

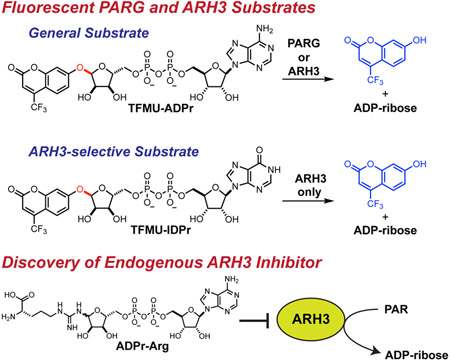
SUMMARY
The post-translational modification (PTM) and signaling molecule poly(ADP-ribose) (PAR) has an impact on diverse biological processes. This PTM is regulated by a series of ADP-ribosyl glycohydrolases (PARG enzymes) that cleave polymers and/or liberate monomers from their protein targets. Existing methods for monitoring these hydrolases rely on detection of the natural substrate, PAR, commonly achieved via radioisotopic labelling. Here we disclose a general substrate for monitoring PARG activity, TFMU-ADPr, which directly reports on total PAR hydrolase activity via release of a fluorophore; this substrate has excellent reactivity, generality (processed by the major PARG enzymes), stability, and usability. A second substrate, TFMU-IDPr, selectively reports on PARG activity only from the enzyme ARH3. Use of these probes in whole-cell lysate experiments has revealed a mechanism by which ARH3 is inhibited by cholera toxin. TFMU-ADPr and TFMU-IDPr are versatile tools for assessing small-molecule inhibitors in vitro and probing the regulation of ADP-ribosyl catabolic enzymes.
Graphical abstract
eTOC Blurb
Drown et al. describe the design and synthesis of fluorescent activity probes for the (ADP-ribosyl)hydrolases PARG and ARH3. These probes are operational in biochemical assays or in cell lysate, and were utilized to identify an endogenous inhibitor of ARH3.
INTRODUCTION
Many biological processes are regulated by post-translational modifications (PTMs), with ADP-ribosylation being one of the most thoroughly studied nucleotide-containing modifications. This modification includes a collection of “writers”, “readers”, and “erasers”. The writers, poly(ADP-ribosyl) polymerases (PARPs), modify protein substrates with adenine diphosphate ribose (ADPr). Nearly all nucleophilic amino acids have been shown to be modified: glutamate, aspartate, cysteine, lysine, and serine (Altmeyer et al., 2009; Barkauskaite et al., 2015; Leidecker et al., 2016; Vyas et al., 2014; Zhen et al., 2017). While the majority of PARPs can only transfer a single ADPr unit from NAD+ (Feijs et al., 2013), the founding member of the family, PARP1 (but also PARP2 and tankyrases), can synthesize large polymers termed poly(ADP-ribose) (PAR) (Vyas et al., 2014). A wealth of literature over several decades has established PARylation as central for proper DNA damage response (Andrabi et al., 2006; Schreiber et al., 2000; Wang et al., 2016), chromatin maintenance (Becker et al., 2016), cell division (Chang et al., 2005), and DNA replication (Illuzzi et al., 2014).
In contrast, much is still unknown about the regulation of ADPr erasure. Several families of enzymes are responsible for removal and degradation of PARylation. Poly(ADP-ribose) glycohydrolase (PARG) hydrolyzes the 2',1" glycosidic linkage (red in Fig. 1A) of polymers mainly in an exo manner (Barkauskaite et al., 2013). PARG modifies PARylated proteins by removing polymers down to the last unit, which it is incapable of removing (Slade et al., 2011). ADP-ribosyl hydrolase 3 (ARH3 or ADPRHL2) is also capable of degrading PAR polymers, albeit at a reduced rate (Mashimo et al., 2014; 2013; Niere et al., 2012; Oka et al., 2006; Ono et al., 2006). Until recently, neither of these proteins were believed capable of removing the last ADPr unit directly bonded to the protein side chain. This hydrolysis is carried out by ARH1 or an enzymatic member of the macrodomain family (terminal ADP-ribosyl protein glycohydrolase (TARG1), MACROD1, and MACROD2 in mammals) (Barkauskaite et al., 2015; Feijs et al., 2013; Jankevicius et al., 2013; Mashimo et al., 2013; Moss et al., 1992; Rosenthal et al., 2013; Sharifi et al., 2013). Recently ARH3 was shown to be the sole enzyme capable of hydrolyzing mono-ADP-ribosyl serine (Abplanalp et al., 2017; Fontana et al., 2017). Beyond substrate recognition, little is known about how these ADPr erasers are regulated or the relative importance of different erasers in various biological contexts. In particular, study of PAR cleavage has been limited by the challenge of separating contributions to PAR degradation by PARG and by ARH3. Several reports suggest that the specific activity of PARG is significantly greater than ARH3, however, the cellular distribution of these two enzymes differ as do their regulatory domains (Mashimo et al., 2014). Genetic knockout of ARH3 does not affect the lifetime of long polymers but does result in longer lived short polymers (Fontana et al., 2017), thus much more detailed information on the relative contribution of PAR-degrading enzymes is needed.
Figure 1.

Design of PARG/ARH3 substrate. A. PAR is cleaved by PARG and ARH3 via hydrolysis of the glycosyl bond (red). B. Synthetic PARG/ARH3 substrates mimic ADP-ribose and release a chromophore or fluorophore upon hydrolysis. Synthesis of these compounds requires a late-stage pyrophosphate formation and 1,2-cis selective glycosylation.
The most widely employed enzyme assays for measuring PARG/ARH3 activity rely on radioisotopically labelled and enzymatically produced PAR (Cortes et al., 2004; Finch et al., 2012; Ménard and Poirier, 2011; Sharifi et al., 2013). The production and isolation of labelled PAR is limited to 0.3-2 mg scale (Ménard and Poirier, 2011). After treatment with PARG, reaction mixtures are separated by TLC or HPLC and quantified by autoradiography or liquid scintillation counting. This technique, while sensitive, is laborious and not scalable. Recent efforts to develop more scalable PARG activity assays have been modestly successful, for example, a four-component FRET system to detect the interaction between a PARylated protein and XRCC1 (Kim et al., 2015; Stowell et al., 2016). While this approach has been implemented in microtiter format and utilized to discover a first-in-class PARG inhibitor, it suffers from an inability to accurately measure enzyme kinetics and cannot operate in cell lysate (James et al., 2016). There clearly is still an unmet need for a facile and continuous activity assay for PAR-degrading enzymatic activity. Herein is described the design, synthesis, and evaluation of fluorescent probes for PAR hydrolyzing enzymes, including a compound that is processed by both ARH3 and PARG, and another that is a selective substrate for ARH3. This latter probe has enabled the first direct measurements of ARH3 activity in cells and was then used to discover the first specific means for cellular ARH3 inhibition.
RESULTS AND DISCUSSION
Design and synthesis of PARG substrates
Reporter substrates for PARG and ARH3, pNP-ADPr and TFMU-ADPr, were designed, inspired by the natural substrate, PAR. The 2',1"-glycosidic bond that is cleaved in PAR (highlighted red in Figure 1A) was replaced with a phenolic glycoside. Initial efforts focused on utilizing 4-nitrophenol (pNP) as a chromaphore reporter but also extended to the fluorophore 4-(trifluoromethyl)umbelliferone (TFMU) for applications requiring greater sensitivity and specificity. Retrosynthetic analysis of pNP-ADPr identified two major synthetic hurdles (Figure 1B): a late-stage pyrophosphate coupling and a 1,2-cis selective glycosylation of a furanoside. Recent work on the synthesis of dimeric PAR (Lambrecht et al., 2015) overcame similar challenges, and it was envisioned that an analogous strategy could be applied to the synthesis of this simplified PARG substrate.
Glycosylation of electron-deficient phenols such as pNP and TFMU is often challenging. Most methods rely of anchimeric assistance leading to the undesired 1,2-trans product (Bordoni et al., 2010). We found that acceptable a-selectivity could be achieved by directly activating a hemi-acetal with bulky protecting groups under Mitsunobu conditions. As outlined in Figure 2, this strategy was employed using orthogonally protected ribose 3, prepared in three steps from D-ribonic γ-lactone (Figure S1A) (Lambrecht et al., 2015); subsequent removal of the trityl group proceeded smoothly under strictly anhydrous conditions to prevent hydrolysis of the glycoside, producing 4 and 5. Although direct phosphorylation with POCl3 was possible, we found phosphoramidite coupling followed by fluorenylmethyl removal more amenable on scale to give phosphates 6 and 7 (Figure 2, Figure S1B, C). Pyrophosphate formation was accomplished via in situ oxidative chlorination of H-phosphonate 8 (Figure 2, Figure S1D), to produce 9 and 10, followed by global desilylation to provide pNP-ADPr and TFMU-ADPr.
Figure 2.

Synthesis of pNP-ADPr and TFMU-ADPr. Abbreviations: ADDP = 1,1’-(azodicarbonyl)dipiperidine, DIAD = diisopropyl azodicarboxylate, Fm = 9H-fluorenylmethyl, DCI = 4,5-dicyanoimidazole, NCS = N-chlorosuccinimide, DBU = 1,8-diazabicycloundec-7-ene, TBS = tert-butyldimethylsilyl.
The late-stage pyrophosphate coupling featured in the synthesis of these compounds also facilitates the construction of other non-natural substrates for PARG. The recently co-crystalized structure of L. chalumnae ARH3 with ADPr (not shown, co-submitted) indicates that the binding modes of hPARG and LchARH3 are dramatically different. In particular, the adenine binding site of LchARH3 is much less organized and more solvent exposed. Molecular docking sugar nucleotides with varied purine bases suggests that PARG is highly discriminatory while ARH3 would accommodate other bases (Table S1). Key interactions between ADPr and hPARG Glu727 and Ile726 cannot be recapitulated with IDPr, and the hPARG binding site is too restrictive to allow for additional favorable interactions (Figure 3A). Conversely, the LchARH3 binding site is predicted to allow IDPr to shift and make additional interactions with Lys132 (Figure 3B). We thus hypothesized that replacing the nucleobase of phenolic substrates, specifically substitution of adenine for hypoxanthine, would provide a substrate selective for ARH3. Therefore the compounds TFMU-IDPr and pNP-IDPr were designed and synthesized from inosine (Figure 3C, and Figure S2).
Figure 3.

Molecular docking sugar nucleotides with hPARG and LchARH3 using Glide XP. A. ADPr (blue) and IDPr (yellow) are docking into hPARG (PDB: 5A7R). B. ADPr (blue) and IDPr (yellow) are docking into LchARH3 (PDB: 6G1Q). C. Synthesis of TFMU-IDPr; for pNP-IDPr synthesis please see Figure S2.
In vitro processing of substrates
Using pNP-ADPr and TFMU-ADPr, the first continuous PARG and ARH3 activity assays were developed. Enzymatic hydrolysis of phenolic glycosides was easily monitored by an increased absorbance or fluorescence (Figure S3A, B). Kinetic parameters for human PARG and ARH3 were determined by non-linear fitting of initial reaction rates (Table 1 and Figure 4A, B). A commonly used ortholog of PARG from T. thermophila (ttPARG) (Barkauskaite et al., 2013; Dunstan et al., 2012; Lambrecht et al., 2015) was also characterized and found to effectively hydrolyze pNP-ADPr and TFMU-ADPr (Table 1 and Figure S3A). The measured KM of ARH3 was similar to the enzyme’s reported activity against O-acetyl-ADP-ribose (Kasamatsu et al., 2011). The kinetic parameters of pNP-ADPr and TFMU-ADPr were quite similar likely due to the leaving groups having similar pKas (7.15 and 7.26). Gratifyingly, the predicted selectivity of TFMU-IDPr for ARH3 over PARG was confirmed (Figure 4C). Further, both PARG and ARH3 selectively hydrolyzed the α anomer of pNP-ADPr over the β anomer as is consistent with their natural substrates (Figure S3C, D). Hydrolysis of substrates depends on catalytically active protein; inactivating mutations in hPARG (the E756N mutant, Lambrecht et al., 2015) and hARH3 (the D77N/D78N mutant, Fontana et al., 2017) abolish substrate processing (Figure S3F, G).
Table 1.
Kinetic parameters derived for ADP-ribosylhydrolase substrates.
| Substate | Enzyme | KM (μM) | Vmax (μmol/min/mg) |
|---|---|---|---|
| TFMU-ADPr | hPARGa | 66.2±15 | 0.84±0.05 |
| ttPARGb | 210±13 | 28.6±0.6 | |
| hARH3c | 6.3±0.2 | 1.61±0.02 | |
| TFMU-IDPr | hPARG | >1500 | n.d. |
| ttPARG | >1500 | n.d. | |
| hARH3 | 312±30 | 1.79±0.06 | |
| pNP-ADPr | ttPARG | 210±10 | 16.9±0.5 |
| hARH3 | 3.2±0.6 | 1.7±0.1 | |
| pNP-IDPr | ttPARG | >1500 | n.d. |
| hARH3 | 410±20 | 5.2±0.2 |
H. sapiens full length PARG
T. thermophila PARG
H. sapiens full length ARH3
Figure 4.

Michaelis-Menton kinetics of recombinantly expressed human PARG and ARH3. A. Kinetics of human PARG processing TFMU-ADPr. B. Kinetics of human ARH3 processing TFMU-ADPr. C. Selectivity of TFMU-IDPr for processing by human ARH3 over human PARG. Error bars indicate SEM, n = 3.
Use of substrates to monitor ARH3 activity assay in cell culture
The ability of TFMU-IDPr to selectively report on ARH3 activity within the cellular milieu would be an important application of these probes. While the experiments in Figure 4 demonstrate the selectivity of this substrate for ARH3 over PARG in a purified enzymatic system, in cells other hydrolases that recognize ADP-ribose could potentially process this substrate, thus two experiments were conducted to assess the activity of ARH3 and PARG with these substrates in cell lysate. First, the hydrolysis of TFMU-IDPr was evaluated in cell lysate from a ARH3 knockout isogenic cell line pair derived from U2OS cells (Fontana et al., 2017). Knockout of ARH3 resulted in near complete loss of TFMU-IDPr activity (Figure 5A) while TFMU-ADPr hydrolysis was only partially decreased (Figure 5B). Second, substrate processing was evaluated in the presence of the selective PARG inhibitor PDD00017273 (James et al., 2016). Hydrolysis of TFMU-ADPr was completely abolished by PDD00017273 in ARH3−/− cell lysate, but TFMU-IDPr was unaffected by PARG inhibitor. Taken together, these experiments indicate that TFMU-IDPr is selectively cleaved by ARH3 in cell lysate, and TFMU-ADPr is cleaved only by ARH3 and PARG.
Figure 5.

Measurement of PARG activity in cell lysate and validation of TFMU-IDPr selectivity. A. Selectivity for ARH3 by TFMU-IDPr validated by CRISPR-Cas9 knockout of ARH3 in U2OS cells. PARG activity was measured in the presence and absence of PARG inhibitor PDD00017273 using TFMU-IDPr at 200 μM. Error bars indicated SEM, n=3. Significance levels are given by asterisks: p<0.05 (*), p<0.01 (**), p<0.001 (***), p>0.05 (n.s.). B. Same as A but with 200 μM TFMU-ADPr. C. Validation of ARH3 knockout in U2OS as measured by Western blotting. D. Western blotting to determine ARH3 expression levels in various indicated cell lines; representative blot of two independent replicates shown, see Figure S4 for all replicates. E. Correlation of ARH3 expression (measured by Western blotting) with ARH3 activity (measured by TFMU-IDPr hydrolysis). Data points reflect average values from different mammalian cell lines. Red indicates MCF10A. ARH3 activity was measured with 200 μM TFMU-IDPr.
Survey of cancer cell lines
As the activity of ARH3 in various cell types is unknown, experiments were conducted to assess ARH3 activity in a variety of cancer cell lines. Twenty cell lines were analyzed for ARH3 activity, measured using TFMU-IDPr, and compared to ARH3 abundance (measured by Western blot, Figure 5D); in general ARH3 activity and abundance are well correlated (Figure 5E, Table S2). Strikingly, no ARH3 activity is observed with MCF10A cells despite the obvious presence of ARH3 by Western blot. Thus, it was hypothesized that in this cell line ARH3 is mutated or being affected by an endogenous inhibitor.
A unique feature of MCF10A cells are the particular additives in the growth media, and most notable is the inclusion of cholera toxin (CTX), a known ADP-ribosyltransferase (Gill and Meren, 1978; Wang and Schultz, 2014). Suspecting that the presence of CTX may impact ARH3 activity, MCF10A cells were passaged five times in the absence of CTX, and ARH3 activity was restored (Figure S5A). This recovery of activity was reversed (in a dose and time-dependent manner) with subsequent addition CTX, and this phenotype was reproduced in other cell lines (MCF7 and MIA PaCa-2) (Figure S5A, B). In all these cell lines, CTX-mediated ARH3 inhibition occurred rapidly (t1/2 < 60 min, Figure S5B). The treatment of cells with CTX appears to be the first example of selective inhibition ARH3 in cells.
Mechanism of ARH3 inhibition by ADP-ribosyl-arginine
Having identified cholera toxin as causing ARH3 inhibition in cell culture, several experiments were performed to elucidate the toxin's mechanism of action. CTX was first found to have no direct effect on ARH3 in vitro (Figure S5C). The canonical mechanism of CTX involves mono-ADP-ribosylation of an active-site arginine on Gsα (G-protein alpha subunit); this post-translational modification results in the constitutive activation of adenylyl cyclase (AC), which synthesizes cAMP (Figure S6) (Inageda et al., 1991; Kato et al., 2007). Thus the role of AC and cAMP in CTX-dependent ARH3 inactivation were investigated. cAMP was found to only weakly inhibit ARH3 (IC50 = 2.96±0.05 mM, Figure S5D). Furthermore, treatment with forskolin, a known activator of AC (Florio et al., 1999; Takeda et al., 1983), did not result in ARH3 inhibition (Figure S5E). Finally, other ADP-ribosyltransferase bacterial ecto-toxins that target other amino acid residues do not inhibit ARH3 activity (Figure S5F). With these experiments pointing upstream of AC, direct interaction with ADP-ribosyl arginine (ADPr-Arg) was considered. To make assessments, ADPr-Arg was synthesized as described previously (Oppenheimer, 1978) and assessed in the in vitro assay. This compound was found to potently inhibit ARH3 in vitro, with a Ki value of 18±2 nM (Figure 6A, B). Interestingly, ADPr-Arg demonstrates markedly reduced activity against PARG (Figure 6C). This differential inhibition is in contrast to the results for the substrate analogue inhibitor of PARG, the compound ADP-HPD (Koh et al., 2003; Slama et al., 1995a; 1995b); analysis of ADP-HPD shows that this compound inhibits both PARG and ARH3 (Figure 6C). Importantly, ADPr-Arg substantially accumulated in MCF7 cells following CTX treatment, from concentrations below the limit of detection in untreated cells to 2.62±0.05 μM in the treated cells (Figure S5G). Changes in related metabolites were also observed: cAMP concentration increased as expected for the canonical activity of CTX, NAD+ concentration decreased (it is a substrate for ADPr-Arg synthesis), but arginine remained unchanged, perhaps due to the presence of exogenous arginine in media. The same trend for these metabolic changes was also observed in U2OS cells (Figure S5H).
