

Abstract
While there are many studies on the subject of hydrogen bonding dynamics in biological systems, few, if any, have investigated this fundamental process in amyloid fibrils. Herein, we seek to add insight into this topic by assessing the dynamics of a hydrogen bond buried in the dry interface of amyloid fibrils. To prepare a suitable model peptide system for this purpose, we introduce two mutations into the amyloid-forming Aβ16-22 peptide. The first one is a lysine analog at position 19, which is used to help form structurally homogeneous fibrils, and the second one is an aspartic acid derivative (DM) at position 17, which is intended (1) to be used as a site-specific infrared probe and (2) to serve as a hydrogen-bond acceptor to lysine so that an inter-β-sheet hydrogen bond can be formed in the fibrils. Using both infrared spectroscopy and atomic force microscopy, we show that (1) this mutant peptide indeed forms well defined fibrils, (2) when bulk solvent is removed, there is no detectable water present in the fibrils, (3) infrared results obtained with the DM probe are consistent with a protofibril structure that is composed of two antiparallel β-sheets stacked in a parallel fashion, leading to formation of the expected hydrogen bond. Using two-dimensional infrared spectroscopy, we further show that the dynamics of this hydrogen bond occur on a timescale of ~2.3 ps, which is attributed to the rapid rotation of the –NH3+ group of lysine around its Cε-Nζ bond. Taken together, these results suggest that (1) DM is a useful infrared marker in facilitating structure determination of amyloid fibrils and (2) even in the tightly packed core of amyloid fibrils certain amino acid sidechains can undergo ultrafast motions, hence contributing to the thermodynamic stability of the system.
Keywords: Amyloid fibril, hydrogen bond, dynamics, linear and two-dimensional infrared spectroscopy, spectral diffusion, hydrophobic core, dry interface
Graphical Abstract

1. INTRODUCTION
Due to its intimate connection with various pathological conditions, amyloid formed by a wide variety of proteins and peptides has been studied.1 In particular, structural characterization of amyloid fibrils derived from different peptide sequences reveals that they share a common backbone architecture, i.e., β-sheet. Another commonality is that amyloid fibrils are intrinsically stable,2 often inert to physical perturbations and hence correspond to the global minimum in their respective folding free energy landscapes. The question is, how is this stability achieved?
Many previous studies have sought to answer this question.3 However, most of them were only focused on the enthalpic contribution to the overall stability of the amyloid system in question, i.e., the stabilizing interactions arising from favorable sidechain packing and interactions,3,4 without addressing, at least not explicitly, the corresponding entropic contribution or the effect of conformational dynamics. Considering the fact that at its core an amyloid fibril consists of highly-ordered and densely packed β-sheets that are interlocked by well-registered sidechains, it is tempting to assume that the system is highly rigid and hence lacks the kind of conformational dynamics seen in folded proteins. Indeed, a solid state NMR study4 on amyloid fibrils formed by Aβ1-40 peptide found that the order parameters of the Cα–H bond vectors in the β-sheet segments are very high (0.8–0.95), suggesting low conformational flexibility of the corresponding backbone units. On the other hand, findings from another NMR study5 indicated that the methyl-bearing sidechains in the hydrophobic core of the Aβ1-40 fibrils are highly dynamic, with μs–ms motions persisting to temperatures of 230–190 K. Such conformational dynamics6,7 are expected to play an important role in stabilizing amyloid fibrils, as in the case of globular proteins.8
To provide further evidence on and insight into the conformational dynamics of sidechains buried in the hydrophobic region of amyloid fibrils, especially those occurring on ultrafast timescales, we seek to assess the dynamics of a hydrogen-bond (H-bond) located in the dry interface of a model amyloid system. To create a peptide that would form amyloid fibrils that carry such a H-bond, we used the sequence (KLVFFAE) of an amyloid-forming peptide,9 Aβ16-22, as the starting point. The crystal structure of Aβ16-21 indicates that two antiparallel Aβ16-21-β-sheets can associate in a parallel manner, leaving half of the Lys sidechains oriented toward the interface region.10 Therefore, we hypothesized that replacing L17 of Aβ16-22 with an amino acid that bears a H-bond acceptor would promote the formation of a H-bond between the Lys residue on one β-sheet and this amino acid on the other in the amyloid fibrils. Based on our previous study,11 we chose an unnatural amino acid, L-aspartic acid 4-methyl ester (DM) for this purpose. This is because the ester C=O group of DM not only provides a H-bond acceptor but can also be used as a site-specific infrared (IR) probe of local H-bonding dynamics. Previously, we have also shown that replacing F19 of Aβ16-22 with an analogue of lysine, lys(Nvoc) (Nvoc = 4,5-dimethoxy-2-nitrobenzyloxycarbonyl),12 can significantly improve the peptide’s ability to self-assemble, hence reducing the conformational and morphological heterogeneity of the resultant fibrils.13 Therefore, in the current study we also chose to use this mutation, taking advantage of the ability of lys(Nvoc) (hereafter referred to as K*) to facilitate formation of structurally homogeneous fibrils. As expected (Figure 1), this double mutant of Aβ16-22 (sequence: KDMVK*FAE, hereafter referred to as Aβ16-22-DMK*) readily self-assembles to form well-defined fibrils, allowing for assessment of amyloid structure and conformational dynamics via linear and two-dimensional (2D) IR spectroscopic methods.
