEM. *p<0.05 and **p<0.01 compared with control, 0 minutes, or non-acidic bile salt treated Scr siRNA. In B &E, cells were transfected with siRNAs for 72 hours. For cell migration assays in B, bar graphs depict the mean ± SEM of three independent experiments.
Increased ZEB1 and ZEB2 and decreased CDH1 mRNA levels are found in the non- neoplastic, columnar-lined esophagus that developed in rats with surgically-induced reflux esophagitis
Having found that acidic bile salts increase ZEB1 and ZEB2 and decrease CDH1 in Barrett’s epithelial cell lines, we sought to confirm that these events occur in vivo using CLE that develops in rats after reflux esophagitis is induced by EJ (Figure 4A).15 CLE at 12 and 16 weeks after EJ exhibited significantly lower Cdh1 and significantly higher Zeb1 mRNA levels than CLE at 6 weeks (Figure 4B). As previously reported, we found that CLE developed in 36 of 52 (69%) rats after EJ. Eleven of the 36 rats with CLE had columnar cells identified in the lamina propria underneath esophageal squamous epithelium (Figure 5A). In 8 of those 11 rats, CLE demonstrated decreased Cdh1 expression focally (2 cases), increased Zeb1 (1 case), Zeb2 (4 cases), or both (1 case) (Figure 5B). These findings demonstrate that rats with surgically-induced reflux develop CLE and “buried” glands underneath squamous epithelium, both with evidence of EMT.
Figure 4.
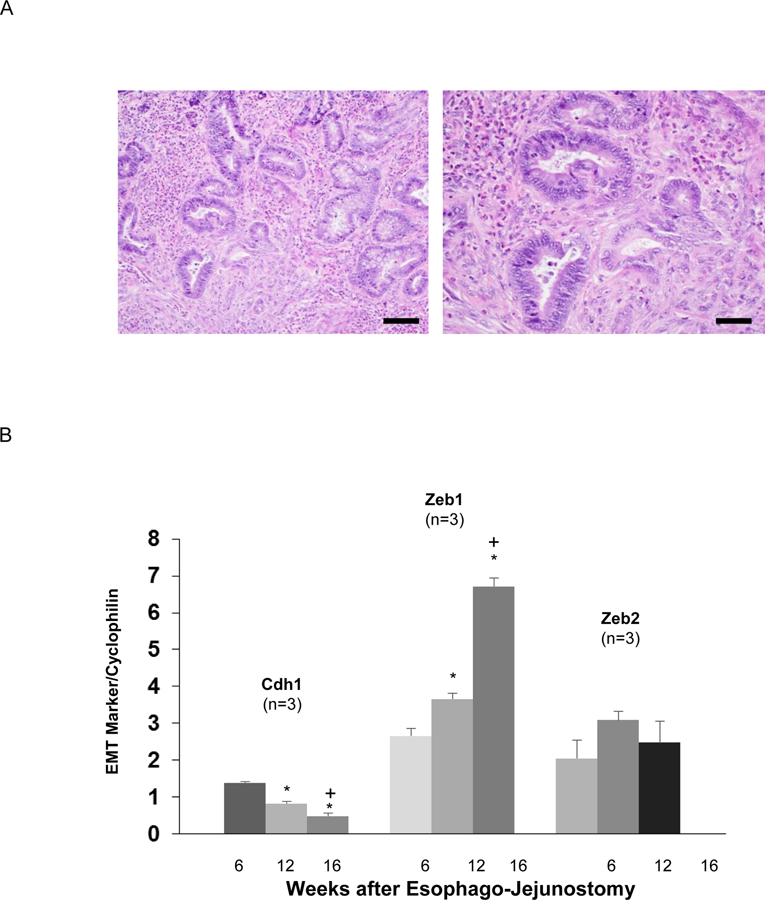
Columnar-lined esophagus (CLE) that develops in rats with surgically-induced reflux esophagitis demonstrates markers of EMT. (A) H&E staining of neoglandular epithelium at medium power (Scale bar = 100 µM) in the left panel and high power (Scale bar = 50 µM) in the right panel (B) Representative qPCR for Cdh1, Zeb1, and Zeb2 mRNA in CLE at week 6, 12, and 16 post-operatively. For each mRNA, relative levels were normalized to cyclophilin. qPCR assays were performed in triplicate in 3 individual animals. Bar graphs depict the mean ± SEM. *p<0.05 compared with week 6; +p<0.05 compared with week 12.
Figure 5.
