

Abstract
BACKGROUND AND AIMS:
In patients with chronic Helicobacter pylori (H pylori) infection, parietal and chief cell atrophy in the gastric corpus, a process known as spasmolytic polypeptide-expressing metaplasia (SPEM), increases the risk for progression to cancer. The relationship between H pylori and these metaplastic changes is unclear. We investigated whether H pylori localizes to regions of SPEM.
METHODS:
We developed an in situ adherence assay in which we incubated H pylori with free-floating tissue sections from the gastric corpora of mice; we assessed H pylori distribution along the gastric unit by immunofluorescence. We analyzed the interactions of H pylori with tissue collected from mice with acute SPEM, induced by high-dose tamoxifen. We also evaluated how adhesin-deficient H pylori strains, chemical competition assays, and epithelial glycosylation affected H pylori adhesion to SPEM glands. Finally, we colonized mice with the mouse-adapted PMSS1 strain and analyzed H pylori colonization in vivo during tamoxifen-induced SPEM or after reduction of stomach acid with omeprazole.
RESULTS:
Compared to uninjured glands, H pylori penetrated deep within SPEM glands, in situ, via interaction of its adhesin, SabA, with sLex, which expanded in SPEM. H pylori markedly increased gastric corpus colonization when SPEM was induced, but this proximal spread reversed in mice allowed to recover from SPEM. Reducing corpus acidity also promoted proximal spread. However, H pylori penetrated deep within corpus glands in vivo only when sLex expanded during SPEM.
CONCLUSIONS:
H pylori differentially binds SPEM glands in situ and in mice, in large part by interacting with sLex. Our findings indicate that H pylori expands its niche into the gastric corpus by promoting and exploiting epithelial metaplastic changes that can lead to tumorigenesis.
Keywords: paligenosis, intestinal metaplasia, patients, stomach cancer
Graphical Abstract
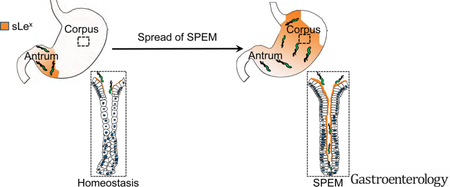
INTRODUCTION
Infection with the Gram-negative bacterium Helicobacter pylori (H pylori) remains the most significant risk factor for the development of gastric adenocarcinoma, one of the leading causes of cancer-related deaths worldwide1. An increased risk for progression to the often fatal endpoint of gastric neoplasia begins in patients who exhibit substantial loss of acid-secreting parietal cells, a process known as oxyntic atrophy, in the setting of chronic inflammation2. This chronic atrophic gastritis is associated with a reorganization of the gastric unit, characterized by increased proliferation of gastric progenitor cells in the isthmus of the gastric unit (between the upper pit/foveolar cells and the deeper cells of the gland) and the reprogramming of post-mitotic chief cells at the base of the gastric gland into a proliferating population of metaplastic cells3. The pattern of gastric unit reorganization during chronic atrophy is often referred to as spasmolytic polypeptide-expressing metaplasia (SPEM)4. It is thought to normally represent an evolutionarily conserved, transient alteration in the gastric unit of the proximal stomach (i.e., corpus) to facilitate repair and restoration of normal architecture in the face of deep glandular injury5. In some cases, however, restoration is blocked, and persistent SPEM can progress to gastric dysplasia6.
SPEM can expand topographically (anatomically) in the stomach via the gradual extension of an atrophic front, or a zone of progressive inflammation and metaplasia7. In humans, it is thought that SPEM first occurs at the transition between the antrum and corpus and moves proximally into the corpus, spreading most rapidly along the lesser curvature7, 8. The extension of the atrophic front into the gastric corpus has clinical significance. In particular, patients with H pylori-induced gastritis and atrophy/SPEM involving the gastric corpus are at a heightened risk of progressing to gastric adenocarcinoma9.
The ability of H pylori to expand its intra-gastric niche into the gastric corpus has often been attributed to a decrease in gastric acidity following chronic inflammation10; while H pylori can survive in acidic conditions, it does not effectively colonize the intensely acidic gastric corpus. At the glandular level, H pylori attaches to gastric epithelium primarily through the binding of two bacterial adhesins, BabA11 and SabA12, to the glycosylated receptors Lewis B (Leb) and sialyl-Lewis X (sLex), respectively, on host epithelium. How the dynamic equilibrium between host and microbial factors determines the topographic (i.e., corpus versus antrum) distribution of H pylori, a critical determinant of neoplastic risk, has not been systematically explored. Notably, in patients demonstrating extensive corpus atrophy/metaplasia, it remains unclear whether H pylori expands into the corpus due to changes in epithelial expression patterns characteristic of SPEM, with parietal cells and chief cells being replaced by metaplastic cells, or whether H pylori simply extends into the relatively less acidic environment resulting from the gradual loss of parietal cells.
In this study, we attempt to address for the first time the mechanism(s) behind H pylori’s expansion into the atrophic corpus and its direct interactions with SPEM glands. We first take advantage of a validated gastric injury model that rapidly, synchronously, and reversibly induces SPEM throughout the corpus13, 14. Using a novel in situ binding assay where we can manipulate host and microbial factors, we find that H pylori differentially binds deep within SPEM glands compared to normal corpus glands and that this binding is in part dependent on the interaction of SabA with sLex. We expand our in situ assay to a mouse model of H pylori colonization to demonstrate that the SabA-sLex interaction also determines H pylori’s ability to extend into the corpus and bind deep within SPEM glands in vivo. Finally, we demonstrate that omeprazole-mediated inhibition of gastric acid allows H pylori to spread to the corpus but without penetrating deeper within corpus glands. Our results elucidate a mechanism by which H pylori exploits the metaplastic changes within the corpus that accompany chronic inflammation to expand its niche at a topographic and glandular level. As the differentiation changes associated with metaplasia pose a risk for the accumulation of mutations8, 15, H pylori’s tropism for SPEM glands could contribute to the establishment of a pre-neoplastic gastric milieu.
MATERIALS AND METHODS
Ethical Permissions
All human gastric specimens were obtained with approval from the Institutional Review Board of Washington University School of Medicine. All experiments involving animals were performed according to protocols approved by the Washington University School of Medicine Animal Studies Committee. Mice were maintained in a specified pathogen-free barrier facility under a 12-hour light cycle.
In Situ H pylori adherence assay
Male and female littermates were given two daily intra-peritoneal injections with either vehicle or high-dose tamoxifen (HD-Tam; 5 mg/20 g body weight), as previously described16. Mice were sacrificed one or fourteen (recovery) days after the last injection. Mouse stomachs were excised, inflated with 4% paraformaldehyde (PFA), and fixed overnight at 4°C with gentle rocking. The next day, the forestomach was removed, and stomachs were cut open along the lesser curvature prior to embedding in 4% low-melting agarose. 100-µm longitudinal sections from the gastric corpus were cut using a vibratome (Leica Biosystems, Vista, CA) and blocked overnight at 4°C in phosphate-buffered saline (PBS) containing 3% bovine serum albumin. Free-floating sections were incubated with overnight H pylori cultures (see Supplemental Methods) diluted in blocking buffer to an OD600 of 0.05, unless otherwise indicated. Sections were incubated in each well of a 48-well plate (Greiner Bio-One, Monroe, NC) for 1 hour at 37°C and 5% CO2 with gentle shaking (30 rpm), then transferred to a new well and washed three times with sterile PBS prior to fixing in 4% PFA for 1 hour at room temperature.