Figure 6.

Selective inhibition of ARH3 by ADP-ribosylated arginine. A. Kinetics of inhibition of ARH3 by ADPr-Arg. B. Lineweaver-Burke plot of ADPr-Arg inhibition of ARH3. C. Selectivity of ADPr-Arg for ARH3 over hPARG is indicated by a shift in dose-response curve. D. Structure of ADPr-Arg.
DISCUSSION
This manuscript describes the first continuous substrates for the glycohydrolases that catabolize PAR. The syntheses of these substrates are robust and scalable (hundreds of milligrams of each were prepared), and as such these compounds should find routine use for the measurement of kinetics, the assessment of inhibitors, and for high-throughput screening applications. In addition, their ability to report on this enzymatic activity in cell lysate will enable a variety of experiments where PARG or ARH3 activity is the needed readout, and in such experiments the ARH3-selective substrate TFMU-IDPr can be used to differentiate cellular ARH3 enzymatic activity from PARG activity.
Selective enzyme assays are powerful for assessment of the differential contribution of enzyme family members to total enzymatic activity. The potential impact such experiments can have is apparent in the widespread use of a variety of compounds as caspase substrates. For caspases, specificity can be imbued through use of peptide sequences known to be specifically recognized by different caspases isozymes, and such tool compounds have been widely employed to understand conditions under which specific caspases are activated, and have been critical to mapping the apoptotic cell death pathway (Berger et al., 2006; Poręba et al., 2013). The lack of analogous reagents for monitoring of PAR processing has necessitated reliance on non-ideal methods such as isozyme-general processing of radiolabeled substrates. In the case of PAR-processing enzymes, the development of specific substrates is considerably more complicated than for a protease (where specificity can be built in though understanding of the endogenous protein substrates). The recently solved X-ray structures of ARH3 and PARG, and subsequent docking studies, has now allowed for the design of reporter substrates, one that is general for ARH3 and PARG, and one that is specific for ARH3.
As a first application, these substrates have been used to discover that CTX suppresses cellular ARH3 activity through the production of ADPr-arginine, an ARH3-selective inhibitor. While the effect of bacterial exotoxins on cAMP synthesis and downstream processes has been extensively described (Figure S6), their effects off-pathway are less understood. CTX, Clostridium difficile binary toxin (CDT), and pertussis toxin (PT) catalyze the mono-ADP-ribosylation of arginine (CTX and CDT) and cysteine (PT) residues. While the background ADP-ribosylation of arginine by CTX has been observed with isolated protein (Oppenheimer, 1978), the activity of CTX has largely been regarded to be highly selective for proteinaceous arginine. We observe and quantify ADP-ribosylation of free arginine for the first time in cell culture at concentrations that rationalize ARH3 inhibition. The absence of ARH3 inhibition following CDT treatment, despite creating the same PTM, may result from differential propensities to produce free and proteinaceous ADPr-arginine. However, the possibility remains that the observed ADPr-arginine results from proteolytic breakdown of ADP-ribosylated G proteins rather than direct ADP-ribosylation. In this case, the differential activities of bacterial toxins can be attributed to stabilities of their respective protein targets. Some of the phenotypes described upon treatment by bacterial toxins (i.e. lose of tight junctions) has also been described for PAR overproduction (Cuzzocrea and Genovese, 2000; Guichard et al., 2013; Mazzon et al., 2002; Nusrat et al., 2001). While the exact mechanism by which these processes operate is yet unknown, inhibition of ARH3 may assist in toxins’ ability to elevate PAR concentrations. The ready availability of these ARH3 and PARG substrates will now facilitate additional discoveries about the regulation of cellular PAR processing.
STAR Methods
Contact for Reagent and Resource Sharing
All requests for reagents and resources should be directed to the lead contact, Paul Hergenrother (hergenro@illinois.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
A549, D54, HCC1937, HCC38, HCT-116, Jurkat, and U937 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (Gemini) and 1% pen/strep. U2OS, Daudi, HEK293TN, HeLa, Hs578t, MEF, MIA PaCa-2, and SK-MEL-5 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% pen/strep. HFF-1 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 15% fetal bovine serum and 1% pen/strep. HepG2, MCF7, T98G, and U87 cells were grown in Eagle's Minimum Essential Medium supplemented with 10% fetal bovin serum and 1% pen/strep. MCF10A cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 2% horse serum, 20 ng/mL EGF, 0.5 μg/mL hydrocortisone, 10 μg/mL insulin, 100 ng/mL cholera toxin, and 1% pen/strep. Phenol red was added to all media except for MCF10A as a pH indicator. All cells were cultured at 37° in a 5% CO2 environment. Media were prepared by the University of Illinois School of Chemical Sciences Cell Media Facility. Sex of cell lines: A549 (Male, 58 years old), CA46 (Male, age unknown), D54 (Female, age unknown), Daudi (Male, 16 years old), HCC1937 (Female, 23 years old), HCC38 (Female, 50 years old), HCT-116 (Male, age unknown), HEK293TN (Female, fetus), HeLa (Female, 31 years old), HepG2 (Male, 15 years old), HFF-1 (Male, newborn), Hs578t (Female, 74 years old), MCF7 (Female, 69 years old), MCF10A (Female, 36 years old), MEF (sex and age unknown), MIA PaCa-2 (Male, 65 years old), Jurkat (Male, 14 years old), SK-MEL-5 (Female, 24 years old), T98G (Male, 61 years old), U2OS (Female, 15 years old), U87 (Male, age unknown), U937 (Male, 37 years old).
METHOD DETAILS
Buffer Composition:
PARG Activity Lysis Buffer: 30 mM Tris (pH 7.5), 500 mM NaCl, 20% glycerol, 1% Triton X-100, 1:500 protease inhibitor cocktail
Lysate Activity Buffer: 50 mM K2HPO4 (pH 7.4), 50 mM KCl, 5 mM MgCl2, 5 mM DTT
T. thermophila PARG reaction buffer: 50 mM K2HPO4, 50 mM KCl, 10 mM β-mercaptoethanol, pH 7.40
Human PARG reaction buffer: 50 mM K2HPO4, 50 mM KCl, 10 mM β-mercaptoethanol, pH 7.40
Human ARH3 reaction buffer: 50 mM Na2HPO4, 10 mM MgCl2, 5 mM DTT, pH 7.40
PARG Purification Lysis buffer: 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 0.5 mg/mL lysozyme, 1 ug/mL leupeptin, 1 ug/mL pepstatin A, 2 ug/mL aprotinin, 1 mM PMSF, pH 8.0
PARG Purification Wash buffer: 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0
PARG Purification Elution buffer: 50 mM NaH2PO4, 300 mM NaCl, 300 mM imidazole, pH 8.0
PARG Purification Dialysis buffer: 25 mM Tris, 50 mM NaCl, 1 mM DTT, 10% glycerol, pH 7.5
Towbin Transfer Buffer: 20% CH3OH, 192 mM glycine, 50 mM Tris, pH 8.3
In vitro Enzyme Kinetics
10X dilutions of substrate were prepared from 10 mM stock solutions in MilliQ H2O into reaction buffer. 5 μL 10X substrate was added to a 384-well plate. 45 μL 2.2 nM enzyme solution in reaction buffer was added to a single column of the 384-well plate via 16-channel Matrix pipet. Plate was placed into plate reader. After shaking for 5 s, absorbance or fluorescence was recorded at 2 s intervals for 5 min. Initial reaction rates were determined by fitting the linear portions of reaction progress curves using SoftMax Pro 6.4. Initial rates were plotted against substrate concentration and fit to the Michaelis-Menton equation using a non-linear curve-fitting algorithm with GraphPad Prism 6. For pNP detection, plate reader was configured: absorbance at 405 nm. For TFMU detection, plate reader was configured: Excitation 385 nm, Cutoff 495 nm, Emission 502 nm, 6 reads/well, Low Gain.
Cell Lysate ARH3 Activity Assay
Cells were cultured in T75 flasks in appropriate medium. Cells were trypsinized, and scrapped (if necessary), and counted. 1×106 cells were harvested and washed with cold PBS 2x. Cell pellet was lysed by addition of activity lysis buffer (150 μL) and incubated on ice for 30 min. Lysate was clarified by centrifugation at 14,300xg at 4° for 10 min. Supernatant was transferred to chilled, empty 500 μL tube. Total protein content of lysate was evaluated by BCA assay. ARH3 expression level was determined by western blotting analysis. 5 μL lysate was added to well of 384-well black plate. 45 μL TFMU-IDPr (200 μM final) in reaction buffer was added to well. Reaction progress was monitored by fluorescent plate reader (ex 385 nm, cutoff 495, em 502 nm, 6 reads/well, high gain).
ADP-ribosylated arginine enzymatic synthesis
CTX (100 μL 500μg/mL, 10μg/mL final) was added to 5 mL buffer (400 mM K2HPO4, 20 mM DTT, pH 7.2) containing 200 mM arginine and 10 mM NAD. Mixture was incubated at 37°C for 16 h. Protein was removed from reaction mixture by filtration through 3k MWCO spin filter. Filtrate was direction subjected to ion-pairing preparative HPLC using Luna C18 21.5×150mm column. Solvent A: 20 mM Et3N•HOAc (pH 7.2), solvent B: acetonitrile. Gradient (A:B, 20 mL/min): 98:2, 0 min; 98:2, 2 min; 75:25, 16 min; 75:25, 23 min; 40:60, 27 min, 40:60, 30 min. ADP-ribosylated arginine eluted at ~5 min as two separate anomers. Individual anomers gradually interconverted in ~1 h, so characterization and inhibition experiments were performed with mixture of anomers. Product-containing fractions were determined by LCMS and lyophilized to yield flocculent white solid (7.8 mg, 21%). 1H NMR was consistent with previous reports (Oppenheimer, 1978).
Metabolic profiling
Six-well plates were seeded with cells at 3×105 cells/well. When cells were ~80% confluent, cells were treated with 100 ng/mL CTX for 6 h. Cells were washed with PBS once and detached with trypsin, quenching trypsin with media with 10% FBS. Cell viability was verified by Trypan Blue exclusion. Cells were centrifuged at 400xg at 4°C for 4 min and washed with PBS twice. Cell pellets were resuspended with cold 70:30 methanol:water and placed on ice. Cell suspension was sonicated on ice (20%, 10 s on, 10 s off, 30 s total) and continued to be incubated on ice for 30 min. Lysate was clarified by centrifugation at 14,300xg at 4°C for 30 min. Supernatent was transferred to an empty 0.5 mL tube and stored at −80°C until analysis. Samples were analyzed with the 5500 QTRAP LC/MS/MS system (Sciex, Framingham, MA) in Metabolomics Lab of Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Software Analyst 1.6.2 was used for data acquisition and analysis. The 1200 series HPLC system (Agilent Technologies, Santa Clara, CA) includes a degasser, an autosampler, and a binary pump. The LC separation was performed on an Agilent SB-Aq (4.6 × 50mm, 5μm) with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetontrile). The flow rate was 0.3 mL/min. The linear gradient was as follows: 0-3min, 100%A; 5-10min, 2%A; 11-15min, 100%A. The autosampler was set at 10°C. The injection volume was 5 μL. Mass spectra were acquired under positive electrospray ionization (ESI) with the ion spray voltage of +5500 V. The source temperature was 450 °C The curtain gas, ion source gas 1, and ion source gas 2 were 36, 65, and 60, respectively. Multiple reaction monitoring (MRM) was used for quantitation: Arg m/z 175.1 --> m/z 70.1; ADP-Arg m/z 716.2 --> m/z 428.1; cAMP m/z 330.2 --> 232.0; ADP-ribose m/z 560.1 --> m/z 348.1; NAD m/z 664.2 --> m/z 136. Limit of quantitation (LOQ) for the compounds were: 10 ng/mL ADPr-Arg, 10 ng/mL ADP-ribose, 10 ng/mL arginine, 20 ng/mL cAMP, and 100 ng/mL NAD+.
Recombinant Protein Expression and Purification
Human ARH3 was expressed as previously described (Barkauskaite et al., 2013; Finch et al., 2012). Briefly, human ARH3 cloned into a pDEST vector with an N-terminal His6-tag was obtained from Hening Lin (Cornell University, Cornell, NY) was transformed into Rosetta2 (DE3) E. coli cells. 500 mL culture of LB (100 μg/mL ampicillin and 20 μg/mL chloramphenicol) was inoculated with 10 mL of overnight culture. The culture was grown at 37°C to mid-log phase (OD 600 = 0.5) and cooled to 18°C. Protein expression was induced with 0.1 mM IPTG for 18 h and harvested. Pellet was resuspended in 13 mL lysis buffer and lysed by sonication. Lysate was clarified by centrifugation (35,000xg, 30 min, 4°C). Supernatent was purified on Ni-NTA beads by batch protocol. Fractions containing pure protein were combined, dialyzed, and stored at −80°C. Protein co ncentration was determined by BCA assay.
T. thermophila PARG enzyme was expressed using a modified protocol a previously reported (Dunstan et al., 2012). Briefly, T. thermophila PARG2 cloned into a pET28a vector was obtained from Ivan Ahel (University of Oxford, Oxford, UK) and transformed into Rosetta2 (DE3) E. coli. 500 mL culture of LB (100 μg/mL kanamycin and 20 μg/mL chloramphenicol) was inoculated with 10 mL of overnight culture. The culture was grown to mid-log phase (OD600 = 0.7). Culture was induced with 0.3 mM IPTG at 30°C for 3 h and harvested. Pellet was resuspended in 13 mL lysis buffer and lysed by sonication. Lysate was clarified by centrifugation. Protein was purified on Ni-NTA beads by batch protocol. Fractions containing pure protein were combined, dialyzed, and stored at −80Ό. Protein concentration was determined by BCA assay.
Human PARG cloned into a pColdTF vector was obtained from Ivan Ahel (University of Oxford, Oxford, UK) and transformed into Rosetta2 (DE3) E. coli. Culture of Terrific Broth supplemented with appropriate antibiotics was grown to an OD600 = 0.6-0.8 at 37°C. Culture was induced with 0.1 mM IPTG at 18°C for 16 h and h arvested. Pellet was resuspended in lysis buffer (40 mM HEPES, 300 mM NaCl, 20 mM imidazole, 10% glycerol, 1 mM TCEP, pH 8.0) supplemented with benzonase, lysozyme and protease inhibitor (Roche Complete EDTA-free protease inhibitor tablet) and lysed by freeze/thaw. Lysate was clarified by centrifugation. Protein was purified on Ni-NTA beads by batch and eluted with lysis buffer supplemented with 500 mM imidazole.
Western blot analysis
Samples loading was normalized based on total protein content as assessed by BCA assay and subjected to SDS-PAGE. Protein was transferred to PVDF membranes (Merck Millipore). Membranes were cut along molecular weight markers and then blocked with TBS-T buffer (25 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.5) supplemented with either 5% BSA (PARP1) or 5% non-fat dried milk (ARH3) for 1 h. Membrane strips were incubated overnight with primary antibodies in blocking buffer at 4°C with gentle rocking. Membranes were washed 3x with TBS-T and incubated with HRP-conjugated secondary antibody in TBS-T for 1 h. Membranes were washed 3x with TBS-T, developed using SuperSignal West Pico and imaged using a ChemiDoc MP system. Membrane strip containing ARH3 bands was stripped and reimaged with anti-actin antibody. Dilutions for antibodies were: anti-PARP1 (1:3000), anti-ARH3 (1:1000), anti-actin (1:3000), anti-rabbit (1:3000), and anti-mouse (1:3000). Band quantification was performed using FIJI.
Molecular docking
Protein and ligand preparation, docking, and scoring was performed using the Schrodinger Suite 2018-1. Proteins were prepared using the Protein Prep Wizard using standard settings with pH set to 7.4. Receptor grids were generated based on bound ADP-ribose. Additional positional constraints were applied so that the nucleobase occupies the adenine binding site and the ribose ring occupies the active site. Sugar nucleotide protonation state and tautomer form was determined using LigPrep. Docking was performed with Glide using both standard precision (SP) and extra precision (XP). Docking poses were exported and rendered using PyMOL.
General chemical synthesis
All reactions were run in flame or oven dried glassware under and atmosphere of dry nitrogen unless otherwise noted. Acetonitrile, tetrahydrofuran, methanol, dimethylformamide, toluene, and methylene chloride using in reactions were obtained from a solvent dispensing system. 4 A molecular sieves were dried at 150°C on high vacuum overnight. Pyridine, diazabicyclo[5.4.0]undec-7-ene, diisopropylethylamine, and trimethylamine were distilled from CaH2 and stored on 4 Å sieves. All other reagents were of standard commercial purity and were used as received. Analytical thin-layer chromatography was performed on EMD Merck silica gel plates with F254 indicator. Plates were visualized with UV light (254 nm) or staining with p-anisaldehyde. Silica gel for column chromatography was purchased from Macherey-Nagel (40-63 μm particle size).
Unless otherwise indicated, 1H, 13C, 19F, and 31P NMR spectra were recorded at 500, 126, 470, and 202 MHz, respectively. 1H and 13C NMR spectra were referenced to the residual solvent peak. 19F NMR spectra were referenced using absolute referencing based on 1H spectra. 31P NMR spectra were externally referenced to 85% H2PO4 (0.00 ppm) in water. Chemical shits are reported in ppm and multiplicities are reported as s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), h (hextet), m (multiplet), and br (broad). For annotated NMR spectra see Data File S1. Mass spectrometry analysis was performed by the University of Illinois Mass Spectrometry Center.
Preparative C18 chromatography was performed using a Teledyne Isco Combiflash Rf system with RediSep Gold columns. Silyl ether protected intermediates were separated using a gradient of H2O/Ch3CN beginning with 95% H2O:5% CH3CN, ramping to 65% H2O:35% CH3CN over 6 min, ramping to 100% CH3CN over 6 min, and holding 100% CH3CN for 5 min. Unprotected substrates were separated using a gradient of 10 mM Et3N•HOAc (pH 7.2)/CH3CN beginning with 100% buffer, ramping to 15% buffer:85% CH3CN over 10 min, ramping to 50% buffer:50% CH3CN over 6 min, and holding 50% buffer:50% CH3CN for 4 min.