Figure 1.
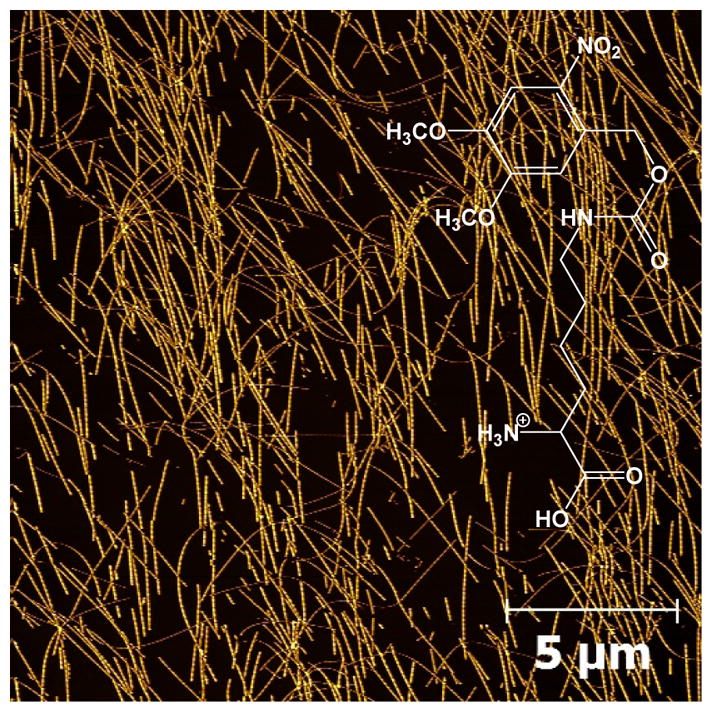
AFM image of Aβ16-22-DMK* fibrils spread over a large area. Also shown in the figure is the structure of Lys(Nvoc).
Using 2D IR spectroscopy and various isotopically labeled Aβ1-40 peptides, Kim et al.14 showed that the amide I′ vibrational transitions of L17, V18, L34 and V36 of Aβ1-40 in the fibrillar form can undergo fast frequency variations (i.e., in 1–2 ps), which was attributed to the existence of mobile water molecules within the inter-β-sheet region of the corresponding fibrils. We found that the ester C=O stretching vibration of DM in hydrated Aβ16-22-DMK* fibrils exhibits similar spectral diffusion dynamics; however, this ultrafast dynamic behavior was found to be present even in dry fibril samples where no fibril-bound water was detected. Thus, this study provides, to the best of our knowledge, the first example showing that the sidechains buried in the hydrophobic core of amyloid fibrils can still undergo conformational fluctuations on the picosecond timescale, which, presumably, contributes toward the stability of the system.
2. EXPERIMENTAL SECTION
2.1. Materials and Sample Preparation
All materials were used as received. Specifically, D2O (D, 99.96%) and deuterium chloride (D, 99.5%) were purchased from Cambridge Isotope Laboratories (Andover, MA). Fmoc-Lys(4,5-dimethoxy-2-nitrobenzyloxycarbonyl)-OH was purchased from Anaspec, Inc. (Fremont, CA) and Fmoc-L-Asp(Me)-OH was purchased from Chem-Impex International Inc. (Wood Dale, IL). All other Fmoc-protected amino acids were purchased from Advanced Chem Tech (Louisville, KY). All peptides were synthesized on a PS3 peptide synthesizer (Protein Technologies, Woburn, MA) and the cleaved peptide samples were purified by reverse-phase high-performance liquid chromatography (HPLC). Matrix-assisted laser desorption ionization (MALDI) mass spectrometry was used to confirm the identity of each peptide. Multiple rounds of lyophilization against a DCl solution were used to remove the residual trifluoroacetic acid (TFA) from peptide synthesis. All peptide samples were prepared by directly dissolving the lyophilized peptide solid in D2O, and the final pH of the resultant peptide solutions was approximately 3, unless explicitly indicated separately. Fibril formation was achieved by incubating an appropriate amount of peptide solution (5 – 15 mM) at 37 °C for at least one week.