Immunofluorescence
Thick (100-µm), fixed sections were blocked for 2 hours in blocking buffer (PBS, 3% bovine serum albumin, 1% Triton X-100, 1% saponin17), then incubated overnight at 4°C in the appropriate primary antibody. Sections were washed and incubated in the appropriate secondary antibodies for 2 hours at room temperature. Sections were then incubated in Hoescht stain (1:20,000; Thermo Fisher Scientific) for 30 minutes at room temperature, transferred to a microscope slide, and mounted using ProLong Gold antifade reagent (Thermo Fisher Scientific). Images were obtained using the Olympus FV1200 confocal microscope (Olympus, Waltham, MA), and z-stacks were reconstructed into three-dimensional images using Amaris software (Thermo Fisher Scientific).
Assessing Depth of Penetration
Intra-glandular bacteria were determined by confocal microscopy. The number of glands containing intra-glandular H pylori bacteria located at or below the level of Griffonia simplicifolia lectin (GSII) staining (below the pit region) was expressed as a percentage of the total colonized glands. Approximately 80–100 H pylori-colonized glands were counted for each of three consecutive, independent experiments. For colonized glands containing more than one bacterium, the location of the deepest bacterium was counted.
Determining Topographic Distribution of H pylori
Male and female littermates aged 6–8 weeks were infected with PMSS1 (~1 × 108 cfu/mouse) for 6 weeks, after which they were intra-peritoneally injected with vehicle or HD-Tam, as described above. For omeprazole treatment, mice were infected for 6 weeks, then gavaged daily for one week with 200 µL of either saline (Hospira Inc., Lake Forest, IL) or an omeprazole slurry (Sigma; 1.5 mg/20 g body weight resuspended in saline) prior to sacrifice. For all treatments, mouse stomachs were excised and cut open along the lesser curvature. The forestomach was removed, food was gently scraped away, and exposed stomachs were cut into three equal horizontal sections: proximal corpus (closest to forestomach), distal corpus, and antrum (closest to intestine). Each gastric section was placed in sterile Brucella broth, weighed, and homogenized for 30 seconds in N°10 Medicons (Beckto n Dickinson, San Jose, CA). Dilutions were plated on selective media plates (tryptic soy agar with 5% sheep blood) containing amphotericin (2 µg/mL), bacitracin (30 µg/mL), nalidixic acid (10 µg/mL), and vancomycin (20 µg/mL). Plates were incubated under micro-aerophilic conditions in Ziploc bags containing GasPak EZ sachets (Becton Dickinson and Company, Franklin Lakes, NJ) at 37°C for 5–7 days. Colonies were counted and expressed as colony-forming units per gram of stomach (cfu/g stomach).
Statistical Analysis
All graphs and statistical analyses were done using GraphPad Prism (GraphPad, La Jolla, CA). Means among multiple (> 2) treatments were compared using one-way analysis of variance with the Dunnett’s test to determine significance. When calculating bacterial densities in infected mice, the adjusted means and standard deviations were calculated for replicates that fell below the level of detection using Cohen’s method18. Means for bacterial densities and glandular distributions in saline- versus omeprazole-treated mice were compared using a one-tailed Student’s t test. P < 0.05 was considered statistically significant. Where indicated, *, p< 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; n.s., not significant (p > 0.05).
RESULTS
H pylori differentially binds to SPEM glands in situ
We first sought to use a controlled in situ system to study how H pylori differentially interacts with atrophic/SPEM-type metaplastic corpus. To generate metaplastic tissue for our assay, we employed an acute, drug-induced gastric injury model that causes nearly all parietal cells to die and corpus units to undergo SPEM within three days of injury13. Equally important, this synchronous injury system is reversible, with near complete recovery to normal corpus within two weeks. As previously reported14, intra-peritoneal administration of high-dose tamoxifen (HD-Tam; 5 mg/20 g body weight16) to wild-type C57BL/6 mice results in a rapid induction of SPEM throughout the mouse stomach, characterized by the co-expression of the murine chief cell marker, gastric intrinsic factor (GIF), and the mucous neck cell marker, GSII, at the gland base (Figures 1A–B). HD-Tam induced similar gastric injury in male and female mice, as has been previously reported13. The acute loss of acid-secreting parietal cells in HD-Tam-treated mice (Supplemental Figure 1A) resulted in mucosal atrophy and pallor, evidenced by a loss in the anatomic distinction between corpus and antrum (Supplemental Figure 1B) as well as by a rise in intra-gastric pH (Supplemental Figure 1C). The histologic, morphologic, and functional changes associated with HD-Tam-induced SPEM were reversible, as glands recovering from HD-Tam injury reverted to the vehicle-treated phenotype at the glandular and gross anatomic levels (Figure 1C and Supplemental Figure 1B).
Figure 1. H pylori differentially binds SPEM glands in situ.
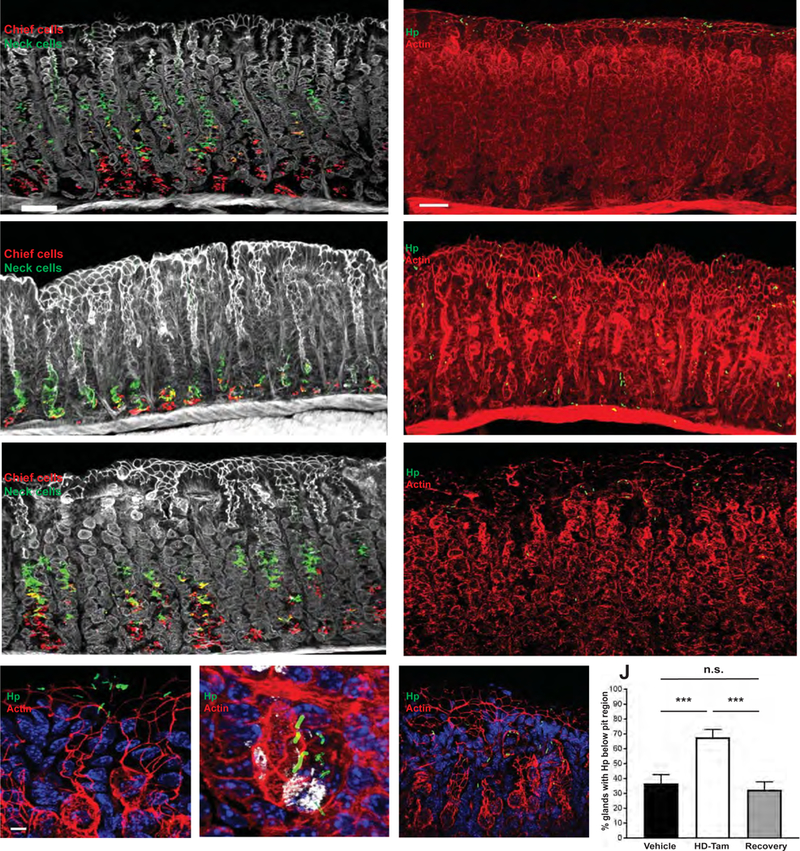
(A-C) HD-Tam treatment rapidly induces co-expression of chief cell (red) and mucous neck cell (green) markers at the base of corpus glands, changes characteristic of SPEM, but this effect is lost following recovery from SPEM. Thick gastric corpus sections from mice intra-peritoneally injected with either vehicle (A) or HD-Tam (B), or allowed to recover (C; see Methods) were imaged by confocal microscopy. White, actin. Scale bar, 50 µm. (D–F) H pylori is able to bind deep within SPEM glands (E) but is largely restricted to the pits of vehicle-treated (D) or recovered (F) glands. Scale bars, 50 µm. (G–I) Magnified, representative images showing H pylori within the pits of vehicle-treated (G) and recovered glands (I) but near the base of HD-Tam-treated glands (H). GSII (white) labels the base of SPEM glands. Scale bars, 3 µm. For (D–I), blue labels nuclei, red labels actin. (J) Data show the mean (±SD) percentage of glands containing H pylori below the pit region under the different treatment conditions from three consecutive, independent experiments. Hp, H pylori.
