

Abstract
Traumatic brain injury (TBI) is a significant public health problem around the world. A promising area of research is the characterization of small, drug-like molecules that have potent clinical properties. One pharmacotherapeutic agent in particular, an aminopropyl carbazole called P7C3, was discovered using an in vivo screen to identify new agents that augmented the net magnitude of adult hippocampal neurogenesis. P7C3 greatly enhanced neurogenesis by virtue of increasing survival rates of immature neurons. The potent neuroprotective efficacy of P7C3 is likely due to enhanced nicotinamide phosphoribosyltransferase (NAMPT) activity, which supports critical cellular processes. The scaffold of P7C3 was found to have favorable pharmacokinetic properties, good bioavailability, and was nontoxic. Preclinical studies have shown that administration of the P7C3-series of neuroprotective compounds after TBI can rescue and reverse detrimental cellular events leading to improved functional recovery. In several TBI models and across multiple species, P7C3 and its analogues have produced significant neuroprotection, axonal preservation, robust increases in the net magnitude of adult neurogenesis, protection from injury-induced LTP deficits, and improvement in neurological functioning. This review will elucidate the exciting and diverse therapeutic findings of P7C3 administration in the presence of a complex and multifactorial set of cellular and molecular challenges brought forth by experimental TBI. The clinical potential and broad therapeutic applicability of P7C3 warrants much needed investigation into whether these remedial effects can be replicated in the clinic. P7C3 may serve as an important step forward in the design, understanding, and implementation of pharmacotherapies for treating patients with TBI.
Keywords: Neuroprotection, P7C3, Neurogenesis, Traumatic Brain Injury, NAMPT, NAD
Graphical Abstract
1.1. TBI Epidemiology and Pathophysiology
Traumatic brain injury (TBI) is a leading cause of death and disability around the globe. Each year in the U.S., 1.7 million people sustain a TBI (Faul et al., 2010). In the absence of mortality, patients with TBI may have significant life-long disability comprised of motor impairment, cognitive deficits, neuropsychiatric complications, and increased risk of suicide (Madsen et al., 2018; DeKosky et al., 2013; Masel and DeWitt, 2010). One of the difficulties encountered when treating TBI in the clinic is the heterogeneity of the injury and of the response of the human brain. No two TBIs are identical, and thus, there is a continued need for preclinical experimental work to mitigate some of the predicable and frequently-observed cellular and molecular challenges that transpire.
The pathophysiology of TBI occurs because of primary and secondary injury mechanisms. The primary injury initiates directly at the time of trauma. Depending on the nature of the insult, primary injury to intracranial contents can occur with rapid acceleration/deceleration forces, direct impact with an external object, object penetration, or blast-induced pressure waves. TBI patients may present with tissue shearing along white matter tracts and diffuse axonal injury (DAI), focal cerebral contusions, hematomas, and cerebral edema. Secondary brain injuries are classified as primary injury-induced ionic and neurochemical cascades resulting in pathophysiological cellular and molecular changes. Secondary injuries, such as mitochondrial dysfunction, generation of free radicals, inflammatory responses, and cell death, occur over a spectrum of hours, days, and weeks, further demonstrating that TBI is a complicated and progressive disease state (Bramlett and Dietrich, 2014).
Currently, there is a paucity of substantiated treatments for TBI patients, and Phase 3 clinical trials have been unsuccessful in showing improvements in neurologic outcomes (Roozenbeek et al., 2012). Neuropharmacotherapies for TBI are an important area of research that warrants extensive investigation. Due to an incomplete understanding of the injured state of the brain, novel treatments that address the dichotomy of both the neuroreparative and pathological responses of the injured brain are of the utmost importance.
1.2. Adult Neurogenesis and TBI
Adult neurogenesis is defined as the birth, maturation, and functional incorporation of newly-generated neurons in the postnatal brain. Postnatal hippocampal neurogenesis initiates in the subgranular zone (SGZ) of the dentate gyrus (DG), and has been detected in virtually all land-born mammals, including humans (Kempermann et al., 2018). The temporal profile of neuronal maturation is species-dependent, taking approximately 4 weeks in the rat (Espósito et al., 2005) and a minimum of 6 months in the nonhuman primate (Kohler et al., 2011). Newborn neurons have distinct electrophysiological profiles and are highly plastic, characteristics that are central to their unique role within the hippocampal network (Schinder and Gage, 2004). Several studies have shown that adult-born hippocampal neurons are involved in contextualization of spatiotemporal information, pattern separation, forgetting, affective behaviors, and endogenous repair responses after injury, across multiple species (Kempermann et al., 2018; Christian et al., 2014; Deng et al., 2010; Lie et al., 2004). Although adult neurogenesis is dynamic, with the rate of newly-added neurons decreasing with age (Galvan and Jin, 2007), several studies have reported continued neurogenesis throughout adulthood in the human hippocampus (Boldrini et al., 2018; Kempermann et al., 2018; Dennis et al., 2016; Ernst et al., 2014; Yang et al., 2014; Spalding et al., 2013; Knoth et al., 2010; Eriksson et al., 1998), however, a recent study argued that adult neurogenesis is extremely rare in adult humans (Sorrells et al., 2018). Additional investigation and optimized techniques to fully assess the phenomenon of human adult neurogenesis are clearly warranted.
Despite the current state of controversy, it has been well established that various stimuli can enhance or downregulate adult neurogenesis. Augmentation occurs with voluntary exercise, cognitive stimulation, and environmental enrichment, as well as after seizures, while reduction ensues with negative external factors such as stress, isolation, and in various CNS disease states (Aimone et al., 2014). TBI in particular has been shown to alter endogenous adult neurogenesis in a biphasic manner. After experimental TBI, there is an acute upregulation of newborn neurons followed by a chronic reduction of baseline neurogenesis (Blaiss et al., 2011; Atkins et al., 2010). Studies suggest this could be the result of a depletion of the neural progenitor pool that is required for sustained neurogenesis as well as the pathological death of immature hippocampal neurons, two adverse events that potentially contribute to chronic cognitive impairment after TBI (Blaiss et al., 2011; Gao et al., 2008). In any event, the significant decrease in the net magnitude of endogenous neurogenesis, together with extensive neuronal cell death and dysfunction throughout the hippocampus and other regions of the brain, significantly contribute to TBI-induced neuropathological outcomes.
TBI may result in neuropsychiatric comorbidities such as the development of depression, anxiety, or posttraumatic stress disorder (Ahmed et al., 2017). TBI-induced alterations of adult neurogenesis have been implicated in the etiology of these neuropsychiatric disorders, as preclinical studies have shown that animal models with impaired hippocampal neurogenesis display more anxiogenic and depressive behaviors (Miller and Hen, 2015). Lack of sufficient adult hippocampal neurogenesis may increase and prolong stress responses, which contribute to clinical manifestation of psychiatric illness (Cameron and Glover, 2015).
2.1. Discovery of the P7C3 Class of Neurotherapeutic Compounds
There is a disruption of adult neurogenesis and increased neuronal cell death in several preclinical models of deleterious neurological conditions, including Alzheimer’s disease (AD), Huntington’s disease, TBI, and other CNS injuries, which affect millions of people around the world. In order to address the unmet need for effective treatments, Pieper and colleagues (2010) utilized a target-agnostic, in vivo screen to identify novel, drug-like small molecules that enhance the net magnitude of adult hippocampal neurogenesis. Intracerebroventricular (ICV) infusion of 1000 drug-like compounds, selected based on their chemical diversity, size, and complexity, was carried out in living mice over 1 week. During the infusion process, mice also received injections of bromodeoxyuridine (BrdU), a thymidine analogue that incorporates into newly-synthesized DNA and labels dividing progenitor cells. By comparing the infused compounds against negative and positive controls, and assessing the net magnitude of hippocampal neurogenesis in the contralateral SGZ, eight proneurogenic compounds were revealed. One in particular, the aminopropyl carbazole Compound #3 (C3) contained within screening Pool #7 (P7) – hence named P7C3 – was found to potently stimulate robust BrdU-positive signal by increasing the survival of newborn neurons, without affecting their rate of proliferation (Figure 1, left). This was determined by comparing short- and long-term incorporation of BrdU, which revealed that P7C3 enhanced the net magnitude of newborn hippocampal neurons through increased survival rates, as opposed to stimulation of mitotic division and subsequent neural stem cell (NSC) proliferation (Pieper et al., 2010).
Figure 1.

Chemical structures of the P7C3 series of drug-like aminopropyl carbazoles. Chemical structure of the P7C3 parent compound (left). Highly-active P7C3–A20 (center) has fluorine substituted for the central hydroxyl and a methoxy group on the aniline ring. In the optimized derivative, (−)-P7C3–S243 (right), the aniline moiety is replaced with an alternative heterocycle conferring increased polarity. This compound can be synthesized as a single enantiomer and currently maintains the most favorable physicochemical properties.
Furthermore, in silico investigation revealed that P7C3 was likely to have favorable brain penetration, and that this molecule could be peripherally administered by oral and intraperitoneal (IP) delivery, eliminating the need for ICV delivery (Pieper et al., 2010). Additional investigations determined bioavailability, half-life, rate of clearance, and magnitude of blood brain barrier (BBB) penetration. P7C3 was found to be 32% orally bioavailable, possess a half-life of 6.7 hours after IP administration, and readily able to cross the BBB. The researchers conducted dose-response experiments and found consistent, graded responses. A subsequent in vivo structure-activity relationship experiment assessed 37 chemical variants of the P7C3 parent compound and revealed one in particular with significantly enhanced neuroprotective activity. This molecule, deemed P7C3–A20, contained a fluorine instead of a hydroxyl group at the chiral center (Pieper et al., 2010; Figure 1, center).
The authors next investigated whether P7C3 increased the net magnitude of neurons alone or also increased numbers of non-neuronal cell types. They found that BrdU immunoreactivity colocalized exclusively with neuronal markers (Pieper et al., 2010). Thus, P7C3 appeared neuron-specific in the context of hippocampal neurogenesis, a process in which a small number of surviving cells sometimes differentiate instead into glia cells (Gage, 2000).