2.2. AFM Measurement
The AFM measurements were performed in air at room temperature on a Bruker Dimension Icon (Billerica, MA) using the tapping-mode and a silicon probe (TESP) from Veeco (Camarillo, CA). Height and deflection images were obtained with a scan rate of 1 Hz, integral gain of 0.4, and a proportional gain of 0.6. Fibril sample was prepared by applying 5 μL of an aggregated peptide solution to a freshly-cleaved mica surface for about 5 s, followed by rinsing the sample surface with 300 μL Millipore water, and subsequently dried with a slow stream of N2 gas. Multiple images were obtained for the same sample at different locations on the mica substrate.
2.3. Fourier Transform Infrared (FTIR) Measurements
FTIR spectra were collected on a Thermo Nicolet Magna 860 FTIR spectrometer equipped with a MCT detector at 25 °C with a spectral resolution of 1 cm−1. A home-made motorized sample holder assembly, which has an optical path-length of 25 μm, was used to allow measurements of the sample and solvent spectra in a single experiment. The final reported result corresponds to an average of 128 spectra and contains the absorbance of the solute only.
The Attenuated Total Reflectance (ATR) IR spectra were collected on a Thermo Nicolet 6700 FTIR spectrometer with a resolution of 1 cm−1 and using a Harrick Horizon multiple-reflection attachment. The fibril dry film was prepared by spreading 10 μL of the aggregated peptide solution in question over the surface area of an EJ2122 Harrick germanium crystal (45°, 50 × 10 × 2 mm), followed by drying under a flow of N2 gas for 7 days.
2.4 Two-Dimensional Infrared (2D IR) Measurements
The detail of the home-built 2D IR spectrometer with a box-CARS geometry has been described elsewhere.15 Briefly, three femtosecond mid-IR pulses (k1, k2, and k3, respectively) were focused onto the sample of interest and the resultant photon echo signal was heterodyned with a local oscillator pulse, dispersed by a monochromator and detected with a 64-element liquid-nitrogen-cooled IR array detector (Infrared Associates, FL). The time delay (τ) between k1 and k2 was scanned from −3.5 to 4.0 ps with a step size of 2 fs. For a specific waiting time (T) between k1/k2 and k3, the absorptive 2D spectrum was obtained by adding up the corresponding rephrasing and nonrephasing spectra. The amyloid fibril dry film used in 2D IR measurements was prepared by depositing an aliquot of a fully aggregated peptide solution at the center of a CaF2 window, followed by drying at room temperature via constant N2 purging for at least 12 hours. Before use, the fibril film thus formed was further dried for another 12 hours.
3. RESULTS AND DISCUSSION
Characterization of the Morphology of Aβ16-22-DMK* Fibrils
As shown (Figure 2), the morphology of Aβ16-22-DMK* fibrils is similar to that of fibrils formed by the wild-type peptide, exhibiting distinct, left-handed twist along the fibril axis.9 A more quantitative assessment (Figure S1 in SI) indicates that the Aβ16-22-DMK* fibrils have an average height of 15 – 20 nm and an average pitch distance of ~150 nm. These morphological parameters are fairly close to those determined for Aβ1-40 fibrils.16 Moreover, those AFM images (Figures 1 and 2) indicate that the fibrils formed by Aβ16-22-DMK* are morphologically rather homogeneous (except the fibril length), making them suitable for spectroscopic studies.
Figure 2.

AFM image of Aβ16-22-DMK* fibrils spread over a smaller area.
FTIR and ATR-FTIR Measurements
As shown (Figure 3A), the FTIR spectrum of an aggregated Aβ16-22-DMK* sample in D2O solution in the amide I′ band region consists of three sharp peaks, centered at about 1624, 1690 and 1724 cm−1, respectively. The first two peaks, with one being approximately 4 times more intense than the other, are characteristic of amyloid fibrils composed of antiparallel β-sheets.17 On the other hand, the 1724 cm−1 peak, which is not present in the FTIR spectrum of Aβ16-22 fibrils (Figure S2 in SI), must arise from the DM sidechain. According to our previous study,11 this peak can be assigned to the stretching vibration of an ester C=O group that interacts with the surroundings through a single hydrogen-bond (H-bond). Interestingly, upon removal of bulk solvent by drying the aggregated Aβ16-22-DMK* sample to form a thin film, the corresponding ester C=O peak becomes much broader. As indicated (Figure 3B), a more quantitative assessment shows that the ester C=O peak obtained in solution can be described by a single Gaussian function (ω0 = 1723.8 cm−1), whereas that measured with the dry film requires two Gaussians (ω10 = 1725.5 cm−1, ω20 = 1739.4 cm−1) of roughly equal intensity to fit. The latter result is particularly interesting as it indicates that even under dry conditions approximately 50% of the DM sidechains are still engaged in certain H-bonding interactions, with either fibril-bound D2O molecules or other H-bond donors (see below). Furthermore, a series of spectra taken at different times of the drying process (Figure 4) demonstrate that the increase of the high-frequency component, which arises from non-H-bonded DM sidechains,11 is concomitant with the decrease of the low-frequency component. Taken together, these results suggest that half of the DM sidechains in the fibrils are exposed to bulk solvent, whereas the other half, despite being H-bonded, are buried in the hydrophobic region between two β-sheets.