Synthesis of PARG substrates
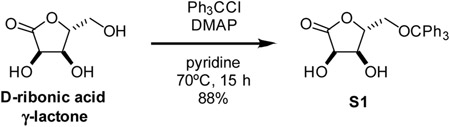
5-O-trityl-D-ribono-1,4-lactone (S1): Synthesized with modification of previous report (Taylor et al., 2002). Specifically, to a 250 mL round bottom flask was added D-ribonic acid γ-lactone (2.15 g, 14.5 mmol, 1 eq), 4-dimethylaminopyridine (360 mg, 2.9 mmol, 0.2 eq), and pyridine (42 mL). Once fully dissolved, triphenylmethyl chloride (4.86 g, 17.4 mmol, 1.2 eq) was added as a solid in one portion to the stirring reaction mixture at room temperature. The solution was stirred at 70°C for 16 h. The cool ed reaction mixture was diluted with dichloromethane and washed with 1 M HCl twice and satd aq NaHCO3 once. The organic layer was dried over MgSO4, filtered through a pad of celite, and concentrated in vacuo. The residue was purified by silica gel chromatography, eluting with 1:1 hexanes-EtOAc, yielding compound S1 as a white solid (5.0 g, 88%).
1H NMR (500 MHz, DMSO-d6) δ 7.41 – 7.24 (m, 15H), 5.93 (d, J = 7.6 Hz, 1H), 5.45 (d, J = 4.0 Hz, 1H), 4.54 (dd, J = 7.6, 5.5 Hz, 1H), 4.39 – 4.34 (m, 1H), 4.02 (ddd, J = 5.3, 4.0, 1.2 Hz, 1H), 3.41 – 3.33 (m, 1H), 3.14 (dd, J = 11.0, 3.8 Hz, 1H).
13C NMR (126 MHz, DMSO-d6) δ 176.2, 143.2, 128.2, 128.1, 127.3, 86.7, 83.4, 69.4, 68.6, 63.0.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C24H22O5Na 413.1365; Found 413.1362
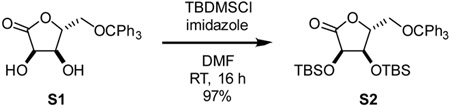
2,3-bis-O-tert-butyldimethylsilyl-5-O-trityl-D-ribono-1,4-lactone (S2): Synthesized with modification of previous reports (Taylor et al., 2002). Specifically, to a 100 mL round bottom flask was added S1 (2.0 g, 5.1 mmol, 1 eq), imidazole (1.9 g, 27 mmol, 5 eq), and dimethylformamide (20 mL). The solution was cooled to 0°C. Tert-butyldimethyls ilyl chloride (2.9 g, 19 mmol, 4 eq) was added as a solid in one portion. Solution was allowed to warm to room temperature and was stirred for 16 h. Reaction mixture was diluted with diethyl ether and quenched with the addition of satd aq NH4Cl. Organic layer was washed with satd aq NaHCO3 thrice and brine once. Organic layer was dried over Na2SO4, filtered, and concentrated. Residue was purified by silica gel chromatography, eluting with 5% Et2O-hexane, to yield compound S2 as a white foam (3.1 g, 97%).
1H NMR (500 MHz, CDCl3) δ 7.40 (d, J = 8.4 Hz, 6H), 7.31 (t, J = 7.6 Hz, 6H), 7.25 (t, J = 7.1 Hz, 3H), 4.68 (d, J = 5.2 Hz, 1H), 4.29 (t, J = 3.0 Hz, 1H), 3.95 (dd, J = 5.2, 1.1 Hz, 1H), 3.62 (dd, J = 11.0, 3.7 Hz, 1H), 3.21 (dd, J = 11.0, 2.8 Hz, 1H), 0.93 (s, 9H), 0.80 (s, 9H), 0.18 (s, 3H), 0.11 (s, 3H), 0.01 (s, 3H), −0.06 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 175.27, 143.19, 128.63, 128.18, 127.49, 84.70, 77.16, 72.16, 70.44, 62.39, 25.96, 25.75, 18.51, 18.20, −3.45, −4.47, −4.70, −5.04.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C36H50O5Si2Na 641.3094; Found 641.3093
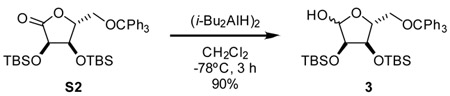
2,3-bis-O-tert-butyldimethylsilyl-5-O-trityl-D-ribose (3): A 1 M solution of (i-Bu2AlH)2 in hexane (17.5 mL, 17.5 mmol, 1.5 eq) was added dropwise via addition funnel over 5 min to a stirring solution of S2 (7.25 g, 11.7 mmol, 1 eq) in CH2Cl2 (120 mL) at −78 °C. The resulting solution was sti rred at −78 °C for 3 h. Reaction was quenched with the addition of CH3OH (4 mL) added dropwise via addition funnel and allowed to warm to 0 °C. After stirring for 30 min, 200 mL 0.5 M potassium sodium tartrate was added and mixture was stirred until aluminum salts were fully dissolved. Extracted mixture with CH2Cl2 three times. The combined organic layers were dried over MgSO4, filtered through a pad of celite, and concentrated. The residue was purified by silica gel chromatography, eluting with 10% Et2O-hexanes, to yield an interconverting mixture of diastereomers of compound 3 as a white foam (6.5 g, 90%).
β anomer (major)
1H NMR (500 MHz CDC3) δ 7.50 – 7.43 (m, 6H), 7.35 – 7.30 (m, 6H), 7.29 – 7.24 (m, 3H), 5.13 (dd, J = 11.4, 4.3 Hz, 1H), 4.24 (d, J = 11.4 Hz, 1H), 4.16 (t, J = 4.5 Hz, 1H), 3.92 (d, J = 4.6 Hz, 1H), 3.26 (dd, J = 10.4, 4.9 Hz, 1H), 3.11 (dd, J = 10.4, 3.3 Hz, 1H), 0.93 (s, 9H), 0.85 (s, 9H), 0.13 (s, 3H), 0.10 (s, 3H), 0.04 (s, 3H), −0.02 (s, 3H)
α anomer (minor)
1H NMR (500 MHz CDCl3) δ 7.50 – 7.43 (m, 6H), 7.35 – 7.30 (m, 6H), 7.29 – 7.24 (m, 3H), 5.19 (d, J = 4.6 Hz, 1H), 4.40 (dd, J = 7.3, 4.0 Hz, 1H), 4.20 (t, J = 4.0 Hz, 1H), 4.11 (ddd, J = 7.0, 3.8, 2.6 Hz, 1H), 3.97 (d, J = 4.1 Hz, 1H), 3.49 (dd, J = 10.3, 2.7 Hz, 1H), 3.10 (dd, J = 10.2, 3.9 Hz, 1H), 2.71 (d, J = 4.6 Hz, 1H), 0.91 (s, 9H), 0.75 (s, 9H), 0.11 (s, 3H), 0.09 (s, 3H), −0.03 (s, 3H), −0.19 (s, 3H)
Both anomers
13C NMR (126 MHz, CDCl3) δ 143.80, 129.01, 128.77, 128.01, 127.95, 127.26, 127.23, 102.19, 97.90, 87.05, 87.01, 84.43, 81.78, 77.06, 74.56, 72.58, 71.74, 63.77, 63.31, 26.06, 25.96, 25.92, 25.86, 18.42, 18.26, 18.11, 18.09, −4.08, −4.29, −4.46, −4.49, −4.55, −4.67, −4.90, −4.94.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C36H52O5NaSi2 643.3251; Found 643.3251
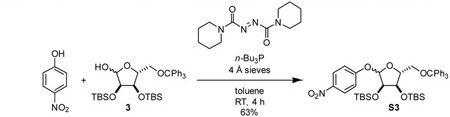
4-nitrophenyl 2,3-bis-O-tert-butyldimethylsilyl-5-O-trityl-D-ribofuranoside (S3): To a 250 mL round bottom flask, were added 3 (3.80 g, 6.11 mmol, 1 eq), 4-nitrophenol (2.51 g, 18.1 mmol, 3 eq), 4 Å molecular sieves (200 mg), and toluene (100 mL). Cooled mixture to 0 °C. Added n-Bu3P (3.80 mL, 0.81 g/mL, 3.1 g, 15 mmol, 2.5 eq) dropwise via syringe. Added 1,1’-(azodicarbonyl)dipiperidine as a solid in one portion. Mixture was stirred at 0 °C for 30 min and then allowed to warm to room temperature and stirred for an additional 4 h. The reaction mixture was cooled to 0 °C, and 30% H 2O2 (5 mL) was added to fully quench phosphine. After stirring for 30 min, the reaction mixture was diluted with hexane (100 mL). The resulting yellow precipitate was removed by filtration through a pad of celite. The filtrate was washed with satd aq NaHcO3 five times, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography, eluting with 5% Et2O-hexanes, to yield compounds S3 (a mixture of anomers, α:β 69:31) as a pale yellow foam (2.87 g, 63%).
α-anomer
1H NMR (500 MHz, CDCl3) δ 8.21 (d, J = 9.0 Hz, 2H), 7.48 – 7.43 (m, 6H), 7.32 (t, J = 7.7 Hz, 6H), 7.29 – 7.24 (m, 3H), 7.13 (d, J = 9.3 Hz, 2H), 5.65 (d, J = 4.1 Hz, 1H), 4.38 (t, J = 4.7 Hz, 1H), 4.18 (q, J = 3.0 Hz, 1H), 4.05 (dd, J = 5.2, 2.2 Hz, 1H), 3.42 (dd, J = 10.5, 3.5 Hz, 1H), 3.12 (dd, J = 10.6, 3.1 Hz, 1H), 0.92 (s, 9H), 0.84 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H), 0.04 (s, 3H), −0.12 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 162.98, 143.82, 142.01, 128.79, 128.04, 127.33, 125.94, 116.35, 100.21, 86.98, 86.66, 73.83, 72.23, 63.43, 26.00, 25.86, 18.44, 18.17, −4.37, −4.39, −4.40, −4.73.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C42H55NO7Si2Na 764.3415; Found 764.3413
β-anomer
1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 9.2 Hz, 2H), 7.38 – 7.33 (m, 6H), 7.20 – 7.13 (m, 11H), 5.59 (d, J = 1.4 Hz, 1H), 4.41 (dd, J = 6.9, 4.1 Hz, 1H), 4.30 (dd, J = 4.1, 1.4 Hz, 1H), 4.23 (ddd, J = 6.9, 4.1, 2.6 Hz, 1H), 3.38 (dd, J = 10.6, 2.7 Hz, 1H), 3.00 (dd, J = 10.6, 4.2 Hz, 1H), 0.92 (s, 9H), 0.76 (s, 9H), 0.13 (s, 3H), 0.13 (s, 3H), 0.02 (s, 3H), −0.16 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 162.09, 143.74, 142.27, 128.82, 127.75, 127.09, 125.87, 116.26, 105.06, 86.55, 83.33, 76.60, 71.64, 62.95, 25.92, 25.89, 18.26, 18.11, −3.99, −4.28, −4.38, −4.86.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C42H55NO7Si2Na 764.3415; Found 764.3414
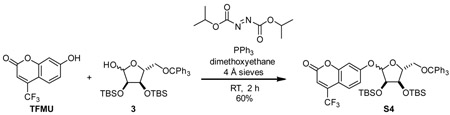
4-(trifluoromethyl)umbellifer-7-yl 2,3-bis-O-tert-butyldimethylsilyl-5-O-trityl-D-ribofuranoside (S4)
A 100 mL round bottom flask was charged with 4-(trifluoromethyl)umbelliferone (1.69 g, 7.36 mmol, 1.5 eq), 2,3-di-(tert-butyldimethylsilyl)-5-(triphenylmethyl)-D-ribofuranose (3) (3.05 g, 4.91 mmol, 1 eq), triphenylphosphine (1.93 g, 7.37 mmol, 1.5 eq), and 4 Å molecular sieves (600 mg). Material was dissolved in dimethoxyethane (35 mL) and stirred for 15 min at room temperature. DlAD (1.45 mL, 7.36 mmol, 1.5 eq) was added dropwise. Reaction mixture was stirred at room temperature for 2 hours and then poured into hexane (400 mL). Resulting precipitate was removed by filtration. Filtrate was washed with satd aq NaHCO3 4x. Organic phase was dried over Na2SO4 and evaporated. Residue was purified by silica gel (5% Et2O:hexane) to give S4 (2.47 g, 60%) as a mixture of anomers. While anomers were could be separated by silica gel chromatography, the mixture of diastereomers was routinely carried into the next step as they were more easily separable with the trityl group removed.
α-anomer
1H NMR (500 MHz, CDCl3) δ 7.55 (dt, J = 9.1, 1.9 Hz, 1H), 7.39 – 7.34 (m, 6H), 7.27 – 7.21 (m, 6H), 7.20 – 7.15 (m, 3H), 7.03 (d, J = 2.3 Hz, 1H), 6.95 (dd, J = 9.0, 2.4 Hz, 1H), 6.54 (s, 1H), 5.55 (d, J = 4.2 Hz, 1H), 4.31 (dd, J = 5.3, 4.3 Hz, 1H), 4.08 (q, J = 3.1 Hz, 1H), 3.96 (dd, J = 5.2, 1.9 Hz, 1H), 3.32 (dd, J = 10.6, 3.4 Hz, 1H), 3.03 (dd, J = 10.6, 3.1 Hz, 1H), 0.83 (s, 9H), 0.75 (s, 9H), −0.00 (s, 6H), −0.05 (s, 3H), −0.21 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 161.79, 159.62, 156.25, 143.83, 141.75 (q, 2JCF = 32.8 Hz), 128.79, 128.07, 127.34, 126.43 (d, 3JCF = 2.3 Hz), 121.79 (q, 1JCF = 275.5 Hz), 114.85, 112.50 (q, 3JCF = 5.7 Hz), 107.61, 104.46, 100.23, 87.03, 86.87, 73.87, 72.25, 63.49, 26.01, 25.85, 18.44, 18.16, −4.37, −4.41, −4.43, −4.72.
19F NMR (471 MHz, CDCl3) δ −64.68.
β-anomer
1H NMR (500 MHz, CDCl3) δ 7.50 (dq, J = 8.9, 1.8 Hz, 1H), 7.25 – 7.20 (m, 6H), 7.06 – 7.01 (m, 10H), 6.99 (dd, J = 9.0, 2.5 Hz, 1H), 6.48 (s, 1H), 5.45 (d, J = 1.3 Hz, 1H), 4.32 (dd, J = 6.9, 4.0 Hz, 1H), 4.17 (dd, J = 4.1, 1.4 Hz, 1H), 4.09 (dt, J = 6.8, 3.3 Hz, 1H), 3.30 (dd, J = 10.6, 2.8 Hz, 1H), 2.86 (dd, J = 10.7, 3.8 Hz, 1H), 0.80 (s, 9H), 0.63 (s, 9H), 0.00 (s, 3H), 0.00 (s, 3H), −0.11 (s, 3H), −0.28 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 160.98, 159.37, 156.10, 143.77, 141.55 (q, 2JCF = 32.7 Hz), 128.85, 127.75, 127.07, 126.43 (q, 3JCF = 2.4 Hz), 121.70 (q, 1JCF = 275.6 Hz), 114.14, 112.96 (q, 3JCF = 5.7 Hz), 107.92, 105.19, 104.79, 86.55, 83.32, 76.62, 71.46, 62.51, 25.93, 25.90, 18.27, 18.11, −3.98, −4.28, −4.37, −4.84.
19F NMR (471 MHz, CDCl3) δ −64.75.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C46H55O7F3NaSi2 855.3336; Found 855.3342.
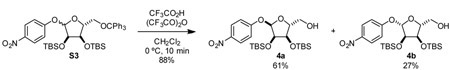
4-nitrophenyl 2,3-bis-O-tert-butyldimethylsilyl-D-ribofuranoside (4): To a stirring solution of compound S3 (5.26 g, 7.09 mmol, 1 eq) in dichloromethane (60 mL) at 0 °C was added trifluoroacetic anhydride (4.00 mL, 1.487 g/mL, 5.95 g, 28.3 mmol, 4 eq) as a 2 M solution in CH2Cl2 dropwise via addition funnel. Trifluoroacetic acid (1.6 mL, 1.489 g/mL, 2.38 g, 20.9 mmol, 3 eq) was added as a 2 M solution in CH2Cl2 dropwise via addition funnel. Reaction was stirred at room temperature for another 10 min. Reaction mixture was quenched with the addition of Et3N (4 mL, 0.7255 g/mL, 2.9 g, 29 mmol, 4.1 eq) followed by CH3OH (30 mL). Mixture was stirred for an additional 15 min at room temperature before being diluted with H2O. Extracted three times with CH2Cl2. Combined organic layers were dried over Na2SO4 and concentrated in vacuo. Resulting residue was purified by silica gel chromatography (5:1 Hexane:EtOAc) to yield compound 4a (2.15 g, 61%) and compound 4b (0.96 g, 27%).
α-anomer (4a)
1H NMR (500 MHz, CDCl3) δ 8.17 (d, J = 9.2 Hz, 2H), 7.10 (d, J = 9.3 Hz, 2H), 5.60 (d, J = 3.6 Hz, 1H), 4.20 – 4.15 (m, 3H), 3.81 (ddd, J = 12.2, 4.5, 2.5 Hz, 1H), 3.67 (ddd, J = 12.3, 7.0, 3.1 Hz, 1H), 1.94 (dd, J = 7.4, 4.5 Hz, 1H), 0.91 (s, 18H), 0.12 (s, 3H), 0.09 (s, 3H), 0.07 (s, 6H).
13C NMR (126 MHz, CDCl3) δ 162.65, 142.13, 125.91, 116.34, 100.21, 86.99, 77.16, 73.96, 71.45, 62.32, 25.96, 25.90, 18.42, 18.20, −4.30, −4.39, −4.40, −4.71.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C23H41NNaO7Si2 522.2314; Found 522.2297
β-anomer (4b)
1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 9.2 Hz, 2H), 7.07 (d, J = 9.2 Hz, 2H), 5.46 (d, J = 1.3 Hz, 1H), 4.36 (dd, J = 7.0, 4.1 Hz, 1H), 4.21 (dd, J = 4.2, 1.3 Hz, 1H), 4.15 (dt, J = 6.7, 3.1 Hz, 1H), 3.80 (ddd, J = 12.5, 4.4, 2.7 Hz, 1H), 3.54 (ddd, J = 12.4, 8.8, 3.5 Hz, 1H), 1.68 (dd, J = 8.8, 4.3 Hz, 1H), 0.91 (s, 9H), 0.91 (s, 9H), 0.13 (s, 6H), 0.10 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 161.52, 142.47, 125.92, 116.23, 105.00, 84.24, 77.16, 76.85, 70.72, 61.21, 25.94, 25.85, 18.20, 18.17, −4.15, −4.45, −4.46, −4.90.
