

Abstract
Bioremediation is an accepted technology for cleanup of soil contaminated with polycyclic aromatic hydrocarbons (PAHs), but it can increase the genotoxicity of the soil despite removal of the regulated PAHs. Although polar biotransformation products have been implicated as causative genotoxic agents, no specific product has been identified. We pursued a non-target analytical approach combining effect-directed analysis (EDA) and metabolite profiling to compare extracts of PAH-contaminated soil from a former manufactured-gas plant site before and after treatment in a laboratory-scale aerobic bioreactor. A compound with the composition C15H8O2 and four methylated homologs were shown to accumulate as a result of bioreactor treatment, and the C15H8O2 compound purified from soil extracts was determined to be genotoxic. Its structure was established by nuclear magnetic resonance and mass spectroscopy as a heretofore unidentified α,β-unsaturated lactone derived from dioxygenation of pyrene at an apical ring, 2H-naphtho[2,1,8-def]chromen-2-one (NCO), which was confirmed by synthesis. The concentration of NCO in the bioreactor was 11 μg g−1 dry soil, corresponding to 13% of the pyrene removed. It also accumulated in aerobically incubated soil from two additional PAH-contaminated sites and was formed from pyrene by two pyrene-degrading bacterial cultures known to be geographically widespread, underscoring its potential environmental significance.
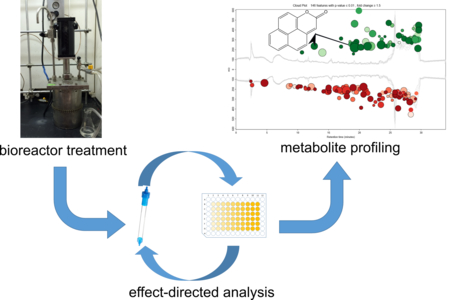
Graphical Abstract
INTRODUCTION
Polycyclic aromatic hydrocarbons (PAHs) are a ubiquitous class of soil contaminants found in high concentrations at industrial sites associated with the former production of manufactured gas, wood preservation, and petroleum processing.1 They are constituents of highly complex mixtures in the primary contamination sources such as coal tar or creosote,1, 2 although only 16 PAHs are regulated by the U.S. Environmental Protection Agency (EPA) and many international agencies.2 Bioremediation is an accepted technology for PAH-contaminated soil,3 but like other remediation methods its efficacy is evaluated only on the basis of removal of the regulated compounds rather than on broader measures of risk. For example, bioremediation of PAH-contaminated soil can have little effect on4, 5 or actually increase6–10 the soil’s genotoxicity, despite extensive removal of the EPA-regulated PAHs.
The source(s) of increased genotoxicity resulting from bioremediation have not been identified in previous studies8, 10, 11, although polar compounds formed during treatment have been implicated.10 Prior efforts have focused on possible microbial metabolites, such as oxygenated PAHs (oxy-PAHs), that can cause DNA damage12 or exhibit mutagenicity.13 However, some commonly observed oxy-PAHs and known bacterial transformation products are not mutagenic13, 14 and have been observed to decrease during bioremediation when increases in genotoxicity have been observed.10, 15
In this study, we used a non-target approach to investigate extracts of contaminated soil from a former manufactured-gas plant (MGP) site whose genotoxicity has been shown to increase following treatment in a lab-scale, aerobic bioreactor.9, 10, 15 The approach involved a two-tiered separation of soil extracts (pre- and post- treatment) in an effect-directed analysis (EDA)16–18 based on the DT40 DNA damage-response assay. A semi-polar fraction of the post-treatment extract exhibiting the greatest cytotoxicity was compared to the corresponding fraction of the pre-treatment extract using metabolite profiling, a method originating in metabolomics19 and more recently used in environmental analysis,20–24 which combines high-resolution analytical techniques with rigorous statistical analysis. Coupling EDA to metabolite profiling can efficiently identify biologically active compounds in complex samples, as has been shown in the field of drug discovery,25 but it has had limited application in environmental studies.
MATERIALS AND METHODS
Chemicals.
PAH standards (EPA 610 PAH mixture), silica gel (high purity, 70–230 mesh), pyrene, methyl methanesulfonate (MMS), dimethylsulfoxide (DMSO), and phosphate buffer solution (PBS) were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.). Anhydrous sodium sulfate and HPLC-grade solvents, including n-hexane, dichloromethane (DCM), acetone, ethyl acetate (EA), and methanol, were purchased from Fisher Scientific (Pittsburgh, PA, U.S.A.).
5H-naphtho[8,1,2-cde]chromen-5-one (4-oxapyrene-5-one) and 4H-cyclopenta[def]phenanthrene-8,9-dione were synthesized according to previously published methods.26, 27 The authentic standard of 2H-naphtho[2,1,8-def]chromen-2-one (NCO) was synthesized de novo and characterized by high-resolution mass spectrometry (HRMS), 1H nuclear magnetic resonance (NMR) spectroscopy, and 2-dimensional NMR methods as described in Supporting Information (SI). Details of the synthetic scheme and characterization of intermediates will be published in a forthcoming manuscript.
Soil and bioreactor treatment.
PAH-contaminated soil treated in the bioreactor was collected from a former manufactured gas plant site in Salisbury, North Carolina, USA. Detailed information on collection, processing and characterization of the soil can be found in a previous publication.9 The concentrations of target PAHs (14 of the 16 EPA-regulated PAHs, excluding acenaphthylene and indeno-[1,2,3-cd]pyrene) were quantified by HPLC with fluorescence detection28 and are shown in Table S1.
The bioremediation process was achieved in a laboratory-scale (2 L), semi-continuous, aerated bioreactor containing a slurry of soil in an aqueous buffer (15% w/w). Every week, 20% of the treated slurry was removed from the bioreactor and replaced with new untreated (feed) soil suspended in fresh buffer (pH 7.5) containing 5 mM phosphate and 2.5 mM ammonium nitrate (“bioreactor buffer”). Bioreactor-treated soil was recovered from the slurry by centrifugation at 3900 rpm for 20 min.
Extraction and Fractionation.
Soil samples were extracted according to the method described in SI. Extracts from both feed and bioreactor-treated soils were fractionated using the two-level EDA method described in Figure S1. In the first level of fractionation, extracts were separated into broad compound classes by column chromatography using silica gel, according to the method of Bandowe et al.29 The second level of fractionation was achieved with solid-phase extraction (SPE) cartridges containing cyanopropyl-bonded silica (SiliCycle, QC, Canada). Details on the fractionation are provided in SI.
Bioassay.