Figure 3.

(A) FTIR spectra of Aβ16-22-DMK* fibrils (in the amide I′ region) measured in solution and in the form of dry film, as indicated. For easy comparison, the spectrum obtained with dry film has been scaled by a factor of 0.25. (B) Fitting of the ester C=O stretching vibrational bands in (A) with one (solution) and two (dry film) Gaussians.
Figure 4.
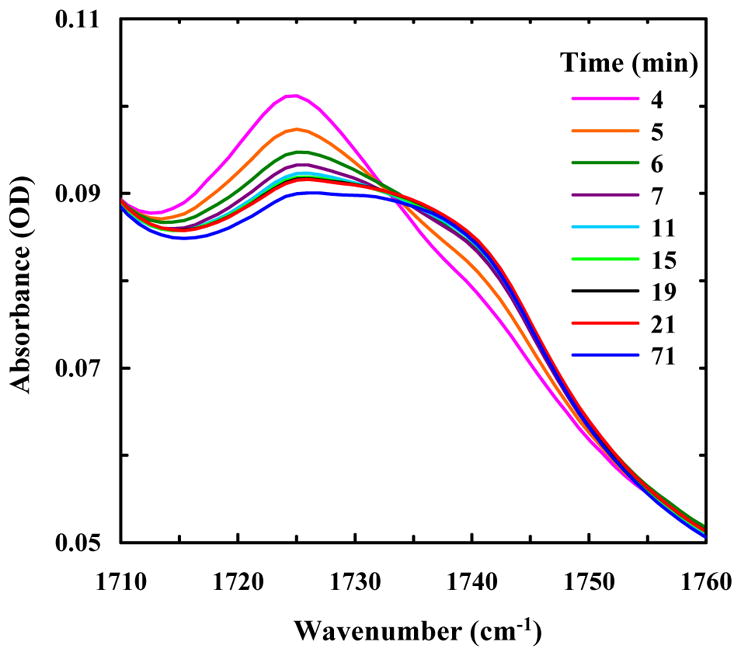
Dependence of the ester C=O stretching vibrational band of Aβ16-22-DMK* fibrils on the drying time, as indicated.
Removing solvent via drying may lead to changes in the structure and morphology of the amyloid fibrils in question. To confirm that this does not happen in the current case, we carried out further polarized ATR measurements on dry films formed by aggregated Aβ16-22-DMK* samples. The rationale of this experiment is based on the following considerations: in a well formed amyloid fibril sample, all individual β-strands in a β-sheet would be aligned perpendicular to the long axis of the fibril and, as a result, all of the backbone carbonyls would point to a direction that is parallel to this axis. Consequently, the direction of the amide I′ vibrational transition that gives rise to the intense and low-frequency component (i.e., the band at ~1624 cm−1 in the current case) would be parallel to the long fibril axis. Therefore, for a peptide dry film consisting of well-formed and long fibrils that can only lay flat on the surface of the ATR crystal, the linear dichroic ratio of this low-frequency amide I′ band should be 1. As shown (Figure 5A), the dichroic ratio of this band for the Aβ16-22-DMK* dry film is 1.08, suggesting that drying does not change in any significant manner the fibril structure and organization. In addition, these ATR-FTIR measurements provide the following important information: (1) while the Aβ16-22-DMK* fibrils were grown under acidic conditions, the band at ~1550 cm−1, which arises mainly from the COO− asymmetric stretching vibration of Glu,18 indicates that the Glu sidechain in the fibrils remains deprotonated and hence does not contribute to the absorption signals observed in the frequency range of 1700 – 1760 cm−1; (2) in comparison to the FTIR spectrum of another K*-containing peptide (sequence: VIKK*) (Figure S3 in SI), the two sharp peaks at ~1528 and ~1584 cm−1 can be assigned to K*; and (3) the ATR-FTIR spectrum in the 2300 – 2600 cm−1 region (Figure 5B) suggests that the dry Aβ16-22-DMK fibrils contain little, if any, D2O molecules, because it has been shown that fibril-bound D2O produces a detectable band at ~2575 cm−1.14,19 This observation is consistent with a molecular dynamics simulation20 on the protofilament assembly of Aβ16-22, which showed that the hydrophobic interior of the protofilament is completely dehydrated and dry. Furthermore, and perhaps more importantly, this result suggests that the 1725.5 cm−1 peak observed for dry Aβ16-22-DMK* fibrils is unlikely to result from interactions between the DM sidechain and residual D2O.
Figure 5.