After affirming prosurvival activity in wild-type mice, Pieper and colleagues (2010) next assessed P7C3 in a mouse model of impaired hippocampal neurogenesis. Neuronal PAS domain protein 3 (NPAS3) is required for appropriate expression of fibroblast growth factor (FGF) receptor 1 in the hippocampus. Transgenic mice lacking npas3, a master regulator of neuropsychiatric function (Michaelson et al., 2017), have nearly complete loss of DG adult neurogenesis by virtue of massively increased cell death in this process (Pickard et al., 2006; Pieper et al., 2005). This knockout model is characterized by aberrant hyperexcitability and morphological abnormalities of DG granular neurons. Prolonged administration of P7C3 to npas3−/− mice restored the dendritic morphology of DG neurons and corrected electrophysiological deficits in the DG, but not in CA1. (Figure 2). Immunohistochemical staining with cleaved caspase-3 (CCSP3), a marker of apoptotic cell death, showed that P7C3-receiving npas3−/− mice had a statistically significant reduction of CCSP3-positive cells in the hippocampus. These findings suggest that P7C3 effectively overcomes the genotypic hippocampal cell death of npas3−/− mice by protecting newborn neurons from death. Apoptotic cell death can be activated by an intrinsic pathway that is initiated in mitochondria (Green and Reed, 1998). Once apoptosis commences, intracellular signaling cascades result in mitochondrial membrane permeabilization (MMP) and degradation of cellular components (Kroemer et al., 2007). Because P7C3-treated animals had reduced levels of CCSP3 in the DG, and MMP plays a significant role in apoptosis, Pieper and colleagues (2010) sought to investigate the effects of P7C3 specifically on mitochondrial membrane integrity. In vitro experiments evaluating calcium-induced mitochondrial dissolution revealed that P7C3 and derivatives significantly protected mitochondrial membrane potential relative to vehicle-treated cells in a dose-dependent manner. These experiments suggested that P7C3 may be protecting vulnerable neurons by acting in a manner that stabilizes mitochondrial function.
Figure 2.

P7C3 Corrects Morphological and Electrophysiological Deficits in npas3−/− DG. A) Golgi-Cox staining of the DG revealed aberrant dendritic arborization in npas3−/− mice relative to wild-type littermates (ten sections from each of five mice in each group). In addition to reduced dendritic length and branching, npas3−/− DG granular neurons also exhibited significantly reduced spine density relative to wild-type littermates (∗p < 0.00001, Student’s t test). The top lower-power micrographs of DG were taken at the same magnification (scale bar, 200 μm). For the bottom two higher-power micrographs of dendritic spines, scale bar is 10 μM. B) In hippocampal slice preparation from npas3−/− mice, synaptic transmission, as assessed by whole-field recordings of fEPSPs, was increased both in the outer molecular layer of the DG and the CA1 region of the hippocampus relative to that of wild-type mice. C) Golgi-Cox staining of DG granular neurons showed that prolonged daily treatment of npas3−/− mice with P7C3 enhanced dendritic arborization. Results shown are representative of ten sections from each of five mice in each group. Higher-power micrographs are shown on top (scale bar, 25 μm), and a lower-power micrograph illustrating the entire granular layer of the DG is shown below (scale bar, 200 μm). D) Prolonged administration of P7C3 normalized whole field recordings of fEPSPs in the DG but not the CA1 region of npas3−/− mice. In all graphs, data are expressed as mean ± SEM. (Reprinted from Cell, 142(1), Pieper et al., Discovery of a Proneurogenic, Neuroprotective Chemical, 39–51, 2010, with permission from Elsevier)
Because adult DG neurogenesis is a putative mediator of hippocampal-dependent learning and memory, the next step was to assess the effect of P7C3 administration in aged rats. A depreciation of hippocampal neurogenesis occurs in biological aging, which is related to greater hippocampal NPC apoptosis in the aged brain, and contributes to cognitive deterioration (Kuhn et al., 1996). The Morris water maze (MWM) is a valuable tool to assess hippocampal-dependent spatial acquisition and retention (Morris, 1981). Rats received daily injections of 10 mg/kg of P7C3 intraperitoneally for 2 months, at which point they were tested for cognitive function in the MWM. Hippocampal-dependent learning in aged animals that were administered P7C3 was significantly enhanced relative to aged control animals (Pieper et al., 2010). In summary, Pieper and colleagues discovered a proneurogenic, neuroprotective compound that was highly active in multiple models of impaired neurogenesis, and maintained significant clinical potential.
2.2. Optimization of the P7C3 Scaffold
In subsequent years since the initial discovery, experiments have been carried out to further characterize and chemically optimize the highly-active scaffold of P7C3. Several derivatives designed from the parent scaffold have been developed and tested in a variety of injury and neurodegenerative disease states in which neuronal survival is compromised. P7C3–A20 was found to have greater potency and stability, was nontoxic at high doses, and, importantly, could be synthesized on a large scale (Naidoo et al., 2013). As discussed briefly above, this improved compound is a result of systematic manipulation and optimization of each subsection of P7C3, which determined that fluorination improved innate neuroprotective efficacy. Using this alteration as a new starting point, Naidoo et al. (2014) further designed an additional P7C3 derivative in which the aniline moiety was replaced with an alternative heterocycle to increase polarity. In this work, they described the discovery and meticulous assessment of a superior P7C3 molecule called (−)-P7C3–S243 with optimal physiochemical and pharmacokinetic properties coupled with greater neuroprotective efficacy (Figure 1, right).
Since this groundbreaking discovery and assessment of P7C3 efficacy in models of impaired neurogenesis (Pieper et al., 2010), multiple experimental models of neurodegenerative diseases have shown robust preclinical evidence for sensitivity to treatment with P7C3 agents. For example, experiments have shown potent neuroprotection of dopaminergic neurons in mouse and rat models of Parkinson’s disease (Jesús-Cortés et al., 2015; Naidoo et al., 2014; Jesús-Cortés et al., 2012), decreased motor neuron cell death in the spinal cord of a mouse model of amyotrophic lateral sclerosis (Tesla et al., 2012), and protection in neonatal rats from nerve cell death and dysfunction in a model of obstetric brachial plexus palsy (Kemp et al., 2015). Research has also shown that P7C3 treatment reversed retinal apoptosis in a zebrafish model of retinal degeneration (Asai-Coakwell et al., 2013), and provided protection from hippocampal cell death in preclinical models of anxiety (Lee et al., 2016) and depression (Walker et al., 2015). In experimental acute ischemic stroke, our laboratory recently reported a significant reduction of cortical and hippocampal atrophy, enhanced neurogenesis, and improved neurological outcomes after treatment with P7C3–A20 (Loris et al., 2018, 2017). More recently, robust protective efficacy of P7C3 compounds for nerve cell death and neurological dysfunction has also been established in preclinical models of AD (Voorhees et al., 2017) and chemotherapy-induced peripheral neuropathy (LoCoco et al., 2017).
3.1. P7C3 Proposed Mechanism: Enhanced NAMPT activity
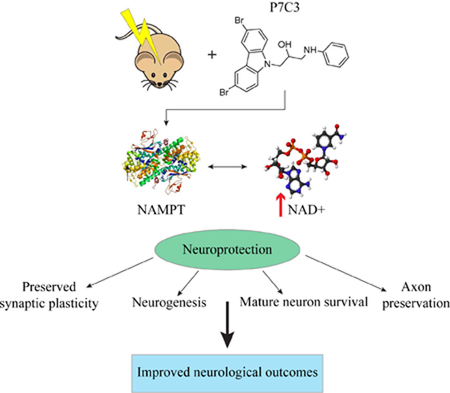
While the proneurogenic efficacy of P7C3 had been reproduced in multiple experimental models of disease and injury in which neuronal survival is compromised, the mechanism by which P7C3 compounds exert these effects remained elusive. However, four years after the initial discovery, Wang and colleagues (2014) reported that P7C3 compounds augmented the activity of a critical enzyme involved in the biosynthesis of nicotinamide adenine dinucleotide (NAD+). NAD+ is an indispensable enzyme that contributes to a broad number of cellular functions and has an essential role in maintaining organism homeostasis. It is a coenzyme in reduction-oxidation reactions and mediates regulation of cellular metabolism and mitochondrial respiration. NAD+ is also a critical cosubstrate for additional pleiotropic enzymes such as sirtuins (SIRTs), poly-adenosine diphosphate (ADP)-ribose polymerases (PARPs), and cyclic ADP-ribose (cADPR) synthases (Verdin, 2015; Figure 3). Using photocrosslinking and click chemistry, Wang et al. (2014) revealed that P7C3 compounds bind to and enhance the activity of nicotinamide phosphoribosyltransferase (NAMPT), which is the rate-limiting enzyme for NAD+ biosynthesis via the salvage pathway. In order to establish whether P7C3-mediated increases in NAMPT activity affected downstream NAD+ levels, the authors utilized doxorubicin (dox), a toxin that damages DNA and activates PARPs (Muñoz-Gámez et al., 2009). PARP proteins contribute to an assortment of cellular processes including DNA repair and cell death pathways. PARPs consume NAD+ to catalyze the synthesis of poly-ADP-ribose (Kim et al, 2005). Thus, activation of PARPs decreases cellular levels of NAD+. Wang et al. (2014) showed that P7C3 treatment significantly reversed dox-mediated decline in intracellular NAD+ stores in a dose-dependent manner. Additional experiments showed that purified, recombinant human NAMPT enzymatic activity was enhanced with multiple active derivatives of P7C3. Thus, the authors concluded that the family of P7C3 aminopropyl carbazoles is neuroprotective and proneurogenic through augmentation of NAMPT activity and subsequent restoration of cellular NAD+ levels (Wang et al., 2014; Figure 3).
Figure 3.