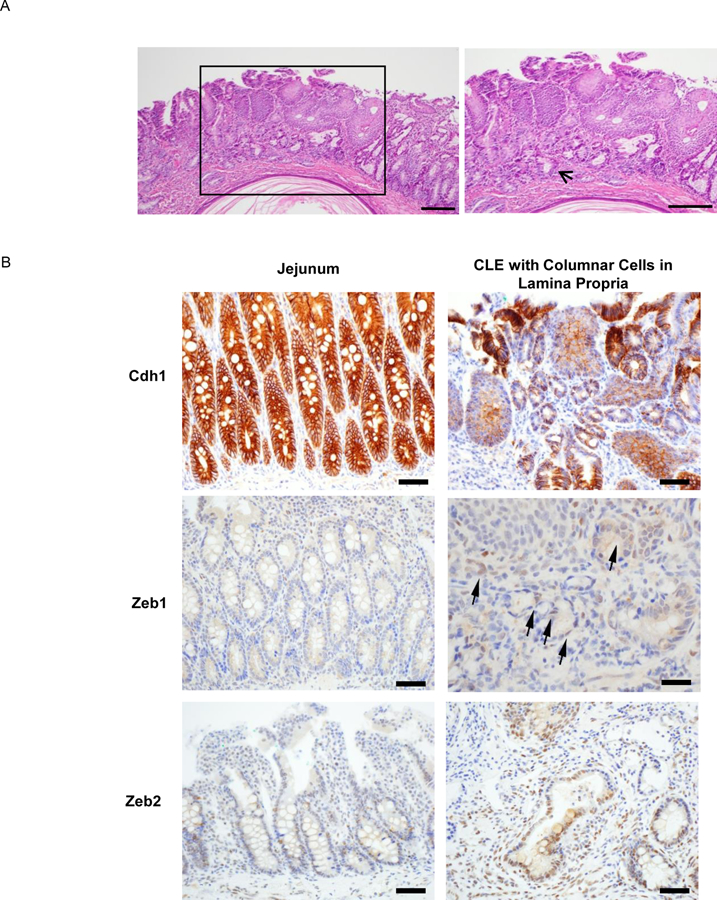
In a rat model of reflux esophagitis, CLE with columnar cells in the lamina propria underneath esophageal squamous epithelium demonstrates markers of EMT. (A) H&E staining of CLE (bracketed region) in the distal esophagus. Right panel showed the enlarged bracketed region, indicating extensive subsquamous glands (arrow; scale bar = 200 µM). (B) Immunostaining for Cdh1, Zeb1, and Zeb2 in native jejunal epithelium (left side of each pair) and CLE with columnar cells in the lamina propria (right side of each pair). Arrows indicate columnar cells in the lamina propria with Zeb1 expression. All scale bars = 50 µM except for Zeb1 whose scale bar = 25 µM.
Epithelial cells in high-grade dysplasia in human Barrett’s esophagus demonstrate increased ZEB1 and decreased CDH1 expression
To determine whether there is an association between ZEB and EMT-related proteins in primary non-neoplastic Barrett’s and Barrett’s-associated neoplastic tissues, we performed immunohistochemistry for ZEB1, CDH1, and FN1 on esophagectomy specimens from patients with neoplastic Barrett’s esophagus. In 28 specimens with adenocarcinomas, we focused on the invasive and non-invasive tumor fronts. Compared to the non-invasive front, epithelial cells at the invasive front of esophageal adenocarcinomas demonstrated increased immunostaining for ZEB1 and FN1; no apparent loss of membranous immunostaining for CDH1 was observed (Figure 6A). We also assessed staining for ZEB1, CDH1, and FN1 in 30 esophagectomy specimens that had areas of both high-grade dysplasia and non-dysplastic Barrett’s metaplasia. Compared to the non-neoplastic epithelial cells, those from areas of high-grade dysplasia demonstrated significantly increased immunostaining for ZEB1 and significantly decreased membranous immunostaining for CDH1 (Figure 6B). FN1 staining was detected in only one case of high-grade dysplasia, and no FN1 staining was found in non-dysplastic Barrett’s metaplasia (data not shown).
Figure 6.