(A) ATR-FTIR spectra of Aβ16-22-DMK* fibrils measured in the form of dry film under different polarization conditions, as indicated. In addition, the origins of the peaks in the amide I′ region are labeled. (B) ATR-FTIR spectrum of Aβ16-22-DMK* fibrils between 2300 – 2600 cm−1, showing the lack of a band at ~2565 cm−1 (in the region highlighted by the dashed cycle) arising from fibril-bound D2O molecules.
Proposed Structural Model
The above discussed IR results provide constraints for us to propose a structure model for the Aβ16-22-DMK* fibrils. For Aβ16-22, there are six possible stacking arrangements between two antiparallel β-sheets (Figure S4 in SI),21 which, we assume, are also applicable for Aβ16-22-DMK*. Since our FTIR results indicate that half of the DM sidechains in the fibril are affected by drying or dehydration, we can rule out stacking patterns that would lead the burial of all DM sidechains in the hydrophobic region of the protofibril, i.e., PAT-3 and PAT-6. Similarly, we can exclude stacking patterns that would allow all DM sidechains to be exposed to solvent, i.e., PAT-2 and PAT-5. For the remaining stacking arrangements, we believe that PAT-1 is more consistent with all of the FTIR data. This is because, as shown (Figure 6), this inter-sheet packing arrangement not only expose half of the DM sidechains to solvent, hence satisfying the dehydration result, but it also brings a Lys sidechain from one sheet to a DM sidechain on another, allowing for H-bonding interactions between the ester C=O group of DM and the positively charged ε-amino group of Lys (the H-bond thus formed is hereafter referred to as Lys-DM H-bond). Moreover, this model is consistent with the crystal structure of Aβ16-21 peptide10 and also a MD simulation22 that indicates that among various Aβ16-22 octamers, the one with antiparallel sheet/parallel layer arrangement is the most stable. While K* also contains a H-bond donor (Figure 1) and could potentially interact with DM via H-bonding interactions, we do not believe that this scenario occurs. This is because every β-strand contains one K*/DM pair and, as a result, formation of an intramolecular H-bond between these two residues would render the ester C=O stretching vibration insensitive to dehydration.
Figure 6.
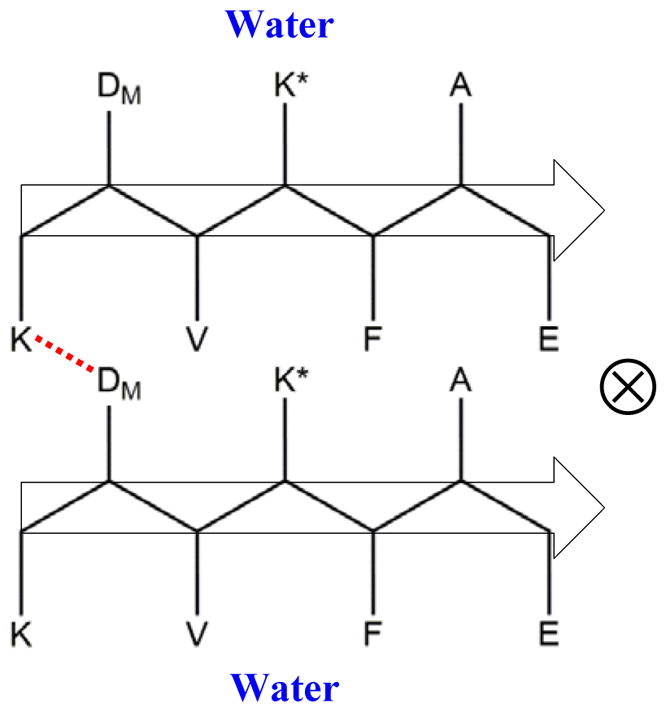
Proposed Aβ16-22-DMK* protofibril structure, which involves stacking of two antiparallel β-sheets in a parallel manner. The red dashed line represents the proposed H-bond formed between the ester C=O group of DM and the ε-amino group of Lys, and ⊗ indicates the long fibril axis.
2D IR Measurements
To access the dynamics of the water-DM H-bond and Lys-DM H-bond, we carried out 2D IR measurements on Aβ16-22-DMK* fibrils in solution and in the format of dry film. As shown (Figure 7), 2D IR spectra at representative waiting times (T) obtained with fully hydrated (left column) and dry (right column) Aβ16-22-DMK fibril samples confirm the FTIR results, i.e., the DM sidechains give rise to one C=O stretching band for the solution sample but two bands for the dry film. For the 2D IR data obtained in solution, we used the center line slope (CLS) method23 to further characterize the frequency variation dynamics of the underlying vibrational transition. As indicated (Figure 8A), the corresponding CLS−1 data can be described by a function composed of an exponential with a decay time constant of 2.3 ± 1.0 ps and an offset of 0.24 ± 0.03. This decay time constant is in the range of values reported for the spectral diffusion dynamics of various vibrational probes that engage in H-bonding interactions with water24–26 and, therefore, supports the proposed model (Figure 6) which allows half of the DM sidechains to form H-bonds with solvent.