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C23H40NO7Si2 498.2343; Found 498.2336
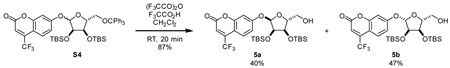
4-(trifluoromethyl)umbellifer-7-yl 2,3-bis-O-tert-butyldimethylsilyl-D-ribofuranoside (5)
To a 100 mL-RBF equipped with addition funnel was added S4 (2.31 g, 2.77 mmol) and dichloromethane (24 mL). Trifluoroacetic anhydride (1.56 mL, 11.0 mmol, 4 eq) diluted with 5 mL of dichloromethane was added to stirring solution via addition funnel at room temperature. Trifluoroacetic acid (0.64 mL, 8.4 mmol, 3 eq) diluted with 5 mL of dichloromethane was added to stirring solution dropwise. Reaction mixture was stirred at room temperature for 20 min. Reaction mixture was neutralized by addition of neat triethylamine (5.0 mL, 36 mmol, 13 eq) via addition funnel followed by methanol (10 mL). Fuming mixture was stirred for 30 min and poured into satd aq NH4Cl. Layers were separated and aqueous layer extracted 3x with dichloromethane. Combined organic layers were dried over Na2SO4, filtered, and evaporated. Residue was purified by silica gel chromatography (6:1 hexane:EtOAc) to give separable anomers 5a (658 mg, 40%) and 5b (767 mg, 47%) as white solids.
α-anomer (5a)
1H NMR (600 MHz, CDCl3) δ 7.63 (dd, J = 9.0, 1.8 Hz, 1H), 7.09 (d, J = 2.4 Hz, 1H), 7.03 (dd, J = 9.0, Hz, 1H), 6.62 (s, 1H), 5.60 (d, J = 3.7 Hz, 1H), 4.22 – 4.14 (m, 3H), 3.81 (dd, J = 12.3, 2.7 Hz, 1H), 3.67 (dd, J = 12.2, 3.1 Hz, 1 Hf), 1.82 (bs, 1H), 0.920 (s, 9H), 0.918 (s, 9H), 0.12 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.08 (s, 3H).
13C NMR (151 MHz, CDCl3) δ 161.44, 159.55, 156.17, 141.73 (q, 2JCF = 32.72 Hz), 126.49 (q, 3JCF = 2.40 Hz), 121.74 (q, 1JCF = 275.62 Hz), 114.80, 112.68 (q, 3JCF = 5.68 Hz), 107.80, 104.46, 100.25, 87.04, 74.01, 71.45, 62.34, 25.98, 25.91, 18.44, 18.21, −4.29, −4.36, −4.37, −4.69.
19F NMR (564 MHz, CDCl3) δ −64.74.
HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C27H42O7F3Si2 591.2421; Found 591.2437.
β-anomer (5b)
1H NMR (500 MHz, CDCl3) δ 7.64 (dd, J = 8.9, 1.9 Hz, 1H), 7.06 (d, J = 2.4 Hz, 1H), 7.01 (dd, J = 9.0, Hz, 1H), 6.65 (s, 1H), 5.47 (d, J = 1.2 Hz, 1H), 4.38 (dd, J = 7.2, 4.1 Hz, 1H), 4.21 (dd, J = 4.1, 1.2 Hz, 1H), 4.17 (dt, J = 7.2, 3.0 Hz, 1H), 3.86 – 3.79 (m, 1H), 3.56 (ddd, J = 12.4, 9.1, 3.3 Hz, 1H), 1.46 (dd, J = 9.1, 4.2 Hz, 1H), 0.93 – 0.92 (s, 18H), 0.14 (s, 9H), 0.12 (s, 3H).
13C NMR (151 MHz, CDCl3) δ 160.36, 159.23, 156.13, 141.54 (q, 2JCF = 32.82 Hz), 126.71 (q, 3JCF = 2.40 Hz), 121.69 (q, 1JCF = 275.34 Hz), 114.34, 113.23 (q, 3JCF = 5.66 Hz), 108.27, 105.14, 104.57, 84.23, 76.90, 70.68, 61.22, 25.99, 25.91, 18.28, 18.23, −4.08, −4.38, −4.41, −4.83.
19F NMR (564 MHz, CDCl3) δ −64.77.
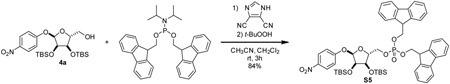
4-nitrophenyl 2,3-bis-O-tert-butyldimethylsilyl-α-D-ribose-5-(difluorenylmethyl phosphate) (S5): To a 20 mL reaction vial, 4a (71.1 mg, 0.142 mmol, 1 eq) and bis-(9H-fluoren-9-ylmethyl)-N,N-diisopropylamidophosphite (Lambrecht et al., 2015) (95.5 mg, 0.183 mmol, 1.3 eq) were added. Dissolved material by addition of CH2Cl2 (1.25 mL). Cooled solution to 0 °C. 4,5-dicyanoimidazole (24.0 mg, 0.203 mmol, 1.4 eq) was added as a solution in acetonitrile (0.25 mL). After stirring at 0 °C for 15 min, mixture was allowed to warm to room temperature and was stirred for an additional 2 h. Once starting material was consumed as indicated by TLC (75:25 hexane:EtOAc), mixture was cooled to 0 °C and subjected to dropwise addition of tert-butyl hydroperoxide (0.14 mL, 0.70 mmol, 5 eq) as a 5 M solution in decane. Mixture was stirred for an additional 1 h and quenched with H2O. Mixture was extracted with CH2Cl2 three times. Combined organic layers were dried over Na2SO4, concentrated, and purified by silica gel chromatography eluting with 60:40 hexane:EtOAc to yield compound S5 as a white foam (111.5 mg, 84%).
1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 9.2 Hz, 2H), 7.77 – 7.70 (m, 4H), 7.57 (dd, J = 18.2, 7.5 Hz, 2H), 7.52 (dd, J = 7.3, 6.3 Hz, 2H), 7.44 – 7.34 (m, 4H), 7.32 – 7.24 (m, 4H), 6.91 (d, J = 9.2 Hz, 2H), 5.26 (m, 1H), 4.38 – 4.26 (m, 4H), 4.22 – 4.12 (m, 3H), 4.06 (ddd, J = 11.4, 5.8, 3.1 Hz, 1H), 4.04 – 4.00 (m, 2H), 3.91 (ddd, J = 11.1, 7.0, 3.8 Hz, 1H), 0.91 (s, 9H), 0.90 (s, 9H), 0.11 (s, 3H), 0.05 (s, 3H), 0.03 (s, 6H).
13C NMR (126 MHz, CDCl3) δ 162.23, 143.16, 143.10, 142.98, 142.94, 142.10, 141.47, 141.44, 128.10, 128.08, 128.06, 127.27, 127.22, 125.78, 125.21, 125.17, 125.13, 125.07, 120.19, 120.18, 120.15, 116.09, 99.76, 83.93, 83.87, 77.16, 73.46, 71.13, 69.56, 69.51, 69.48, 69.43, 66.55, 66.51, 48.03, 48.00, 47.97, 47.93, 25.90, 25.86, 18.38, 18.12, −4.25, −4.37, −4.47, −4.80.
31P NMR (202 MHz, CDCl3) δ −0.41.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C51H62NO10NaSi2P 958.3548; Found 958.3554.
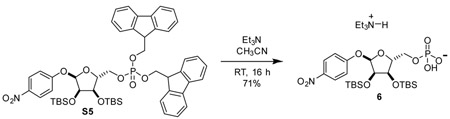
Triethylammonium 4-nitrophenyl 2,3-bis-O-tert-butyldimethylsilyl-α-D-ribose-5-phosphate (6): 20 mL reaction vial was charged with compound S5 (464 mg, 0.50 mmol, 1 eq). Added acetonitrile (5 mL) and triethylamine (freshly distilled from CaH2, 1 mL) successively. Stirred at room temperature for 16 h. Added 1 mL toluene to stirring solution and concentrated in vacuo. Residue was redissolved in methanol (0.5 mL) and purified by C-18 chromatography to yield the triethylammonium salt of compound 6 as a tan foam (241 mg, 71%).
1H NMR (500 MHz, CD3OD) δ 8.20 (d, J = 9.3 Hz, 2H), 7.17 (d, J = 9.3 Hz, 2H), 5.71 (d, J = 4.2 Hz, 1H), 4.39 (dd, J = 5.4, 4.2 Hz, 1H), 4.31 (dd, J = 5.4, 2.3 Hz, 1H), 4.20 (dq, J = 3.9, 1.9 Hz, 1H), 3.99 (ddd, J = 10.1, 4.4, 3.1 Hz, 1H), 3.95 (ddd, J = 9.1, 4.6, 3.4 Hz, 1H), 3.17 (q, J = 7.3 Hz, 6H), 1.31 (t, J = 7.3 Hz, 9H), 0.94 (s, 9H), 0.93 (s, 9H), 0.16 (s, 3H), 0.13 (s, 3H), 0.13 (s, 3H), 0.10 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 164.14, 143.13, 126.62, 117.36, 101.35, 87.69 (d, J = 8.9 Hz), 74.75, 73.14, 65.98 (d, J = 5.2 Hz), 47.40, 26.49, 26.46, 19.15, 18.96, 9.11, −4.10, −4.31 (2C), −4.43.
31P NMR (202 MHz, CD3OD) δ 0.77
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C23H41NO10PSi2 578.2012; Found 578.2017.
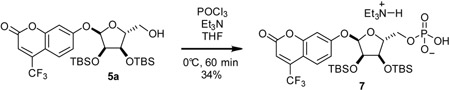
Triethylammonium 4-(trifluoromethyl)umbellifer-7-yl 2,3-bis-O-tert-butyldimethylsilyl-α-D-ribose-5-phosphate (7) (method A): To a stirring solution of 5a (507 mg, 0.858 mmol) in THF (8 mL) at 0 °C was added triethylamine (1.4 mL, 10 mmol, 1 2 eq) followed by phosphorus oxychloride (0.16 mL, 1.7 mmol, 2 eq). Reaction mixture was stirred at 0 °C for 40 min then warmed to room temperature and stirred for an additional 20 min. After cooling to 0 °C once again, reaction was quenched with addition of water (1 mL). Mixture was stirred for an additional 30 min at room temperature then solvent was removed by rotary evaporator. Residue was purified by C18 chromatography to give 7 (225 mg, 34%) as a white solid.
1H NMR (500 MHz, CD3OD) δ 7.68 (dd, J = 8.8, 2.0 Hz, 1H), 7.12 – 7.06 (m, 2H), 6.71 (s, 1H), 5.72 (d, J = 4.3 Hz, 1H), 4.41 (dd, J = 5.4, 4.2 Hz, 1H), 4.32 (dd, J = 5.4, 2.2 Hz, 1H), 4.21 (dq, J = 3.9, 1.9 Hz, 1H), 4.01 – 3.91 (m, 2H), 3.19 (q, J = 7.3 Hz, 6H), 1.31 (t, J = 7.3 Hz, 9H), 0.95 (s, 9H), 0.93 (s, 9H), 0.16 (s, 3H), 0.14 (s, 6H), 0.11 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 163.04, 160.83, 157.41, 142.36 (q, 2JCF = 32.30 Hz) 127.32 (q, 3JCF = 1.72 Hz), 123.22 (q, 1JCF = 274.75 Hz), 115.81, 113.88 (q, 3JCF = 5.81 Hz), 108.56, 105.05, 101.51, 87.94 (d, 3JCP = 9.1 Hz), 74.81, 73.20, 65.99 (d, 2JCP = 5.2 Hz), 47.56, 26.48f, 26.46, 19.16, 18.99, 9.15, −4.12, −4.34 (2C), −4.43.
19F NMR (470 MHz, CD3OD) δ −66.10.
31P NMR (202 MHz, CD3OD) δ 0.83.
HRMS (ESI-TOF) m/z: [M-H]− Cald for C27H41F3O10PSi2 669.1933; Found 669.1918.
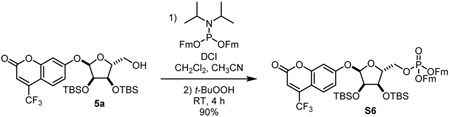
4-(trifluoromethyl)umbellifer-7-yl 2,3-bis-O-tert-butyldimethylsilyl-α-D-ribose-5-(difluorenylmethyl phosphate) (S6): To a stirring solution of compound 5a (373.9 mg, 0.633 mmol) and N,N-diisopropyl bis(9-methylfluorenyl)phosphoramidite (403.8 mg, 0.774 mmol) in CH2Cl2 (12 mL). Dicyanoimidazole (151 mg, 1.3 mmol) was added as a solution in acetonitrile (4.8 mL). Stirred at room temperature for 3 h. Reaction mixture was cooled to 0 °C added t-butylperoxide as a 5 M solution in decane (0.6 mL). Stirred at 0 °C for 1 h. Reaction mixture was quenched with water and extracted with CH2Cl2. Combined organic layers were dried over Na2SO4 and evaporated. Residue was purified by silica gel chromatography (1:1 hexane:EtOAc) to give compound S6 as a yellow foam (579.5 mg, 90%).
1H NMR (500 MHz, CDCl3) δ 7.76 – 7.68 (m, 4H), 7.58 – 7.46 (m, 5H), 7.43 – 7.32 (m, 4H), 7.31 – 7.22 (m, 5H), 6.95 (d, J = 2.4 Hz, 1H), 6.86 (dd, J = 9.0, 2.4 Hz, 1H), 6.61 (s, 1H), 5.34 – 5.30 (m, 1H), 4.34 (td, J = 6.3, 1.4 Hz), 4.29 (ddt, J = 9.9, 6.8, 3.7 Hz), 4.20 – 4.12 (m, 3H), 4.05 – 4.00 (m, 3H), 3.90 (ddd, J = 11.1, 7.1, 3.8 Hz, 1H), 0.90 (s, 9H), 0.89 (s, 9H), 0.10 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H), 0.02 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 161.13, 159.34, 156.03, 143.16 (d, J = 11.6 Hz), 143.04 (d, J = 9.6 Hz), 141.53 (q, 2JCF = 32.8 Hz), 141.52, 141.47, 128.07 (d, J = 1.7 Hz), 127.28 (d, J = 1.2 Hz), 127.26, 126.45 (q, 3JCF = 2.2 Hz), 125.22 (d, J = 6.8 Hz), 125.16 (d, J = 4.2 Hz), 121.72 (q, 1JCF = 275.5 Hz), 120.19 (d, J = 1.9 Hz), 120.16, 114.20, 112.80 (q, 3JCF = 5.7 Hz), 107.81, 104.59, 99.82, 84.30 (d, J = Hz), 73.54, 71.26, 69.54 (t, J = 5.7 Hz), 66.66 (d, J = 5.8 Hz), 48.04 (dd, J = 7.9, 5.7 Hz), 25.94, 25.88, 18.42, 18.15, −4.25, −4.36, −4.42, −4.74.
19F NMR (471 MHz, CDCl3) δ −64.71.
31P NMR (203 MHz, CDCl3) δ −1.55.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C55H62O10F3NaSi2P 1049.3469; Found 1049.3500.
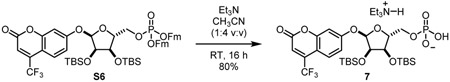
Triethylammonium 4-(trifluoromethyl)umbellifer-7-yl 2,3-bis-O-tert-butyldimethylsilyl-α-D-ribose-5-phosphate (7) (method B): To a stirring solution of compound S6 (1.18 g, 1.00 mmol) in acetonitrile (10 mL) was added Et3N (freshly distilled from CaH2, 2.5 mL). Mixture was stirred at room temperature for 16 h. Reaction mixture was co-evaporated with toluene to dryness. Residue was purified by C18 reverse phase chromatography to give compound 7 as a white solid (80%).
1H NMR (500 MHz, CDCl3) δ 7.57 (dt, J = 9.0, 1.8 Hz, 1H), 7.04 (d, J = 2.4 Hz, 1H), 7.00 (dd, J = 9.0, Hz, 1H), 6.56 (s, 1H), 5.54 (d, J = 4.1 Hz, 1H), 4.25 (dd, J = 5.4, 4.1 Hz, 1H), 4.22 (dd, J = 5.4, 1.9 Hz, 1H), 4.16 (dt, J = 4.0, 2.2 Hz, 1H), 3.98 (ddd, J = 11.3, 5.4, 3.8 Hz, 1H), 3.93 (ddd, J = 11.2, 5.6, 4.1 Hz, 1H), 3.06 (q, J = 7.3 Hz, 6H), 1.30 (t, J = 7.3 Hz, 9H), 0.86 (s, 9H), 0.86 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 161.76, 159.58, 156.06, 141.72 (q, 2JCF = 32.6 Hz), 126.33 (q, 3JCF = 1.70 Hz), 121.66 (q, 1JCF = 275.6 Hz), 114.93, 112.20 (q, 3JCF = 5.7 Hz), 107.32, 104.07, 100.00, 86.72 (d, 3JCP = 8.6 Hz), 73.52, 71.66, 64.78 (d, 2JCP = 4.8 Hz), 45.47, 25.90, 25.84, 18.29, 18.08, 8.62, −4.46, −4.47, −4.49, −4.66.
19F NMR (471 MHz, CDCl3) δ −64.74.
31P NMR (203 MHz, CDCl3) δ 1.69.
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C27H42O10NaPF3Si2 693.1904; Found 693.1901.
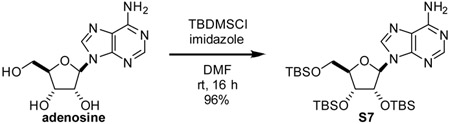
2’,3’,5’-tris-O-(tert-butyldimethylsNyl)-adenosme (S7): To a 100 mL round-bottom flask, adenosine (5.0 g, 19 mmol, 1 eq), imidazole (6.9 g, 102 mmol, 5.4 eq), and dimethylformamide (40 mL) were added. Once fully dissolved, solution was cooled to 0 °C. Tert-butyldimethylsilyl chloride (12.2 g, 81 mmol, 4.2 eq) was added to the stirring solution. Once fully dissolved, reaction vessel was allowed to warm to room temperature. Stirred at room temperature for 16 h. Reaction was quenched by the addition of satd aq NH4Cl. Extracted three times with CH2Cl2. Combined organic layers were dried over MgSO4, filtered through a pad of celite, and evaporated. Residue was purified by silica gel chromatography eluting with 50:50 hexane:EtOAc to yield S7 as a white foam (12.5 g, 96%).