The rapidity and reversibility of the HD-Tam injury system allowed us to assess the direct interactions between H pylori and the reconfigured gastric epithelium by examining H pylori’s interaction with gastric tissue harvested both during SPEM and after recovery from injury. Previous attempts at studying H pylori adherence to gastric epithelium largely relied on incubating slides containing thin, deparaffinized sections of murine or human gastric tissue with H pylori19, 20. We developed a novel in situ adherence assay using thick gastric sections. Compared to previous studies, the use of thicker, free-floating, 100-µm sections facilitated analysis of depth of penetration and enhanced spatial resolution of H pylori binding to gastric epithelium. While in situ binding of the mouse-adapted PMSS1 isolate of H pylori was largely restricted to the pit region of vehicle-treated glands, H pylori were able to bind more deeply along the gastric unit axis in SPEM glands, and this phenotype was lost when H pylori were incubated with gastric glands from mice that had recovered from SPEM (Figures 1D–I).
To quantify the differences in the depth of H pylori binding to normal versus metaplastic glands, we determined the number of glands containing H pylori adherent to the gastric unit deep to the pit region (i.e., within the glandular region of the corpus unit) and normalized to the total number of glands containing bound H pylori. Immunofluorescent images were analyzed by confocal microscopy to ensure that H pylori deep within a gland were indeed within the glandular lumen (Supplemental Movie 1). For glands containing more than one H pylori bacterium, the location of the intra-glandular bacterium found deepest within the gastric unit was scored. H pylori more frequently bound below the pit/foveolar region of SPEM glands (HD-Tam) compared to vehicle-treated glands (68% vs 37%, respectively; p < 0.001; Figure 1J). The depth of binding of H pylori to glands that had recovered from SPEM was not significantly different from the pattern seen with vehicle-treated glands (32% vs 37%, respectively; p > 0.05; Figure 1J). Taken together, these findings demonstrate that H pylori binds deeper within SPEM glands in situ and that this binding is reversible following recovery from HD-Tam-induced glandular injury.
Sialyl-Lewis X (sLex) expression is expanded in SPEM
We next hypothesized that H pylori’s differential binding to SPEM glands was the result of the enhanced expression of a receptor for H pylori on host gastric epithelium following HD-Tam injury. H pylori has been shown to adhere to gastric epithelium through the binding of two of its adhesins, BabA and SabA, to the blood group antigen Lewis b (Leb) and sialylated Lewis X (sLex), respectively21. No difference in the pattern of Leb expression was seen between vehicle- and HD-Tam-treated glands, suggesting that HD-Tam-induced SPEM did not alter glandular Leb expression (Figure 2A). SPEM induction, however, drastically enhanced the expression of sLex, which extended deep into the metaplastic neck and gland base, identified by GIF and GSII co-staining (Figure 2B). This staining pattern was lost following recovery from HD-Tam injury, with the scant sLex expression in recovered glands resembling that of vehicle treatment.
Figure 2. SPEM induces a reversible expansion of sLex along the corpus gland axis.
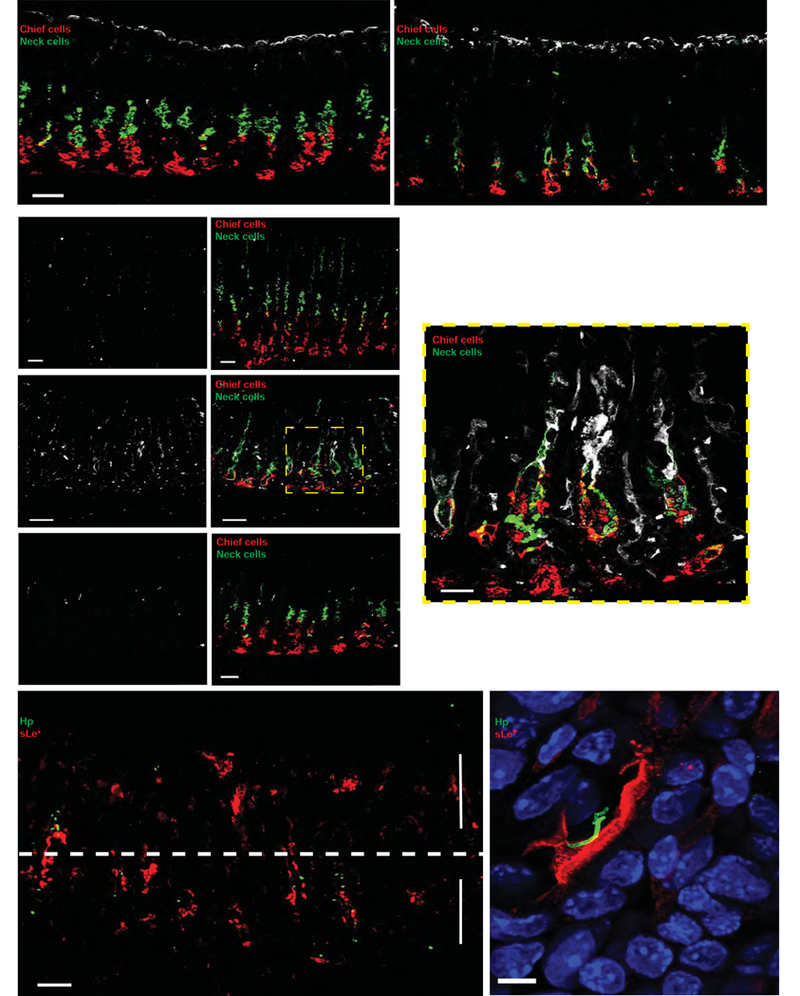
(A) Leb expression (white) is unchanged following HD-Tam treatment. Representative 100-µm sections of mouse stomachs treated with either vehicle (left) or HD-Tam (right). Scale bars, 50 µm. (B) sLex expression (white) extends along the length of SPEM glands (HD-Tam) and co-localizes with metaplastic cells co-expressing chief cell (red) and mucous neck cell (green) markers. This pattern of expression is lost following recovery from HD-Tam injury. Left panels show isolated sLex signal from the corresponding, merged right panels. Scale bars, 50 µm (20 µm for boxed image). (C) H pylori co-localizes with expanded sLex in SPEM glands in situ. H pylori (green) penetrate below the pits of SPEM glands, with sLex (red) extending along the gland length. The pit and gland regions are highlighted by arrows and separated by a dotted line. Scale bar, 50 µm. (D) Representative image of H pylori (green) co-localizing with sLex-expressing epithelium (red). Blue, nuclei. Scale bar, 5 µm. Hp, H pylori.