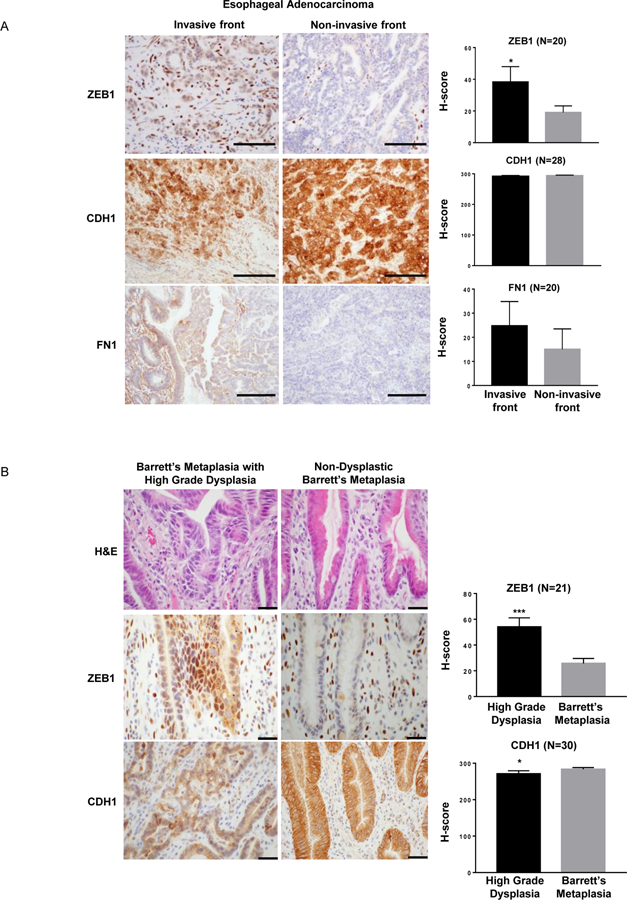
Staining for ZEB1, CDH1 and FN1 in (A) the invasive and non-invasive fronts of esophageal adenocarcinomas (Scale bar = 200 μm) and (B) high-grade dysplastic and non- dysplastic Barrett’s metaplasia (Scale bar = 1000 μm). In non-dysplastic Barrett’s metaplasia, ZEB1 is expressed primarily by mesenchymal cells in the stroma and not by epithelial cells. Bar graphs depict the mean ± SEM. *p≤0.05; ***p<0.0001.
Acidic bile salts increase VEGF/VEGFR2 signaling to induce EMT features in non- neoplastic Barrett’s cells
Having found that ZEBs regulate CDH1 expression in Barrett’s cells in vitro, with supporting evidence in human tissues, we investigated upstream mechanisms whereby acidic bile salts might increase ZEB expression. Noting that growth factors acting through transmembrane receptor tyrosine kinases (RTKs) often stimulate EMT,11 we compared RTK protein phosphorylation levels in BAR-T, BEC-20W, and CP-D cells using a phospho-RTK array panel of 49 RTKs. Compared with BAR-T, BEC-20W and CP-D cells showed a marked increase in phosphorylation of VEGFR2; this was confirmed by Western blotting (Supplemental Figure 4 A&B). To link VEGFR2 signaling with EMT in non-neoplastic Barrett’s cells, we stably overexpressed VEGFR2 in BAR-T cells. The VEGFR2-overexpressing BAR-T cells exhibited an increase in phospho-VEGFR2, an increase in ZEB1 and ZEB2, a decrease in CDH1, and an increase in cell migration (Figure 7 A&B). Knockdown of VEGFR2 with a specific shRNA (Supplemental Figure 4C) decreased ZEB1 and ZEB2 mRNA expression (Supplemental Figure 4D), and significantly reduced cell migration in BAR-T cells (Supplemental Figure 4E). We next explored whether VEGFR2 signaling was involved in acidic bile salt-induced EMT in Barrett’s epithelial cells. In BAR-T cells, acidic bile salt exposure increased levels of phospho-VEGFR2 and its downstream pathway proteins phospho-PLCγ1 and phospho-PKCα/β (Figure 7C). Following a single 15- or 30-minute exposure to acidic bile salts, we found a rapid increase in VEGF-A secretion, which remained elevated 24 hours later (Supplemental Figure 5A). VEGF-A mRNA expression also increased at 6 and 24 hours after exposure to acidic bile salts (Supplemental Figure 5B). Treatment with a VEGF-A neutralizing antibody (VEGF-NA) prevented the acidic bile salt-induced increase in VEGFR2 phosphorylation (Supplemental Figure 5C), whereas treatment with a VEGFR2-specific inhibitor (SU1498) prevented the acidic bile salt-induced increases in ZEB1 and ZEB2 expression and the decrease in CDH1 protein (Supplemental Figure 5D). VEGFR2 inhibition with a specific shRNA (Figure 7D) prevented the acidic bile salt-induced increases in ZEB1 and ZEB2 expression and the decrease in CDH1 protein. Genetic deletion of VEGF-A using CRISPR-Cas9n technology (Supplemental Figure 6A) significantly decreased the acidic bile salt-induced increase in VEGF-A secretion (Supplemental Figure 6B), reduced VEGFR2 phosphorylation (Figure 7E), and significantly decreased the acidic bile salt-induced increase in cell migration of BAR-T cells (Figure 7F and Supplemental Figure 6C). A schematic model summarizing our findings is provided in Figure 7G.