To avoid toxicity effects related to the extraction or elution solvents, extracts were dried and re-dissolved in DMSO prior to the bioassay. Aliquots of fractions and subfractions were evaporated in pre-weighed vials, and dry masses of residues were determined on an analytical balance. Mass balances of residues in the fractions were calculated according to the dry masses and the proportions relative to the mass of residue in the whole extract. The residues were then dissolved in DMSO under sterile conditions and stored at −80 °C until analysis.
Toxicity and genotoxicity of the soil extracts and fractions were evaluated using a 96-well plate-based DT40 chicken lymphocyte DNA-damage response assay.9, 30 The DT40 isogenic cell line, and its mutants knocked out in specific DNA repair pathways, were exposed to residues dissolved in DMSO and serially diluted with PBS. The concentration of DMSO was adjusted to 0.3% for all cell exposures. To verify the consistency of cells across different batches, methyl methanesulfonate (MMS) was used as positive control while the vehicle blank (DMSO diluted in PBS) was used as negative control.
LD50 values were calculated by fitting the dose-response curve (survival percentage versus log concentration) using GraphPad Prism 6.05 for Windows. The mass of residue in each dose was converted to the equivalent mass of soil from which that mass would have been obtained 9 for the purpose of comparing doses between samples. The LD50 for the DT40 parental cell line represents the cytotoxicity, and the relative LD50 (LD50 of mutant cell line divided by LD50 of parental cell line) was calculated as the measure of genotoxicity.9, 31
Instrumental analysis.
For the selected fractions, analysis was performed using an Agilent 1100 series HPLC coupled to a 6520 series high-resolution quadrupole time-of-flight MS (HR-QToF-MS) equipped with an electrospray ionization (ESI) source (Agilent Technologies, Palo Alto, CA, USA). HPLC-MS analysis using an atmospheric pressure photoionization (APPI) source was performed on a Thermo TSQ Quantum Ultra triple quadrupole mass spectrometer (QqQ MS; Thermo Fisher Scientific). Details of HPLC-MS methods are described in SI.
Quantification of NCO was accomplished on the triple quadrupole mass spectrometer using the synthetic compound as an analytical standard. The samples were analyzed in selected reaction monitoring (SRM) mode using the transition m/z 221.06 ([M+H]+) to m/z 165.06 ([M+H-2(CO)]+) and the optimized collision energy, 29 V.
Purification of NCO and structure identification.
To obtain enough material for NMR analysis, 150 g (dry weight) of bioreactor-treated soil samples were extracted and combined for purification. NCO formed during bioreactor treatment was purified by a three-level separation method: low-pressure silica gel-column chromatography, medium-pressure column chromatography, and HPLC. Details of the purification and structural identification are provided in SI.
Other biotransformation experiments.
To demonstrate that the isolated product was derived from pyrene, microcosm incubations were performed with the bioreactor-treated slurry in the presence of pyrene. To confirm that NCO is a bacterial metabolite, transformation of pyrene by selected bacterial strains was evaluated in resting-cell incubations. To test the potential prevalence of NCO formation in PAH-contaminated soils, microcosm incubations were performed with soil samples from two independent and geographically distant PAH-contaminated sites: creosote-contaminated soil from a wood-treatment facility in Andalucía, Spain, which had a 100-year history of soil pollution32 and soil from the Holcomb Creosote Superfund Site in Yadkinville, North Carolina, USA. Incubation details are provided in SI.
Data Analysis.
The HPLC-QToF profiles of extract fractions from feed soil and bioreactor-treated soil were compared with the metabolomics platform XCMS Online (https://xcmsonline.scripps.edu/). Data for the treated and feed soil samples were set as the test and control groups, respectively. Data files from three replicate samples were obtained for each group, and the input parameters were based on the default method optimized for HPLC-QToF (details in SI). It has been shown that three replicates are sufficient for metabolite profiling of complex samples that have been simplified through EDA.25 Independent t-tests (Welch’s t-tests) were used to test for statistically significant differences of features between the two groups (feed and treated soils).
The R language and environment for statistical computing (version 3.2.3) was used for analysis and hypothesis testing on toxicological data. Paired t-tests were employed to test for statistically significant differences of LD50 or relative LD50 values between samples. Significance level was set at α = 0.05.
RESULTS AND DISCUSSION
Fractionation of soil extracts and selection of genotoxic fraction.
Two-level fractionation of soil extracts was directed by cytotoxicity and genotoxicity measured with the DT40 DNA damage-response bioassay, which is based on DT40 isogenic chicken lymphocytes and mutants that are each knocked out in a specific DNA repair mechanism.30 The Rad54−/− and Rev1−/− mutants were selected for this study because of their sensitivity to the increased genotoxicity of bioreactor-treated soil.9 The Rad54−/− mutant is deficient in Rad54 proteins and therefore is sensitive to genotoxicants that cause double-strand breaks and replication-fork blockage. The Rev1−/− mutant is deficient in Rev1 proteins, resulting in vulnerability to chemicals causing DNA depurination or stable covalent adducts.
The first level of fractionation using deactivated silica gel is a classical adsorption-based method to separate the extracts into broad classes,29 in this case a nonpolar fraction (containing compounds such as PAHs and alkylated PAHs), a semi-polar fraction (containing compounds such as oxy-PAHs and azaarenes), and a polar fraction (containing acids and phenols) designated A, B, and C, respectively. As indicated in Figure S1, mass balance was achieved in the separation, with the greatest mass in Fraction B. Complete recovery of the toxic compounds was demonstrated by comparison of the cytotoxicity and genotoxicity of recombined fractions and whole-soil extracts (Figures S2 A and B, respectively). Consistent with earlier work,9, 10, 15 whole-soil extracts indicated that the bioreactor-treated soil was more genotoxic than the untreated feed soil (Figure S2B).
Fraction C did not cause toxicity and is not discussed further. Nonpolar fraction A from the treated soil had a significantly higher LD50 than for feed soil for all three cell lines (Figure 1A), suggesting detoxification of the nonpolar compounds; this would be consistent with removal of the PAHs in the bioreactor (Table S1). Semi-polar fraction B showed the opposite trend (Figure 1B); the LD50 for all three cell lines was significantly lower for treated soil than for feed soil, indicating greater toxicity of treated soil. The relative LD50 values of fraction B, however, were not significantly different between treated soil and feed soil (Figure S3A). Given that fractionation did not appear to influence the recovery of cytotoxic or genotoxic compounds when the fractions were recombined (Figures S2 A and B), we inferred that the high cytotoxicity of the semi-polar compounds in fraction B for all three cell lines could have masked a genotoxic effect. On this basis, we selected fraction B for further fractionation.
Fig 1.