NAD+ biosynthesis via the salvage pathway, circadian regulation, and P7C3-mediated NAMPT enhancement. NAMPT (green) catalyzes the conversion of nicotinamide to NMN, which is then converted to NAD+ (dark blue) through the activity of NMNATs (yellow). NAD+ is a critical cosubstrate for the downstream enzymatic activity of PARPs, SIRTs, and cADPR synthases, which have extensive influence on cellular function, viability, and organism homeostasis. The circadian clock and NAD+-dependent SIRT1 (purple) regulate the transcription of NAMPT (right inset). Oscillations in NAMPT levels rely on SIRT1 transcription, maintaining a positive feedback cycle between NAMPT, NAD+, and SIRT1. P7C3 compounds (red triangle) bind to and enhance the activity of neuronal NAMPT thereby increasing cellular NAD+ levels. NAD+-mediated neuroprotection implicates SIRT1 signaling cascades, critical prosurvival pathways governing a multitude of cellular processes associated with viability, repair, inflammation, and neurogenesis. BMAL1, brain muscle ARNT-like protein 1; cADPR, cyclic ADP (adenosine diphosphate) ribose; CLOCK, circadian locomotor output cycles kaput; NAD, nicotinamide adenine dinucleotide; NAMPT, nicotinamide phosphoribosyltransferase; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide mononucleotide adenylyltransferease; PARP, poly ADP ribose polymerase; SIRT1, sirtuin-1.
3.2. NAMPT-NAD+ Axis in TBI
NAD+ is disproportionately depleted after TBI (Clark et al 2007; Satchell et al 2003). Secondary injury mechanisms contribute to the pathological activation of PARPs, which consume NAD+ and deplete ATP, creating cellular energy failure and initiating mitochondria-mediated apoptosis (Alano et al., 2010). Although the temporal expression of NAMPT after TBI remains to be fully delineated, our laboratory and others have shown that neuronal NAMPT in the hippocampus is acutely upregulated after TBI (Wang et al., 2011; Figure 4). It is suggested that this upregulation is part of a natural stress response to injury and initiation of a neuroreparative program. However, due to excessive TBI-induced depletion of NAD+ and subsequent energy failure, this endogenous reparative process is insufficient and unable to inhibit neuronal death. Because neuronal NAMPT transcription relies on NAD-dependent sirtuin 1 (SIRT1; Figure 3), NAMPT levels may become depleted in the chronic period after TBI when NAD+ is pathologically consumed. Because of the importance of NAMPT-mediated NAD+ production to the survival of the cell, and because it is compromised after TBI, it is an enticing neuropharmaceutical target.
Figure 4.

6 hours after moderate FPI, Western blot analysis showed increased hippocampal DG NAMPT compared to sham-operated controls.
3.3. NAMPT and Neurogenesis
NAMPT is also highly expressed in both immature and mature neurons in the hippocampus. NAD+ is essential for maintaining the stemness of pluripotent stem cells and for their unique bioenergetic requirements (Zhang et al., 2012). Stem cell metabolic plasticity requires adequate availability of NAD+ for participation in critical pathways governing proliferation and fate determination (Folmes et al., 2012). When the NAMPT-regulated salvage pathway is disrupted and neuronal NAD+ is depleted, the resulting imbalance alters cellular metabolism that can result in premature stem cell death (Wang et al., 2016).
NAMPT involvement in adult neurogenesis has also been reported by Stein and Imai (2014) who showed that ablation of NAMPT in adult NSCs impaired self-renewal, proliferation, and neuronal maturation, and mimicked age-related cognitive impairment. Aged animals have deficient neurogenesis and cognitive deterioration (Kuhn et al., 1996). These age-related changes coincide with temporal depletion of hippocampal NAMPT and NAD+. Thus, injury-induced alterations in NAMPT/NAD+ levels are considered one of the contributing factors underlying disruption and chronic depletion of hippocampal neurogenesis after TBI.
3.4. NAMPT-NAD+ Axis as a Therapeutic Target: Role for Sirtuins
Several studies have evaluated interventions to enhance NAMPT/NAD+ pools and have provided evidence to support a role for the NAMPT-NAD+ axis in neuroprotection. In experiments manipulating NAMPT activity, there was significant aggravation of ischemia-induced cerebral infarct volumes and neuronal injury when NAMPT was inhibited, while overexpression of NAMPT resulted in a considerable reduction of neuronal injury as well as increased numbers of NSCs, greater adult neurogenesis, and neurological recovery (Zhao et al., 2015; Wang P. et al., 2011). Won and colleagues (2012) reported that intranasal administration of NAD+ after concussive TBI protected hippocampal neurons from death and attenuated proinflammatory microglia activation. The ability of neuronal NAMPT/NAD+ to exert neuroprotective effects and positively mediate cell survival makes this axis an alluring target to combat the deleterious consequences of brain injury. The activity by which neuroprotection occurs has been attributed to NAMPT/NAD+-dependent prosurvival, antiapoptotic signaling cascades.
Sirtuins are NAD+-dependent deacetylases that interact with an assortment of proteins involved in cell survival, DNA repair, and apoptosis (Haigis and Sinclair, 2010). Importantly, NAMPT is under transcriptional control of SIRT1 as an essential part of the mammalian circadian clock feedback cycle (Haigis and Sinclair, 2010; Ramsey et al., 2009; Figure 3). Thus, NAMPT cosynthesizes NAD+, which activates SIRT1, which then transcribes NAMPT, over a 12-hour cycle.
SIRT1 signaling may be an important neurotherapeutic target in several neurodegenerative diseases, CNS injury, and age-related decline (Donmez and Outeiro, 2013). NAMPT/NAD+-mediated neuroprotection in experimental TBI and other CNS injury studies has been attributed to prosurvival SIRT1 signaling cascades (Zhao et al., 2012). Under ischemic stress, NAMPT exhibited substantial neuroprotection in vivo through SIRT1/AMP-activated protein kinase (AMPK) signaling, which is critical for cell growth, metabolism, and senescence (Mihaylova and Shaw, 2012; Wang Y. et al., 2011). Furthermore, SIRT1 has been shown to suppress inflammation in several tissues (Haigis and Sinclair, 2010), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a principal regulator in the innate and adaptive immune responses activated after TBI, was effectively repressed by SIRT1 through blockade of NF-κB proinflammatory transcriptional activity (Salminen et al., 2008). Inhibition of SIRT1 after TBI exacerbated mitochondrial impairment and neuronal injury (Yang et al., 2016). Furthermore, knockout experiments concluded that SIRT1 was essential for hippocampal-dependent cognition and synaptic plasticity in mice (Michán et al., 2010). SIRT1 is a positive regulator of Wnt signaling, a critical pathway for NPC proliferation and adult neurogenesis (Holloway et al 2010; Lie et al., 2005). These findings support acute NAMPT-NAD+-SIRT activation as an important inhibitor in TBI-mediated secondary injury and a promoter of survival and proneurogenic activity. The ability of P7C3 to target and enhance NAMPT activity may enhance signaling of neuroprotective SIRT1 through restoration of NAD+ levels (Figure 3).
4. P7C3 Provides Neuroprotection, Enhances Hippocampal Neurogenesis, and Restores Function after Moderate TBI
Our laboratory first investigated P7C3 in experimental moderate parasagittal fluid percussion injury (FPI) characterized by clinically-relevant histopathological changes and long-term functional impairment (Blaya et al., 2014). Because of the vulnerability of select populations of adult neurons and of the depression of hippocampal neurogenesis in this model, we hypothesized that administration of the highly-active agent P7C3–A20 would stimulate reparative neurogenic responses and promote neuroprotection, which would translate to histological and functional improvement after TBI.
Sprague Dawley rats received a moderate fluid percussion-induced TBI that was generated over the right parietal cortex. Animals were injected IP with 10 mg/kg of P7C3–A20 or vehicle 30 minutes after insult followed by subsequent injections twice daily for the next 7 days. Animals also received IP injections of a concentrated aqueous solution of a BrdU labeling reagent 24 hours post TBI and daily thereafter for 7 days. Sham-designated animals underwent identical procedures and treatment except for the physical fluid percussion-induced insult.
One week after TBI, volumetric and stereological analyses revealed that P7C3–A20 significantly reduced contusion volumes at multiple bregma levels and prevented cortical neuronal loss (Figure 5).
Figure 5.

P7C3–A20 administration was neuroprotective in vulnerable ipsilateral cortical regions 1 week post TBI. (A and B) Representative NeuN-stained images of vehicle- (A) and P7C3–A20-treated (B) animals. Selective neuronal loss was observed in the cortical region overlying the contusion site. Treatment with P7C3–A20 significantly reduced NeuN-positive cell loss. Boxes in A, B enclose the areas of neuronal loss. Arrows indicate subcortical lesion. C) Quantification of NeuN-positive cells between treatment groups revealed a significant neuroprotective effect of P7C3–A20 1 week post injury (Student’s t-test: p = 0.015, t = 2.695; *p < 0.05; scale bar, 500 m); n=20. (Blaya et al., 2014).
Doublecortin (DCX) is a microtubule-associated protein that is transiently expressed in immature neurons prior to maturation. At 1 week post TBI, we observed an increase in the magnitude of both BrdU and DCX-immunoreactive cells in the ipsilateral DG, indicative of early stage neurogenesis. P7C3–A20 treatment significantly enhanced the number of surviving nerve cells expressing these markers (Blaya et al., 2014). Five weeks after TBI, sufficient time for neuronal maturation, we quantified dentate granular cells that were immunoreactive for both BrdU and neuronal nuclei (NeuN), a marker for mature neurons. Cells coexpressing both markers indicated newly-born dentate granular neurons. Stereological quantification revealed a TBI-induced increase in hippocampal neurogenesis, which is consistent with the literature, while injury coupled with administration of P7C3–A20 significantly enhanced neurogenesis as demonstrated by a more robust number of double-labeled cells relative to controls. Furthermore, these newly-generated neurons appeared to migrate outside of the SGZ into the granular cell layer, which is consistent with maturation profiles in adult hippocampal neurogenesis (Figure 6).
Figure 6.

Confocal micrographs showing BrdU- and NeuN-immunoreactive cells within the DG 5 weeks after TBI. A) Representative fluorescent images showing cells that colabeled for BrdU and NeuN in the ipsilateral DG (scale bars, 200 μm). Boxed regions outline the areas of higher magnification shown in bottom row of (A). B) Representative micrographs from a TBI-P7C3–A20 animal showing several cells coexpressing markers for BrdU and NeuN (arrows) in the ipsilateral DG. Double-labeled cells in P7C3–A20 animals appeared to migrate out of the SGZ into more superficial layers of the DG, consistent with neuronal maturation profiles (GCL, granular cell layer; SGZ, subgranular zone; scale bar, 10 μm). (Blaya et al., 2014).