H pylori binding to SPEM glands is α-linkage-specific and is mediated by binding of SabA to sLex
In our in situ adherence assay, H pylori co-localized with epithelial cells showing increased sLex expression within SPEM glands (Figures 2C–D). To demonstrate that the binding of H pylori to SPEM glands in situ was mediated by the direct interaction of SabA with its cognate receptor, sLex, we incubated SPEM glands with H pylori in the presence of one of two glycoconjugates that differentially binds terminal sialic acid residues on gastric epithelium. SabA has been previously shown to bind α−2,3-linked terminal sialic acid residues on sLex 22. Consistent with this, increasing concentrations of the glycoconjugate 3’-sialyllactose, which preferentially binds α−2,3-linked terminal sialic acid residues, significantly inhibited the ability of H pylori to bind deep within SPEM glands in a dose-dependent manner (Figure 3). However, an enantiomeric glycoconjugate, 6’-sialyllactose, which preferentially binds α−2,6-linked terminal sialic acid residues, had no effect on the ability of H pylori to bind deep within SPEM glands, suggesting that the binding of H pylori to SPEM glands in situ is α-linkage-specific. Similarly, the ability of H pylori to penetrate deep within SPEM glands was abolished if the tissue sections were pre-treated with neuraminidase, which specifically cleaves terminal sialic acid residues; accordingly, neuraminidase pre-treatment also significantly reduced binding of Maackia amurensis lectin, which recognizes α−2,3-linked terminal sialic acid residues on sLex, to SPEM glands (Supplemental Figure 2).
Figure 3. The binding of H pylori to SPEM glands in situ is α-linkage-specific.
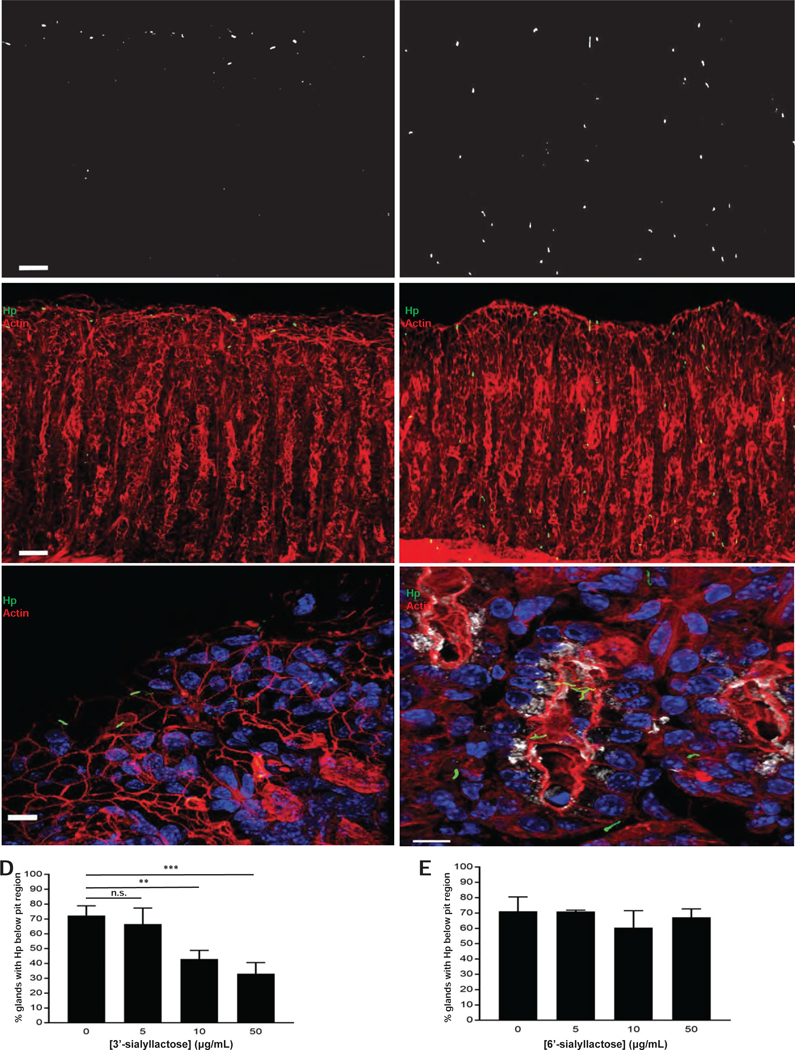
(A–C) 3’-sialyllactose (left), but not 6’-sialyllactose (right), prevents penetration of H pylori (green) into SPEM glands. Images in (A) represent isolated H pylori signal from the corresponding, merged images in (B). Magnified images in (C) demonstrate that H pylori is restricted to the pit in the presence of 3’- sialyllactose but able to access deeper portions of SPEM glands, highlighted by GSII (white), in the presence of 6’-sialyllactose. Blue, nuclei; red, actin. Scale bars, 50 µm for (A) and (B), 5 µm for (C). (D–E) Data show the mean (±SD) percentage of glands containing H pylori below the pit region under the different treatment conditions from three consecutive, independent experiments. For (E), there was no statistically significant difference among any of the tested concentrations relative to the untreated condition. Hp, H pylori.
Because sLex availability on host gastric tissue affected H pylori binding to SPEM glands, we wanted to further confirm the role of the H pylori adhesin, SabA, in binding metaplastic gastric tissue. We assessed the in situ binding of a wild-type clinical H pylori strain, NSH57, or isogenic mutants lacking either SabA (∆sabA), BabA (∆babA), or both SabA and BabA (∆sabA/∆babA). These strains have been previously characterized23, and no differences were seen among the strains in terms of in vitro growth or motility (not shown). Similar to the PMSS1 strain (Figure 1E), the wild-type NSH57 strain adhered deep within SPEM glands (Figure 4A), though the frequency at which NSH57 was found below the pit region (53%; Figure 4E) was slightly lower than that observed with the PMSS1 strain (68%; Figure 1J). Nonetheless, the ∆sabA mutant could not bind deep within SPEM glands as effectively as its isogenic wild-type counterpart (35% vs 53%, respectively; p < 0.05; Figures 4B and 4E). Notably, the ∆babA mutant bound deep within SPEM glands to a similar extent as the wild-type, isogenic NSH57 strain (Figures 4C and 4E), while the mutant lacking both adhesins (∆sabA/∆babA) showed no appreciable binding to SPEM glands in situ (i.e., fewer than 5% of glands showed bound H pylori anywhere along the gland axis; Figures 4D–E). Taken together, these results suggest that H pylori binding to SPEM glands is an active and specific process that depends on both of the H pylori adhesins, BabA and SabA, but the ability to penetrate deep within SPEM glands depends in large part on the ability of SabA to bind sLex.
Figure 4. The ability of H pylori to bind deep within SPEM glands is largely mediated by SabA.