Figure 7.
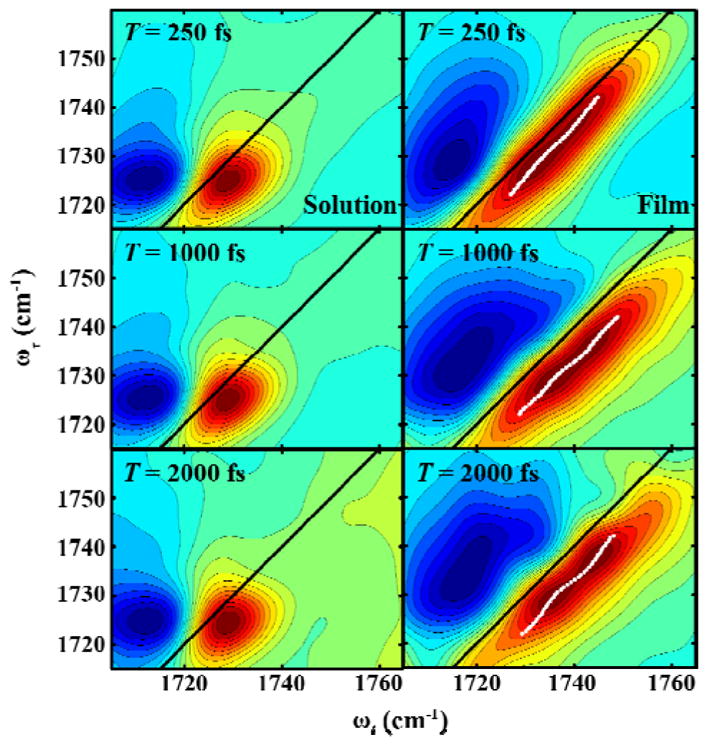
2D IR spectra at representative waiting times (T), as indicated, obtained with Aβ16-22-DMK* fibrils in solution (left) and in the form of dry film (right). The locust of points in the right column is to help visualizing the center lines of the two overlapping vibrational transitions.
Figure 8.

Spectral diffusion dynamics of the ester C=O stretching vibration of Aβ16-22-DMK* fibrils as measured by the CLS method for data collected in solution (A) and by the integrated peak shift method for data collected with dry film (B). In each case, the smooth line corresponds to the best fit of the data to the following equation: S(T) = A · exp(−T/τ) + B.
The 2D IR spectra measured with dry β16-22-DMK* fibrils cannot be directly analyzed by the CLS method due to spectral overlapping. Therefore, to estimate the spectral diffusion dynamics of the two ester C=O stretching vibrational transitions, we resorted to the commonly used integrated peak shift (IPS) approch.27,28 Specifically, we selected a 10 cm−1 spectral window centered at the 2D IR peak of interest along the ωτ axis. We then inverse Fourier transformed the selected data from the frequency domain (ωτ) to the time domain (τ). The integrated peak shift was obtained by measuring the peak position in the plot of the sum of data over the ωt axis versus τ. As expected (Figure 8B), for the high-frequency peak the corresponding signal does not show any appreciable change in the time window of the experiment, due to the removal of bulk solvent. Interestingly, however, the signal arising from the low-frequency peak shows a measureable decay that can be fit to an exponential function with a time constant of 2.1 ± 0.9 ps. This result is somewhat surprising as spectral diffusion dynamics occurring on this timescale are typically assigned to mobile water14 which is lacking in our case (Figure 5B). Therefore, based on our structure model (Figure 6), this dynamic process must manifest conformational motions that affect the Lys-DM H-bonding interactions and hence the C=O stretching vibrational frequency of the respective ester group. Because no 2D IR cross-peaks are observed in the time window of the experiment, we can rule out the possibility that these motions lead to complete disruption of the Lys-DM H-bond. Instead, we believe that the observed picosecond spectral diffusion dynamics are caused by ultrafast exchange between different Lys-DM H-bond configurations, due to the existence of three degenerate H-bond donors on the protonated ε-amino group (i.e., –NH3+) of Lys. In other words, ultrafast rotation around the Cε-Nζ bond of Lys would result in transient breakage of the Lys-DM H-bond, leading to spectral diffusion of the stretching vibration of the ester C=O group (the H-bond acceptor in this case) to occur on the timescale of the bond rotation. This picture is consistent with several NMR studies,29–31 which showed that in proteins the Cε-Nζ bond of H-bonded Lys sidechains can rotate on the sub-nanosecond timescale and that H-bonds involving –NH3+ groups are highly dynamic and transient in nature. Thus, taken together these 2D IR results not only validate the conclusions reached based on the FTIR measurements, but also demonstrate that amino acid sidechains buried in the hydrophobic region of amyloid fibrils, which is often described as crystal-like, can exhibit ultrafast conformational motions. We expect that such motions are important to the stability of the very rigid fibril structure.