Figure 7.
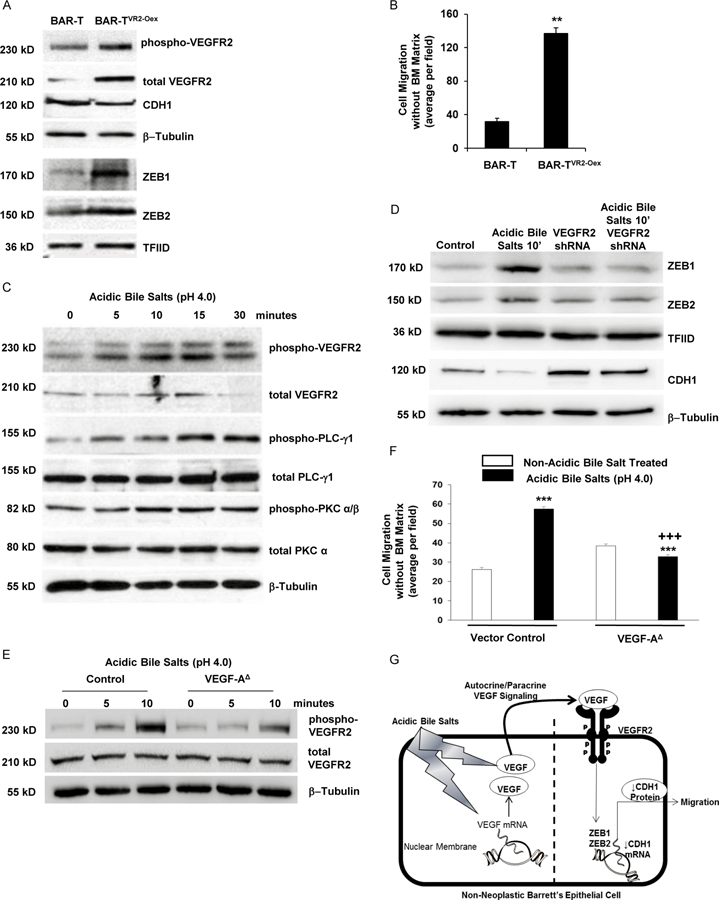
Acidic bile salts-induce VEGF/VEGFR2 signaling that mediates EMT features in non- neoplastic Barrett’s cells. (A) Western blot for phopho-VEGFR2, CDH1, ZEB1, and ZEB2 in BAR-T cells with and without VEGFR2 overexpression (VR2-Oex) and (B) cell migration in BAR-T cells with and without VEGFR2 overexpression. Bar graphs depict the mean ± SEM of three independent experiments. **p<0.01 compared with control. (C) Western blot for phopho- VEGFR2 and its downstream pathway proteins phospho-PLC-γ1 and –PKC α/β after exposure to acidic bile salts. (D) Western blot of ZEB1, ZEB2 and CDH1 with and without VEGFR2 knockdown by shRNA following acidic bile salt exposure; empty vector-containing cells served as control. β-tublin and TFIID served as loading controls for whole cell and nuclear lysates, respectively. (E) Western blots for phospho-VEGFR2 with and without VEGF-A deletion (VEGF- A∆) following acidic bile salt exposure; empty vector-containing cells served as control. (F) Cell migration in BAR-T cells with VEGF-A∆ following acidic bile salt exposure. Bar graphs depict the mean ± SEM of three independent experiments. ***p<0.001 compared with corresponding control; +++p<0.001 compared with acidic bile salt treated vector control. (G) Schematic model demonstrating the EMT-inducing effects of acidic bile salts in non-neoplastic Barrett’s cells. Exposure to acidic bile salts causes a rapid increase in VEGF secretion along with a delayed increase in VEGF mRNA expression. The secreted VEGF binds in an autocrine/paracrine fashion to VEGFR2, resulting in increased expression of ZEB1 and ZEB2, which decrease the expression of CDH1 and increase cell migration.