While histopathological improvement was promising and neurogenic responses were robust, our laboratory next sought to assess functional outcome measures to fully investigate the therapeutic capacity of P7C3–A20 after experimental TBI. A behavioral test assessing sensorimotor function at 1 week showed rescue of contralateral forelimb placement with P7C3–A20, which is compromised in our TBI model. Testing in the MWM 4 weeks after FPI, a time point selected based on the temporal maturation and electrophysiological profiles of newborn neurons, revealed that P7C3–A20-treated TBI animals performed significantly better in several parameters relative to TBI-controls (Blaya et al., 2014). We proposed that improved sensorimotor function was attributed to preservation of the cortical cytoarchitecture, as observed with reduced contusion volume and decreased cortical neuron cell death at this 1-week time point. We suggested that improved cognitive capacity however, was a result of augmented hippocampal neurogenesis. We tested a separate group of animals 2 weeks post TBI, before neuronal maturation is fully complete, and found no P7C3–A20-mediated effect in hippocampal-dependent memory (Blaya et al., 2014).
Taken together, P7C3–A20 administration improved histopathological consequences and rescued pericontusional neurons. Furthermore, we observed a significant increase in the magnitude of adult hippocampal proliferation and neurogenesis, changes that were associated with attenuation of hippocampal-dependent memory impairment and sensorimotor deficits. Injury-induced neurogenesis is considered an initiation of an endogenous neuroreparative strategy aimed at correcting disproportionate cell loss (Blaiss et al., 2011). However, this acute upregulation appears insufficient and dysregulated as it is followed by a persistent depression of neurogenesis in the chronic posttraumatic period. The ability of P7C3–A20 to mitigate TBI-induced death of vulnerable newborn and adult neurons, enhance endogenous neurogenic responses, and promote clinically-relevant functional recovery further supports the P7C3 class of aminopropyl carbazoles as novel translational agents to preserve mature brain structures and help promote endogenous reparative processes after brain injury.
5. Administration of P7C3–A20 following FPI Prevents a Reduction in Long-Term Potentiation in the Medial Perforant Path-Middle Molecular Layer Synapse
In the aforementioned study, we reported improvement in spatial memory capacity with P7C3–A20 administration after moderate FPI (Blaya et al., 2014). In order to further expand on these findings, we next sought to assess potential changes in electrophysiological correlates of learning and memory with and without P7C3–A20 treatment. As discussed previously, traumatic insults to the brain result in attentional, motivational, learning, and memory impairments, due in part to damage to the hippocampus. The DG gates information from the superficial layers of the entorhinal cortex into hippocampal areas CA1 and CA3 (Winson and Abzug 1997). Electrographic changes in excitability in the hippocampus following FPI manifest in a biphasic fashion with time (Reeves et al. 1997). Acute extracellular recordings from CA1 revealed increased synaptic strength after electrical stimulation of Schaffer collaterals two days following FPI (Reeves et al., 1995). At a one-week post FPI time point, however, injured animals had reduced CA1 excitability relative to sham controls, while increased excitability persisted in the DG (Zhang et al. 2011; Santhakumar et al. 2001). Furthermore, long-term potentiation (LTP), a proposed mechanism for information storage in the brain (Bliss and Lomo 1973), was reduced in CA1, but increased in the DG one week following moderate FPI (Zhang et al, 2011). Evaluation at 2 and 4 weeks after injury also showed a continued reduction of CA1 LTP (Sanders et al. 2000; Reeves et al. 1995). However, less is known about DG LTP at more chronic time points following FPI, and P7C3–A20 administration had yet to be investigated in this context. We sought to answer these questions using acute hippocampal slice recordings (prepared as described in Raval et al., 2012) acquired 4 weeks after moderate FPI from Sprague Dawley rats receiving twice-daily IP injections of PC73-A20 (10 mg/kg) 7 days post injury. In this study, P7C3–A20 treatment was initiated at either 30 min or 3 hours post surgery in order to investigate whether a temporal effect was present with delayed onset of administration.
Following high-frequency stimulation of the medial perforant path (MPP; as described by Colino and Malenka, 1993) in the presence of 100 uM picrotoxin, there was a modest 20% post tetanic potentiation, which approached 10% and persisted until the end of the recording (Figure 7). In sham animals, this modest degree of LTP in the dorsal DG was consistent with findings comparing the magnitude of LTP along the dorsal-ventral axis in the DG (Schreurs et al. 2017). One hour after high-frequency stimulation, the slope of the evoked field excitatory postsynaptic potential (fEPSP) from the MPP-middle molecular layer (MML) synapse was significantly less after FPI than that observed in sham animals. P7C3–A20 administration at 30 min and 3 hours post injury, however, was associated with significantly greater evoked responses from the MPPMML synapse relative to TBI control animals, and comparable to sham groups. No significant differences were observed in the input/output relationships for the slope of each fEPSP (data not shown). The fact that no differences were observed between experimental groups during paired pulses suggests that FPI did not impair short-term plasticity in the MPP-MML synapse. These results indicate that at 4 weeks after FPI, synaptic plasticity is reduced in the DG, but basal synaptic transmission is unaltered.
Figure 7.

Administration of P7C3–A20 at 30 min or 3 hours following FPI prevented a reduction in long-term potentiation in the medial perforant path-middle molecular synapse in vitro. Two-way repeated measures ANOVA (p = 0.029) showed a significant interaction between groups and time (p = 0.021). A student Newman Keuls post-hoc comparison revealed significance (p < 0.05) between sham and TBI control groups at time points (in min) 52, 55, 58, 64, 69, 71, 72, and 73. TBI+A20–30 min was significantly different (p < 0.05) from the TBI control group at time points 19, 21, 24, 25, 36, 37, 38, 39, 40, 44, 49, 51, 52, 53, 55, 56, 57, 58, 64, 65, 66, 68, 69, 70, 71, 72, and 73. TBI+A20–3 hr was significantly different (p < 0.05) from the TBI control group at time points 21, 39, 40, 41, 43, 44, 45, 49, 51, 52, 53, 55, 57, 58, 62, 64, 65, 66, 68, 69, 70, 71, 72, and 73. n = 2–5/group. Sham+Veh and Sham+A20 recordings were not significantly different from each other (p > 0.05), so these groups were pooled into a single sham group.
Taken together, we found that P7C3–A20 administration initiated at both 30 min and 3 hours prevented an injury-induced reduction of LTP in the MPP-MML 4 weeks after moderate FPI. Interestingly, we observed the greatest degree of LTP in 3 hour-TBI-A20-treated animals relative to all other groups (Figure 7). This finding, however, was not significantly different from 30 min-TBI-A20 animals. Given the low n in the 3 hour group (n = 2), further experimentation into the temporal differences of P7C3–A20 administration in this FPI model are warranted.
6.1. P7C3 Efficacy in Additional Models of TBI
An imperative quality of an effective and translational pharmacological intervention is therapeutic outcomes in an assortment of species and experimental models. After having positive histological, neurobehavioral, and electrophysiological outcomes with P7C3–A20 in our FPI model in rats, we evaluated P7C3–A20 in a mouse model of controlled cortical impact (CCI). This injury is most commonly generated using a device that rapidly accelerates a rod to impact the exposed surface of the brain, which produces histological and neurobehavioral alterations comparable to those observed clinically (Osier and Dixon, 2016). CD-1 mice were subjected to an actuator velocity of 4 m/s and a deformation depth of 0.5 mm resulting in a moderate TBI. 10 mg/kg of P7C3–A20 and a BrdU labeling reagent were administered IP as in our previous treatment paradigm (Blaya et al., 2014). Seven days after TBI, mice were transcardially perfused and immunostained for DCX and BrdU. Immunoreactive double-labeled cells in the ipsilateral DG were quantified using non-biased stereological methods. P7C3–A20-treated CCI mice exhibited significantly greater numbers of DCX/BrdU double-labeled cells indicative of elevated adult hippocampal early-stage neurogenesis (Figure 8). These results were consistent with the proneurogenic outcomes observed in our rat FPI model.
Figure 8.

Confocal micrographs double-labeled with BrdU (green) and DCX (red) at 7 days post CCI in the mouse with P7C3–A20 administration. Double-labeled cell bodies and processes are visible in the ipsilateral DG (top panels). Quantification of cells coexpressing BrdU/DCX are shown (bottom). P7C3–A20 administration coupled with moderate CCI significantly increased the estimated population of BrdU/DCX cells relative to sham controls at this early posttraumatic time point. n = 4–5/group, one-way ANOVA, *p = 0.012, DF, 2. Tukey post hoc **p = 0.0095, DF, 10.
6.2. P7C3 after Blast-Mediated TBI
Another important rodent model of TBI is the blast injury model. This model reproduces some of the behavioral and histological changes that are frequently observed in military personnel exposed to explosions (Hoge et al., 2008). While multitrauma to several tissues and organs may occur, blast injury elicits a unique type of TBI characterized by DAI and neuronal dysfunction, which may result in cognitive and motor impairment as well as psychiatric symptoms (Hicks et al., 2010). As in other incidents of brain trauma, amnesia is a common comorbidity among patients with blast-induced TBI (Hicks et al., 2010). Hippocampal-dependent memory behavioral paradigms, such as the Barnes maze, can evaluate learning and memory disruption in rodent models of injury (Goldstein et al., 2012).
Yin and colleagues (2016, 2014) first demonstrated the unique susceptibility of neuronal axons to blast injury and neuroaxonal protective capacity of P7C3 compounds in a mouse model of blast-induced TBI. After blast exposure, they assessed the efficacy of the highly-active compound, (−)-P7C3–S243. P7C3–S243 was administered 24 hours post TBI at various doses and Barnes maze testing commenced 7 days after injury. The authors reported that all doses (3, 10, and 30 mg/kg/day) of P7C3–S243 injected IP for 11 days offered complete protection of hippocampal-dependent spatial memory while vehicle-treated TBI mice had marked spatial memory deficits. To expand on these findings, the authors investigated whether the efficacy of P7C3–S243 was directly related to synaptic plasticity and hippocampal function. Using both LTP and paired pulse facilitation (PPF), Yin et al. (2014) reported that blast injury impaired both LTP and PPF, and that treatment with P7C3–S243 reversed these deficits.