Results of DT40 DNA-damage response bioassays on solvent extracts and fractionated extracts from PAH-contaminated soil used to feed a lab-scale bioreactor (“feed soil”, FS) and bioreactor-treated soil (TS). Assays were conducted with the parental DT40 cell line and two mutant cell lines deficient in different DNA-repair mechanisms, Rad54−/− and Rev1-/−. (A and B) LD50 values from the two fractions exhibiting measurable cytotoxicity (over the dose range tested) after the first level of extract fractionation. (C and D) LD50 values from the two subfractions of primary fraction B that exhibited cytotoxicity. Error bars represent standard deviations from replicates at each dose (n = 5). Asterisks indicate a significant difference between feed soil and bioreactor-treated soil for a given cell line. Plots of relative LD50 values corresponding to data in panels A-D are in Figure S2.
The second level of fractionation on fraction B was achieved by cyanopropyl solid-phase extraction (SPE), which retains semi-polar to polar compounds based on partitioning and has been used previously to separate mutagens without affecting activity.33 Three subfractions (B1, B2, and B3) were obtained for each soil (Figure S1). Subfractions B1 from each sample (feed and treated soil) accounted for the greatest dry mass of residue, but the cytotoxicity in treated soil was lower than in feed soil (Figure 1C). Subfractions B3 did not present any detectable toxicity for either soil. For subfraction B2, treated soil had significantly lower LD50 than feed soil (Figure 1D), implying greater cytotoxicity. The relative LD50 values for treated soil and feed soil were not significantly different at α = 0.05 (Figure S3B); nevertheless the relative LD50 values indicate genotoxicity. As a result, subfractions B2 were selected for metabolite profiling.
Metabolite profiling.
Metabolite profiling was achieved by HPLC-MS and data analysis with XCMS Online, a widely used metabolomics platform.34 Assuming that the compounds responsible for increased genotoxicity would be significantly more abundant in bioreactor-treated soil than in feed soil, we focused on what the software categorizes as “upregulated” analytes and infer that they were formed in the bioreactor from constituents of feed soil. Although these analytes were likely to be transformation products of soil constituents, they would not necessarily be microbial metabolites.
Because the sensitivity of different ionization sources in mass spectrometry varies for different compound classes and functional groups,35, 36 we employed electrospray ionization in positive (ESI+) and negative (ESI-) ionization modes, and atmospheric pressure photoionization in positive (APPI+) and negative (APPI-) ionization modes. The composition of ions of interest was determined from exact mass measurements on the HR-QToF-MS instrument. Few upregulated ions, none with notable intensities, were detected by ESI operated in the negative ionization mode or by APPI in negative ionization mode. The greatest number of upregulated features was observed in the ESI+ mode, a small subset of which were also detected by APPI+ (Table S3). As a result, we focused on the ions detected in ESI+ mode for further investigation.
Of the ions detected in ESI+ mode, XCMS Online prioritized 146 with absolute intensity ≥ 1000 and fold-change ≥ 1.5, with p ≤ 0.01 (Figure 2A); 54 of them had higher concentrations in bioreactor-treated soil (“upregulated”). Application of more-restrictive criteria (absolute intensity ≥ 5000 and fold-change ≥ 2, p ≤ 0.01) yielded 14 significantly upregulated ions (Table S2). Two of the upregulated ions can be attributed to the neutral composition C15H8O2: m/z 221.0595, [M+H]+ (Figure 2B) and m/z 243.0416, [M+Na]+. A third ion, at m/z 463.0934, can be assigned as the sodium adduct of the dimer, [2M+Na]+. Intensity at m/z 235.0753, corresponding to the composition C16H10O2, was resolved into three ions in the extracted ion chromatogram (EIC; Figure 2C); because two of the three ions co-eluted, only two were distinguished by the XCMS Online software (Table S2). The composition C16H10O2 corresponds to addition of a methyl group to the parent C15H8O2 framework, suggesting the presence of three methyl congeners of the C15H8O2 parent. Similarly, an upregulated ion at m/z 249.0912, with the composition C17H12O2, may represent either a dimethyl or ethyl congener of C15H8O2.
Fig.2.

Non-target analysis of extract subfractions B2. (A) Cloud plot from metabolite profiling comparing the high-resolution mass spectra (ESI+ mode) of subfraction B2 from bioreactor-treated soil to subfraction B2 from feed soil. Mass spectral features with significantly higher intensities in the treated soil than in the feed soil (upregulated) are in green and those significantly lower in the treated soil are in red. Bubble size is proportional to peak intensity; the darker the shading, the higher fold change. All 146 features with intensity ≥ 1,000, fold-change ≥ 1.5 in either direction, and p ≤ 0.01 are shown. (B) and (C) Extracted ion chromatograms for m/z 221.0596 and m/z 235.0753, respectively.
Based on the summed intensities of the protonated and sodiated adduct ions, C15H8O2 was the most abundant of the upregulated compounds in the bioreactor-treated soil extract, with the mono- and dimethylated (or ethylated) congeners also present at high levels. This group of compounds showed exclusively high values in fold-change between feed soil and bioreactor-treated soil, 861 for C15H8O2 and 196–347 for the remaining compounds (Table S2). Among the limited number of upregulated ions detected in APPI+ mode that met the input criteria, m/z 221.06 and m/z 235.08 were also prominent (Table S3), confirming that these compounds were not artifacts of the ionization source. In a preliminary EDA study on our bioreactor-treated soil using one level of fractionation,10 genotoxic fractions subjected to non-target analysis by HPLC/(ESI+)-MS were also observed to contain analytes corresponding to C15H8O2 and C16H10O2, although the compounds were not identified.37
The relevance of the putative biotransformation product C15H8O2 to our previous studies on increased genotoxicity of treated soil9 was determined by analysis of eight archived extracts of bioreactor-treated soil samples collected over a four-year period. The compound and its methylated homologs were detected in all samples at levels comparable to those in this study. Consequently the most abundant product, C15H8O2, was considered the most likely source of increased genotoxicity resulting from bioreactor treatment of the soil and was targeted for structural characterization.
Isolation and structural characterization of C15H8O2.
The fragmentation pattern obtained from MS/MS of the parent ion at m/z 221.0595 was characterized by a major product ion at m/z 165.0691 (rel. intensity 100), corresponding to [M+H-2(CO)]+, and a second product ion at m/z 193.0640 (rel. intensity ~0.3) corresponding to [M+H-(CO)]+ (Figure S4F). Such a fragmentation pattern is typical of PAH-derived quinones and lactones.35, 38
Based on current knowledge of PAH biotransformation39–41 and database searches (SciFinder and ChemSpider), two structures were deduced as candidates for C15H8O2. 5H-naphtho[8,1,2-cde]chromen-5-one (also called 4-oxapyrene-5-one; Figure S4A) has been found in particulate matter from diesel exhaust and verified by comparison with an authentic standard.42 Lundstedt et al.40 reported that 4-oxapyrene-5-one accumulated in an aerobic bioreactor treating a creosote-contaminated soil; to our knowledge, however, this assignment was based solely on mass spectrometry, which does not allow a definitive structural assignment in the absence of an authentic standard. We synthesized 5H-naphtho[8,1,2-cde]chromen-5-one and definitively ruled it out as the C15H8O2 product observed in this study by HPLC/HR-QToF-MS (Figure S4).