Discussion
Our studies have demonstrated EMT features (decreased CDH1 expression, increased FN1 and vimentin expression, spindled-shaped cell morphology, and migratory ability) in neoplastic Barrett’s cells, as well as in non-neoplastic Barrett’s cells treated with acidic bile salts for 20 weeks. Non-neoplastic Barrett’s cells exposed only briefly to acidic bile salts (≤30 minutes) also develop EMT markers and, when these cells are exposed to acidic bile salts for 5 minutes once daily for 7 days, they exhibit increased migration in transwell and 3D organotypic culture systems. We have found that acidic bile salts induce ZEB transcription factors that reduce CDH1 expression in Barrett’s cells and, in rats that develop CLE as a result of surgically- induced reflux esophagitis, we have found increased Zebs and decreased Cdh1 in their CLE as well as in columnar cells in the lamina propria underneath adjacent esophageal squamous epithelium. We have demonstrated robust staining for ZEB1 with decreased staining for CDH1 in epithelial cells in high-grade dysplastic Barrett’s tissues, further supporting a link between ZEBs and EMT in human Barrett’s esophagus. Finally, in non-neoplastic Barrett’s cells, we have found that acidic bile salts activate VEGF/VEGFR2 signaling to induce ZEB upregulation with reciprocal CDH1 downregulation, and that inhibition of the VEGF/VEGFR2 axis abolishes these acidic bile salt effects on gene expression. These observations establish that acidic bile salts can directly induce non-neoplastic Barrett’s epithelial cells to undergo EMT, and support our hypothesis that EMT leads to the development of SSIM in patients with Barrett’s esophagus.
It is well established that EMT occurs as part of normal wound healing processes triggered by inflammatory cytokines generated during tissue injury and inflammation,30 and that neoplastic cells can usurp features of this normal EMT molecular program to enable cancer progression and metastasis.31 However, relatively little has been published on the role of EMT in non-neoplastic metaplasias. In a mouse model of pancreatic cancer that used a Cre-lox based system to generate pancreas-specific mutations in Kras and p53 genes, Rhim et al. found markers of EMT in areas of acinar-to-ductal metaplasia.32 Before development of malignancy, pre-neoplastic pancreatic cells with EMT markers were noted to break through the basement membrane and migrate into the stroma, especially in areas of pancreatic inflammation.32 Our studies show that inflammation and neoplasia are not required to trigger EMT in metaplastic Barrett’s epithelial cells.
As expected, our neoplastic Barrett’s cells did exhibit EMT features spontaneously (i.e. without exposure to acidic bile salts). Our BEC-20W cells (non-neoplastic Barrett’s cells treated with acidic bile salts for 20 weeks) also showed evidence of EMT, and earlier studies showing that BEC-20W cells are not transformed suggest that neoplasia did not contribute to their EMT features.20 Furthermore, inflammation clearly did not underlie the EMT of BEC-20W cells in cultures that are devoid of inflammatory cells. Nevertheless, we cannot exclude the possibility that EMT in BEC-20W cells is caused by their protracted stressful culture conditions rather than direct effects of acid and bile salts. However, our observation that non-neoplastic Barrett’s cells exposed very briefly to acidic bile salts develop an EMT gene expression pattern and the ability to migrate in stromal equivalent culture systems (transwell and 3D organotypic) is strong evidence that acidic bile salts can trigger EMT directly in Barrett’s metaplasia, in the absence of neoplasia and inflammation.
Loss of the adherens junction protein CDH1, which plays a key role in binding epithelial cells to one another, is a fundamental EMT event that is mediated by repressors of CDH1 transcription including ZEB1, ZEB2, SNAIL, SLUG, and TWIST.33, 34 Through a series of knockdown and overexpression experiments, we found that only ZEB1 and ZEB2 were linked with CDH1 expression and cell migration in Barrett’s epithelial cells. Smith et al. used qPCR to show that, compared to non-neoplastic Barrett’s metaplasia, esophageal adenocarcinomas exhibit significantly greater expression of both ZEB1 and ZEB2 mRNA, with the greatest increase noted for ZEB1.34 Robust ZEB1 protein expression has been found in cells at the invasive front of human tumor tissues (e.g. esophageal squamous cell, breast, colorectal and pancreatic carcinomas), with less or no ZEB1 detected in areas of well-differentiated tumor cells.35 Using human Barrett’s-associated neoplastic tissues, we found increased immunostaining for nuclear ZEB1 in epithelial cells at the invasive front of esophageal adenocarcinomas but, for reasons that are not clear, we did not find decreased immunostaining for membranous CDH1 in those cells. In contrast, cells from areas of high-grade dysplasia in Barrett’s esophagus demonstrated both significantly increased immunostaining for nuclear ZEB1 and significantly decreased immunostaining for membrane CDH1 (compared to non- neoplastic Barrett’s epithelial cells). The finding that our Barrett’s cell lines recapitulate the inverse association between ZEB and CDH1 proteins observed in non-cancerous patient tissue specimens supports the validity of these cell lines as models for the study of acidic bile salt- induced regulation of EMT in Barrett’s esophagus.