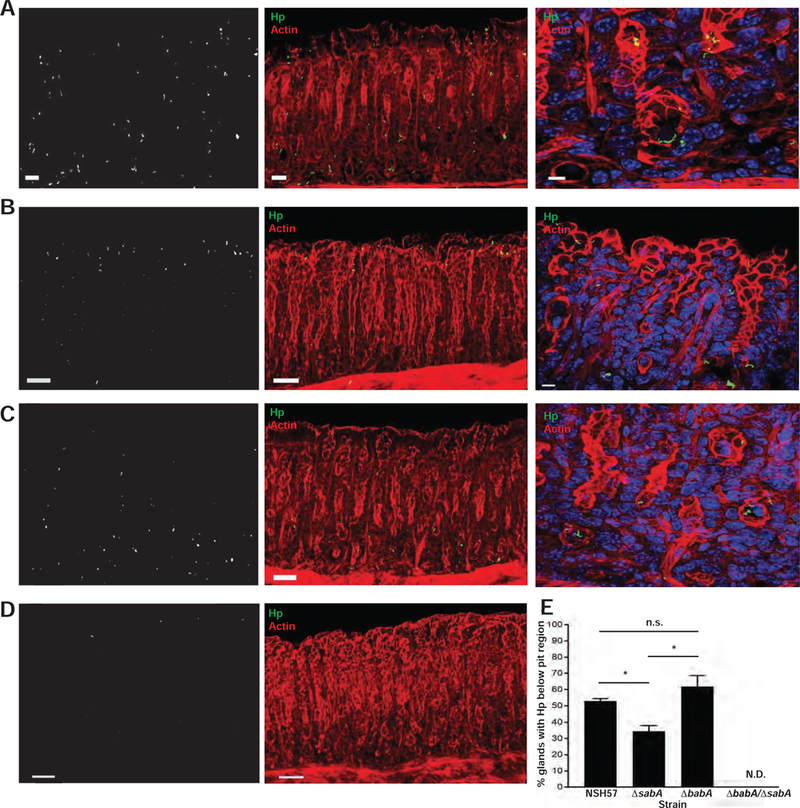
(A–D) The NSH57 strain (A), or corresponding isogenic mutants (B–D) were incubated with thick gastric corpus sections from mice treated with HD-Tam. The absence of sabA, but not babA, impairs the ability of H pylori to bind deep within SPEM glands. The double mutant lacking both babA and sabA is unable to effectively bind SPEM glands. Left panels show the isolated H pylori signal from the corresponding middle panels. Right panels show magnified images of the corresponding H pylori strains (NHS57 and ∆babA) binding deep within the gland (labeled base) or confined to the pit (∆sabA). Blue, nuclei; green, H pylori; red, actin. Scale bars, 50 µm (left and middle panels), 5 µm (right panels). (E) Data show the mean (±SD) percentage of glands containing H pylori below the pit region for each H pylori strain from two consecutive, independent experiments. The ∆babA/∆sabA double mutant bound fewer than 5% of glands and was not scored (N.D., not determined). Hp, H pylori.
SPEM affects the topographic and glandular distribution of H pylori in vivo
To demonstrate that the interactions observed in situ were important for H pylori pathogenesis in vivo, we first examined mice that had been chronically infected with PMSS1 and noticed an expansion of sLex expression in glands undergoing SPEM (Figures 5B–C), similar to the expression pattern seen in HD-Tam-induced SPEM (Figure 2B). Moreover, this pattern of sLex expression was restricted to injured corpus glands undergoing SPEM and was not a non-specific mucosal response to chronic H pylori infection, as sLex expression was not enhanced in uninjured, non-colonized corpus mucosa within the same infected mouse stomach (Figure 5A). Of note, HD-Tam-induced SPEM did not cause the expansion of foveolar/pit cells observed in SPEM induced by chronic inflammation; hence, H pylori -infected gastric units with SPEM showed longer spans of sLex-expressing foveolar regions.
Figure 5. Gastric epithelial expression of sLex is expanded in chronic inflammatory conditions in which SPEM occurs.
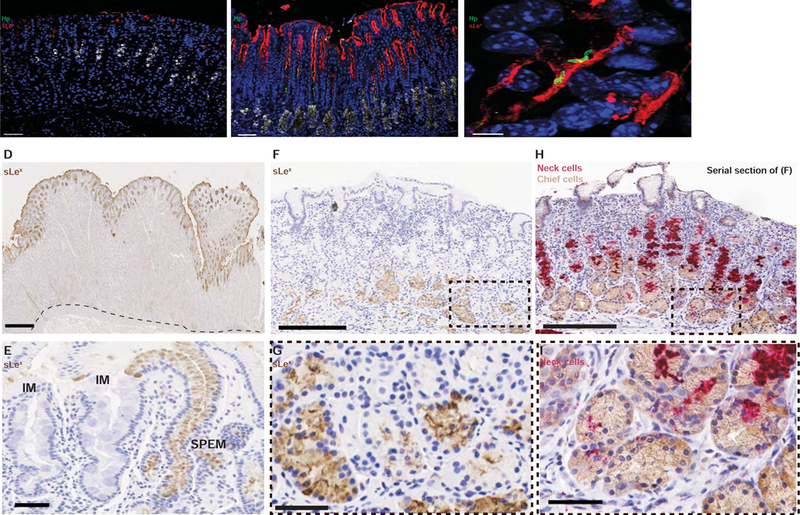
(A–B) Representative images of a mouse infected with H pylori (green) for 6 weeks show enhanced sLex expression (red) in colonized glands (B) but scant expression in areas of non-colonized mucosa (A). GSII (white) is in its usual distribution in non-colonized mucosa but at the base in colonized mucosa, indicating SPEM. Scale bars, 50 µm. (C) H pylori (green) co-localizes with sLex-expressing gastric epithelium (red) in vivo. Scale bar, 5 µm. For (A–C), blue marks nuclei. Hp, H pylori. (D–I) Histologic sections from human gastric specimens. (D) Normal, H pylori-negative gastric tissue from a 36 year-old patient who underwent a bariatric sleeve gastrectomy, stained against sLex (brown). The dotted line shows the interface between the gastric epithelium and the muscularis mucosa. Scale bar, 500 µm. (E) Representative image from a gastric biopsy of a 71 year-old H pylori-infected patient with chronic atrophic gastritis and focal intestinal metaplasia. sLex staining (brown) highlights an area of foveolar hyperplasia in a SPEM gland but is excluded from areas of intestinal metaplasia (IM). Scale bar, 50 µm. (F–I) Representative images from a gastric biopsy of a 67 year-old H pylori-infected patient with chronic atrophic gastritis, demonstrating sLex staining (F,G) restricted to the bases of SPEM glands, marked by the co-expression of the chief cell (brown) and mucous neck cell (red) markers (panels H and I). (G) and (I) represent magnified images of (F) and (H), respectively. For (F) and (H), scale bars 200 µm; for (G) and (I), scale bars 50 µm.
Similar to the mouse, we also consistently observed that in regions of human SPEM/atrophy, sLex expanded into the bases of SPEM glands (Figures 5F–I and Supplemental Figures 5B–D). In uninjured human glands, sLex was confined to the surface and shallow foveolar regions without extending deep within the gastric unit (Figure 5D and Supplemental Figure 5A). sLex was also not expressed in regions of intestinal metaplasia (Figures 5E and Supplemental Figure 5C). A summary of the sLex staining pattern in human gastric specimens with varying histopathology is presented in Supplemental Table 1.
Given that glandular sLex is expanded in SPEM glands and that H pylori reversibly binds SPEM glands through its interactions with sLex, we reasoned that the presence of SPEM could determine H pylori’s gastric colonization patterns during chronic infection. Taking advantage of the fact that HD-Tam induces synchronous SPEM throughout the gastric corpus and that this injury is reversible, we could assess H pylori colonization patterns following manipulation of the gastric landscape. We developed a method for assessing H pylori colonization of distinct topographic regions of infected mouse stomachs in the presence or absence of SPEM. Male and female littermate mice were infected with PMSS1 for 6 weeks, intra-peritoneally injected with vehicle or HD-Tam for 2 days, then sacrificed either one or fourteen days after the last injection. Excised stomachs were cut open along the lesser curvature, and H pylori was isolated and quantified from three distinct regions of the mouse stomach (see Methods). Treatment with HD-Tam allowed H pylori to more effectively colonize the proximal and distal regions of the gastric corpus compared to vehicle-treated mice (Figures 6A–B). HD-Tam injury appeared to have no effect on H pylori density within the gastric antrum (Figure 6C), consistent with SPEM representing an injury response of the gastric corpus8. More importantly, this pattern of colonization was reversible, with H pylori redistributing away from the proximal corpus following recovery from HD-Tam injury. These findings suggest that the presence of SPEM dictates H pylori’s ability to colonize more proximal regions of the gastric corpus and that H pylori redistributes away from the corpus following recovery from SPEM.