A second known structure with the composition C15H8O2 is 4H-cyclopenta[def]phenanthrene-8,9-dione, a quinone derived from 4H-cyclopenta[def]phenanthrene. We also synthesized this compound and while the MS/MS spectrum was similar to that of the target product in this study (Figure S4D), it was ruled out based on a different HPLC retention time. Since the target product did not correspond to any known structure, structural assignment required isolation and characterization by an unambiguous technique. Hence, we purified a sufficient quantity for NMR studies from extracts of bioreactor-treated soil using bulk extraction and three-level chromatographic separation (SI).
The 1D 1H NMR spectrum (Figure S5) along with 2D homonuclear coherence (COSY; Figure S6C), heteronuclear multiple bond coherence (HMBC, Figure S6A), and heteronuclear single quantum coherence (HSQC; Figure S6B) spectra of the purified compound were acquired. The expected eight proton signals observed in the 1H NMR spectrum appeared as two pairs of doublets and two doublets coupling into a triplet in the aromatic region along with a singlet at higher field than generally observed for aromatic protons. As inferred from the 1H NMR spectrum, the 13C NMR spectrum indicated 8 carbon signals with intensities expected for H-bearing carbons and 7 low-intensity signals characteristic of quaternary carbons. Importantly, only one quaternary carbon signal appeared at a chemical shift associated with carbonyl carbon (162 ppm), ruling out a quinone structure. A second quaternary carbon signal at 154 ppm appeared in a region characteristic of an aromatic carbon bearing an oxygen atom. These two observations were consistent with a PAH-derived lactone structure. The COSY spectrum confirmed two sets of coupled proton signals with two additional protons coupled into the triplet. This pattern is typical for the K-region and apical ring signals of pyrene. The pattern of proton signals suggested 2H-naphtho[2,1,8-def]chromen-2-one (NCO; Figure 3 inset), an apparent pyrene-derived structure in which an apical ring had been oxidized to a lactone.
Fig. 3.

DT40 bioassay results on 2H-naphtho[2,1,8-def]chromen-2-one (NCO, inset) purified from extracts of bioreactor-treated soil.
Connectivities observed in the HSQC and HMBC spectra were key to the structural determination. A high-field carbon signal (106 ppm) in the HSQC spectrum was coupled to the high-field proton, confirming the high-field proton signal as non-aromatic. No C-H coupling was detected for the carbon signals at 162 and 154 ppm, in line with expectations for carbonyl and quaternary aromatic oxygen-substituted lactone carbons, respectively. In the HMBC spectrum, couplings predicted for NCO were observed between the non-aromatic singlet and the carbonyl carbon, and between a K-region doublet and the oxygen-substituted carbon assigned as part of the lactone structure. Thus, the NMR data are uniquely satisfied by the NCO structure.
Finally, NCO synthesized by an unambiguous route was identical in all respects to the product isolated from extracts of bioreactor-treated soil (Figures S5 and S7), establishing the NCO structure at the highest level of confidence for identifying unknown compounds.43 The synthetic standard was subsequently used as an analytical standard for quantification of NCO in samples.
The cytotoxicity and genotoxicity of NCO were confirmed by the DT40 bioassay. The LD50 of NCO for the parental cell line was 16.8 μM (Figure 3), lower than that of the positive control methyl methanesulfonate (MMS; 58.5 μM), suggesting stronger cytotoxicity. The LD50 was 7.36 μM for the Rad54−/− mutant and 6.49 μM for the Rev1−/− mutant, corresponding to relative LD50 values of 0.44 and 0.28, respectively, clearly demonstrating the genotoxicity of this compound. For comparison, the relative LD50 values for MMS were 0.31 and 0.07 for the Rad54−/− and Rev1−/− mutants, respectively. In a recently published study44 the relative LD50 values for hydrogen peroxide for these same two mutants were 0.54 and 0.32, respectively, comparable to that of NCO.
One possible mechanism for the observed genotoxicity of NCO might be through DNA alkylation, as α,β-unsaturated lactones have been demonstrated to modify DNA bases45 and formation of bulky adducts is consistent with the type of damage detected by both DT40 mutant cell lines used in this study. While biotransformation products other than NCO might contribute to the increased genotoxicity resulting from bioreactor treatment of the soil, the high concentration of NCO and its potent genotoxicity support its significant contribution to the overall genotoxicity of the treated soil. As a newly discovered biotransformation product in an environmentally relevant system, further studies on its toxicological behavior would be warranted.
Source, putative formation pathway, and potential prevalence.
The structure of NCO is consistent with its proposed origin as a transformation product of pyrene. This hypothesis was verified in microcosm incubations of the bioreactor-treated soil with or without added pyrene. After 3 days of incubation, the average concentration of NCO in the pyrene-spiked microcosms increased from 0.27 μg·mL−1 to 2.05 μg·mL−1, while the concentration in the control group was essentially unchanged (Figure S8). This experiment unambiguously demonstrated that NCOwas a transformation product of pyrene.