1H NMR (500 MHz, CDCl3) δ 8.32 (s, 1H), 8.15 (s, 1H), 6.11 (bs, 2H), 6.02 (d, J = 5.2 Hz, 1H), 4.68 (t, J = 4.8 Hz, 1H), 4.31 (t, J = 3.9 Hz, 1H), 4.12 (q, J = 3.5 Hz, 1H), 4.02 (dd, J = 11.4, 4.2 Hz, 1H), 3.77 (dd, J = 11.3, 2.9 Hz, 1H), 0.94 (s, 9H), 0.92 (s, 9H), 0.78 (s, 9H), 0.13 (s, 3H), 0.12 (s, 3H), 0.09 (s, 3H), 0.09 (s, 3H), −0.06 (s, 3H), −0.24 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 155.70, 153.03, 150.05, 139.69, 120.19, 88.41, 85.58, 75.89, 72.11, 62.66, 26.20, 25.98, 25.81, 18.65, 18.22, 17.99, −4.28, −4.57, −4.59, −4.95, −5.24 (2C).
HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C28H56N5O4Si3 610.3640; Found 610.3647
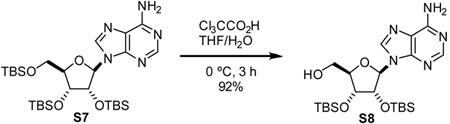
2’,3’-bis-O-(tert-butyldimethylsilyl)-adenosine (S8): A 500 mL round-bottom flask was charged with S7 (4.4 g, 7.2 mmol, 1 eq). Compound was dissolved in wet tetrahydrofuran (120 mL). Solution was cooled to 0 °C. To vigorously stirring solution, trichloroacetic acid (56 g, 340 mmol, 47 eq) was added as an ice-cold solution in H2O (25 mL). Stirred reaction mixture at 0 °C for 3 h. Quenched reaction by slowly cannulating reaction mixture into ice cold satd aq NaHCO3. Once evolution of gas ceased, extracted three times with EtOAc. Dried organic layer over Na2SO4, filtered, and evaporated. Residue was purified by silica gel chromatography eluting with 95:5 CH2Cl2:CH3OH to yield compound S8 as a white solid (3.3 g, 92%).
1H NMR (500 MHz, 1:1 CDCl3:DMSO (v:v)) δ 8.24 (s, 1H), 8.11 (s, 1H), 7.29 (s, 2H), 6.08 (dd, J = 9.3, 3.3 Hz, 1H), 5.86 (d, J = 7.1 Hz, 1H), 4.87 (dd, J = 7.2, 4.5 Hz, 1H), 4.26 (dd, J = 4.4, 1.4 Hz, 1H), 4.01 (q, J = 2.1 Hz, 1H), 3.75 (dt, J = 12.6, 3.2 Hz, 1H), 3.58 (ddd, J = 12.1, 9.4, 2.6 Hz, 1H), 0.90 (s, 9H), 0.69 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H), −0.18 (s, 3H), −0.52 (s, 3H).
13C NMR (126 MHz, 1:1 CDCl3:DMSO (v:v)) δ 156.22, 151.89, 148.40, 140.12, 119.86, 88.45, 87.40, 73.99, 73.06, 61.60, 25.52, 25.33, 17.65, 17.36, −4.88, −4.97, −5.07, −6.02.
HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C22H42O4Si2 496.2775; Found 496.2774
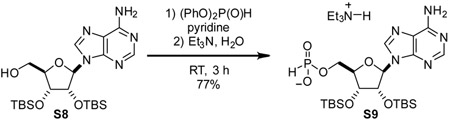
Triethylammonium 2’,3’-bis-O-(tert-butyldimethylsilyl)-adenosin-5’-yl H-phosphonate (S9): To a 20 mL reaction vial was added compound S8 (324 mg, 0.654 mmol, 1 eq). Added dry pyridine (6.5 mL). Once fully dissolved, diphenyl phosphite (0.65 mL, 1.22 g/mL, 795 mg, 3.4 mmol, 5 eq) was added, and reaction mixture was stirred at room temperature for 1.5 h. Then, water (0.5 mL) was added followed by triethylamine (0.5 mL). After stirring for an additional 30 min, the reaction mixture was concentrated and purified by C-18 chromatography to yield the triethylammonium salt of compound S9 as a white foam (331 mg, 77%).
1H NMR (500 MHz, CD3OD) δ 8.51 (s, 1H), 8.22 (s, 1H), 6.83 (d, 1JHP = 618.5 Hz, 1H), 6.10 (d, J = 6.7 Hz, 1H), 4.87 (dd, J = 6.7, 4.4 Hz, 1H), 4.41 (dd, J = 4.4, 1.9 Hz, 1H), 4.21 (td, J = 3.9, 2.1 Hz, 1H), 4.14 (ddd, J = 11.0, 6.7, 4.2 Hz, 1H), 4.08 (ddd, J = 11.4, 6.7, 3.8 Hz, 1H), 3.15 (q, J = 7.3 Hz, 6H), 1.27 (t, J = 7.3 Hz, 9H), 0.97 (s, 9H), 0.74 (s, 9H), 0.18 (s, 3H), 0.16 (s, 3H), −0.02 (s, 3H), −0.31 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 157.20, 153.66, 151.00, 141.41, 120.32, 88.64, 86.71 (d, 3JCP = 8.1 Hz), 77.07, 74.49, 64.22 (d, 2JCP = 4.5 Hz), 47.61, 26.44, 26.24, 18.90, 18.68, 9.16, −4.17, −4.28 (2C), −5.08.
31P NMR (202 MHz, CD3OD) δ 4.14
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C22H41N5O6PSi2 558.2338; Found 558.2323
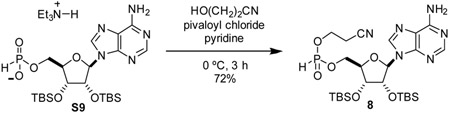
2’,3’-bis-O-(tert-butyldimethylsilyl)-adenosin-5’-yl 2-cyanoethyl phosphonate (8): To a 25 mL round-bottom flask was added compound S9 (238 mg, 0.360 mmol, 1 eq). Dried material by co-azeotroping with dry acetonitrile three times. Pyridine was added (4 mL) and solution was cooled to 0 °C. 3-hydroxypropionitrile (0.070 mL, 1.04 g/mL, 73 mg, 1.0 mmol, 3 eq) was added to stirring solution. Pivaloyl chloride (0.09 mL, 0.98 g/mL, 88 mg, 0.73 mmol, 2 eq) was added dropwise. After stirring at 0 °C for 2.5 h, reaction mixture was concentrated, azeotroping with toluene. Resulting residue was redissolved in acetonitrile and purified by C-18 chromatography to yield compound 8 as a white solid as a mixture of diastereomers (159 mg, 72%).
1H NMR (500 MHz, CDCl3) δ 8.30 (s, 1H), 8.30 (s, 1H), 8.00 (s, 1H), 7.98 (s, 1H), 6.91 (d, J = 720.9 Hz, 1H), 6.83 (d, J = 723.5 Hz, 1H) 6.50 (d, J = 7.8 Hz, 2H), 5.87 (dd, J = 5.8, 4.2 Hz, 2H), 4.89 (q, J = 4.1 Hz, 2H), 4.52 – 4.14 (m, 10H), 2.72 (t, J = 6.3 Hz, 2H), 2.69 (t, J = 6.2 Hz, 2H), 0.90 (s, 9H), 0.90 (s, 9H), 0.80 (s, 18H), 0.10 (s, 3H), 0.09 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H), −0.03 (s, 6H), −0.19 (s, 3H), −0.19 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 156.00, 155.96, 153.01, 149.52, 149.49, 140.14, 139.98, 120.54, 116.33, 89.94, 89.83, 82.60, 82.57, 82.55, 82.52, 77.16, 74.42, 74.33, 71.68, 71.60, 64.90, 64.85, 64.77, 64.73, 60.20, 60.16, 60.12, 60.08, 25.87, 25.76, 19.98, 19.93, 18.09, 17.94, −4.30, −4.31, −4.61, −4.63, −4.75, −4.76, −4.83, −4.85.
31P NMR (202 MHz, CDCl3) δ 8.70, 8.23.
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C25H44N6O6PSi2 611.2604; Found 611.2594
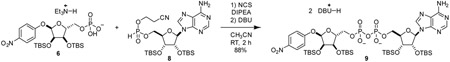
α-1”-0-(4-nitrophenyl)-2’,2”,3’,3”-O-tetrakis-(tert-butyldimethylsilyl)-ADP-ribose (9): To a 20 mL reaction vial was added 6 (158.6 mg, 0.233 mmol, 1.0 eq) and 8 (138.3 mg, 0.226 mmol, 1.0 eq). Mixture was dried by co-azeotroping with dry acetonitrile three times and placing under vacuum over P2O5 for 12 h. The mixture was dissolved in acetonitrile (3.0 mL). Then, (i-Pr2)2NEt (129 mg, 1.00 mmol, 4.4 eq) and N-chlorosuccinimide (90.8 mg, 0.68 mmol, 3 eq) were sequentially added as 1 M solutions in acetonitrile and stirred at room temperature for 1 h. 1,8-diazabicycloundec-7-ene (350 mg, 2.3 mmol, 10 eq) was added as a 1 M solution in acetonitrile. After stirring for 30 min, reaction mixture was evaporated in vacuo and purified by C18 chromatography to yield compound 9 as a white foam (287 mg, 88%).
1H NMR (500 MHz, CD3OD) δ 8.67 (s, 1H), 8.18 (s, 1H), 8.14 (d, J = 9.2 Hz, 2H), 7.11 (d, J = 9.2 Hz, 2H), 6.15 (d, J = 7.5 Hz, 1H), 5.64 (d, J = 4.3 Hz, 1H), 4.85 (dd, J = 7.5, 4.5 Hz, 1H), 4.46 (d, J = 4.4 Hz, 1H), 4.37 (dd, J = 5.4, 4.2 Hz, 1H), 4.35 - 4.27 (m, 3H), 4.21 (ddt, J = 16.3, 4.3, 2.3 Hz, 2H), 4.11 (hept, J = 5.7, 5.0 Hz, 2H), 3.58 - 3.53 (m, 4H), 3.50 (t, J = 6.5, 1.7 Hz, 4H), 3.36 (t, J = 5.6 Hz, 4H), 2.73 - 2.67 (m, 4H), 1.99 (pd, J = 5.4, 1.3 Hz, 4H), 1.79 - 1.62 (m, 12H), 1.34 (d, J = 6.5 Hz, 2H), 0.98 (s, 9H), 0.93 (s, 9H), 0.91 (s, 9H), 0.70 (s, 9H), 0.20 (s, 3H), 0.17 (s, 3H), 0.15 (s, 3H), 0.13 (s, 3H), 0.11 (s, 3H), 0.08 (s, 3H), −0.01 (s, 3H), −0.38 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 167.42, 164.19, 157.30, 153.85, 151.17, 143.00, 141.66, 126.58, 120.04, 117.32, 101.23, 87.89, 87.86, 87.82, 87.64, 87.56, 77.60, 74.87, 74.76, 73.28, 66.76, 66.71, 66.66, 55.23, 49.53, 49.00, 39.34, 33.60, 30.01, 27.54, 26.49, 26.44, 26.21, 24.99, 20.43, 19.59, 19.14, 18.97, 18.93, 18.65, −4.07, −4.09, −4.19, −4.26, −4.29, −5.26.
31P NMR (202 MHz, CD3OD) δ −11.30 (d, J = 21.8 Hz), −11.67 (d, J = 21.6 Hz).
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C45H81N6O16P2Si4 1135.4267; Found 1135.4273
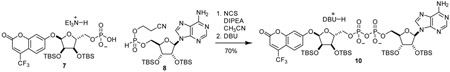
α-1”-O-(4-(trifluoromethyl)umbellifer-7-yl)-2’,2”,3’,3”-O-tetrakis-(tert-butyldimethylsilyl)-ADP-ribose (10): To a stirring solution of 7 (95.8 mg, 0.124 mmol) and 8 (76.3 mg, 0.124 mmol) in dichloromethane (5 mL) was added DIPEA as a 1 M solution in CH3CN (0.74 mL, 0.74 mmol, 6 eq) and N-chlorosuccinimide as a 1 M solution in CH3CN (0.62 mL, 0.62 mmol, 5 eq). Mixture was stirred for 30 min at room temperature and then 1,8-diazabicycloundec-7-ene as a 1 M solution in THF (1 mL, 1 mmol, 8 eq) was added. After stirring for an additional 30 min, solvent was removed by rotavap. Residue was purified by C18 chromatography to give 10 (124 mg, 70%) as a white solid.
1H NMR (500 MHz, CD3OD) δ 8.29 (s, 1H), 7.81 (s, 1H), 7.24 (dd, J = 8.9, 2.0 Hz, 1H), 6.65 (m, 2H), 6.29 (s, 1H), 5.73 (d, J = 7.2 Hz, 1H), 5.27 (d, J = 4.3 Hz, 1H), 4.42 (dd, J = 7.2, 4.4 Hz, 1H), 4.05 (dd, J = 4.4, 1.3 Hz, 1H), 3.99 (dd, J = 5.4, 4.3 Hz, 1H), 3.95 – 3.87 (m, 3H), 3.85 – 3.80 (m, 2H), 3.75 – 3.70 (m, 2H), 3.18 – 3.14 (m, 1H), 3.11 (t, J = 6.0 Hz, 2H), 2.96 (dd, J = 10.1, 4.4 Hz, 2H), 2.91 (p, J = 1.6 Hz, 1H), 2.80 (q, J = 7.4 Hz, 3H), 2.33 – 2.27 (m, 2H), 1.60 (pd, J = 5.5, 1.1 Hz, 2H), 1.38 – 1.24 (m, 4H), 0.97 (ddt, J = 14.4, 9.2, 5.2 Hz, 22H), 0.57 (s, 9H), 0.54 (s, 9H), 0.51 (s, 9H), 0.31 (s, 9H), - 0.21 (s, 3H), −0.24 (s, 3H), −0.25 (s, 3H), −0.28 (s, 3H), −0.29 (s, 3H), −0.31 (s, 3H), −0.41 (s, 3H), −0.77 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 163.03, 160.75, 157.36, 153.02, 151.00, 142.21, 141.85, 127.28, 120.00, 115.77, 113.82, 108.45, 105.03, 101.34, 88.03, 87.46, 87.39, 77.72, 74.77, 73.27, 66.76, 66.63, 55.43, 55.25, 43.49, 39.35, 33.62, 30.00, 27.54, 26.49, 26.48, 26.22, 24.99, 20.43, 19.57, 19.14, 18.98, 18.92, 18.86, 18.65, 17.44, 13.05, 9.12, −4.09, −4.18, −4.20, −4.24, −4.28, −4.31, −5.21.
19F NMR (470 MHz, CD3OD) δ −66.37.
31P NMR (202 MHz, CD3OD) δ −10.38 (d, J = 21.2 Hz), −10.80 (d, J = 21.2 Hz).
HRMS (ESI-TOF) m/z: [M-H]− Cald for C49H81F3N5O16P2Si4 1226.4188. Found 1226.4139.
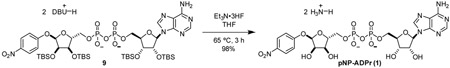
pNP-ADPr (1): To a 25 mL round-bottom flask was added 9 (109.7 mg, 0.0761 mmol, 1 eq) and THF (3.0 mL). Once starting material was fully dissolve, triethylamine trihydrofluoride (0.5 mL) was added neat dropwise. Reaction vessel was sealed and heated to 65 °C. Reaction was closely monitored by TLC (80:20 2-propanol:0.2% NH4OH(aq)) Once reaction had gone to completion, reaction mixture was cooled to room temperature and concentrated in vacuo. Remaining acid was quenched by dropwise addition of satd aq NaHCO3. Aqueous solution of crude product was purified by C18 chromatography utilizing ion-pairing reagent in the mobile phase (10 mM Et3N-HOAc, pH 7.0) to yield compound 1 as a triethylammonium salt. The triethylammonium salt was eluted through Dowex 50W-8X (ammonium-form) to give the ammonium salt of compound 1 as a white powder (53.8 mg, 98%).
1H NMR (500 MHz, D2O) δ 8.36 (s, 1H, Ade-2), 8.03 (s, 1H, Ade-8), 7.92 (d, J = 9.2 Hz, 2H, pNP-3,5), 6.92 (d, J = 9.2 Hz, 2H, pNP-2,6), 6.04 (d, J = 5.7 Hz, 1H, Ade-1’), 5.67 (d, J = 4.4 Hz, 1H, Rib-1”), 4.62 (t, J = 5.7 Hz, 1H, Ade-2’), 4.46 (dd, J = 5.1, 3.6 Hz, 1H, Ade-3’), 4.41 (dd, J = 6.0, 4.6 Hz, 1H, Rib-2”), 4.36 (m, 2H, Ade-4’, Rib-4”), 4.27 (dd, J = 6.2, 2.6 Hz, 1H, Rib-3”), 4.23 (m, 2H, Ade-5’), 4.08 (m, 2H, Rib-5”).
13C NMR (126 MHz, D2O) δ 161.8, 155.2, 152.5, 148.7, 141.5, 139.6, 125.6, 118.3, 116.3, 100.4, 86.8, 84.7, 83.8, 74.5, 71.3, 70.4, 69.7, 65.7, 65.3.
31P NMR (202 MHz, D2O) δ −11.2
HRMS (ESI-TOF) m/z: [M-H]− Calcd for 679.0808; Found 679.0805
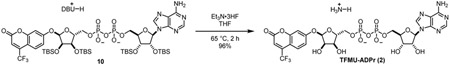
TFMU-ADPr (2)
To a stirring solution of 10 (67.2 mg, 0.044 mmol) in THF (1 mL) was added triethylamine trihydrofluoride (0.3 mL). Mixture was heated to 65 °C and stirred for 2 h. Once cooled to room temperature, reaction was quenched with addition of satd aq NaHCO3. Aqueous mixture was purified by ion-pairing chromatography (10 mM Et3N-HOAc, C18) followed by ion exchange (Dowex 50W-8, ammonium form) to provide TFMU-ADPr (2) as a white solid (33.8 mg, 96%).
1H NMR (500 MHz, D2O) δ 8.11 (s, 1H), 7.71 (s, 1H), 7.25 (dd, J = 9.1, 1.8 Hz, 1H), 6.70 (dd, J = 9.0, Hz, 1H), 6.58 (d, J = 2.5 Hz, 1H), 6.50 (s, 1H), 5.76 (d, J = 5.6 Hz, 1H), 5.56 (d, J = 4.5 Hz, 1H), 4.41 (t, J = 5.3 Hz, 1H), 4.33 – 4.28 (m, 2H), 4.28 – 4.25 (m, 1H), 4.22 (q, J = 4.1 Hz, 1H), 4.15 (dd, J = 6.2, 2.8 Hz, 1H), 4.15 – 4.05 (m, 2H), 4.01 (ddd, J = 11.5, 5.0, 3.0 Hz, 1H), 3.94 (dt, J = 11.4, 4.4 Hz, 1H).