To evaluate the treatment window for P7C3 compound administration, Yin and colleagues (2014) found that spatial memory was intact with 30 mg/kg/day of P7C3–S243 when started 36 hours after injury. When treatment was initiated 48 hours post TBI, however, no protective effect was observed at any of the doses. The authors also reported that P7C3–S243 was efficacious with daily oral administration, strengthening clinical relevancy of P7C3 compounds.
DAI and axonal degradation are some of the principle consequences of blast TBI. This study demonstrated significant axonal degeneration in the hippocampus and cerebellum 12 days post injury. However, P7C3–S243 administration initiated at 24 hours post TBI prevented axonal pathology. Transmission electron microscopy revealed a dose-dependent preservation of myelin integrity and axonal mitochondria structures in the hippocampus (Figure 9). Blast-injured animals exhibited disruption of myelin sheath, abnormal mitochondrial membranes and internal cristae, fewer hippocampal dendrites, and accumulation of pyknotic neurons. These adverse microstructural outcomes were not observed in P7C3–S243-treated TBI animals. Furthermore, oral treatment of P7C3–S243 augmented motor function in balance and coordination tasks relative to injured-control groups (Yin et al., 2014). Taken together, these findings further extend credence to the potent neuroprotective capacity of the P7C3 series of compounds, which maintain efficacious outcomes across various injury models of experimental TBI.
Figure 9.

Toluidine blue staining and transmission electron microscopy visualization of hippocampal protection by orally administered (−)-P7C3–S243 after blast injury. Daily oral administration of the highly active enantiomer (−)-P7C3–S243 for 14 days, starting 24 hr after injury, dose-dependently preserved CA1 morphology as well as myelin and mitochondrial structures in the hippocampus after blast injury. Two weeks after either sham or blast injury, animals were perfused and processed for ultrastructural pathology. Toluidine-blue-stained semithin sections (left panel) of sham-injured mice treated with vehicle or (−)-P7C3–S243 showed normal CA1 histology, with densely packed neurons in the stratum pyramidale (1) and profuse dendritic profiles in the stratus radiatum (2; black arrows). Blast-injured animals treated with vehicle showed accumulation of chromatolytic and pyknotic neurons (white arrow) throughout the stratum pyramidale as well as fewer dendrites in the stratum radiatum. There is no protection in CA1 morphology at the lowest concentration of blast-injured animals treated with 0.3 mg/kg/day of (−)-P7C3–S243. However, treatment with 3 mg/kg/day (−)-P7C3–S243 lowered the abundance of chromatolytic and pyknotic neurons and resulted in a more densely packed stratum pyramidale. At the highest concentration (30 mg/kg/day) of (−)-P7C3–S243, there was complete preservation of CA1 morphology after blast-mediated TBI. Transmission electron micrographs (TEM; right panel) of immediately adjacent ultrathin sections showed normal myelin and axonal mitochondrial structures in the stratum radiatum of sham-injury mice treated with vehicle or (−)-P7C3–S243. Blast-injured mice treated with vehicle or 0.3 mg/kg/day of (−)-P7C3–S243 showed degeneration of myelin sheath (red arrows), along with abnormal outer membrane and internal cristae structures within neuronal mitochondria (blue arrows). At 3 and 30 mg/kg/day doses, however, both myelin and neuronal mitochondria were preserved. Pictures shown are representative of four animals per condition. Scale bars, 50 μM in Toluidine blue; 500 nm in TEM. (Reprinted from Cell Reports, 8(6), Yin et al., P7C3 Neuroprotective Chemicals Block Axonal Degeneration and Preserve Function after Traumatic Brain Injury, 1731–1740, 2014, with permission from Elsevier).
7. Neuropsychiatric Comorbidity and P7C3
According to a large cohort study, the prevalence of any psychiatric illness in the first year after moderate or severe TBI is 49%, and 34% after mild TBI (Fann et al., 2004). Psychiatric comorbidities include depression, generalized anxiety disorder, posttraumatic stress disorder, and agoraphobia (Bryant et al., 2010). As discussed previously, experimental studies have shown that aberrant hippocampal neurogenesis may contribute to depressive- and anxiety-like behavior in animal models of TBI (Miller and Hen, 2015).
Walker et al. (2015) investigated the effects of P7C3 compounds in ghrelin receptor (Ghsr)-null mice, which exhibit depressive-resembling behaviors when exposed to subclinical levels of psychosocial stress. The authors reported that P7C3 treatment reduced the prevalence of depressive behaviors in chronic stress-exposed transgenic mice, and that rescued hippocampal neurogenesis and neuroprotection were contributing factors. Walker and colleagues (2015) also showed that P7C3–A20 in particular conferred an antidepressant-like effect on wild-type mice not genetically prone to depressive-like behaviors under these experimental conditions. Of note, P7C3–A20 exhibited significantly greater neurogenic activity than currently marketed antidepressant therapies. Taken together, in addition to augmentation of cell survival and neurogenesis in CNS injury and neurodegenerative diseases, P7C3 compounds may also represent a novel approach for treating patients with neuropsychiatric disease, a common comorbidity of TBI.
8. P7C3 and Reactive Glia
Posttraumatic neuroinflammation can be both advantageous and deleterious. In the acute period after insult, cellular and molecular changes are initiated as protective measures, however prolonged overactivation is detrimental to recovery and repair (Simon et al., 2017). Microglia and astrocytes respond to environmental changes. Microglia can change along an activation spectrum spanning from an activated M1-like phenotype to an alternative M2-like phenotype. M1-like microglia generate free radicals and proinflammatory cytokines, while M2-like microglia secrete anti-inflammatory agents and mitigate inflammation. TBI-induced inflammation causes a disproportionate number of microglia to become activated and resemble M1 morphology, which is characterized by short, or lack of, neurites and small ameboid-like cell bodies (Kettenmann et al., 2011). Prolonged overactivation of microglia is detrimental to the injured milieu and a significant contributor to secondary injury. In early preliminary studies, our laboratory has observed beneficial alterations in neuroglia in the posttraumatic environment after P7C3 treatment. Seven days of P7C3–A20 administration after moderate FPI appeared to reduce the reactive phenotype of Iba1-positive cells, a putative marker for activated microglia and macrophages, in the ipsilateral hippocampal hilus, with a greater proportion exhibiting resting-state morphology with highly-ramified branches (Figure 10A).
Figure 10.

Preliminary findings indicated P7C3–A20 administration altered neuroglia profiles in the hippocampal hilus 6 weeks after FPI. A) Qualitative observations showed Iba1-positive activated microglia and macrophage cells in the ipsilateral hippocampal hilus had varying activation states according to treatment paradigm. P7C3–A20 administration 30 min post TBI was associated with a greater proportion of Iba1-postive cells exhibiting the alternative M2-like phenotype relative to the TBI control group, and comparable to phenotypic observations in sham animals. Yellow arrows show highly-ramified resting state cells (M2-like). Yellow arrowheads indicate ameboid, hyperactivated cells (M1-like). B) GFAP-immunoreactivity was used to visualize astrogliosis 6 weeks after moderate FPI. GFAP-positive cells in the TBI-vehicle group appeared to have greater staining intensity (area demarcated by white box) and increased branching (red arrows). TBI P7C3–A20-treated animals, however, had astroglia morphology resembling that of sham animals.
The role of reactive astrocytes after TBI is not fully understood. Posttraumatic astrocytic alterations include changes in proliferation, hypertrophy, and gene expression. In glia fibrillary-associated protein (GFAP)-immunoreactive sections, TBI-control animals exhibited classic astrocytic reactivity with greater GFAP staining intensity and increased number and length of GFAP-positive processes, which are morphological changes indicative of reactive gliosis (Wilhelmsson et al., 2006; Figure 10B). GFAP-positive astrocytes in TBI P7C3–A20-treated animals, however, exhibited morphology resembling that of sham animals.
While it has not yet been delineated how P7C3 specifically may be affecting neuroglia in the posttraumatic environment, Chen and colleagues (2015) showed that NAD+-dependent SIRT1 modulates immune responses by influencing M1-like microglia to adopt an alternative M2-like phenotype. Thus, P7C3-enhanced NAMPT/NAD+ levels may alleviate injury-induced inflammatory responses and direct the posttraumatic environment towards homeostatic, resting state conditions via anti-inflammatory signaling cascades.
9. Concluding Remarks
Although preclinical and clinical research report enthusiastic findings on potential treatment strategies for patients with TBI, there has not been an improvement in overall neurological outcomes or a decrease in TBI-related death over the past two decades (Roozenbeek et al., 2013). With an increasing incidence of TBI worldwide, there has never been more urgency to tackle this significant global health problem.
There is an unmet need for pharmacotherapies that can be administered safely for prolonged periods and effectively target ongoing pathomechanisms that contribute to poor neurological outcomes in the chronic phases of TBI. Contemporary drug discovery utilizes high-throughput screening against specific molecular targets. However, in TBI there is a lack of consensus regarding which molecular entities should be targeted. Unbiased, in vivo screening assays are an ideal route to uncover novel therapeutics given the ability to assess bioavailability, potency, stability, and toxicity directly in live animals.
Using this optimal method of unbiased drug discovery, Pieper and colleagues (2010) unveiled a novel neurotherapeutic compound. The P7C3-series of drug-like chemicals exerts potent neuroprotection in an assortment of CNS disease and injury states in which neurodegeneration and pathological cell death are hallmarks. P7C3 improved outcomes in several maladies with distinct pathologies, which suggests utilization as a neuroprotectant that may act on a central aspect of neuronal cell death pathways.
Some additional questions remain to be answered. First, TBI frequently results in polytrauma of multiple organ systems. Does systemic P7C3 administration have the potential to confer protection in other tissues? P7C3 protected neurons in regions outside of the brain (LoCoco et al., 2017; Kemp et al., 2015; Asai-Coakwell, et al., 2013; Tesla et al., 2012) and it would be interesting to investigate whether there is any effect on the peripheral or enteric nervous systems, or in other organs in which NAD+ levels play an important role in vitality after injury. Secondly, investigation of the treatment window for P7C3 administration after blast injury revealed lack of efficacy when treatment was initiated 48 hours after TBI (Yin et al., 2014). What is the significance of this treatment window with regard to the mechanisms underlying the neuroprotective effects of P7C3? Is P7C3 mediating some early-occurring event in cell death pathways? Furthermore, what downstream signaling cascades are P7C3-mediated NAMPT/NAD+ activating that implement the behavioral effect? Wang and colleagues (2014) made the groundbreaking revelation that P7C3 enhanced the prosurvival NAMPT-NAD+ axis in neurons. Parallel investigation of the contribution of NAMPT/NAD+ to positive outcomes in CNS injury implicates SIRT1 involvement in neuroprotection and neurogenic processes. Lastly, preliminary findings in our laboratory showed qualitative changes in posttraumatic neuroglia after P7C3–A20 treatment that may result from neuronal SIRT1 signaling cascades mitigating the pro-inflammatory environment after TBI. Additional investigations assessing downstream signaling of P7C3-mediated NAMPT/NAD+ enhancement, as well as changes in non-neuronal cell types in the posttraumatic environment, are crucial.