The concentration of pyrene in the untreated soil used to feed the bioreactor was 108 ± 8.5 μg·g−1 dry soil, second highest among the EPA-regulated PAHs monitored (Table S1). The estimated yield of NCO in the bioreactor was ~13%, based on its concentration in treated soil samples (11.2 ± 2.5 μg·g−1 dry soil, n=8) compared to the removal of pyrene in the bioreactor (84.0 ± 9.0 μg·g−1 dry soil). We assume that the detected alkylated congeners of NCO (C16H10O2, C17H12O2) are likewise biotransformation products from the corresponding methylated and dimethylated pyrene isomers also present at relatively high concentrations (53.5 ± 2.0 μg·g−1 and 21.4 ± 1.0 μg·g−1 dry soil, respectively) in the soil used to feed the bioreactor.31
To further confirm NCO as a bacterial metabolite of pyrene, the potential biotransformation to NCO was screened with three bacteria known to grow on pyrene as a sole carbon and energy source: Mycobacterium vanbaalenii strain PYR-1T, Rugisobacter aromaticivorans strain Ca6 T,46 and Immundisolibacter cernigliae strain TR3.2 T.47 M. vanbaalenii PYR-1T is the most comprehensively studied39, 48 bacterial isolate capable of growing on pyrene but is not known to be present in our bioreactor. R. aromaticivorans strain Ca6 T and I. cernigliae strain TR3.2 T (previously referred to as PG1 and PG2, respectively) were identified by stable-isotope probing as major contributors to pyrene degradation in several PAH-contaminated soils49–51 and possess 16S rRNA gene sequences highly similar to those recovered from geographically widespread contaminated soils;46, 47. Rugosibacter sequences are present in the soil that was treated in the bioreactor in this study,28 but are not present in high relative abundance in the bioreactor. Strain TR3.2 T, recently isolated from bioreactor-treated soil46, 52, and uncultivated Immundisolibacter strains are significant members of the bioreactor microbial community53. Both strains Ca6 T and TR3.2 T accumulated NCO after 24 hours of resting-cell incubation in the presence of pyrene, whereas strain PYR-1 did not. The yield of NCO from pyrene was 1.3 ± 0.4% for strain Ca6 T and 0.3 ± 0.1% for strain TR3.2 T. These results confirmed that NCO is a bacterial metabolite of pyrene. The low yields of NCO with the pure cultures compared to the yield observed in the bioreactor could have resulted from significant differences between bioreactor conditions and incubations of the pure cultures; for example, inocula were uninduced for pyrene metabolism, the incubation was short-term, and there were no other substrates that would be present in the bioreactor that could influence metabolic flux or differential transcription of the multiple ring-hydroxylating dioxygenases in both organisms. It is also possible that other, undetermined PAH-degrading bacteria in the bioreactor contributed to NCO accumulation.
The proposed pathway for formation of NCO is shown in Figure 4. The first three steps are consistent with the canonical steps for aerobic bacterial metabolism of PAHs:48 cis-dihydroxylation by a ring-hydroxylating dioxygenase (RHD), dehydrogenation, and ring cleavage by an extradiol dioxygenase. Lactonization of the α-ketohydroxycarboxylic acid resulting from extradiol ring cleavage has been reported for the bacterial oxidation of naphthalene,54 phenanthrene55 and anthracene;56, 57 in most cases, however, the lactone was considered to be a minor product relative to further metabolism of the ring-cleavage product by an aldolase-catalyzed release of pyruvate.
Fig. 4.

Proposed pathway for the formation of NCO from pyrene by PAH-degrading bacteria.
Initial ring hydroxylation would have to occur at the C1-C2 position to form NCO from pyrene, whereas complete (growth-related) metabolism of pyrene involves initial attack at the C4-C5 position, or K-region.39 M. vanbaalenii PYR-1 is known to initiate C1,2 oxidation of pyrene39, 58, 59 via Nid-type dioxygenases,39 although as a minor side reaction relative to dihydroxylation at C4-C5 59. Dead-end products retaining three58 or four39 rings have been found to accumulate from initial C1,2-dioxygenation of pyrene by strain PYR-1, but NCO was not reported as a product in either case. Given the low yield of NCO from the two pyrene-degrading pure cultures shown to produce it, it may be a dead-end metabolite initiated by a dioxygenase whose dominant regiospecificity is for C4-C5. Alternatively, given the differential substrate specificity of RHDs in strain Ca6 T,60 it is possible that pyrene was oxidized in the pure cultures via an RHD with higher specificity for a different PAH substrate in which pyrene would align differently at the active site than in an RHD highly specific for pyrene. After ring hydroxylation, the next two steps would have to be carried out by enzymes that can accommodate the substitutions at C1-C2. It appears that the hydratase-aldolase that would normally be involved in releasing pyruvate from an extradiol ring-cleavage product48 of other PAHs is either not present in the organism(s) that produce NCO or is unable to accommodate the product resulting from oxidation of pyrene at C1-C2.
To verify that NCO could be produced in PAH-contaminated soils other than the soil we treated in our bioreactor, we prepared aerobic microcosms of creosote-contaminated soil samples from two other sites, to which no additional pyrene was added: the Holcomb Superfund site in North Carolina, USA, and a wood treatment facility in Andalucía, Spain.32 After six weeks of incubation, NCO was detected in both samples (no NCO was present in either sample initially). In the Holcomb sample, 0.5 ± 0.02 μg NCO ·g−1 dry soil was produced, and in the Andalucía sample 10.3 ± 0.54 μg NCO ·g−1 dry soil was produced.
Environmental relevance.
We have identified a genotoxic bacterial metabolite of pyrene, one of the most abundant of the EPA-regulated PAHs in contaminated systems,61 produced during lab-scale bioremediation of field-contaminated soil from a former MGP site. Its yield and concentration in the lab-scale bioreactor are high enough to be of potential concern, because oxy-PAHs such as NCO and quinones are likely to be more mobile in the environment than their parent PAHs.13 Furthermore, we have shown that NCO formation from pyrene in our bioreactor is not likely to be an isolated observation. We observed its formation in soils from three different sites, two in North Carolina (USA) and one in Europe, with different contamination sources (coal tar and creosote). It is also produced by two major pyrene-degrading bacteria that are closely related phylogenetically to uncultivated bacteria found in geographically diverse contaminated environments.46, 47 In addition, it is possible that NCO has been observed in previous studies but not recognized as such. A metabolite of pyrene from two different Mycobacterium strains, including M. vanbaalenii PYR-1, was reported41, 62 to have an electron-ionization mass spectrum with a molecular ion at m/z 220 and fragment ions at m/z 192 [M+ - (CO)] and 164 [M+ - 2(CO)] or 163 [M+ - COOCH], consistent with the composition C15H8O2. However, the metabolite was not further characterized in either study; in the case of strain PYR-1, the authors assumed that the metabolite was 4-oxapyrene-5-one.62 Although we did not observe NCO production (or any other product with m/z 220) by strain PYR-1 in the present study, this may have been due to differences in incubation conditions. As noted above, Lundstedt et al.40 reported the accumulation of a compound assumed to be 4-oxapyrene-5-one in an aerobic bioreactor treating a PAH-contaminated soil. In these earlier studies it would have been reasonable to assume that an analyte with the same molecular formula as NCO was 4-oxapyrene-5-one; it had already been identified definitively in particulate matter from diesel exhaust,42 and it would be reasonable to predict a bacterial metabolite of pyrene via oxidation at C4-C5, the predominant site of attack in aerobic bacterial metabolism.