In esophageal squamous cell carcinoma, a receptor tyrosine kinase (RTK) has been found to contribute to EMT by upregulating ZEB transcription factors.36 We used a phospho- RTK array that included 49 RTKs to compare our Barrett’s cell lines, and found that our BEC- 20W and dysplastic Barrett’s cells exhibited far greater phosphorylation of VEGFR2 RTK than our non-neoplastic BAR-T cells. We found this especially interesting because, in earlier studies, we had shown that benign and malignant Barrett’s cells secrete VEGF-A that initiates proliferative signaling via autocrine/paracrine activation of VEGFR2.37 When we treated our non- neoplastic Barrett’s cells with acidic bile salts, we found a rapid increase in VEGF-A secretion, and a later increase in VEGF-A mRNA expression. The increase in VEGF-A was accompanied by increased VEGFR2 phosphorylation with activation of downstream signaling pathways leading to upregulation of ZEBs, downregulation of CDH1, and increased cell migration. Inhibition of VEGF/VEGFR2 signaling (by VEGF blocking antibody, VEGF-A genetic deletion, pharmacologic VEGFR2 inhibition, or VEGFR2 shRNA) eliminated these EMT features. These findings show that acidic bile salts induce EMT features in non-neoplastic Barrett’s cells by activating VEGF/VEGFR2 signaling.
Our findings support our hypothesis that refluxed acid and bile salts directly induce Barrett’s epithelial cells to undergo EMT that results in SSIM, a potential cause of Barrett’s cancers that develop despite endoscopic surveillance, and of recurrences of Barrett’s metaplasia after apparent complete eradication by RFA. Cotton et al. reported that the large majority of recurrent intestinal metaplasia after RFA develops at or within 1 cm proximal to the top of the gastric folds.38 Such recurrences could well be due to residual foci of intestinal metaplasia, located at the distal margin of the ablated esophageal segment, that are stimulated to undergo EMT by ongoing acid and bile reflux and by the acute inflammation that accompanies RFA. Recurrences resulting from overlying squamous epithelium “shielding” SSIM from RFA destruction might be expected to occur most frequently at the proximal margin of the ablation segment (the pre-ablation squamo-columnar junction), which appears to be a less common site for recurrence. However, Barrett’s mucosa typically contains multiple “squamous islands” throughout its length and, consequently, has multiple squamo-columnar junctions where EMT could form SSIM. Although residual, shielded SSIM might account for some metaplasia recurrences after RFA, the finding that such recurrences usually occur close to the new squamo-columnar junction suggests that EMT of residual intestinal metaplasia at the distal margin of the ablation segment might be the primary cause.
In conclusion, we have demonstrated that acid and bile salts can directly induce non- neoplastic Barrett’s cells to reduce epithelial gene expression, increase mesenchymal gene expression, and migrate through a basement membrane-like matrix into an underlying stromal equivalent. Moreover, acidic bile salt-induced EMT is a unique property of Barrett’s epithelial cells, as similar treatment conditions did not induce EMT in esophageal squamous cells. We have shown that acidic bile salts cause the secretion and production of VEGF-A, which activates VEGFR2 signaling, leading to increases in the transcriptional repressors ZEB1 and ZEB2 that decrease the expression of CDH1. Thus, acid and bile salts trigger molecular events that induce the conversion of epithelial cells into motile cells by EMT, a process that may underlie the pathogenesis of SSIM. These observations provide a rationale for research on interventions that might reduce EMT by Barrett’s cells as a way to prevent recurrent Barrett’s metaplasia after RFA.