Figure 6. SPEM dictates the topographic and glandular distribution of H pylori in vivo.
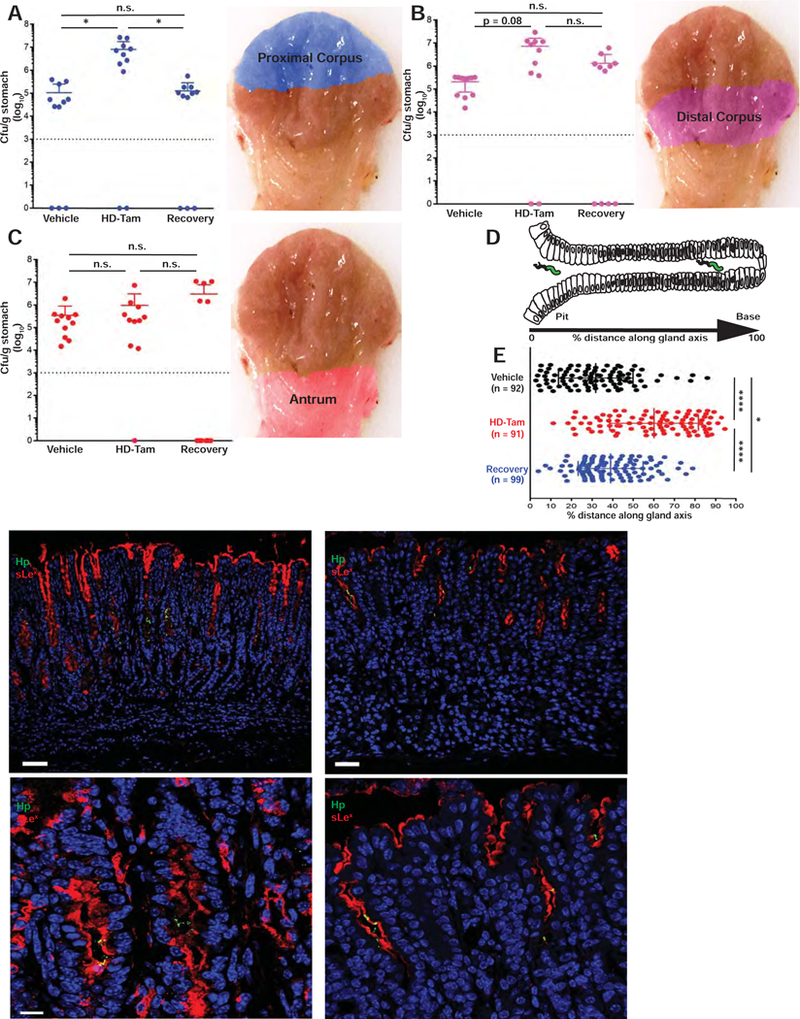
(A-C) Induction of SPEM allows H pylori to more effectively colonize proximal regions of the gastric corpus, and H pylori redistributes away from the corpus following recovery from injury. Data represent the mean (±SD) bacterial densities from three pooled, consecutive, independent experiments. Each data point represents a biological replicate. Pseudo-colored anatomic regions used for analysis are shown on right. Dotted line represents the limit of detection. The difference in colonization of the distal corpus between vehicle and HD-Tam treatments shows a trend toward statistical significance (p = 0.08). n.s., not significant (p > 0.1). (D–E) The onset of SPEM alters the glandular distribution of H pylori in vivo. The distance of H pylori along the gland axis was expressed as a percentage of the total gland length (D). (E) Data represent the mean (±SD) glandular distribution of H pylori from three infected littermates/cagemates for each treatment condition. Each data point represents a gland colonized with H pylori, with the total numbers of scored glands indicated in parentheses. (F–G) The expansion of sLex (red) and ability of H pylori (green) to bind deep within gastric glands are lost following recovery from SPEM. Scale bars, 50 µm. (H–I) Magnified representative images showing H pylori co-localizing with sLex-expressing gastric epithelium, with H pylori binding deeper within SPEM glands following HD-Tam treatment but restricted to the pit following recovery from HD-Tam. Scale bars, 5 µm. For (F–I), blue highlights nuclei. Hp, H pylori.
While the topographic, or anatomic, distribution of H pylori within the infected stomach has clinical significance, the distribution of H pylori within colonized glands may be equally relevant for the host epithelial response and the establishment of a pre-neoplastic micro-environment. To assess how SPEM altered the glandular distribution of H pylori in vivo, we analyzed paraffin-embedded gastric corpus sections from chronically infected, vehicle- or HD-Tam-treated mice (see Supplemental Methods). The distance of H pylori along the gland axis was expressed as a percentage of the total gland length under the different treatment conditions (Figure 6D). For the recovery experiment, mice were sacrificed fourteen days after the last HD-Tam injection. Similar to our in situ findings (Figures 1D–J), H pylori was able to access deeper portions of metaplastic corpus glands following the induction of SPEM with HD-Tam. However, the H pylori foray deep within injured corpus glands was reversed in mice allowed to recover from SPEM (Figure 6E). In further corroboration of our in situ findings, sLex expression expanded along the gland unit in H pylori -colonized SPEM glands following HD-Tam treatment, while in stomachs that had recovered from SPEM and were no longer colonized deep within the gland, sLex staining reverted to the vehicle control pattern (Figures 6F–I).
SPEM, and not inhibition of acid secretion, determines H pylori’s glandular distribution in vivo
Treatment with HD-Tam induces a rapid loss of parietal cells (Supplemental Figure 1A) and an expected rise in intra-gastric pH (Supplemental Figure 1C). It has previously been hypothesized that it is the change in intra-gastric pH that allows for the proximal expansion of H pylori from the antrum into the corpus24. Thus, we decided to formally test the relative contribution of metaplasia and acid to H pylori colonization. To test the effect of gastric pH on H pylori colonization patterns, PMSS1-infected littermates were gavaged daily with either saline or omeprazole. Regional bacterial densities were determined, as previously described. Similar to HD-Tam treatment, omeprazole treatment significantly enhanced H pylori colonization of the proximal and distal corpus compared to saline controls (Figures 7A–B), with no significant effect on colonization of the antrum (Figure 7C). However, omeprazole alone failed to induce SPEM (Supplemental Figure 3). Accordingly, unlike HD-Tam (Figure 7E), omeprazole treatment did not stimulate an expansion of sLex expression deep within the corpus gland axis (Figure 7D). Moreover, consistent with the pit-restricted sLex expression pattern following omeprazole treatment, the glandular distribution of H pylori in omeprazole-treated mice was not significantly different from saline-treated littermates (Figure 7G). These findings indicate that H pylori’s colonization of the gastric corpus may be impeded by acid, but it is specifically the presence of SPEM that dictates H pylori’s ability to penetrate deep within corpus units.