While the prospect of enhancing endogenous neuroprotective pathways such as NAMPTNAD+-SIRT1 axis in CNS injury is enticing, it is vital to recognize the full extent of how systemic administration of a novel exogenous compound might affect these tightly-regulated interactions, especially for extended periods of time. Furthermore, increased neurogenesis resulting from greater NSC survival could potentially have negative consequences. After TBI there is aberrant mossy fiber sprouting in the DG and reorganization of the hippocampal circuit, events that may contribute to the etiology of posttraumatic epilepsy (Neuberger et al., 2017; Atkins et al., 2010). Interestingly, Wang and colleagues (2014) showed that P7C3 administration had no effect on NAD+ levels in naive cells in which there was no dox-induced depletion of NAD+. In our experiments, there were no statistically significant differences between sham vehicle-treated groups and sham P7C3–A20-treated groups in any outcome measures. Why there is no detectable difference of NAMPT/NAD+ influence on naïve cells remains unclear.
Although preclinical work revealed a broad spectrum of encouraging findings with P7C3 administration after TBI (Table 1), there are limitations to the interpretation of these results. First, the majority of the aforementioned studies investigating P7C3 were carried out in rodent models of brain injury, or, in the case of LTP assessment, in hippocampal slice preparations in vitro. While there are significant overlaps in neuropathology, the rodent brain is different from the human brain, and there are challenges that need to be accounted for when investigating clinical therapies in this context (Marklund, 2016). Additional higher-order animal studies are imperative to establish therapeutic efficacy and translational potential. To this end, very recent work by Bauman and colleagues (in press) assessed long-term administration of P7C3–A20 in the nonhuman primate brain. They reported robust hippocampal neuroprotection and complete lack of toxicity in the nervous system and peripheral organs after 9 months of oral P7C3–A20 treatment, further extending credence to the translational potential of P7C3. Future directions in P7C3-TBI studies will need to ascertain whether functional neurogenesis occurs with P7C3 administration after TBI. Assessment of specific synaptic markers, immediate early genes, and/or evaluating the electrophysiological profiles of newborn hippocampal cells would illuminate whether P7C3-mediated survival fosters an increase in the number of newborn neurons actively participating in hippocampal neurocircuitry.
| TBI Model | Species | Treatment Paradigm | Outcomes | Reference |
|---|---|---|---|---|
| FPI | Rat | Twice daily 10 mg/kg P7C3–A20 IP 30 min post TBI for 7 days | Decreased contusion volume; protected pericontusional mature neurons; augmented adult hippocampal neurogenesis; improved neurobehavioral outcomes | Blaya et al., 2014 |
| FPI | Rat | Twice daily 10 mg/kg P7C3–A20 IP 30 min or 3 hr post TBI for 7 days | Preserved injury-induced reduction in LTP | Unpublished |
| CCI | Mouse | Twice daily 10 mg/kg P7C3–A20 IP 30 min post TBI for 7 days | Increased early-stage adult hippocampal neurogenesis | Unpublished |
| Blast | Mouse | 3, 10, and 30 mg/kg/day P7C3–S243 IP or oral 24 hrs post TBI for 11 days | Blocked axonal degeneration in brain; improved learning, memory, motor coordination; preserved synaptic plasticity | Yin et al, 2014 |
| FPI | Rat | Twice daily 10 mg/kg P7C3–A20 IP 30 min post TBI for 7 days | Reduced reactive phenotype of activated microglia, macrophages, astrocytes (qualitative) | Unpublished |
Replication studies are imperative for successful translation of preclinical findings to the clinic. Replication and reproducibility in translational biomedical research can be accomplished when independent groups obtain similar results and draw the same conclusions using near identical procedures (Macleod et al., 2014). Furthermore, the U.S. Food and Drug Administration requires at least two positive randomized controlled trials to demonstrate clinical efficacy of a new drug (U.S. Department of Health and Human Services, 1998). In this regard, P7C3–A20 is actively being tested by Operation Brain Trauma Therapy (OBTT), a multicenter, preclinical drug-screening consortium with the goal of parsing out candidates with the greatest therapeutic potential and streamlining them towards clinical trial (Kochanek et al., 2016). OBTT utilizes several TBI injury models across multiple, experienced laboratories to show efficacy and reproducibility in various neurological outcome measures.
While not having yet run the gauntlet of randomized controlled clinical trials to determine efficacy and safety for human consumption, toxicology studies and preclinical experimental work have been encouraging, most recently in the nonhuman primate. Thus, the highly potent scaffold and enhancement of neuroprotective NAMPT-NAD+-axis by P7C3 establishes two new compelling neurotherapeutic targets for the treatment of TBI.
Highlights.
Aminopropyl carbazole P7C3 was discovered via non-biased, in vivo screening
P7C3 compounds are nontoxic, orally available, and highly brain penetrant
P7C3 compounds enhance endogenous adult neurogenesis and protect mature neurons
In TBI, P7C3 compounds blocked neurodegeneration and improved neurological outcomes
P7C3 increases neuronal NAD+ levels and may promote prosurvival signaling
Acknowledgements:
The authors wish to acknowledge Mr. Yoandy Ferrer and Ms. Jessie Truettner for their immunohistochemical assistance.
Funding:
This work was supported by the NIH NINDS NS089443 and the Department of Defense W18XWH-14-1-0119.
Abbreviations
- AD
Alzheimer’s disease
- ALS
Amyotrophic lateral sclerosis
- BBB
Blood brain barrier
- BMAL1
Brain muscle ARNT-like protein 1
- BrdU
Bromodeoxyuridine
- cADPR
Cyclic ADP (adenosine diphosphate) ribose
- CCI
Controlled cortical impact
- CCSP3
Cleaved caspase 3
- CLOCK
Circadian locomotor output cycles kaput
- CNS
Central nervous system
- DCX
Doublecortin
- DG
Dentate gyrus
- fEPSPs
Field excitatory postsynaptic potential
- FGF
Fibroblast growth factor
- FPI
Fluid percussion injury
- GCL
Granular cell layer
- GFAP
Glia fibrillary-associated protein
- Iba1
Ionized calcium-binding adapter molecule 1
- ICV
Intracerebroventricular
- IP
Intraperitoneal
- IV
Intravenous
- LTP
Long-term potentiation
- MMP
Mitochondrial membrane permeabilization
- MPP-MML
Medial perforant path-medial molecular layer
- MWM
Morris water maze
- NAD
Nicotinamide adenine dinucleotide
- NAMPT
Nicotinamide phosphoribosyltransferase
- NeuN
Neuronal nuclei
- NMN
Nicotinamide mononucleotide
- NMNAT
Nicotinamide mononucleotide adenylyltransferease
- NPAS3
Neuronal PAS domain-containing protein 3
- NSC
Neural stem cell
- PARP
Poly ADP ribose polymerase
- SGZ
Subgranular zone
- SIRT1
Sirutin-1
- TEM
Transmission electron microscopy
- TBI
Traumatic brain injury
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ahmed S, Venigalla H, Mekala HM, Dar S, Hassan M, Ayub S. 2017. Traumatic Brain Injury and Neuropsychiatric Complications. Indian J Psychol Med. 39(2), 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH. 2014. Regulation and Function of Adult Neurogenesis: From Genes to Cognition. Physiol Rev. 94(4), 991–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. 2010. NAD+ Depletion Is Necessary and Sufficient for Poly (ADP-Ribose) Polymerase-1-Mediated Neuronal Death. J Neurosci. 30(8), 2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asai-Coakwell M, March L, Dai XH, Duval M, Lopez I, French CR, Famulski J, De Baere E, Francis PJ, Sundaresan P, Sauve Y, Koenekoop RK, Berry FB, Allison WT, Waskiewicz AJ, Lehmann OJ. 2013. Contribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophies. Hum Mol Genet. 22(7), 1432–42. [DOI] [PubMed] [Google Scholar]
- 5.Atkins CM, Truettner JS, Lotocki L, Sanchez-Molano J, Kang Y, Alonso OF, Sick TJ, Dietrich WD, Bramlett HM. 2010. Post-traumatic seizure susceptibility is attenuated by hypothermia therapy. Eur J Neurosci. 2, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauman MD, Schumann CM, Carlson EL, Taylor SL, Vázquez-Rosa E, Cintrón-Pérez CL, Shin MK, Williams NS, Pieper AA. (in press). Neuroprotective efficacy of P7C3 compounds in primate hippocampus. Transl Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaiss CA, Yu TS, Zhang G, Chen J, Dimchev G, Parada LF, Powell CM, Kernie SG. 2011. Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. Journal Neurosci. 31(13), 4906–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD. Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. 2014. J Neurotraum. 31(5), 476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. 1973. J Physiol. 232(2), 331–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, Hen R, Mann JJ. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell. 2018;22(4):589–99.e5. doi: 10.1016/j.stem.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bramlett HM, Dietrich DW. 2014. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J Neurotraum. 32(23), 1834–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryant RA, O’Donnell ML, Creamer M, McFarlane AC, Clark CR, Silove D. 2010. The psychiatric sequelae of traumatic injury. Am J Psychiat. 167(3), 312–20. [DOI] [PubMed] [Google Scholar]
- 13.Cameron HA, Glover LR. Adult Neurogenesis: Beyond Learning and Memory. 2015. Annu Rev Psychol. 66(1), 53–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Lu Y, Zhang Z, Wang J, Yang H, Liu G. 2015. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology. 145(4), 455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christian KM, Song H, Ming G-l. Functions and Dysfunctions of Adult Hippocampal Neurogenesis. Annual Review of Neuroscience. 2014;37(1):243–62. doi: 10.1146/annurev-neuro-071013-014134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark RS, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabo C. 2007. Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD+ depletion and improves water maze performance after traumatic brain injury in mice. J Neurotraum. 24(8), 1399–405. [DOI] [PubMed] [Google Scholar]
- 17.Colino A, Malenka RC.1993. Mechanisms underlying induction of long-term potentiation in rat medial and lateral perforant paths in vitro. J Neurophysiol. 69(4), 1150–9. [DOI] [PubMed] [Google Scholar]
- 18.DeKosky ST, Blennow K, Ikonomovic MD, Gandy S. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nature Reviews Neurology. 2013;9:192. doi: 10.1038/nrneurol.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng W, Aimone JB, Gage FH. 2010. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 11(5), 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dennis CV, Suh LS, Rodriguez ML, Kril JJ, Sutherland GT. 2016. Human adult neurogenesis across the ages: An immunohistochemical study. Neuropath Appl Neuro. 42(7), 621–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donmez G, Outeiro TF. 2013. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Molecular Medicine. 5(3), 344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. 1998. Neurogenesis in the adult human hippocampus. Nat Med. 4(11), 1313–7. [DOI] [PubMed] [Google Scholar]
- 23.Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, Possnert G, Druid H, Frisen J. 2014. Neurogenesis in the striatum of the adult human brain. Cell. 156(5), 1072–83. [DOI] [PubMed] [Google Scholar]
- 24.Espósito MS, Piatti VC, Laplagne DA, Morgenstern NA, Ferrari CC, Pitossi FJ, Schinder AF. 2005. Neuronal Differentiation in the Adult Hippocampus Recapitulates Embryonic Development. Journal Neurosci. 25(44), 10074–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fann JR, Burington B, Leonetti A, Jaffe K, Katon WJ, Thompson RS. 2004. Psychiatric illness following traumatic brain injury in an adult health maintenance organization population. Arch Gen Psychiatry. 61(1), 53–61. [DOI] [PubMed] [Google Scholar]
- 26.Faul M, Xu L, Wald MM, Coronado VG. 2010. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Death 2002–2006.