4. CONCLUSIONS
The dry interface of amyloid fibrils consists of interdigitated and hence densely packed sidechains. Therefore, it is unclear whether sidechains buried in this region would undergo any ultrafast fluctuations. To help answer this question, herein we study the dynamics of a H-bond located in the hydrophobic region of amyloid fibrils formed by a model peptide (Aβ16-22-DMK*). This peptide is a double mutant of Aβ16-22 and has the following sequence: KDMVK*AE, where K* (lys(Nvoc)) is introduced to improve the fibril-forming ability of the peptide and DM (L-aspartic acid 4-methyl ester) is employed to provide a H-bond acceptor (i.e., its ester C=O group) and also serve as an IR probe (through its ester C=O stretching vibration) of the dynamics of any resulting H-bond(s). Our results show that this peptide indeed self-assembles to form well defined amyloid fibrils composed of antiparallel β-sheets. Interestingly, for fibrils in solution, the DM IR probe exhibits one peak centered at ~1723.8 cm−1, whereas for fibrils in dry film this probe gives rise to two peaks of roughly equal intensity, centered at ~1725.5 and ~1739.4 cm−1, respectively. Taken together, these results indicate that in fully hydrated fibrils all the DM sidechains are H-bonded, whereas in fully dehydrated fibrils only half of the DM sidechains are involved in H-bonding interactions. Therefore, these findings support a fibril structure model wherein two antiparallel β-sheets are brought together in a parallel manner, promoting formation of an inter-sheet H-bond between the ester C=O group of DM and the –NH3+ group of Lys in the dry interface of the fibril. Further 2D IR measurements show that the spectral diffusion dynamics of the vibrational band arising from the H-bonded ester C=O group in dry fibrils occur on a timescale of ~2.3 ps, indicating that the –NH3+ group of Lys can still undergo ultrafast rotational motions in a crowded and tightly packed environment. This type of ultrafast motions, we believe, is important to the stability amyloid fibrils and, hence, it would be interesting to study whether such H-bonds in disease-causing fibrils afford similar property.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (P41GM104605).
Footnotes
ASSOCIATED CONTENT
Supporting Information: Additional FTIR spectra and AFM image analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Meersman F, Dobson CM. Probing the pressure–temperature stability of amyloid fibrils provides new insights into their molecular properties. Biochim Biophys Acta - Proteins Proteomics. 2006;1764:452–460. doi: 10.1016/j.bbapap.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemkul JA, Bevan DR. Assessing the stability of Alzheimer’s amyloid protofibrils using molecular dynamics. J Phys Chem B. 2010;114:1652–1660. doi: 10.1021/jp9110794. [DOI] [PubMed] [Google Scholar]
- 5.Vugmeyster L, Clark MA, Falconer IB, Ostrovsky D, Gantz D, Qiang W, Hoatson GL. Flexibility and solvation of amyloid-β hydrophobic core. J Biol Chem. 2016;291:18484–18495. doi: 10.1074/jbc.M116.740530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler J, Scheidt HA, Krüger M, Thomas L, Huster D. Local interactions influence the fibrillation kinetics, structure and dynamics of Aβ(1–40) but leave the general fibril structure unchanged. Phys Chem Chem Phys. 2014;16:7461–7471. doi: 10.1039/c3cp54501f. [DOI] [PubMed] [Google Scholar]
- 7.Linser R, Sarkar R, Krushelnitzky A, Mainz A, Reif B. Dynamics in the solid-state: perspectives for the investigation of amyloid aggregates, membrane proteins and soluble protein complexes. J Biomol NMR. 2014;59:1–14. doi: 10.1007/s10858-014-9822-6. [DOI] [PubMed] [Google Scholar]
- 8.Lee AL, Wand AJ. Microscopic origins of entropy,heat capacity and the glass transition in proteins. Nature. 2001;411:501–504. doi: 10.1038/35078119. [DOI] [PubMed] [Google Scholar]
- 9.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid fibril formation by Aβ16-22, a seven-residue fragment of the Alzheimer’s β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 10.Colletier JP, Laganowsky A, Landau M, Zhao M, Soriaga AB, Goldschmidt L, Flot D, Cascio D, Sawaya MR, Eisenberg D. Molecular basis for amyloid-β polymorphism. Proc Natl Acad Sci. 2011;108:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pazos IM, Ghosh A, Tucker MJ, Gai F. Ester carbonyl vibration as a sensitive probe of protein local electric field. Angew Chemie. 2014;126:6194–6198. doi: 10.1002/anie.201402011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rusiecki VK, Warne SA. Synthesis of Nα-Fmoc-Nε-Nvoc-Lysine and use in the preparation of selectively functionalized peptides. Bioorg Med Chem Lett. 1993;3:707–710. [Google Scholar]