Figure 7. Inhibition of gastric acid alters the topographic, but not the glandular, distribution of H pylori in vivo.
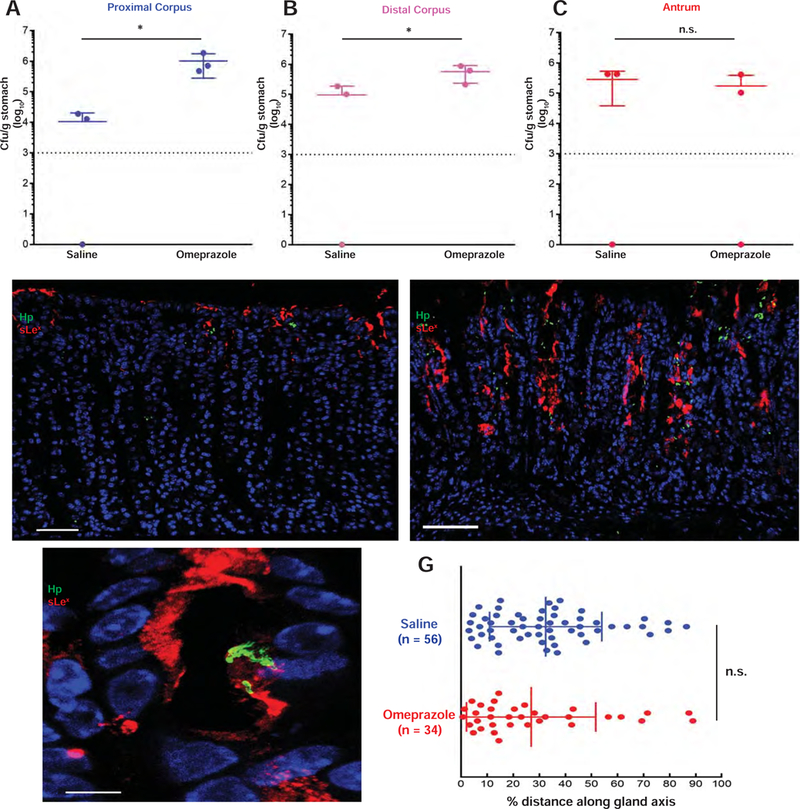
(A–C) A representative experiment showing the mean (±SD) bacterial densities from distinct anatomic regions of mice treated with saline or omeprazole. Each data point represents a biological replicate. The dotted line represents the limit of detection. (D–E) Representative images showing that H pylori (green) are restricted to the pits in infected, omeprazole-treated mice (D) and that inhibition of gastric acid secretion with omeprazole does not expand sLex (red) expression. The onset of SPEM following HD-Tam treatment allows H pylori to penetrate deep within SPEM glands demonstrating expanded sLex expression (E). Blue, nuclei. Scale bars, 50 µm. (F) Magnified, representative image shows H pylori (green) co-localizing with sLex (red) within the pit of an omeprazole-treated mouse. Blue, nuclei. Scale bar, 5 µm. (G) The distance of H pylori along the gland axis was expressed as a percentage of total gland length. Data represent the mean (±SD) distribution from three infected littermates/cagemates for each treatment condition. Each data point represents a gland colonized with H pylori, with total numbers of scored glands indicated in parentheses. Hp, H pylori.
DISCUSSION
In this study, we used a combination of in situ and in vivo models to systematically examine the interaction of H pylori with a changing corpus landscape. The atrophic and metaplastic changes that H pylori both induces and leverages are what allows it, in some patients, to cause gastric cancer, so the questions that we have addressed in this study are critical for understanding this clinically relevant aspect of H pylori pathogenesis. We brought new tools to bear on this problem, with the acute, reversible metaplastic injury caused by HD-Tam allowing us to alter the gastric environment in a synchronous manner in the absence of chronic inflammation.
The in situ H pylori adherence assay allowed us to directly manipulate and compare H pylori’s interactions with uninjured and metaplastic glands in the absence of active acid secretion. However, we caution that this interaction is not the sole determinant of H pylori’s interaction with SPEM glands, as the ∆sabA mutant was still able to deeply penetrate approximately one third of SPEM glands. The in situ adherence assay is thus an oversimplification of H pylori’s binding to gastric epithelium. Fixed tissue lacks active acid secretion, local pH gradients, and infiltrating host immune cells, all relevant factors that dynamically influence H pylori pathogenesis in vivo9, 25.
Nonetheless, the in vivo experiments validated the significance of H pylori’s interactions with SPEM glands observed in situ. While it has been previously reported that chronically inflamed gastric tissue manifests expanded sLex 26, the cellular context of this expansion has not been studied – namely, that it specifically occurs in SPEM glands. Furthermore, it has not been clear how altering the gastric landscape by inducing SPEM and expanded sLex affects H pylori pathogenesis directly. The ability of H pylori to directly and reversibly bind SPEM glands through SabA’s interaction with expanded sLex represents a dynamic adaptation of H pylori to a chronic inflammatory environment that has significant implications for H pylori colonization in vivo.
Previous analysis of the host determinants that dictate H pylori’s topographic distribution and oncogenic risk has largely been correlative27. Indeed, chronic acid suppression through the use of proton pump inhibitors had been shown to promote a multi-focal gastritis in humans24, 28 and mice29 infected with H pylori, though the pre-neoplastic implications of gastritis distribution patterns were not pursued. We demonstrate that H pylori’s colonization of the metaplastic corpus is fluid, with the bacteria redistributing away from the most proximal regions of the corpus following recovery from HD-Tam injury. One could argue that these changes in H pylori distribution within the stomach are simply the result of changes in gastric acidity, as HD-Tam treatment raises intra-gastric pH within the range of omeprazole treatment. Indeed, omeprazole-induced acid suppression, like HD-Tam treatment, promoted a more efficient colonization of the gastric corpus in mice, and a similar phenomenon has been observed in humans on chronic acid suppression24.
However, our findings equally indicate that H pylori’s distribution within the depths of the gastric corpus unit depends on the injury state of the gland, not simply on loss of gastric acidity. Inhibition of gastric acid secretion in vivo did not alter the distribution of H pylori within each gland; specifically, it did not promote binding deep within corpus glands. Instead, it was the presence of SPEM, presumably in large part because of the concomitant expansion of sLex toward the gland base, that dictated whether H pylori could access deeper regions along the gastric corpus unit.
Distinguishing the topographic distribution of H pylori from its glandular distribution may be relevant from a pathophysiologic standpoint. The glandular changes that characterize the gastric epithelial injury response that is SPEM involve cellular reprogramming and proliferation along the metaplastic gland axis30; lectins have been shown to mark various cells and differentiation patterns in gut epithelial lineages31. We demonstrate that the pattern of sLex expression in human gastric specimens is specific to SPEM but does not mark another gastric pre-neoplastic lesion, intestinal metaplasia6. The role of increased sLex expression during SPEM remains unclear, though it may mark a transient return to an oncofetal state as part of a conserved injury response in the gastric corpus32. Our evolving understanding of SPEM as a glandular injury response implicates cellular plasticity as a key underlying mechanism governing the response of the damaged corpus unit8. Indeed, we have previously shown that there is a conserved cellular program, paligenosis, whereby post-mitotic, differentiated cells can revert to an oncofetal state by first scaling down mature cellular features, then re-expressing more primitive cell surface markers (e.g., SOX9 and, presumably, sLex), and finally proliferating in order to efficiently repair the gland33.