Because of its cytotoxicity, genotoxicity, and potential occurrence in other PAH-contaminated environmental systems, we believe that attention should be paid to NCO during active bioremediation efforts or as a result of natural attenuation in situ. This study adds to the increasing awareness of the potential toxicity and persistence of transformation products of environmental contaminants63–67 and reinforces the need to include such products in risk analysis. Traditional environmental analysis has focused on target compounds that could be identified and quantified by commercial standards, overlooking many possibly important contaminants.68 As illustrated in this study, combining non-target analysis with EDA methods allows greater insights into the chemicals that can pose substantial threats to human health.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) under its Superfund Research Program, grant number P42ES005948. Partial support for LBC and for MS analysis in the UNC Biomarker Mass Spectrometry Facility was provided through the UNC Center for Environmental Health and Susceptibility, NIEHS grant number P30ES010126. JV was supported by a Marie Skłodowska Curie Individual Fellowship from the European Commission (Grant Agreement 661361 – NETPAC – H2020-MSCA-IF-2014). We thank Joe Alfaro (US EPA, Atlanta, Georgia), Doug Peters (US EPA, Athens, Georgia) and Lauren Redfern (Duke University) for their assistance in obtaining the soil sample from the Holcomb Creosote Superfund Site.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental details; PAH concentrations in feed and bioreactor-treated soils; supplemental genotoxicity data; HPLC-MS data on synthesized compounds; NMR data on NCO; results of bioreactor-treated soil incubation with spiked pyrene; supplemental data from metabolite profiling.
The authors declare no competing financial interest.
References
- 1.Agency for Toxic Substances and Disease Registry Toxicological Profile for Polycyclic Aromatic Hydrocarbons; U.S Department of Health and Human Services, Public Health Service: Atlanta, Georgia, 1995. [PubMed] [Google Scholar]
- 2.Andersson JT; Achten C, Time to say goodbye to the 16 EPA PAHs? Toward an up-to-date use of PACs for environmental purposes. Polycyclic Aromat. Compd 2015, 35, (2/4), 330–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.USEPA Office of Solid Waste and Emergency Response Superfund Remedy Report, Fourteenth Edition; EPA 542-R-13–016; U.S. Environmental Protection Agency: Washington, DC, 2013. [Google Scholar]
- 4.Mueller JG; Middaugh DP; Lantz SE; Chapman PJ, Biodegradation of creosote and pentachlorophenol in contaminated groundwater: chemical and biological assessment. Appl. Environ. Microbiol 1991, 57, (5), 1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillespie AM; Wang S; McDonald T; Garcia SG; Cosgriff D; He L-Y; Huebner H; Donnelly KC, Genotoxicity assessment of wood-preserving waste-contaminated soil undergoing bioremediation. Biorem. J 2007, 11, (4), 171–182. [Google Scholar]
- 6.Hughes TJ; Claxton LD; Brooks L; Warren S; Brenner R; Kremer F, Genotoxicity of bioremediated soils from the Reilly Tar Site, St. Louis Park, Minnesota. Environ. Health Perspect 1998, 106, Supp. 6, 1427–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belkin S; Steiber M; Tiehm A; Frimmel F; Abeliovich A; Werner P; Ulitzur S, Toxicity and genotoxicity enhancement during polycyclic aromatic hydrocarbons’ degradation. Environ. Toxicol. Water Qual 1994, 9, 303–309. [Google Scholar]
- 8.Lemieux CL; Lynes KD; White PA; Lundstedt S; Öberg L; Lambert IB, Mutagenicity of an aged gasworks soil during bioslurry treatment. Environ. Mol. Mutag 2009, 50, (5), 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J; Nakamura J; Richardson SD; Aitken MD, Evaluating the effects of bioremediation on genotoxicity of polycyclic aromatic hydrocarbon-contaminated soil using genetically engineered, higher eukaryotic cell lines. Environ. Sci. Technol 2012, 46, (8), 4607–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chibwe L; Geier MC; Nakamura J; Tanguay RL; Aitken MD; Simonich SLM, Aerobic bioremediation of PAH contaminated soil results in increased genotoxicity and developmental toxicity. Environ. Sci. Technol. 2015, 49, (23), 13889–13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brooks LR; Hughes TJ; Claxton LD; Austern B; Brenner R; Kremer F, Bioassay-directed fractionation and chemical identification of mutagens in bioremediated soils. Environ. Health Perspect 1998, 106, (Suppl 6), 1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zielinska-Park J; Nakamura J; Swenberg JA; Aitken MD, Aldehydic DNA lesions in calf thymus DNA and HeLa S3 cells produced by bacterial quinone metabolites of fluoranthene and pyrene. Carcinogenesis 2004, 25, 1727–1733. [DOI] [PubMed] [Google Scholar]
- 13.Lundstedt S; White PA; Lemieux CL; Lynes KD; Lambert IB; Öberg L; Haglund P; Tysklind M, Sources, fate, and toxic hazards of oxygenated polycyclic aromatic hydrocarbons (PAHs) at PAH-contaminated sites. Ambio 2007, 36, (6), 475–485. [DOI] [PubMed] [Google Scholar]
- 14.Park J; Ball LM; Richardson SD; Zhu H-B; Aitken MD, Oxidative mutagenicity of polar fractions from polycyclic aromatic hydrocarbon-contaminated soils. Environ. Toxicol. Chem 2008, 27, (11), 2207–2215. [DOI] [PubMed] [Google Scholar]
- 15.Hu J; Adrion AC; Nakamura J; Shea D; Aitken MD, Bioavailability of (geno)toxic contaminants in polycyclic aromatic hydrocarbon–contaminated soil before and after biological treatment. Environ. Eng. Sci 2014, 31, (4), 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brack W, Effect-directed analysis: a promising tool for the identification of organic toxicants in complex mixtures? Anal. Bioanal. Chem 2003, 377, (3), 397–407. [DOI] [PubMed] [Google Scholar]
- 17.Vughs D; Baken KA; Kolkman A; Martijn AJ; de Voogt P, Application of effect-directed analysis to identify mutagenic nitrogenous disinfection by-products of advanced oxidation drinking water treatment. Environ. Sci. Pollut. Res 2016, 24, (212), 1–14. [DOI] [PubMed] [Google Scholar]
- 18.Yue S; Ramsay BA; Brown RS; Wang J; Ramsay JA, Identification of estrogenic compounds in oil sands process waters by effect directed analysis. Environ. Sci. Technol 2015, 49, (1), 570–577. [DOI] [PubMed] [Google Scholar]