13C NMR (126 MHz, D2O) δ 161.71, 159.94, 154.33, 154.18, 151.56, 147.93, 141.37 (q, 2JCF = 32.8 Hz), 139.31, 125.98, 121.09 (q, 1JCF = 275.1 Hz), 117.67, 114.65, 112.35 (q, 3JCF = 4.3 Hz), 107.33, 103.50, 100.16, 86.90, 84.51 (d, 3JCP = 8.2 Hz), 83.42 (d, 3JCP = 8.3 Hz), 74.46, 71.13, 70.10, 69.57, 65.66 (d, 2JCP = 4.0 Hz), 65.11 (d, 2JCP = 4.2 Hz).
19F NMR (471 MHz, D2O) δ −64.43.
31P NMR (203 MHz, D2O) δ −11.08 (d, J = 21.9 Hz), −11.28 (d, J = 21.5 Hz).
HRMS (ESI-TOF) m/z: [M-H]− Cald for C25H25F3N5O16P2 770.0729; found 770.0723.
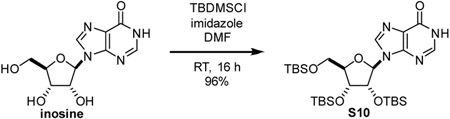
2’,3’,5’-tris-O-(tert-butyldimethylsilyl)-inosine (S10): Intermediate was prepared according to a modified version of a previous report (Höbartner and Silverman, 2005). Briefly, a 100 mL round-bottom flask was charged with inosine (1.0 g, 3.7 mmol, 1 eq) and imidazole (1.7 g, 25 mmol, 6.7 eq). Material was dissolved in dimethylformamide (4 mL). Tert-butyldimethylsilyl chloride (2.8 g, 18.7 mmol, 5 eq) was added in one portion. The solution was stirred at room temperature for 16 h. Reaction mixture was diluted with CH2Cl2 and quenched with the addition of satd aq NaHCO3. Organic layer was washed two times with satd aq NaHCO3, dried over Na2SO4, filtered through a pad of celite, and evaporated to give S10 as a white solid (2.2 g, 96%) which was used in subsequent reactions without further purification. 1H NMR spectrum was consistent with literature.
1H NMR (500 MHz, CDCl3) δ 12.94 (bs, 1H), 8.24 (s, 1H), 8.10 (s, 1H), 6.02 (d, J = 4.9 Hz, 1H), 4.50 (t, J = 4.6 Hz, 1H), 4.30 (t, J = 4.0 Hz, 1H), 4.13 (q, J = 3.4 Hz, 1H), 4.00 (dd, J = 11.4, 3.7 Hz, 1H), 3.79 (dd, J = 11.4, 2.5 Hz, 1H), 0.96 (d, J = 0.7 Hz, 9H), 0.93 (d, J = 0.7 Hz, 9H), 0.81 (d, J = 0.7 Hz, 9H), 0.15 (s, 3H), 0.14 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H), −0.02 (s, 3H), −0.18 (s, 3H).
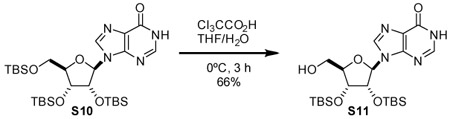
2’,3’-bis-O-(tert-butyldimethylsilyl)-inosine (S11): A 100 mL round-bottom flask was charged with S10 (1.2 g, 2.0 mmol, 1 eq). Compound was dissolved in tetrahydrofuran (31 mL). Solution was cooled to 0 °C. To vigorously stirring solution, trichloroacetic acid (15 g, 92 mmol, 47 eq) was added as an ice-cold solution in H2O (6.8 mL). Stirred reaction mixture at 0 °C for 3 h. Quenched reaction by slowly cannulating reaction mixture into satd aq NaHCO3. Once evolution of gas ceased, extracted three times with EtOAc. Dried organic layer over Na2SO4, filtered, and evaporated. Residue was purified by silica gel chromatography eluting with 93:7 CH2Cl2:CH3OH to yield compound S11 as a white solid (641 mg, 66%).
1H NMR (500 MHz, CDCl3) δ 13.34 (bs, 1H), 8.46 (s, 1H), 7.95 (s, 1H), 5.80 (d, J = 7.7 Hz, 1H), 5.68 (bs, 1H), 4.87 (dd, J = 7.8, 4.6 Hz, 1H), 4.31 (d, J = 4.5 Hz, 1H), 4.16 (s, 1H), 3.93 (dd, J = 12.9, 2.1 Hz, 1H), 3.72 (d, J = 13.0 Hz, 1H), 0.94 (d, J = 0.9 Hz, 9H), 0.75 (d, J = 0.9 Hz, 9H), 0.12 (s, 3H), 0.11 (s, 3H), −0.12 (s, 3H), −0.52 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 158.90, 147.87, 145.79, 141.04, 126.72, 91.05, 89.33, 74.79, 73.84, 62.91, 25.93, 25.80, 18.20, 17.92, −4.42, −4.46, −4.49, −5.70.
HRMS (ESI-TOF) m/z: [M+H]+ Calc for C22H40N4O5Si2 497.2610; Found 497.2614
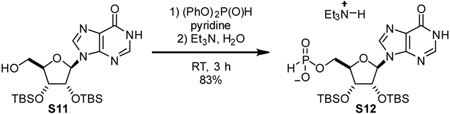
Triethylammonium 2’,3’-bis-O-(tert-butyldimethylsilyl)-inosin-5’-yl H-phosphonate (S12): A 100 mL round-bottom flask was charged with S11 (420 mg, 0.85 mmol, 1 eq) and pyridine (8.5 mL). Diphenylphosphite (0.81 mL, 1.223 g/mL, 990 mg, 4.2 mmol, 5 eq) was added dropwise. After stirring reaction mixture at room temperature for 1.5 h, H20 (0.5 mL) and triethylamine (0.5 mL) were added sequentially. Reaction was stirred for 30 min, then concentrated in vacuo. Remaining pyridine was removed by repeated azeotropic evaporation with toluene. Residue was purified by 018 chromatography to provide compound S12 as a white foam (464 mg, 83%).
1H NMR (500 MHz, CD3OD) δ 8.48 (s, 1H), 8.16 (s, 1H), 6.84 (d, Jhp = 620.8 Hz, 1H), 6.07 (d, J = 6.6 Hz, 1H), 4.83 (dd, J = 6.5, 4.4 Hz, 1H), 4.41 (dd, J = 4.4, 2.1 Hz, 1H), 4.24 - 4.20 (m, 1H), 4.20 - 4.14 (m, 1H), 4.12 - 4.05 (m, 1H), 3.23 (q, J = 7.3 Hz, 6H), 1.30 (t, J = 7.3 Hz, 9H), 0.96 (s, 9H), 0.77 (s, 9H), 0.17 (s, 3H), 0.15 (s, 3H), 0.00 (s, 3H), −0.27 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 158.78, 150.22, 146.99, 140.81, 125.52, 89.00, 86.60(d, JCP = 8.0 Hz), 77.09, 74.17, 64.05 (d, J = 4.4 Hz), 47.49, 26.44, 26.26, 18.88, 18.67, 9.19, −4.18, −4.29, −4.31, −5.10.
31P NMR (202 MHz, CD3OD) δ 4.11.
HRMS (ESI-TOF) m/z: [M-H]− Calc for C22H41N4O7PSi2 559.2179; Found 559.2164.
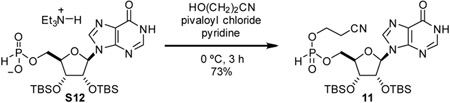
2’,3’-bis-O-(tert-butyldimethylsilyl)-inosin-5’-yl 2-cyanoethyl phosphonate (11): To a 20 mL reaction vial was added compound S12 (246 mg, 0.372 mmol, 1 eq) and dry pyridine (4 mL). Solution was cooled to 0 °C. 3-hydroxypropionitrile (0.070 mL, 1.04 g/mL, 73 mg, 1.0 mmol, 2.8 eq) was added to stirring solution. Pivaloyl chloride (0.09 mL, 0.98 g/mL, 88 mg, 0.73 mmol, 2 eq) was added dropwise. After stirring at 0 °C for 2.5 h, reaction mixture was concentrated, azeotroping with toluene. Resulting residue was redissolved in acetonitrile and purified by C-18 chromatography to yield compound 11 as a white solid as a mixture of diastereomers (166 mg, 73%).
1H NMR (500 MHz, CDCl3) δ 8.40 (s, 0.5H), 8.36 (s, 0.5H), 8.03 (s, 0.5H), 8.01 (s, 0.5H), 6.98 (d, J = Hz, 0.5H), 6.94 (d, J = 722.1 Hz, 0.5H), 5.90 (dd, J = 4.7, 3.2 Hz, 1H), 4.79 (t, J = 4.5 Hz, 1H), 4.76 (t, J = 4.0 Hz, 1H), 4.52 – 4.23 (m, 7H), 2.87 – 2.75 (m, 2H), 0.92 (s, 9H), 0.91 (s, 9H), 0.805 (s, 9H), 0.799 (s, 9H), 0.12 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H), −0.017 (s, 3 H), −0.022 (s, 3H), −0.19 (s, 3H), −0.21 (s, 3H)
13C NMR (126 MHz, CDCl3) δ 159.03, 148.66, 148.64, 145.75, 145.68, 139.73, 139.53, 125.75, 125.69, 116.73, 116.60, 89.71, 89.64, 83.15, 83.10, 83.03, 82.97, 77.36, 74.94, 74.74, 71.73, 71.66, 64.55, 60.60, 60.56, 60.53, 25.87, 25.75, 20.17, 20.12, 20.08, 18.10, 17.94, −4.30, −4.56, −4.58, −4.69, - 4.70, −4.90, −4.93.
31P NMR (202 MHz, CDCl3) δ 8.66, 8.41.
HRMS (ESI-TOF) m/z: [M-H]− Calc for C25H44N5O7PSi2 612.2444; Found 612.2448.
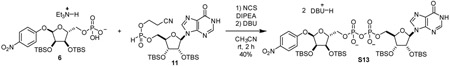
α-1”-O-(4-nitrophenyl)-2’,2”,3’,3”-O-tetrakis-(tert-butyldimethylsilyl)-IDP-ribose (S13): To a 20 mL reaction vial was added 6 (41.1 mg, 0.0604 mmol, 1.0 eq) and 11 (38.7 mg, 0.0630 mmol, 1.04 eq). Mixture was dried by co-azeotroping with dry acetonitrile three times and placing under vacuum over P2O5 for 12 h. The mixture was dissolved in acetonitrile (1.0 mL). Then, (i-Pr2)2NEt (31 mg, 0.24 mmol, 4.0 eq) and N-chlorosuccinimide (24 mg, 0.18 mmol, 3 eq) were sequentially added as 1 M solutions in acetonitrile and stirred at room temperature for 1 h. 1,8-diazabicycloundec-7-ene (92 mg, 0.6 mmol, 10 eq) was added as a 1 M solution in acetonitrile. After stirring for 30 min, reaction mixture was evaporated in vacuo and purified by C18 chromatography to yield compound S13 as a white foam (34.6 mg, 40%).
1H NMR (500 MHz, CD3OD) δ 8.55 (s, 1H), 8.14 (d, J = 9.2 Hz, 2H), 8.08 (s, 1H), 7.12 (d, J = 9.3 Hz, 2H), 6.08 (d, J = 7.5 Hz, 1H), 5.66 (d, J = 4.3 Hz, 1H), 4.46 (d, J = 4.3 Hz, 1H), 4.39 (dd, J = 5.4, 4.3 Hz, 1H), 4.35 - 4.28 (m, 3H), 4.23 (t, J = 4.8 Hz, 1H), 4.19 (dt, J = 4.3, 2.6 Hz, 1H), 4.15 - 4.07 (m, 2H), 3.64 - 3.56 (m, 4H), 3.53 (t, J = 6.1 Hz, 4H), 3.36 (t, J = 5.6 Hz, 4H), 2.74 - 2.65 (m, 4H), 2.02 (tdd, J = 6.9, 5.2, 4.2 Hz, 4H), 1.82 - 1.66 (m, 14H), 0.97 (s, 9H), 0.93 (s, 9H), 0.91 (s, 9H), 0.73 (s, 9H), 0.20 (s, 3H), 0.17 (s, 3H), 0.15 (s, 3H), 0.13 (s, 3H), 0.11 (s, 3H), 0.08 (s, 3H), −0.00 (s, 3H), −0.36 (s, 3H).
31P NMR (202 MHz, CD3OD) δ −11.3 (d, J = 21.9 Hz), −11.6 (d, J = 21.8 Hz).
13C NMR (126 MHz CD3OD) δ 167.50, 164.28, 159.27, 150.61, 147.02, 143.00, 141.44, 126.59, 125.58, 117.36, 101.30, 88.57, 88.02 (d, J = 9.2 Hz), 87.70 (d, J = 9.3 Hz), 77.11, 74.81, 73.33, 66.71 (d, J = 5.5 Hz), 66.52 (d, J = 5.4 Hz), 55.33, 49.57, 39.37, 33.70, 30.01, 27.53, 26.50 (3C), 26.48, 26.22, 24.97, 20.44, 19.15, 18.99, 18.93, 18.68, −4.04, −4.11, −4.20, −4.23, −4.26, −4.30, −4.32, −5.32.
HRMS (ESI-TOF) m/z: [M-H]− Calc for C45H80N5O17P2Si4 1136.4107; Found 1136.4115.
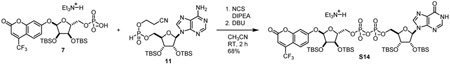
α-1”-O-(4-(trifluoromethyl)umbellifer-7-yl)-2’,2”,3’,3”-O-tetrakis-(tert-butyldimethylsilyl)-IDP-ribose (S14): To a stirring solution of 7 (93.6 mg, 0.121 mmol) and 11 (75.5 mg, 0.123 mmol) in dichloromethane (1 mL) was added DIPEA as a 1 M solution in CH3CN (0.73 mL, 0.73 mmol, 6 eq) and N-chlorosuccinimide as a 1 M solution in CH3CN (0.61 mL, 0.61 mmol, 5 eq). Mixture was stirred for 30 min at room temperature and then DBU as a 1 M solution in THF (1 mL, 1 mmol, 8 eq) was added. After stirring for an additional 30 min, solvent was removed by rotary evaporator. Residue was purified by C18 chromatography to give S14 (126 mg, 68%)
1H NMR (500 MHz, CD3OD) δ 8.56 (s, 1H), 8.09 (s, 1H), 7.65 (dd, J = 9.6, 2.0 Hz, 1H), 7.05 (dd, J = 6.8, 2.4 Hz, 3H), 6.70 (s, 1H), 6.07 (d, J = 7.4 Hz, 1H), 5.70 (d, J = 4.3 Hz, 1H), 4.89 (dd, J = 7.4, 4.4 Hz, 1H), 4.44 (d, J = 4.5 Hz, 1H), 4.40 (dd, J = 5.4, 4.3 Hz, 1H), 4.35 – 4.25 (m, 6H), 4.22 (q, J = 3.9 Hz, 4H), 4.12 (t, J = 4.7 Hz, 3H), 3.72 (hept, J = 6.7 Hz, 2H), 3.58 (m, 2H), 3.52 (t, J = 5.9 Hz, 2H), 3.37 (t, J = 5.7 Hz, 2H), 3.20 (q, J = 7.4 Hz, 2H), 2.76 – 2.68 (m, 2H), 2.01 (pd, J = 5.6, 1.3 Hz, 2H), 1.80 – 1.64 (m, 6H), 1.38 (t, J = 7.2 Hz, 14H), 0.96 (s, 9H), 0.94 (s, 9H), 0.91 (s, 9H), 0.73 (s, 9H), 0.19 (s, 3H), 0.16 (s, 3H), 0.15 (s, 3H), 0.13 (s, 3H), 0.12 (s, 3H), 0.09 (s, 3H), −0.00 (s, 3H), −0.36 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 167.45, 163.08, 160.79, 158.93, 157.38, 150.54, 146.83, 142.33 (q, 2JCF = 32.6 Hz), 141.41, 129.48, 127.29, 125.52, 123.23 (q, 1JCF = 274.9 Hz), 115.77, 113.81 (q, 3JCF = 6.4 Hz), 108.44, 105.04, 101.36, 88.59, 88.12 (d, 3JCP = 9.2 Hz), 87.55 (d, 3JCP = 9.4 Hz), 77.15, 74.79, 74.74, 73.31, 66.79 (d, 2JCP = 5.5 Hz), 66.54 (d, 2JCP = 5.4 Hz), 55.40, 55.27, 43.49, 39.36, 33.64, 30.04, 27.56, 26.49, 26.49, 26.47, 26.22, 25.00, 20.44, 19.57 (d, J = 1.7 Hz), 19.15, 19.00, 18.92, 18.67, 13.11, −4.06, −4.10, −4.20, −4.22, −4.24, −4.30, −4.32, −5.29.
31P NMR (203 MHz, CD3OD) δ −11.41 (d, J = 21.3 Hz), −11.81 (d, J = 21.9 Hz).
HRMS (ESI-TOF) m/z: [M-H]− Cald for C49H80F3N4O17P2Si4 1227.4028; Found 1227.4056.
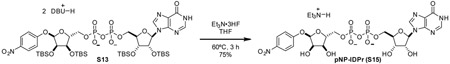
pNP-IDPr (S15): To a 7 mL reaction vial was added S13 (17.4 mg, 0.0121 mmol, 1 eq) and THF (1.0 mL). Once starting material was fully dissolve, triethylamine trihydrofluoride (0.2 mL) was added neat dropwise. Reaction vessel was sealed and heated to 60 °C. Reaction was closely monitored by TLC (80:20 2-propanol:0.2% NH4OH(aq)). Once reaction had gone to completion, reaction mixture was cooled to room temperature and concentrated in vacuo. Remaining acid was quenched by dropwise addition of satd aq NaHCO3. Aqueous solution of crude product was purified by C-18 chromatography utilizing ion-pairing reagent in the mobile phase (10 mM Et3N-HOAc) to yield compound pNP-lDPr (S15) as a triethylammonium salt. The triethylammonium salt was eluted through Dowex 50W-8X (ammonium-form) to give the ammonium salt of compound pNP-IDPr (S15) as a white powder (6.2 mg, 75%).