Supplementary Material
Acknowledgments:
We thank Raphael Bueno, MD, Chief, Division of Thoracic Surgery, Brigham and Women’s Hospital for his contribution to the creation of the esophageal adenocarcinoma tissue microarray; Elizabeth Cook, H.T. (ASCP), Histopathology Technician at the Center for Esophageal Research, Baylor University Medical Center and Baylor Scott and White Research Institute for assistance in preparation of tissue samples; Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core and core technologists Ashley Pelton and Benjamin Ferland, who provided immunohistochemical staining service. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NIH 5 P30 CA06516.
Grant Support: This work was supported by the National Institutes of Health (R01-DK63621, R01-DK103598, R21-DK11139 to R.F.S. and S.J.S.) and Baylor Scott and White Research Institute.
Abbreviations
- CDH1
E-Cadherin
- CLE
columnar lined esophagus
- EJ
esophagojejunostomy
- ELISA
enzyme-linked immunosorbent assay
- EMT
epithelial mesenchymal transition
- FN1
Fibronectin 1
- GERD
gastroesophageal reflux
- IF
immunofluorescence
- IHC
immunohistochemistry
- MET
mesenchymal epithelial transition
- MMP
matrix metalloproteinase
- PLC
protein lipase C
- PKC
protein kinase C
- qPCR
quantitative polymerase chain reaction
- RTK
receptor tyrosine kinase
- RFA
radiofrequency ablation
- SEM
standard error of the mean
- SSIM
subsquamous intestinal metaplasia
- VEGF
vascular endothelial growth factor
- VEGF-NA
vascular endothelial growth factor neutralization antibody
- VEGFR
vascular endothelial growth factor receptor
- ZEB
Zinc Finger E-Box-Binding
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: No disclosures exist.
VA/US Government Disclaimer: The contents do not represent the views of the U.S. Department of Veterans Affairs or the United State Government.
Author names in bold designate shared co-first authorship
References
- 1.Shaheen NJ, Sharma P, Overholt BF, et al. Radiofrequency ablation in Barrett’s esophagus with dysplasia. N Engl J Med 2009;360:2277–88. [DOI] [PubMed] [Google Scholar]
- 2.Small AJ, Sutherland SE, Hightower JS, et al. Comparative risk of recurrence of dysplasia and carcinoma after endoluminal eradication therapy of high-grade dysplasia versus intramucosal carcinoma in Barrett’s esophagus. Gastrointest Endosc 2015;81:1158–66.e1–4. [DOI] [PubMed] [Google Scholar]
- 3.Guthikonda A, Cotton CC, Madanick RD, et al. Clinical Outcomes Following Recurrence of Intestinal Metaplasia After Successful Treatment of Barrett’s Esophagus With Radiofrequency Ablation. Am J Gastroenterol 2017;112:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Titi M, Overhiser A, Ulusarac O, et al. Development of subsquamous high-grade dysplasia and adenocarcinoma after successful radiofrequency ablation of Barrett’s esophagus. Gastroenterology 2012;143:564–6.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray NA, Odze RD, Spechler SJ. Buried metaplasia after endoscopic ablation of Barrett’s esophagus: a systematic review. Am J Gastroenterol 2011;106:1899–908; quiz 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou C, Tsai TH, Lee HC, et al. Characterization of buried glands before and after radiofrequency ablation by using 3-dimensional optical coherence tomography (with videos). Gastrointest Endosc 2012;76:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anders M, Lucks Y, El-Masry MA, et al. Subsquamous extension of intestinal metaplasia is detected in 98% of cases of neoplastic Barrett’s esophagus. Clin Gastroenterol Hepatol 2014;12:405–10. [DOI] [PubMed] [Google Scholar]
- 8.Bartel MJ, Srivastava A, Gordon S, et al. Subsquamous intestinal metaplasia is common in treatment-naive Barrett’s esophagus. Gastrointest Endosc 2018;87:67–74. [DOI] [PubMed] [Google Scholar]
- 9.Corley DA, Mehtani K, Quesenberry C, et al. Impact of endoscopic surveillance on mortality from Barrett’s esophagus-associated esophageal adenocarcinomas. Gastroenterology 2013;145:312–9.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganz RA, Utley DS, Stern RA, et al. Complete ablation of esophageal epithelium with a balloon-based bipolar electrode: a phased evaluation in the porcine and in the human esophagus. Gastrointest Endosc 2004;60:1002–10. [DOI] [PubMed] [Google Scholar]
- 11.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 2009;119:1429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai M, Liu J, Geng X, et al. Repeated exposure to acid and bile selectively induces colonic phenotype expression in a heterogeneous Barrett’s epithelial cell line. Lab Invest 2008;88:643–51. [DOI] [PubMed] [Google Scholar]