- 27.Folmes Clifford DL, Dzeja Petras P, Nelson Timothy J, Terzic A. 2012. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell. 11(5), 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gage FH. Mammalian neural stem cells. 2000. Science. 287(5457), 1433–8 [DOI] [PubMed] [Google Scholar]
- 29.Galvan V, Jin K. Neurogenesis in the aging brain. Clinical interventions in aging. 2007;2(4):605–10. Epub 2008/01/30. PubMed PMID: ; PMCID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao X, Deng-Bryant Y, Cho W, Carrico KM, Hall ED, Chen J. 2008. Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J Neurosci Res. 86(10), 2258–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green DR, Reed JC. 1998. Mitochondria and apoptosis. Science. 281(5381), 1309–12. [DOI] [PubMed] [Google Scholar]
- 32.Goldstein LE, Fisher AM, Tagge CA, Zhang X-L, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC. 2012. Chronic Traumatic Encephalopathy in Blast-Exposed Military Veterans and a Blast Neurotrauma Mouse Model. Sci Transl Med. 4(134), 134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haigis MC, Sinclair DA. 2010. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu Rev Pathol. 5(1):253–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hicks RR, Fertig SJ, Desrocher RE, Koroshetz WJ, Pancrazio JJ. 2010. Neurological Effects of Blast Injury. J Trauma Acute Care. 68(5), 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild Traumatic Brain Injury in U.S. Soldiers Returning from Iraq. 2008. N Engl J Med. 358(5), 453–63. [DOI] [PubMed] [Google Scholar]
- 36.Holloway KR, Calhoun TN, Saxena M, Metoyer CF, Kandler EF, Rivera CA, Pruitt K. 2010. SIRT1 regulates Dishevelled proteins and promotes transient and constitutive Wnt signaling. Proc Natl Acad Sci USA. 107(20), 9216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jesús-Cortés HD, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, Melito LM, Wang G, Williams NS, Ready JM, McKnight SL, Pieper AA. 2012. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc Natl Acad Sci USA. 109(42), 17010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Jesus-Cortes H, Miller AD, Britt JK, DeMarco AJ, De Jesus-Cortes M, Stuebing E, Naidoo J, Vazquez-Rosa E, Morlock L, Williams NS, Ready JM, Narayanan NS, Pieper AA. 2015. Protective efficacy of P7C3–S243 in the 6-hydroxydopamine model of Parkinson’s disease. NPJ Parkinsons Dis. 1, pii: 15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kemp SW, Szynkaruk M, Stanoulis KN, Wood MD, Liu EH, Willand MP, Morlock L, Naidoo J, Williams NS, Ready JM, Mangano TJ, Beggs S, Salter MW, Gordon T, Pieper AA, Borschel GH. 2015. Pharmacologic rescue of motor and sensory function by the neuroprotective compound P7C3 following neonatal nerve injury. Neuroscience. 284, 202–16. [DOI] [PubMed] [Google Scholar]
- 40.Kempermann G, Gage FH, Aigner L, Song H, Curtis MA, Thuret S, Kuhn HG, Jessberger S, Frankland PW, Cameron HA, Gould E, Hen R, Abrous DN, Toni N, Schinder AF, Zhao X, Lucassen PJ, Frisén J. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell. 2018;23(1):25–30. doi: 10.1016/j.stem.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. 2011. Physiology of microglia. Physiol Rev. 91(2), 461–553. [DOI] [PubMed] [Google Scholar]
- 42.Kim MY, Zhang T, Kraus WL. 2005. Poly(ADP-ribosyl)ation by PARP-1: `PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 19(17), 1951–67. [DOI] [PubMed] [Google Scholar]
- 43.Knoth R, Singec I, Ditter M, Pantazis G, Capetian P, Meyer RP, Horvat V, Volk B, Kempermann G. 2010. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One. 5(1):e8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kochanek PM, Bramlett HM, Shear DA, Dixon CE, Mondello S, Dietrich WD, Hayes RL, Wang KK, Poloyac SM, Empey PE, Povlishock JT, Mountney A, Browning M, Deng-Bryant Y, Yan HQ, Jackson TC, Catania M, Glushakova O, Richieri SP, Tortella FC. 2016. Synthesis of Findings, Current Investigations, and Future Directions: Operation Brain Trauma Therapy. J Neurotrauma. 33(6), 606–14. [DOI] [PubMed] [Google Scholar]
- 45.Kohler SJ, Williams NI, Stanton GB, Cameron JL, Greenough WT. Maturation time of new granule cells in the dentate gyrus of adult macaque monkeys exceeds six months. Proc Natl Acad Sci U S A. 2011;108(25):10326–31. Epub 2011/06/08. doi: 10.1073/pnas.1017099108. PubMed PMID: ; PMCID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kroemer G, Galluzzi L, Brenner C. 2007. Mitochondrial Membrane Permeabilization in Cell Death. Physiol Rev. 87(1), 99–163. [DOI] [PubMed] [Google Scholar]
- 47.Kuhn HG, Dickinson-Anson H, Gage FH. 1996. Neurogenesis in the Dentate Gyrus of the Adult Rat: Age-Related Decrease of Neuronal Progenitor Proliferation. J Neurosci. 16(6), 2027–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee AS, De Jesus-Cortes H, Kabir ZD, Knobbe W, Orr M, Burgdorf C, Huntington P, McDaniel L, Britt JK, Hoffmann F, Brat DJ, Rajadhyaksha AM, Pieper AA. 2016. The Neuropsychiatric Disease-Associated Gene cacna1c Mediates Survival of Young Hippocampal Neurons. eNeuro. 3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lie D-CC, Colamarino SA, Song H-JJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. 2005. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 437(7063), 1370–5. [DOI] [PubMed] [Google Scholar]
- 50.Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. 2004. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 44(1), 399–421. [DOI] [PubMed] [Google Scholar]
- 51.LoCoco PM, Risinger AL, Smith HR, Chavera TS, Berg KA, Clarke WP. 2017. Pharmacological augmentation of nicotinamide phosphoribosyltransferase (NAMPT) protects against paclitaxel-induced peripheral neuropathy. eLife. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loris ZB, Pieper AA, Dietrich WD. 2017. The neuroprotective compound P7C3–A20 promotes neurogenesis and improves cognitive function after ischemic stroke. Exp Neurol. 290, 63–73. [DOI] [PubMed] [Google Scholar]
- 53.Loris ZB, Hynton JR, Pieper AA, Dietrich WD. Beneficial Effects of Delayed P7C3–A20 Treatment After Transient MCAO in Rats. 2018. Transl Stroke Res. 9(2), 146–56 [DOI] [PubMed] [Google Scholar]
- 54.Macleod MR, Michie S, Roberts I, Dirnagl U, Chalmers I, Ioannidis JPA, Salman RA-S, Chan A-W, Glasziou P. Biomedical research: increasing value, reducing waste. The Lancet. 2014;383(9912):101–4. [DOI] [PubMed] [Google Scholar]
- 55.Madsen T, Erlangsen A, Orlovska S, Mofaddy R, Nordentoft M, Benros ME. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580–8. doi: 10.1001/jama.2018.10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marklund N Rodent Models of Traumatic Brain Injury: Methods and Challenges. Methods in molecular biology (Clifton, NJ). 2016;1462:29–46. Epub 2016/09/09. [DOI] [PubMed] [Google Scholar]
- 57.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27(8):1529–40. Epub 2010/05/28. doi: 10.1089/neu.2010.1358. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 58.Michaelson JJ, Shin MK, Koh JY, Brueggeman L, Zhang A, Katzman A, McDaniel L, Fang M, Pufall M, Pieper AA. 2017. Neuronal PAS Domain Proteins 1 and 3 Are Master Regulators of Neuropsychiatric Risk Genes. Biol Psychiatry. 82(3), 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michán S, Li Y, Chou MM-H, Parrella E, Ge H, Long JM, Allard JS, Lewis K, Miller M, Xu W, Mervis RF, Chen J, Guerin KI, Smith LEH, McBurney MW, Sinclair DA, Baudry M, de Cabo R, Longo VD. 2010. SIRT1 is essential for normal cognitive function and synaptic plasticity. J Neurosci. 30(29), 9695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mihaylova MM, Shaw RJ. 2011. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy, & metabolism. Nat Cell Biol. 13(9), 1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller BR, Hen R. 2015. The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol. 30, 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morris RGM. 1981. Spatial localization does not require the presence of local cues. Learn Motiv. 12(2), 239–60. [Google Scholar]
- 63.Muñoz-Gámez JA, Rodríguez-Vargas JM, Quiles-Pérez R, Aguilar-Quesada R, Martín-Oliva D, de Murcia G, de Murcia JM, Almendros A, de Almodóvar MR, Oliver FJ. 2009. PARP-1 is involved in autophagy induced by DNA damage. Autophagy. 5(1), 61–74. [DOI] [PubMed] [Google Scholar]
- 64.Naidoo J, Bemben CJ, Allwein SR, Liang J, Pieper AA, Ready JM. 2013. Development of a scalable synthesis of P7C3–A20, a potent neuroprotective agent. Tetrahedron Lett. 54(33), 4429–31. [Google Scholar]