1H NMR (500 MHz, D2O) δ 8.37 (s, 1H), 8.09 (s, 1H), 8.06 (d, J = 9.1 Hz, 2H), 7.06 (d, J = 9.0 Hz, 2H), (d, J = 5.9 Hz, 1H), 5.79 (d, J = 4.5 Hz, 1H), 4.70 (t, J = 5.5 Hz, 1H), 4.50 (dd, J = 5.2, 3.5 Hz, 1H), 4.45 (dd, J = 6.2, 4.5 Hz, 1H), 4.42 - 4.37 (m, 2H), 4.31 (dd, J = 6.0, 2.5 Hz, 1H), 4.29 – 4.22 (m, 2H), 4.16 - 4.05 (m, 2H)
31P NMR (202 MHz, D2O) δ −11.14 (d, J = 21.8 Hz, 1P), −11.36 (d, J = 21.6 Hz, 1P)
13C NMR (from 1H,13C-HMBC, 500 MHz, D2O) 162.04, 158.43, 148.37, 146.14, 141.63,139.60, 125.85, 123.59, 116.73, 100.32, 87.36, 84.78, 83.97, 74.53, 71.21, 70.38, 69.64, 65.57, 64.86
HRMS (ESI-TOF) m/z: [M-H]− Calc for C21H24N5O17P2 680.0648; Found 680.0650.
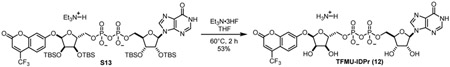
TFMU-IDPr (12)
To a stirring solution of S13 (119.9 mg, 0.78 mmol) in THF (1.0 mL) was added triethylamine trihydrofluoride (0.5 mL, 3.1 mmol, 40 eq). Reaction mixture was sealed with Teflon cap and heated to 60 °C. After stirring at 60 °C for 2 h, solvent was removed by rotavap. Residue was neutralized with satd aq NaHCO3 and aqueous mixture was purified by ion-pairing chromatography (10 mM Et3NHOAc, C18) followed by ion exchange (Dowex 50W-8, ammonium form) to provide TFMU-IDPr (12) (33.6 mg, 53%) as a white solid.
1H NMR (500 MHz, D2O) δ 8.27 (s, 1H), 7.96 (s, 1H), 7.48 (dd, J = 9.1, 1.9 Hz, 1H), 6.93 (dd, J = 9.0, Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 6.70 (s, 1H), 5.95 (d, J = 5.6 Hz, 1H), 5.77 (d, J = 4.4 Hz, 1H), 4.61 (t, J = 5.4 Hz, 1H), 4.49 – 4.42 (m, 2H), 4.44 – 4.39 (m, 1H), 4.37 (q, J = 3.8 Hz, 1H), 4.30 (dd, J = 6.3, 2.8 Hz, 1H), 4.28 – 4.21 (m, 2H), 4.16 (ddd, J = 11.5, 5.1, 3.2 Hz, 1H), 4.09 (dt, J = 11.3, 4.6 Hz, 1H).
13C NMR (126 MHz, D2O) δ 161.91, 160.21, 157.82, 154.77, 148.22, 145.93, 141.55 (q, 2JCF = 32.9 Hz), 139.48, 126.22, 123.33, 121.31 (q, 1JCF = 276.4, 275.8 Hz), 115.03, 112.72 (q, 3JCF = 8.2 Hz), 107.67, 103.87, 100.30, 87.54, 84.77 (d, 3JCP = 8.2 Hz), 83.86 (d, 3JCP = 8.5 Hz), 74.62, 71.32, 70.36, 69.71, 65.81 (d, 2JCP = 5.1 Hz), 65.29 (d, 2JCP = 5.3 Hz).
31P NMR (202 MHz, D2O) δ −11.09 (d, J = 21.5 Hz), −11.31 (d, J = 21.3 Hz).
19F NMR (470 MHz, D2O) δ −64.55.
HRMS (ESI-TOF) m/z: [M-H]− Cald for C25H24F3N4O17P2 771.0569; Found 771.0566.
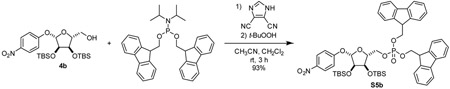
4-nitrophenyl 2,3-bis-O-tert-butyldimethylsilyl-β-D-ribose-5-(difluorenylmethyl phosphate) (S5b): To a 20 mL reaction vial, 4b (360.4 mg, 0.721 mmol, 1 eq) and bis-(9H-fluoren-9-ylmethyl)-N,N-diisopropylamidophosphite (Lambrecht et al., 2015) (488 mg, 0.936 mmol, 1.3 eq) were added. Dissolved material by addition of CH2Cl2 (11 mL). Cooled solution to 0 °C. 4,5-dicyanoimidazole (129 mg, 1.1 mmol, 1.5 eq) was added as a solution in acetonitrile (2 mL). After stirring at 0 °C for 15 min, mixture was allowed to warm to room temperature and was stirred for an additional 2 h. Once starting material was consumed as indicated by TLC (75:25 hexane:EtOAc), mixture was cooled to 0 °C and subjected to dropwise addition of tert-butyl hydroperoxide (0.5 mL, 2.5 mmol, 3.5 eq) as a 5 M solution in decane. Mixture was stirred for an additional 1 h and quenched with H2O. Mixture was extracted with CH2Cl2 three times. Combined organic layers were dried over Na2SO4, concentrated, and purified by silica gel chromatography eluting with 80:20 hexane:EtOAc to yield compound S5b as a white foam (628 mg, 93%).
1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 9.2 Hz, 2H), 7.72 (dd, J = 11.9, 7.5 Hz, 3H), 7.50 – 7.33 (m, 9H), 7.30 – 7.20 (m, 4H), 6.76 (d, J = 9.2 Hz, 2H), 5.31 (m, 1H), 4.28 (dd, J = 7.0, 4.0 Hz, 1H), 4.21 – 3.98 (m, 9H), 3.92 (dt, J = 11.3, 4.0 Hz, 1H), 0.91 (s, 9H), 0.90 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H).
13C NMR (126 MHz, CDCl3) δ 161.50, 143.09, 143.07, 143.04, 142.22, 141.43, 141.40, 141.37, 128.07, 128.04, 127.22, 127.20, 127.18, 125.64, 125.20, 125.14, 125.12, 120.19, 120.16, 120.12, 116.01, 105.07, 81.77, 81.70, 76.53, 70.84, 69.47, 69.45, 69.42, 69.41, 65.74, 65.70, 48.02, 47.98, 47.96, 47.92, 25.95, 25.86, 18.21, 18.14, −4.10, −4.43, −4.44, −4.86.
31P NMR (202 MHz, CDCl3) δ −1.68
HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C51H62NO10NaSi2P 958.3548; Found 958.3540.
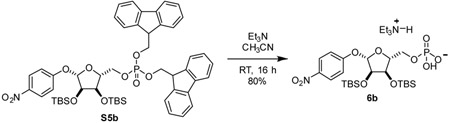
Triethylammonium 4-nitrophenyl 2,3-bis-O-tert-butyldimethylsilyl-β-D-ribose-5-phosphate (6b): 20 mL reaction vial was charged with compound S5b (551 mg, 0.59 mmol, 1 eq). Added acetonitrile (6 mL) and triethylamine (1.5 mL) successively. Stirred at room temperature for 16 h. Added 1 mL toluene to stirring solution and concentrated in vacuo. Residue was redissolved in methanol (0.5 mL) and purified by C-18 chromatography to yield the triethylammonium salt of compound 6b as a tan foam (322 mg, 80%).
1H NMR (500 MHz, CD3OD) δ 8.22 (d, J = 9.2 Hz, 2H), 7.19 (d, J = 9.3 Hz, 2H), 5.55 (d, J = 3.0 Hz, 1H), 4.40 (dd, J = 4.3, 3.0 Hz, 1H), 4.36 (t, J = 4.3 Hz, 1H), 4.23 (dt, J = 5.7, 4.4 Hz, 1H), 4.00 (dt, J =10.0, 4.7 Hz, 1H), 3.87 – 3.80 (m, 1H), 3.15 (q, J = 7.3 Hz, 6H), 1.28 (t, J = 7.3 Hz, 9H), 0.95 (s, 9H), 0.93 (s, 9H), 0.19 (s, 3H), 0.17 (s, 3H), 0.16 (s, 3H), 0.16 (s, 3H).
13C NMR (126 MHz, CD3OD) δ 163.55, 143.73, 126.70, 117.67, 106.77, 85.92 (d, J = 9.2 Hz), 78.04, 73.84, 66.50 (d, J = 4.8 Hz), 47.53, 26.46, 26.38, 19.05, 18.97, 9.11, −4.09, −4.17, −4.33, −4.52.
31P NMR (202 MHz, CD3OD) δ 0.66.
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C23H41NO10Si2P 578.2007; Found 578.2009.
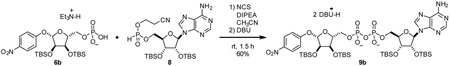
β-1”-O-(4-nitrophenyl)-2’,2”,3’,3”-O-tetrakis-(tert-butyldimethylsilyl)-ADP-ribose (9b): To a 20 mL reaction vial was added 6b (113.7 mg, 0.167 mmol, 1.0 eq) and 8 (110.0 mg, 0.179 mmol, 1.1 eq). Mixture was dried by co-azeotroping with dry acetonitrile three times and placing under vacuum over P2O5 for 12 h. The mixture was dissolved in acetonitrile (2.0 mL). Then, (i-Pr2)2NEt (64.6 mg, 0.5 mmol, 3 eq) and N-chlorosuccinimide (53.4 mg, 0.4 mmol, 2.4 eq) were sequentially added as 1 M solutions in acetonitrile and stirred at room temperature for 1 h. 1,8-diazabicycloundec-7-ene (350 mg, 2.3 mmol, 10 eq) was added as a 1 M solution in THF. After stirring for 30 min, reaction mixture was evaporated in vacuo and purified by C18 chromatography to yield compound 9b as a white foam (149.6 mg, 60%).
1H NMR (500 MHz, CD3OD) δ 8.64 (s, 1H), 8.173 (s, 1H), 8.172 (d, J = 9.2 Hz, 2H), 7.15 (d, J = 9.2 Hz, 2H), 6.10 (d, J = 7.5 Hz, 1H), 5.51 (d, J = 4.1 Hz, 1H), 4.80 (dd, J = 7.5, 4.4 Hz, 1H), 4.45 (t, J = 4.2 Hz, 1H), 4.42 (d, J = 4.5, 1H), 4.38 (dd, J = 4.4, 2.7 Hz, 1H), 4.26 (td, J = 5.9, 2.8 Hz, 1H), 4.23 (m, 2H), 4.16 (m, 1H), 4.08 (qt, J = 10.9, 5.5 Hz, 2H), 3.55 (m, 4H), 3.49 (t, J = 5.9 Hz, 4H), 3.35 (m, 9H), 2.68 (m, 4H), 1.99 (m, 4H), 1.70 (m, 12H), 0.97 (s, 9H), 0.94 (s, 9H), 0.90 (s, 9H), 0.69 (s, 9H), 0.18 (s, 3H), 0.17 (s, 3H), 0.16 (s, 3H), 0.15 (s, 6H), 0.14 (s, 3H), −0.03 (s, 3H), −0.40 (s, 3H)
13C NMR (126 MHz, CD3OD) δ 167.45, 163.85, 157.31, 153.80, 151.17, 143.64, 141.65, 126.69, 120.02, 117.69, 107.14, 87.75, 87.63 (d, J = 8.6 Hz), 86.75 (d, J = 9.0 Hz), 78.21, 77.67, 74.85, 74.26, 66.86 (d, J = 5.2 Hz), 66.68 (d, J = 5.1 Hz), 55.25, 49.85, 49.54, 39.34, 33.62, 30.02, 27.55, 26.51, 26.48, 26.44, 26.21, 24.98, 20.43, 19.08, 18.99, 18.92, 18.64, −4.08, −4.13, −4.19, −4.21, −4.26, −4.29, −5.29.
31P NMR (202 MHz, CD3OD) δ −11.44 (d, J = 21.5 Hz), −11.65 (d, J = 21.5 Hz).
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C45H82N6O16P2Si4 1135.4267; Found 1135.4272.
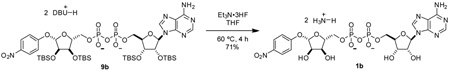
β-pNP-ADPr (1b): To a 20 mL reaction vial was added 9b (24.1 mg, 0.0167 mmol, 1 eq) and THF (1 mL). Triethylamine trihydrofluoride (0.1 mL, 0.989 g/mL) was added dropwise to stirring solution. Reaction vessel was sealed and heated to 60 C for 4 h. Reaction was closely monitored by TLC (80:20 2-propanol:0.2% NH4OH(aq)) Once reaction had gone to completion, reaction mixture was cooled to room temperature and concentrated in vacuo. Remaining acid was quenched by dropwise addition of satd aq NaHCO3. Aqueous solution of crude product was purified by C18 chromatography utilizing ion-pairing reagent in the mobile phase (10 mM Et3N-HOAc, pH 7.0) to yield compound 1b as a triethylammonium salt. The triethylammonium salt was eluted through Dowex 50W-8X (ammonium-form) to give the ammonium salt of compound 1b as a white powder (8.5 mg, 71%).
1H NMR (500 MHz, D2O) δ 8.33 (s, 1H), 8.11 (s,1H), 7.94 (d, J = 9.3 Hz, 2H), 6.93 (d, J = 9.4 Hz, 2H), 5.99 (d, J = 5.4, 1H), 5.64 (d, J = 1.0 Hz, 1H), 4.62 (t, J = 5.2 Hz, 1H), 4.46 (m, 2H), 4.34 (m, 2H), 4.29 (td, J = 5.9, 4.1 Hz, 1H), 4.19 (m, 2H), 4.16 (q, J = 3.9 Hz, 1H), 4.08 (m, 1H)
13C NMR (126 MHz, D2O) δ 181.63, 161.38, 155.43, 152.74, 148.68, 141.68, 139.53, 125.67, 118.51, 116.45, 105.24, 87.17, 83.80, 82.78, 74.68, 70.70, 70.32, 66.69, 65.14.
31P NMR (202 MHz, D2O) δ −10.19.