- 15.Agoston AT, Pham TH, Odze RD, et al. Columnar-Lined Esophagus Develops via Wound Repair in a Surgical Model of Reflux Esophagitis. Cell Mol Gastroenterol Hepatol 2018;6:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaiswal KR, Morales CP, Feagins LA, et al. Characterization of telomerase-immortalized, non-neoplastic, human Barrett’s cell line (BAR-T). Dis Esophagus 2007;20:256–64. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Yu C, Wilson K, et al. Malignant transformation of non-neoplastic Barrett’s epithelial cells through well-defined genetic manipulations. PLoS One 2010;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang HY, Zhang X, Chen X, et al. Differences in activity and phosphorylation of MAPK enzymes in esophageal squamous cells of GERD patients with and without Barrett’s esophagus. Am J Physiol Gastrointest Liver Physiol 2008;295:G470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huo X, Zhang HY, Zhang XI, et al. Acid and bile salt-induced CDX2 expression differs in esophageal squamous cells from patients with and without Barrett’s esophagus. Gastroenterology 2010;139:194–203.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das KM, Kong Y, Bajpai M, et al. Transformation of benign Barrett’s epithelium by repeated acid and bile exposure over 65 weeks: a novel in vitro model. Int J Cancer 2011;128:274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bajpai M, Aviv H, Das KM. Prolonged exposure to acid and bile induces chromosome abnormalities that precede malignant transformation of benign Barrett’s epithelium. Mol Cytogenet 2012;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng E, Zhang X, Wilson KS, et al. JAK-STAT6 Pathway Inhibitors Block Eotaxin-3 Secretion by Epithelial Cells and Fibroblasts from Esophageal Eosinophilia Patients: Promising Agents to Improve Inflammation and Prevent Fibrosis in EoE. PLoS One 2016;11:e0157376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palanca-Wessels MC, Barrett MT, Galipeau PC, et al. Genetic analysis of long-term Barrett’s esophagus epithelial cultures exhibiting cytogenetic and ploidy abnormalities. Gastroenterology 1998;114:295–304. [DOI] [PubMed] [Google Scholar]
- 24.Kalabis J, Wong GS, Vega ME, et al. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc 2012;7:235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu T, Zhang X, So CK, et al. Regulation of Cdx2 expression by promoter methylation, and effects of Cdx2 transfection on morphology and gene expression of human esophageal epithelial cells. Carcinogenesis 2007;28:488–96. [DOI] [PubMed] [Google Scholar]
- 26.Kauer WK, Peters JH, DeMeester TR, et al. Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery 1997;122:874–81. [DOI] [PubMed] [Google Scholar]
- 27.Nehra D, Howell P, Williams CP, et al. Toxic bile acids in gastro-oesophageal reflux disease: influence of gastric acidity. Gut 1999;44:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirsch FR, Varella-Garcia M, Bunn PA Jr., et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 2003;21:3798–807. [DOI] [PubMed] [Google Scholar]
- 29.John T, Liu G, Tsao MS. Overview of molecular testing in non-small-cell lung cancer: mutational analysis, gene copy number, protein expression and other biomarkers of EGFR for the prediction of response to tyrosine kinase inhibitors. Oncogene 2009;28 Suppl 1:S14–23. [DOI] [PubMed] [Google Scholar]
- 30.Stone RC, Pastar I, Ojeh N, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res 2016;365:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabbah M, Emami S, Redeuilh G, et al. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat 2008;11:123–51. [DOI] [PubMed] [Google Scholar]
- 32.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012;148:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jethwa P, Naqvi M, Hardy RG, et al. Overexpression of Slug is associated with malignant progression of esophageal adenocarcinoma. World J Gastroenterol 2008;14:1044–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith CM, Watson DI, Leong MP, et al. miR-200 family expression is downregulated upon neoplastic progression of Barrett’s esophagus. World J Gastroenterol 2011;17:1036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohashi S, Natsuizaka M, Naganuma S, et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res 2011;71:6836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi S, Natsuizaka M, Wong GS, et al. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 2010;70:4174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Yu C, Peng S, et al. Autocrine VEGF signaling promotes proliferation of neoplastic Barrett’s epithelial cells through a PLC-dependent pathway. Gastroenterology 2014;146:461–72.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cotton CC, Wolf WA, Pasricha S, et al. Recurrent intestinal metaplasia after radiofrequency ablation for Barrett’s esophagus: endoscopic findings and anatomic location. Gastrointest Endosc 2015;81:1362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.