- 19.Fernie AR; Trethewey RN; Krotzky AJ; Willmitzer L, Metabolite profiling: from diagnostics to systems biology. Nat. Rev. Mol. Cell Biol 2004, 5, (9), 763–769. [DOI] [PubMed] [Google Scholar]
- 20.LeFevre GH; Müller CE; Li RJ; Luthy RG; Sattely ES, Rapid phytotransformation of benzotriazole generates synthetic tryptophan and auxin analogs in Arabidopsis. Environ. Sci. Technol 2015, 49, (18), 10959–10968. [DOI] [PubMed] [Google Scholar]
- 21.Men Y; Han P; Helbling DE; Jehmlich N; Herbold C; Gulde R; Onnis-Hayden A; Gu AZ; Johnson DR; Wagner M, Biotransformation of two pharmaceuticals by the ammonia-oxidizing archaeon Nitrososphaera gargensis. Environ. Sci. Technol 2016, 50, (9), 4682–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schollée JE; Schymanski EL; Avak SE; Loos M; Hollender J, Prioritizing unknown transformation products from biologically-treated wastewater using high-resolution mass spectrometry, multivariate statistics, and metabolic logic. Anal. Chem 2015, 87, (24), 12121–12129. [DOI] [PubMed] [Google Scholar]
- 23.Singh RR; Lester Y; Linden KG; Love NG; Atilla-Gokcumen GE; Aga DS, Application of metabolite profiling tools and time-of-flight mass spectrometry in the identification of transformation products of iopromide and iopamidol during advanced oxidation. Environ. Sci. Technol 2015, 49, (5), 2983–2990. [DOI] [PubMed] [Google Scholar]
- 24.Strynar M; Dagnino S; McMahen R; Liang S; Lindstrom A; Andersen E; McMillan L; Thurman M; Ferrer I; Ball C, Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environ. Sci. Technol 2015, 49, (19), 11622–11630. [DOI] [PubMed] [Google Scholar]
- 25.Kellogg JJ; Todd DA; Egan JM; Raja HA; Oberlies NH; Kvalheim OM; Cech NB, Biochemometrics for Natural Products Research: Comparison of Data Analysis Approaches and Application to Identification of Bioactive Compounds. J. Nat. Prod 2016, 79, (2), 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillis RG; Porter QN, 5-Methoxyphenanthrene-4-carboxylic acid. Aust. J. Chem 1989, 42, (6), 1007–1010. [Google Scholar]
- 27.Trost BM; Kinson PL, Perturbed [12] annulenes. Derivatives of dibenzo [cd, gh] pentalene. J. Am. Chem. Soc 1975, 97, (9), 2438–2449. [Google Scholar]
- 28.Richardson SD; Lebron BL; Miller CT; Aitken MD, Recovery of phenanthrene-degrading bacteria after simulated in situ persulfate oxidation in contaminated soil. Environ. Sci. Technol 2010, 45, (2), 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandowe BAM; Shukurov N; Kersten M; Wilcke W, Polycyclic aromatic hydrocarbons (PAHs) and their oxygen-containing derivatives (OPAHs) in soils from the Angren industrial area, Uzbekistan. Environ. Pollut 2010, 158, (9), 2888–2899. [DOI] [PubMed] [Google Scholar]
- 30.Ji K; Kogame T; Choi K; Wang X; Lee J; Taniguchi Y; Takeda S, A novel approach using DNA-repair-deficient chicken DT40 cell lines for screening and characterizing the genotoxicity of environmental contaminants. Environ. Health Perspect 2009, 117, (11), 1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adrion AC; Nakamura J; Shea D; Aitken MD, Screening nonionic surfactants for enhanced biodegradation of polycyclic aromatic hydrocarbons remaining in soil after conventional biological treatment. Environ. Sci. Technol 2016, 50, (7), 3838–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tejeda-Agredano M; Gallego S; Vila J; Grifoll M; Ortega-Calvo J; Cantos M, Influence of the sunflower rhizosphere on the biodegradation of PAHs in soil. Soil Biol. Biochem 2013, 57, 830–840. [Google Scholar]
- 33.Durant JL; Lafleur AL; Plummer EF; Taghizadeh K; Busby WF; Thilly WG, Human lymphoblast mutagens in urban airborne particles. Environ. Sci. Technol 1998, 32, (13), 1894–1906. [Google Scholar]
- 34.Tautenhahn R; Patti GJ; Rinehart D; Siuzdak G, XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem 2012, 84, (11), 5035–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connell SG; Haigh T; Wilson G; Anderson KA, An analytical investigation of 24 oxygenated-PAHs (OPAHs) using liquid and gas chromatography–mass spectrometry. Anal. Bioanal. Chem 2013, 405, (27), 8885–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grosse S; Letzel T, Liquid chromatography/atmospheric pressure ionization mass spectrometry with post-column liquid mixing for the efficient determination of partially oxidized polycyclic aromatic hydrocarbons. J. Chromatogr. A 2007, 1139, (1), 75–83. [DOI] [PubMed] [Google Scholar]
- 37.Chibwe L; Davie-Martin CL; Aitken MD; Hoh E; Simonich SLM, (In Press) Identification of Polar Transformation Products and High Molecular Weight Polycyclic Aromatic Hydrocarbons (PAHs) in Contaminated Soil Following Bioremediation. Sci. Total Environ 2017. [DOI] [PubMed] [Google Scholar]
- 38.Gallampois CM; Schymanski EL; Krauss M; Ulrich N; Bataineh M; Brack W, Multicriteria approach to select polyaromatic river mutagen candidates. Environ. Sci. Technol 2015, 49, (5), 2959–2968. [DOI] [PubMed] [Google Scholar]
- 39.Kim S-J; Kweon O; Jones RC; Freeman JP; Edmondson RD; Cerniglia CE, Complete and integrated pyrene degradation pathway in Mycobacterium vanbaalenii PYR-1 based on systems biology. J. Bacteriol 2007, 189, (2), 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lundstedt S; Haglund P; Öberg L, Degradation and formation of polycyclic aromatic compounds during bioslurry treatment of an aged gasworks soil. Environ. Toxicol. Chem 2003, 22, (7), 1413–1420. [PubMed] [Google Scholar]
- 41.Vila J; López Z; Sabaté J; Minguillón C; Solanas AM; Grifoll M, Identification of a novel metabolite in the degradation of pyrene by Mycobacterium sp. strain AP1: actions of the isolate on two-and three-ring polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol 2001, 67, (12), 5497–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pitts JN; Lokensgard DM; Harger W; Fisher TS; Mejia V; Schuler JJ; Scorziell GM; Katzenstein YA, Mutagens in diesel exhaust particulate identification and direct activities of 6-nitrobenzo [a] pyrene, 9-nitroanthracene, 1-nitropyrene and 5H-phenanthro [4, 5-bcd] pyran-5-one. Mutat. Res. Lett 1982, 103, (3–6), 241–249. [DOI] [PubMed] [Google Scholar]
- 43.Schymanski EL; Jeon J; Gulde R; Fenner K; Ruff M; Singer HP; Hollender J, Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol 2014, 48, (4), 2097–2098. [DOI] [PubMed] [Google Scholar]