- 65.Naidoo J, Jesus-Cortes HD, Huntington P, Estill S, Morlock LK, Starwalt R, Mangano TJ, Williams NS, Pieper AA, Ready JM. 2014. Discovery of a Neuroprotective Chemical, (S)-N-(3-(3,6-Dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(−)-P7C3–S243], with Improved Druglike Properties. J Med Chem. 57(9), 3746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osier ND, Dixon CE. 2016. The Controlled Cortical Impact Model: Applications, Considerations for Researchers, and Future Directions. Front Neurol. 7, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pickard BS, Pieper AA, Porteous DJ, Blackwood DH, Muir WJ. 2006. The NPAS3 gene--emerging evidence for a role in psychiatric illness. Annals of medicine. 38(6), 439–48. [DOI] [PubMed] [Google Scholar]
- 68.Pieper AA, Wu X, Han TW, Estill SJ, Dang Q, Wu LC, Reece-Fincanon S, Dudley CA, Richardson JA, Brat DJ, McKnight SL. 2005. The neuronal PAS domain protein 3 transcription factor controls FGF-mediated adult hippocampal neurogenesis in mice. Proc Natl Acad Sci USA. 102(39), 14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen C-H, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J, De Brabander J, Ready JM, McKnight SL. 2010. Discovery of a Proneurogenic, Neuroprotective Chemical. Cell. 142(1), 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pieper AA, McKnight SL, Ready JM. 2014. P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chemical Society reviews. 43(19), 6716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong H-K, Chong JL, Buhr ED, Lee C, Takahashi JS, Imai S-i, Bass J. 2009. Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+ Biosynthesis. Science. 324(5927), 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raval AP, Sick JT, Gonzalez GJ, Defazio RA, Dong C, Sick TJ. 2012. Chronic nicotine exposure inhibits estrogen-mediated synaptic functions in hippocampus of female rats. Neurosci Lett. 517(1), 41–6. [DOI] [PubMed] [Google Scholar]
- 73.Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, Povlishock JT. 1997. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 757(1), 119–32. [DOI] [PubMed] [Google Scholar]
- 74.Reeves TM, Lyeth BG, Povlishock JT. 1995. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 106(2), 248–56. [DOI] [PubMed] [Google Scholar]
- 75.Roozenbeek B, Lingsma HF, Maas AIR. 2012. New considerations in the design of clinical trials for traumatic brain injury. Clin Investig (Lond). 2(2), 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roozenbeek B, Maas AIR, Menon DK. 2013. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol. 9(4), 231–6. [DOI] [PubMed] [Google Scholar]
- 77.Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. 2008. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev. 7(2), 83–105. [DOI] [PubMed] [Google Scholar]
- 78.Santhakumar V, Ratzliff AD, Jeng J, Toth Z, Soltesz I. 2001. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol. 50(6), 708–17. [DOI] [PubMed] [Google Scholar]
- 79.Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick J, Szabo C, Clark RS. 2003. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem. 85(3), 697–708. [DOI] [PubMed] [Google Scholar]
- 80.Sanders MJ, Sick TJ, Perez-Pinzon MA, Dietrich WD, Green EJ. 2000. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 861(1), 69–76. [DOI] [PubMed] [Google Scholar]
- 81.Schinder AF, Gage FH. A Hypothesis About the Role of Adult Neurogenesis in Hippocampal Function. Physiology. 2004;19(5):253–61. doi: 10.1152/physiol.00012.2004 [DOI] [PubMed] [Google Scholar]
- 82.Simon DW, McGeachy MJ, Bayır H, Clark RSB, Loane DJ, Kochanek PM. 2017. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 13(3), 171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, Chang EF, Gutierrez AJ, Kriegstein AR, Mathern GW, Oldham MC, Huang EJ, Garcia-Verdugo JM, Yang Z, Alvarez-Buylla A. 2018. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature.555(7696), 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisen J. 2013. Dynamics of hippocampal neurogenesis in adult humans. Cell. 153(6), 1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stein LR, Imai S. 2014. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 33(12), 1321–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, Starwalt R, Morlock L, Naidoo J, Williams NS, Ready JM, McKnight SL, Pieper AA. 2012. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 109(42), 17016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Department of Health U.S. and Human Services, Food and Drug Administration. 1998. Guidance for Industry: Providing Clinical Evidence of Effectiveness for Human Drugs and Biological Products Office of the Federal Register, National Archives and Records Administration, Rockville, MD, pp. 27093–27094 [Google Scholar]
- 88.Verdin E 2015. NAD+ in aging, metabolism, and neurodegeneration. Science. 350(6265), 1208–13. [DOI] [PubMed] [Google Scholar]
- 89.Voorhees JR, Remy MT, Cintron-Perez CJ, El Rassi E, Khan MZ, Dutca LM, Yin TC, McDaniel LN, Williams NS, Brat DJ, Pieper AA. 2017. (−)-P7C3–S243 Protects a Rat Model of Alzheimer’s Disease From Neuropsychiatric Deficits and Neurodegeneration Without Altering Amyloid Deposition or Reactive Glia. Biol Psychiatry. doi: 10.1016/j.biopsych.2017.10.023 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Walker AK, Rivera PD, Wang Q, Chuang JC, Tran S, Osborne-Lawrence S, Estill SJ, Starwalt R, Huntington P, Morlock L, Naidoo J, Williams NS, Ready JM, Eisch AJ, Pieper AA, Zigman JM. 2015. The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatr. 20, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper Andrew A, Ready Joseph M, McKnight Steven L. 2014. P7C3 Neuroprotective Chemicals Function by Activating the Rate-Limiting Enzyme in NAD Salvage. Cell. 158(6), 1324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, Zhai QW, Su DF, Miao CY. 2011. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1‐dependent adenosine monophosphate–activated kinase pathway. Ann Neurol. 69(2), 360–74. [DOI] [PubMed] [Google Scholar]
- 93.Wang SN, Xu TY, Li WL, Miao CY. 2016. Targeting Nicotinamide Phosphoribosyltransferase as a Potential Therapeutic Strategy to Restore Adult Neurogenesis. CNS Neurosci Ther. 22(6), 431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, Liang Y, Vanhoutte PM. 2011. SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett. 585(7), 986–94. [DOI] [PubMed] [Google Scholar]
- 95.Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M. 2006. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci USA. 103(46), 17513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Winson J, Abzug C. 1977. Gating of neuronal transmission in the hippocampus: efficacy of transmission varies with behavioral state. Science. 196(4295), 1223–5. [DOI] [PubMed] [Google Scholar]
- 97.Won SJ, Choi BY, Yoo BH, Sohn M, Ying W, Swanson RA, Suh SW. 2012. Prevention of traumatic brain injury-induced neuron death by intranasal delivery of nicotinamide adenine dinucleotide. J Neurotraum. 29(7), 1401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang H, Gu Z-t, Li L, Maegele M, Zhou B-y, Li F, Zhao M, Zhao KS. 2016. SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway. Acta Pharmacol Sin. 38(2), 168–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang P, Zhang J, Shi H, Zhang J, Xu X, Xiao X, Liu Y. 2014. Developmental profile of neurogenesis in prenatal human hippocampus: an immunohistochemical study. Int J Dev Neurosci. 38:1–9 [DOI] [PubMed] [Google Scholar]
- 100.Yin TC, Britt JK, De Jesús-Cortés H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, Wassink C, McDaniel L, Newell EA, Dutca LM, Naidoo J, Cui H, Bassuk AG, Harper MM, McKnight SL, Ready JM, Pieper AA. 2014. P7C3 Neuroprotective Chemicals Block Axonal Degeneration and Preserve Function after Traumatic Brain Injury. Cell Rep. 8(6), 1731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yin TC, Voorhees JR, Genova RM, Davis KC, Madison AM, Britt JK, Cintron-Perez CJ, McDaniel L, Harper MM, Pieper AA. Acute Axonal Degeneration Drives Development of Cognitive, Motor, and Visual Deficits after Blast-Mediated Traumatic Brain Injury in Mice. eNeuro. 2016;3(5). Epub 2016/11/09. doi: 10.1523/eneuro.0220-16.2016. PubMed PMID: ; PMCID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang B, Chen X, Lin Y, Tan T, Yang Z, Dayao C, Liu L, Jiang R, Zhang J. 2011. Impairment of synaptic plasticity in hippocampus is exacerbated by methylprednisolone in a rat model of traumatic brain injury. Brain Res. 1382, 165–72. [DOI] [PubMed] [Google Scholar]
- 103.Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. 2012. Metabolic Regulation in Pluripotent Stem Cells during Reprogramming and Self-Renewal. Cell Stem Cell. 11(5), 589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao Y, Guan YF, Zhou XM, Li GQ, Li ZY, Zhou CC, Wang P, Miao CY. 2015. Regenerative Neurogenesis After Ischemic Stroke Promoted by Nicotinamide Phosphoribosyltransferase–Nicotinamide Adenine Dinucleotide Cascade. Stroke. 46(7), 1966–74. [DOI] [PubMed] [Google Scholar]
- 105.Zhao Y, Luo P, Guo Q, Li S, Zhang L, Zhao M, Xu H, Yang Y, Poon W, Fei Z. 2012. Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Exp Neurol. 237(2), 489–98. [DOI] [PubMed] [Google Scholar]