HRMS (ESI-TOF) m/z: [M-H]− Calcd for C21H26N6O16P2 679.0808; Found 679.0809.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the data were presented as mean ± standard error of mean from at least three independent trials. All data fitting and statistical analysis was performed using GraphPad Prism (version 6).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-ARH3 | Santa Cruz Biotech | Cat# sc-374162 |
| Rabbit anti-beta-actin | Cell Signaling | Cat# 4970 |
| Rabbit anti-PARP1 | Cell Signaling | Cat# 9542 |
| HRP-conjugated goat anti-rabbit | Cell Signaling | Cat# 7074 |
| HRP-conjugated horse anti-mouse | Cell Signaling | Cat# 7076 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TFMU-ADPr | This paper | n/a |
| TFMU-IDPr | This paper | n/a |
| pNP-ADPr | This paper | n/a |
| pNP-IDPr | This paper | n/a |
| ADPr-Arg | This paper | n/a |
| ADP-HPD | Calbiochem | Cat# 118415 |
| ADP-ribose | Sigma-Aldrich | Cat# A0752 |
| PDD 00017273 | Tocris | Cat# 5952 |
| 4-(trifluoromethyl)umbelliferone | Enamine Building Blocks | Cat# EN300–05279 |
| 4-nitrophenol | Sigma-Aldrich | Cat# 241326 |
| 2,3-di-(tert-butyldimethylsilyl)-5-(triphenylmethyl)-D-ribofuranose | (Lambrecht et al.,2015) | n/a |
| 1,1’-(azodicarbonyl)dipiperidine | Accela ChemBio Inc. | Cat# SY001006 |
| Diisopropyl azodicarboxylate | Sigma-Aldrich | Cat# 225541 |
| Triphenylphosphine | Sigma-Aldrich | Cat# T84409 |
| Tributylphosphine | Sigma-Aldrich | Cat# 90827 |
| 1,2-dimethoxyethane | GFS Chemicals | Cat# 2510 |
| N,N-diisopropyl bis(9-methylfluorenyl)phosphoramidite | (Lambrecht et al., 2015) | n/a |
| 4,5-Dicyanoimidazole | Sigma-Aldrich | Cat# 554030 |
| N-chlorosuccinimide | Sigma-Aldrich | Cat# 109681 |
| Triethylamine trihydrofluoride | Oakwood Chemical | Cat# 003029 |
| L-Arginine | Sigma-Aldrich | Cat# A3784 |
| NAD+ | Sigma-Aldrich | Cat# N0632 |
| Silicagel 60M | Macherey-Nagel | Cat# 815381.25P |
| Dowex 50Wx8 | Sigma-Aldrich | Cat# 217514 |
| Acetic Acid | Fisher Scientific | Cat# A38–212 |
| Sodium Chloride | Fisher Scientific | Cat# S271–500 |
| DTT | Sigma Aldrich | Cat# DTT-RO ROCHE |
| EDTA | Fisher Scientific | Cat# S311–100 |
| Triton-X-100 | Fisher Scientific | Cat# BP151–100 |
| Sodium phosphate dibasic | EMD | Cat# SX0715–1 |
| Potassium phosphate dibasic | VWR Analytical | Cat# BDH9266.500 |
| Magnesium chloride | Fisher Scientific | Cat# M33 |
| Glycine | Bio-Rad | Cat# 161–0718 |
| 2-mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Glycerol | Fisher Scientific | Cat# G33 |
| Tween 20 | Fisher Scientific | Cat# BP337 |
| 0.05% Trypsin | Corning | Cat# 25–052-Cl |
| Protease Inhibitor Cocktail Set III, EDTA-free | Calbiochem | Cat# 539134 |
| Fetal Bovine Serum | Gemini | Cat# 100–106 |
| BSA | Research Products International Corp. | Cat# A30075–100.0 |
| INTERFERin Transfection Agent | Polyplus | Cat# 409–10 |
| Opti-MEM Media | ThermoFisher | Cat# 11058–021 |
| Cholera Toxin from Vibrio cholerae | List Biological Labs | Cat# 100B |
| Binary Toxin from Clostridium difficile, Subunit A | List Biological Labs | Cat# 157A |
| Binary Toxin from Clostridium difficile, Subunit B | List Biological Labs | Cat# 157B |
| Pertussis Toxin from B. pertussis | List Biological Labs | Cat# 180 |
| Ni-NTA | Qiagen | Cat# 30230 |
| IPTG | Gold Biotechnology | Cat# 12481C100 |
| Kanamycin | Research Products International | Cat# K22000 |
| Ampicillin | Fisher Scientific | Cat# BP1760 |
| Chloramphenicol | Sigma-Aldrich | Cat# C0378 |
| Tris base | Fisher Scientific | Cat# BP152 |
| Imidazole | Sigma-Aldrich | Cat# I202 |
| Leupeptin | Sigma-Aldrich | Cat# L2884 |
| Pepstatin A | Sigma-Aldrich | Cat# P4265 |
| Aprotinin | Sigma-Aldrich | Cat# A1153 |
| PMSF | Sigma-Aldrich | Cat# 78830 |
| Human full-length PARG | (Barkauskaite et al., 2013; Tucker et al., 2012) | n/a |
| T. thermophila PARG protein | (Dunstan et al., 2012) | n/a |
| Human ARH3 protein | (Barkauskaite et al., 2013; Finch et al., 2012) | n/a |
| Human ARH3 D77N/D78N protein | (Fontana et al., 2017) | n/a |
| Human PARG catalytic domain wild type protein | (Tucker et al., 2012) | PARG26 |
| Human PARG catalytic domain E756N protein | (Tucker et al., 2012) | n/a |
| Critical Commercial Assays | ||
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat# 23227 |
| SuperSignal West Pico | ThermoFisher | Cat# 34577 |
| Penicillin/Streptomycin | Lonza | Cat# 17–602E |
| Mini-PROTEAN TGX 4–20% Precast Gel | Bio-Rad | Cat# 456–1093 |
| SDS (Tris/Glycine) Buffer (10x) | Bio-Rad | Cat# 161–0732 |
| Laemmili Sample Buffer | Bio-Rad | Cat# 161–0737 |
| Restore Western Blot Stripping Buffer | ThermoFisher | Cat# 21059 |
| Immobilon-P PVDF Transfer Membrane | EMD Millipore | Cat# IPV00010 |
| Deposited Data | ||
| Structure of Human PARG | (Lambrecht et al., 2015) | PDB:5A7R |
| Structure of Latimeria chalumnae ARH3 | Unpublished | PDB:6G1Q |
| Experimental Models: Cell Lines | ||
| Human: A549 | ATCC | Cat# CCL-185, RRID:CVCL_0023 |
| Human: CA46 | From Tim Fan (University of Illinois, Urbana) | RRID:CVCL_1101 |
| Human: HCT-116 | ATCC | Cat# CCL-247, RRID:CVCL_0291 |
| Human: HEK293TN | System Biosciences | Cat# LV900A-1 |
| Human: HeLa | ATCC | Cat# CCL-2, RRID:CVCL_0030 |
| Human: HepG2 | ATCC | Cat# HB-8065, RRID:CVCL_0027 |
| Mouse: MEF | From Navdeep Chandel (Northwestern University) | n/a |
| Human: MIA PaCa-2 | From David Boothman (Indiana University School of Medicine) | RRID:CVCL_0428 |
| Human: U2OS | ATCC | Cat# HTB-96, RRID:CVCL_0042 |
| Human: U2OS ARH3 KO | (Fontana et al., 2017) | |
| Human: U87 | ATCC | Cat# HTB-14, RRID:CVCL_0022 |
| Human: U937 | ATCC | Cat# CRL-1593, RRID:CVCL_0007 |
| Human: D54 | From Gregory Riggins (Johns Hopkins) | RRID:CVCL_7185 |
| Human: Daudi | ATCC | ATCC Cat# CCL-213, RRID:CVCL_0008 |
| Human: HCC1937 | ATCC | Cat# CRL-2336, RRID:CVCL_0290 |
| Human: HCC38 | ATCC | Cat# CRL-2314, RRID:CVCL_1267 |
| Human: HFF-1 | ATCC | Cat# SCRC-1041, RRID:CVCL_3285 |
| Human: Hs578t | ATCC | Cat# HTB-126, RRID:CVCL_0332 |
| Human: Jurkat | From Richard Hotchkiss (Washington University, St. Louis) | n/a |
| Human: MCF7 | ATCC | Cat# HTB-22, RRID:CVCL_0031 |
| Human: MCF10A | ATCC | Cat# CRL-10317, RRID:CVCL_0598 |
| Human: SK-MEL-5 | ATCC | Cat# HTB-70, RRID:CVCL_0527 |
| Human: T98G | ATCC | Cat# CRL-1690, RRID:CVCL_0556 |
| Experimental Models: Organisms/Strains | ||
| E. coli: Rosetta 2 (DE3) | Novagen | Cat# 71400 |
| Recombinant DNA | ||
| Plasmid: hARH3-pDEST | Hening Lin (Cornell University) | n/a |
| Plasmid: hPARG-pColdTF | Ivan Ahel (Oxford University) | n/a |
| Plasmid: ttPARG-pET28a | Ivan Ahel (Oxford University) | n/a |
| Software and Algorithms | ||
| Prism 6 | GraphPad | |
| SoftMax Pro 6.4 | Molecular Devices | Build 204720 |
| ImageJ (FIJI) | NIH | http://imagej.net/Fiji/Downloads |
| Image Lab 4.1 | Bio-Rad | |
| PyMOL | PyMOL Molecular Graphics System (built from source) | https://www.pymol.org/ |
| Small Molecule Drug Discovery Suite | Schrödinger | Version 2018-1 |
| Other | ||
| CombiFlash Rf+ | Teledyne Isco | Cat# 68-5230-022 |
| SpectraMax Multi-mode Microplate Reader | Molecular Devices | Model# M3 |
| ChemiDoc MP Imaging System | Bio-Rad | Cat# 170-8280 |
| RediSep Rf C18 Gold 5.5g | Teledyne Isco | Cat# 69-2203-328 |
| RediSep Rf C18 Gold 150g | Teledyne Isco | Cat# 69-2203-338 |
| Zorbax Eclipse Plus C18 1.8μm 2.1×50mm | Agilent | Cat# 959741-902 |
| Luna C18 5 μm 21.2×150mm | Phenomenex | Cat# 00F-4252-P0-AX |
| Digital Sonifier | Branson Ultrasonics | Model# S-450 |
Highlights.
Synthesis of fluorescent substrates that are enzymatically cleaved by PARG and ARH3
Synthesis of a fluorescent substrate that is enzymatically cleaved by ARH3 only
Use of these substrates in cell lysate to measure PARG and ARH3 activity
Discovery that ARH3 is inhibited by the metabolite ADP-ribosyl arginine
SIGNIFICANCE.
ADP-ribosylation has long captured interest due to its prevalence in normal biological processes and in disease states, but study of this PTM has traditionally relied on imprecise and laborious methods for measuring the activity of ADPr erasers. As such, the conditions and mechanisms by which PAR is regulated are poorly understood. Here we introduce a rapid and precise technology for measuring poly(ADP-ribose) glycohydrolase activity. Recent advances in pyrophosphate couplings have also improved our capability to construct sugar nucleotides in a modular and scalable fashion, leading to robust and scalable syntheses of the target substrates. These substrates are useful for the detection and quantitation of endogenous cellular PARG and ARH3 activity, and the utility of this tool is increased with the discovery that replacement of the nucleobase provides selectivity for ARH3 over PARG. Together this toolset is expected to greatly enhance the ability to interrogate PAR-cleaving enzymes.
ACKNOWLEDGMENTS
We express our thanks to Prof. Hening Lin (Cornell U.) for ARH3 plasmid. We thank David Boothman (Indiana University School of Medicine) for the MIA-PaCa cell line, Timothy Fan (University of Illinois, Urbana) for the CA46 cell line, Navdeep Chandel (Northwestern U.) for the MEF cell line, Gregory Riggins (Johns Hopkins) for the D54 cell line, and Richard Hotchkiss (Washington University, St. Louis) for the Jurkat cell line. We are grateful for the University of Illinois and the NIH (R21 CA212732) for funding this work. The work in I.A. laboratory is funded by the Wellcome Trust (grant number 101794) and Cancer Research United Kingdom (grant number C35050/A22284).
Footnotes
SUPPLEMENTAL DATAFILES
Data S1: Related to Figure 2. NMR spectra of PARG substrates, inhibitors, and synthetic intermediates.
DECLARATIONS OF INTERESTS
The University of Illinois has filed a patent on materials related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abplanalp J, Leutert M, Frugier E, Nowak K, Feurer R, Kato J, Kistemaker HVA, Filippov DV, Moss J, Caflisch A, et al. (2017). Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun 8, 2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer M, Messner S, Hassa PO, Fey M, and Hottiger MO (2009). Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res 37, 3723–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu S-W, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al. (2006). Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 103, 18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskaite E, Brassington A, Tan ES, Warwicker J, Dunstan MS, Banos B, Lafite P, Ahel M, Mitchison TJ, Ahel I, et al. (2013). Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nat. Commun 4, 2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkauskaite E, Jankevicius G, and Ahel I (2015). Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol. Cell 58, 935–946. [DOI] [PubMed] [Google Scholar]
- Becker A, Zhang P, Allmann L, Meilinger D, Bertulat B, Eck D, Hofstaetter M, Bartolomei G, Hottiger MO, Schreiber V, et al. (2016). Poly(ADP-ribosyl)ation of Methyl CpG Binding Domain Protein 2 Regulates Chromatin Structure. J. Biol. Chem 291, 4873–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AB, Witte MD, Denault J-B, Sadaghiani AM, Sexton KMB, Salvesen GS, and Bogyo M (2006). Identification of Early Intermediates of Caspase Activation Using Selective Inhibitors and Activity-Based Probes. Mol. Cell 23, 509–521. [DOI] [PubMed] [Google Scholar]
- Chang P, Coughlin M, and Mitchison TJ (2005). Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol 7, 1133–1139. [DOI] [PubMed] [Google Scholar]
- Cortes U, Tong W-M, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, Herceg Z, Jacobson EL, Jacobson MK, and Wang Z-Q (2004). Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell. Biol 24, 7163–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzocrea S, and Genovese T (2000). Role of Free Radicals and Poly(ADP-ribose) Synthetase in Intestinal Tight Junction Permeability. Mol. Med 6, 766–778. [PMC free article] [PubMed] [Google Scholar]
- Dunstan MS, Barkauskaite E, Lafite P, Knezevic CE, Brassington A, Ahel M, Hergenrother PJ, Leys D, and Ahel I (2012). Structure and mechanism of a canonical poly(ADP-ribose) glycohydrolase. Nat. Commun 3, 878. [DOI] [PubMed] [Google Scholar]
- Feijs KLH, Forst AH, Verheugd P, and Luscher B (2013). Macrodomain-containing proteins: regulating new intracellular functions of mono(ADP-ribosyl)ation. Nat. Rev. Mol. Cell Biol 14, 443453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch KE, Knezevic CE, Nottbohm AC, Partlow KC, and Hergenrother PJ (2012). Selective small molecule inhibition of poly(ADP-ribose) glycohydrolase (PARG). ACS Chem. Biol 7, 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio C, Frausin F, Vertua R, and Gaion RM (1999). Amplification of the cyclic AMP response to forskolin in pheochromocytoma PC12 cells through adenosine A(2A) purinoceptors. J. Pharmacol. Exp. Ther 290, 817–824. [PubMed] [Google Scholar]
- Fontana P, Bonfiglio JJ, Palazzo L, Bartlett E, Matic I, and Ahel I (2017). Serine ADP- ribosylation reversal by the hydrolase ARH3. eLife 6, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill DM, and Meren R (1978). ADP-ribosylation of membrane proteins catalyzed by cholera toxin: basis of the activation of adenylate cyclase. Proc. Natl. Acad. Sci. USA 75, 3050–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard A, Cruz-Moreno B, Cruz-Moreno BC, Aguilar B, van Sorge NM, Kuang J, Kurkciyan AA, Wang Z, Hang S, Pineton de Chambrun GP, et al. (2013). Cholera toxin disrupts barrier function by inhibiting exocyst-mediated trafficking of host proteins to intestinal cell junctions. Cell Host Microbe 14, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höbartner C, and Silverman SK (2005). Modulation of RNA tertiary folding by incorporation of caged nucleotides. Angew. Chem. Int. Ed. Engl 44, 7305–7309. [DOI] [PubMed] [Google Scholar]
- Illuzzi G, Fouquerel E, Amé J-C, Noll A, Rehmet K, Nasheuer H-P, Dantzer F, and Schreiber V (2014). PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic Acids Res 42, 7776–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inageda K, Nishina H, and Tanuma S-I (1991). Mono-ADP-ribosylation of Gs by an eukaryotic arginine-specific ADP-ribosyltransferase stimulates the adenylate cyclase system. Biochem. Biophys. Res. Commun 176, 1014–1019. [DOI] [PubMed] [Google Scholar]
- James DI, Smith KM, Jordan AM, Fairweather EE, Griffiths LA, Hamilton NS, Hitchin JR, Hutton CP, Jones S, Kelly P, et al. (2016). First-in-Class Chemical Probes against Poly(ADP-ribose) Glycohydrolase (PaRg) Inhibit DnA Repair with Differential Pharmacology to Olaparib. ACS Chem. Biol 11, 3179–3190. [DOI] [PubMed] [Google Scholar]
- Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, and Ladurner AG (2013). A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol 20, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasamatsu A, Nakao M, Smith BC, Comstock LR, Ono T, Kato J, Denu JM, and Moss J (2011). Hydrolysis of O-Acetyl-ADP-ribose Isomers by ADP-ribosylhydrolase 3. J. Biol. Chem 286, 21110–21117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J, Zhu J, Liu C, and Moss J (2007). Enhanced sensitivity to cholera toxin in ADP- ribosylarginine hydrolase-deficient mice. Mol. Cell. Biol 27, 5534–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I-K, Stegeman RA, Brosey CA, and Ellenberger T (2015). A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem 290, 3775–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DW, Coyle DL, Mehta N, Ramsinghani S, Kim H, Slama JT, and Jacobson MK (2003). SAR Analysis of Adenosine Diphosphate (Hydroxymethyl)pyrrolidinediol Inhibition of Poly(ADP-ribose) Glycohydrolase. J. Med. Chem 46, 4322–4332. [DOI] [PubMed] [Google Scholar]
- Lambrecht MJ, Brichacek M, Barkauskaite E, Ariza A, Ahel I, and Hergenrother PJ (2015). Synthesis of Dimeric ADP-Ribose and Its Structure with Human Poly(ADP-ribose) Glycohydrolase. J. Am. Chem. Soc 137, 3558–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Žaja R, Palazzo L, Stockum A, Ahel I, and Matic I (2016). Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol 12, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo M, Kato J, and Moss J (2013). ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP- ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. USA 110, 18964–18969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo M, Kato J, and Moss J (2014). Structure and function of the ARH family of ADP-ribosyl- acceptor hydrolases. DNA Repair 23, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzon E, De Sarro A, Caputi AP, and Cuzzocrea S (2002). Role of tight junction derangement in the endothelial dysfunction elicited by exogenous and endogenous peroxynitrite and poly(ADP-ribose) synthetase. Shock 18, 434–439. [DOI] [PubMed] [Google Scholar]
- Ménard L, and Poirier GG (2011). Rapid assay of poly(ADP-ribose) glycohydrolase. Biochem. Cell Biol 65, 668–673. [DOI] [PubMed] [Google Scholar]
- Moss J, Stanley SJ, Nightingale MS, Murtagh JJ, Monaco L, Mishima K, Chen HC, Williamson KC, and Tsai SC (1992). Molecular and immunological characterization of ADP-ribosylarginine hydrolases. J. Biol. Chem 267, 10481–10488. [PubMed] [Google Scholar]
- Nusrat A, Eichel-Streiber, von C, Turner JR, Verkade P, Madara JL, and Parkos CA (2001). Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun 69, 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer NJ (1978). Structural determination and stereospecificity of the choleragen-catalyzed reaction of NAD+ with guanidines. J. Biol. Chem 253, 4907–4910. [PubMed] [Google Scholar]
- Poręba M, Stróżyk A, Salvesen GS, and Drąg M (2013). Caspase substrates and inhibitors. Cold Spring Harb. Perspect Biol 5, a008680–a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal F, Feijs KLH, Frugier E, Bonalli M, Forst AH, Imhof R, Winkler HC, Fischer D, Caflisch A, Hassa PO, et al. (2013). Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol 20, 502–507. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Murcia JM-D, La Rubia, de G, Menissier-De Murcia J, Hostomsky Z, and de Murcia G (2000). Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 39, 7559–7569. [DOI] [PubMed] [Google Scholar]
- Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, et al. (2013). Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. Embo J. 32, 1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, and Ahel I (2011). The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slama JT, Aboul-Ela N, and Jacobson MK (1995a). Mechanism of Inhibition of Poly(ADP-ribose) Glycohydrolase by Adenosine Diphosphate (Hydroxymethyl)pyrrolidinediol. J. Med. Chem 38, 4332–4336. [DOI] [PubMed] [Google Scholar]
- Slama JT, Aboul-Ela N, Goli DM, Cheesman BV, Simmons AM, and Jacobson MK (1995b). Specific Inhibition of Poly(ADP-ribose) Glycohydrolase by Adenosine Diphosphate (Hydroxymethyl)pyrrolidinediol. J. Med. Chem 38, 389–393. [DOI] [PubMed] [Google Scholar]
- Stowell AIJ, James DI, Waddell ID, Bennett N, Truman C, Hardern IM, and Ogilvie DJ (2016). A high-throughput screening-compatible homogeneous time-resolved fluorescence assay measuring the glycohydrolase activity of human poly(ADP-ribose) glycohydrolase. Analytical Biochemistry 503, 58–64. [DOI] [PubMed] [Google Scholar]
- Takeda J, Adachi K, Halprin KM, Itami S, Levine V, and Woodyard C (1983). Forskolin Activates Adenylate Cyclase Activity and Inhibits Mitosis in In Vitro in Pig Epidermis. J. Investig. Dermatol 81, 236–240. [DOI] [PubMed] [Google Scholar]
- Taylor CM, Barker WD, Weir CA, and Park JH (2002). Toward a General Strategy for the Synthesis of 3,4-Dihydroxyprolines from Pentose Sugars. J. Org. Chem 67, 4466–4474. [DOI] [PubMed] [Google Scholar]
- Tucker JA, Bennett N, Brassington C, Durant ST, Hassall G, Holdgate G, McAlister M, Nissink JWM, Truman C, and Watson M (2012). Structures of the human poly (ADP-ribose) glycohydrolase catalytic domain confirm catalytic mechanism and explain inhibition by ADP-HPD derivatives. PLoS oNe 7, e50889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Matic I, Uchima L, Rood J, Žaja R, Hay RT, Ahel I, and Chang P (2014). Familywide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun 5, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, and Schultz BD (2014). Cholera toxin enhances Na+ absorption across MCF10A human mammary epithelia. Am. J. Physiol. Cell Physiol 306, C471–C484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354, aad6872–aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen Y, Zhang Y, and Yu Y (2017). A Cell-Line-Specific Atlas of PARP-Mediated Protein Asp/Glu- ADP-Ribosylation in Breast Cancer. Cell Rep 21, 2326–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.