- 44.Sharma V; Collins LB; Chen T.-h.; Herr N; Takeda S; Sun W; Swenberg JA; Nakamura J, Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, (18), 25377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfenning C; Esch HL; Fliege R; Lehmann L, The mycotoxin patulin reacts with DNA bases with and without previous conjugation to GSH: implication for related α, β-unsaturated carbonyl compounds? Arch. Toxicol 2016, 90, (2), 433–448. [DOI] [PubMed] [Google Scholar]
- 46.Corteselli EM; Aitken MD; Singleton DR, Rugosibacter aromaticivorans gen. nov., sp. nov., a novel bacterium within the family Rhodocyclaceae isolated from contaminated soil, capable of degrading aromatic compounds. Int. J. Syst. Evol. Microbiol 2016, 67, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corteselli EM; Aitken MD; Singleton DR, Description of Immundisolibacter cernigliae gen. nov., sp. nov., a high-molecular-weight polycyclic aromatic hydrocarbon-degrading bacterium within the class Gammaproteobacteria, and proposal of Immundisolibacterales ord. nov. and Immundisolibacteraceae fam. nov. Int. J. Syst. Evol. Microbiol 2016, 67, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kweon O; Kim S-J; Holland RD; Chen H; Kim D-W; Gao Y; Yu L-R; Baek S; Baek D-H; Ahn H; Cerniglia CE, Polycyclic aromatic hydrocarbon metabolic network in Mycobacterium vanbaalenii PYR-1. J. Bacteriol 2011, 193, (17), 4326–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones MD; Crandell DW; Singleton DR; Aitken MD, Stable-isotope probing of the polycyclic aromatic hydrocarbon‐degrading bacterial guild in a contaminated soil. Environ. Microbiol. 2011, 13, (10), 2623–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones MD; Singleton DR; Carstensen DP; Powell SN; Swanson JS; Pfaender FK; Aitken MD, Effect of incubation conditions on the enrichment of pyrene-degrading bacteria identified by stable-isotope probing in an aged, PAH-contaminated soil. Microb. Ecol 2008, 56, (2), 341–349. [DOI] [PubMed] [Google Scholar]
- 51.Singleton DR; Sangaiah R; Gold A; Ball LM; Aitken MD, Identification and quantification of uncultivated Proteobacteria associated with pyrene degradation in a bioreactor treating PAH‐contaminated soil. Environ. Microbiol. 2006, 8, (10), 1736–1745. [DOI] [PubMed] [Google Scholar]
- 52.Singleton DR; Dickey AN; Scholl EH; Wright FA; Aitken MD, Complete genome sequence of a bacterium representing a deep uncultivated lineage within the Gammaproteobacteria associated with the degradation of polycyclic aromatic hydrocarbons. Genome announcements 2016, 4, (5), e01086–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singleton DR; Richardson SD; Aitken MD, Pyrosequence analysis of bacterial communities in aerobic bioreactors treating polycyclic aromatic hydrocarbon-contaminated soil. Biodegradation 2011, 22, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davies J; Evans W, Oxidative metabolism of naphthalene by soil pseudomonads. The ring-fission mechanism. Biochem. J 1964, 91, (2), 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans WC; Fernley HN; Griffiths E, Oxidative metabolism of phenanthrene and anthracene by soil pseudomonads. Biochem. J 1965, 95, 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Herwijnen R; Springael D; Slot P; Govers HAJ; Parsons JR, Degradation of anthracene by Mycobacterium sp. strain LB501T proceeds via a novel pathway, through o-phthalic acid. Appl. Environ. Microbiol 2003, 69, (1), 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moody JD; Freeman JP; Doerge DR; Cerniglia CE, Degradation of phenanthrene and anthracene by cell suspensions of Mycobacterium sp. strain PYR-1. Appl. Environ. Microbiol 2001, 67, (4), 1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heitkamp MA; Freeman JP; Miller DW; Cerniglia CE, Pyrene degradation by a Mycobacterium sp.: identification of ring oxidation and ring fission products. Appl. Environ. Microbiol 1988, 54, (10), 2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kweon O; Kim S-J; Kim D-W; Kim JM; Kim H.-l.; Ahn Y; Sutherland JB; Cerniglia CE, Pleiotropic and epistatic behavior of a ring-hydroxylating oxygenase system in the polycyclic aromatic hydrocarbon metabolic network from Mycobacterium vanbaalenii PYR-1. J. Bacteriol 2014, 196, (19), 3503–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singleton DR; Hu J; Aitken MD, Heterologous expression of polycyclic aromatic hydrocarbon ring-hydroxylating dioxygenase genes from a novel pyrene-degrading betaproteobacterium. Appl. Environ. Microbiol 2012, 78, (10), 3552–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haeseler F; Blanchet D; Druelle V; Werner P; Vandecasteele J-P, Analytical characterization of contaminated soils from former manufactured gas plants. Environ. Sci. Technol 1999, 33, (6), 825–830. [Google Scholar]
- 62.Kim Y-H; Freeman JP; Moody JD; Engesser K-H; Cerniglia CE, Effects of pH on the degradation of phenanthrene and pyrene by Mycobacterium vanbaalenii PYR-1. Appl. Microbiol. Biotechnol 2005, 67, (2), 275–285. [DOI] [PubMed] [Google Scholar]
- 63.Fenner K; Canonica S; Wackett LP; Elsner M, Evaluating pesticide degradation in the environment: blind spots and emerging opportunities. Science 2013, 341, (6147), 752–758. [DOI] [PubMed] [Google Scholar]
- 64.Mohler RE; O’Reilly KT; Zemo DA; Tiwary AK; Magaw RI; Synowiec KA, Non-Targeted Analysis of Petroleum Metabolites in Groundwater Using GC× GC–TOFMS. Environ. Sci. Technol 2013, 47, (18), 10471–10476. [DOI] [PubMed] [Google Scholar]
- 65.Qu S; Kolodziej EP; Long SA; Gloer JB; Patterson EV; Baltrusaitis J; Jones GD; Benchetler PV; Cole EA; Kimbrough KC, Product-to-parent reversion of trenbolone: unrecognized risks for endocrine disruption. Science 2013, 342, (6156), 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cwiertny DM; Snyder SA; Schlenk D; Kolodziej EP, Environmental designer drugs: when transformation may not eliminate risk. Environ. Sci. Technol 2014, 48, (20), 11737–11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Reilly KT; Mohler RE; Zemo DA; Ahn S; Tiwary AK; Magaw RI; Espino Devine C; Synowiec KA, Identification of ester metabolites from petroleum hydrocarbon biodegradation in groundwater using GC× GC‐TOFMS. Environ. Toxicol. Chem 2015, 34, (9), 1959–1961. [DOI] [PubMed] [Google Scholar]
- 68.Krauss M; Singer H; Hollender J, LC–high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal. Bioanal. Chem 2010, 397, (3), 943–951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.