From a microbial perspective, one could make the teleological argument that H pylori has adapted to exploit the host’s glandular repair mechanisms by specifically, and perhaps preferentially, binding SPEM glands. Colonization deep within SPEM glands could serve to protect H pylori from the harsher, more acidic environment near the gastric lumen. As sLex expression deep within the gland is lost during injury repair, H pylori is forced out of this micro-environment and must seek to establish a new niche, perhaps in adjacent injured glands. The changes in the pattern of sLex expression that accompany SPEM provide a means by which the spread of H pylori parallels that of SPEM. H pylori may encourage the spread of SPEM throughout the stomach, either indirectly by inducing chronic inflammation that causes this metaplastic repair response, or directly by injecting toxins like CagA that affect epithelial differentiation. Thus, H pylori can advance throughout the stomach by following, and binding, an advancing front of inflammation and SPEM, marked in part by expanded sLex.
From a clinical perspective, the ability of H pylori to bind deep within and interact with injured corpus epithelium, characterized by enhanced sLex expression, could potentially lead to an accumulation of mutations within metaplastic cells undergoing cycles of proliferation and differentiation changes as part of glandular repair, as we and others have proposed8, 34. Indeed, enhanced sLex expression is a marker of poor prognosis in gastric adenocarcinoma35. Overall, H pylori’s interaction with sLex in SPEM glands during chronic inflammation would not only be a crucial determinant of its natural history within a host but would also have important implications for the establishment of a pre-neoplastic gastric milieu that contributes to the patient’s overall oncogenic risk.
Supplementary Material
ACKNOWLEDGMENTS:
We thank James Fitzpatrick and Dennis Oakley (Washington University Center for Cellular Imaging) for technical assistance with confocal imaging. We would also like to thank Hei-Yong (Grant) Lo for help with the intra-gastric pH measurements.
Grant support: J.C.M. is supported by National Institute of Diabetes and Digestive and Kidney Diseases awards DK094989, DK105129, and DK110406, by the Alvin J. Siteman Cancer Center–Barnes Jewish Hospital Foundation Cancer Frontier Fund, NIH National Cancer Institute P30 CA091842, and by the Barnard Trust. J.B.S. holds a Postdoctoral Enrichment Program Award from the Burroughs Wellcome Fund and is also supported by the American Gastroenterology Association Gastric Cancer Foundation Research Scholar Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None of the authors have any financial disclosures.
Conflict(s) of Interest: None to declare.
REFERENCES:
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–86. [DOI] [PubMed] [Google Scholar]
- 2.Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis 2012;13:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010;139:2028–2037 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt PH, Lee JR, Joshi V, et al. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest 1999;79:639–46. [PubMed] [Google Scholar]
- 5.Willet SG, Mills JC. Stomach Organ and Cell Lineage Differentiation: from Embryogenesis to Adult Homeostasis. Cell Mol Gastroenterol Hepatol 2016;2:546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldenring JR, Nam KT, Wang TC, et al. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology 2010;138:2207–10, 2210 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham DY. History of Helicobacter pylori, duodenal ulcer, gastric ulcer and gastric cancer. World J Gastroenterol 2014;20:5191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saenz JB, Mills JC. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol 2018;15:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amieva M, Peek RM Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016;150:64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee A, Dixon MF, Danon SJ, et al. Local acid production and Helicobacter pylori: a unifying hypothesis of gastroduodenal disease. Eur J Gastroenterol Hepatol 1995;7:461–5. [PubMed] [Google Scholar]
- 11.Boren T, Falk P, Roth KA, et al. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993;262:1892–5. [DOI] [PubMed] [Google Scholar]
- 12.Mahdavi J, Sonden B, Hurtig M, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002;297:573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huh WJ, Khurana SS, Geahlen JH, et al. Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 2012;142:21–24 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leushacke M, Tan SH, Wong A, et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol 2017. [DOI] [PubMed] [Google Scholar]
- 15.Mills JC, Sansom OJ. Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 2015;8:re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saenz JB, Burclaff J, Mills JC. Modeling Murine Gastric Metaplasia Through Tamoxifen-Induced Acute Parietal Cell Loss. Methods Mol Biol 2016;1422:329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pentecost M, Otto G, Theriot JA, et al. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog 2006;2:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen AC. Simplified Estimators for the Normal Distribution When Samples Are Singly Censored or Truncated. Technometrics 1959;1:217–237. [Google Scholar]
- 19.Falk P, Roth KA, Boren T, et al. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. a 1993;90:2035–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magalhaes A, Rossez Y, Robbe-Masselot C, et al. Muc5ac gastric mucin glycosylation is shaped by FUT2 activity and functionally impacts Helicobacter pylori binding. Sci Rep 2016;6:25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juge N Microbial adhesins to gastrointestinal mucus. Trends Microbiol 2012;20:30–9. [DOI] [PubMed] [Google Scholar]
- 22.Hirmo S, Kelm S, Schauer R, et al. Adhesion of Helicobacter pylori strains to alpha-2,3-linked sialic acids. Glycoconj J 1996;13:1005–11. [DOI] [PubMed] [Google Scholar]
- 23.Talarico S, Whitefield SE, Fero J, et al. Regulation of Helicobacter pylori adherence by gene conversion. Mol Microbiol 2012;84:1050–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logan RP, Walker MM, Misiewicz JJ, et al. Changes in the intragastric distribution of Helicobacter pylori during treatment with omeprazole. Gut 1995;36:12–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bugaytsova JA, Bjornham O, Chernov YA, et al. Helicobacter pylori Adapts to Chronic Infection and Gastric Disease via pH-Responsive BabA-Mediated Adherence. Cell Host Microbe 2017;21:376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syder AJ, Guruge JL, Li Q, et al. Helicobacter pylori attaches to NeuAc alpha 2,3Gal beta 1,4 glycoconjugates produced in the stomach of transgenic mice lacking parietal cells. Mol Cell 1999;3:263–74. [DOI] [PubMed] [Google Scholar]
- 27.Peterson WL. Helicobacter pylori and gastric adenocarcinoma. Aliment Pharmacol Ther 2002;16 Suppl 1:40–6. [DOI] [PubMed] [Google Scholar]
- 28.Kuipers EJ, Lundell L, Klinkenberg-Knol EC, et al. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N Engl J Med 1996;334:1018–22. [DOI] [PubMed] [Google Scholar]
- 29.Danon SJ, O’Rourke JL, Moss ND, et al. The importance of local acid production in the distribution of Helicobacter felis in the mouse stomach. Gastroenterology 1995;108:1386–95. [DOI] [PubMed] [Google Scholar]
- 30.Weis VG, Goldenring JR. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer 2009;12:189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falk P, Roth KA, Gordon JI. Lectins are sensitive tools for defining the differentiation programs of mouse gut epithelial cell lineages. Am J Physiol 1994;266:G987–1003. [DOI] [PubMed] [Google Scholar]
- 32.Karam SM, Li Q, Gordon JI. Gastric epithelial morphogenesis in normal and transgenic mice. Am J Physiol 1997;272:G1209–20. [DOI] [PubMed] [Google Scholar]
- 33.Willet SG, Lewis MA, Miao ZF, et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koeppel M, Garcia-Alcalde F, Glowinski F, et al. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep 2015;11:1703–13. [DOI] [PubMed] [Google Scholar]
- 35.Futamura N, Nakamura S, Tatematsu M, et al. Clinicopathologic significance of sialyl Le(x) expression in advanced gastric carcinoma. Br J Cancer 2000;83:1681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.