- 13.Measey TJ, Gai F. Light-Triggered disassembly of amyloid fibrils. Langmuir. 2012;28:12588–12592. doi: 10.1021/la302626d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YS, Liu L, Axelsen PH, Hochstrasser RM. 2D IR provides evidence for mobile water molecules in β-amyloid fibrils. Proc Natl Acad Sci. 2009;106:17751–17756. doi: 10.1073/pnas.0909888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding B, Hilaire MR, Gai F. Infrared and fluorescence assessment of protein dynamics: from folding to function. J Phys Chem B. 2016;120:5103–5113. doi: 10.1021/acs.jpcb.6b03199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt M, Sachse C, Richter W, Xu C, Fändrich M, -Grigorieff N. Comparison of Alzheimer Aβ (1–40) and Aβ (1–42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufrêne YF, Narayanaswami V, Goormaghtigh E, Ruysschaert JM, Raussens V. Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem J. 2009;421:415–423. doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 18.Barth A. The infrared absorption of amino acid side chains. Prog Biophy Mol Biol. 2000;74:141–173. doi: 10.1016/s0079-6107(00)00021-3. [DOI] [PubMed] [Google Scholar]
- 19.Hilaire MR, Ding B, Mukherjee D, Chen J, Gai F. Possible existence of α-Sheets in the amyloid fibrils formed by a TTR105–115 mutant. J Am Chem Soc. 2018;140:629–635. doi: 10.1021/jacs.7b09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krone MG, Hua L, Soto P, Zhou R, Berne BJ, Shea JE. Role of water in mediating the assembly of Alzheimer amyloid-β Aβ16–22 protofilaments. J Am Chem Soc. 2003:11066–11072. doi: 10.1021/ja8017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Röhrig UF, Laio A, Tantalo N, Parrinello M, Petronzio R. Stability and structure of oligomers of the alzheimer peptide Aβ16–22: from the dimer to the 32-Mer. Biophys J. 2006;91:3217–3229. doi: 10.1529/biophysj.106.088542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma B, Nussinov R. Stabilities and conformations of alzheimer’s beta -amyloid peptide oligomers (Abeta 16-22, Abeta 16-35, and Abeta 10-35): sequence effects. Proc Natl Acad Sci U S A. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwak K, Park S, Finkelstein IJ, Fayer MD. Frequency-frequency correlation functions and apodization in two-dimensional infrared vibrational echo spectroscopy: A new approach. J Chem Phys. 2007;127:124503–124519. doi: 10.1063/1.2772269. [DOI] [PubMed] [Google Scholar]
- 24.Hamm P, Lim M, Hochstrasser RM. Non-Markovian dynamics of the vibrations of ions in water from femtosecond infrared three-pulse photon echoes. Phys Rev Lett. 1998;81:5326–5329. [Google Scholar]
- 25.Urbanek DC, Vorobyev DY, Serrano AL, Gai F, Hochstrasser RM. The two-dimensional vibrational echo of a nitrile probe of the villin HP35 protein. J Phys Chem Lett. 2010;1:3311–3315. doi: 10.1021/jz101367d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung JK, Thielges MC, Fayer MD. Dynamics of the folded and unfolded villin headpiece (HP35) measured with ultrafast 2D IR vibrational echo spectroscopy. Proc Natl Acad Sci. 2011;108:3578–3583. doi: 10.1073/pnas.1100587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh A, Wang J, Moroz YS, Korendovych IV, Zanni M, DeGrado WF, Gai F, Hochstrasser RM. 2D IR spectroscopy reveals the role of water in the binding of channel-blocking drugs to the influenza M2 channel. J Chem Phys. 2014;140:235105–235114. doi: 10.1063/1.4881188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamm P, Zanni M. Concepts and methods of 2D infrared spectroscopy. Cambridge University Press; 2011. [Google Scholar]
- 29.Esadze A, Li DW, Wang T, Brüschweiler R, Iwahara J. Dynamics of lysine side-chain amino groups in a protein studied by heteronuclear 1H– 15N NMR spectroscopy. J Am Chem Soc. 2010;133:909–919. doi: 10.1021/ja107847d. [DOI] [PubMed] [Google Scholar]
- 30.Zandarashvili L, Li DW, Wang T, Brüschweiler R, Iwahara J. Signature of mobile hydrogen bonding of lysine side chains from long-range 15N–13C scalar J-couplings and computation. J Am Chem Soc. 2011;133:9192–9195. doi: 10.1021/ja202219n. [DOI] [PubMed] [Google Scholar]
- 31.Baird-Titus JM, Thapa M, Doerdelmann T, Combs KA, Rance M. Lysine side-chain dynamics in the binding Site of homeodomain/DNA complexes as observed by NMR relaxation experiments and molecular dynamics simulations. Biochemistry. 2018 doi: 10.1021/acs.biochem.8b00195. Article ASAP. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.