

Abstract
We combined de novo mutation (DNM) data from 10,927 cases of developmental delay and autism to identify 253 candidate neurodevelopmental disease genes with an excess of missense and/or likely gene-disruptive mutations. Of these genes, 124 reach exome-wide significance (p < 5 × 10−7) for DNM. Intersecting these results with copy number variation morbidity data shows an enrichment for genomic disorder regions (30/253, LR+ 1.85, p = 0.0017). We identify genes with an excess of missense DNMs overlapping deletion syndromes (e.g., KIF1A and the 2q37 deletion) as well as duplication syndromes, such as recurrent MAPK3 missense mutations within the chromosome 16p11.2 duplication, recurrent CHD4 missense DNMs in the 12p13 duplication region, and recurrent WDFY4 missense DNMs in the 10q11.23 duplication region. Network analyses of genes showing an excess of DNMs highlights functional networks, including cell-specific enrichments in the D1+ and D2+ spiny neurons of the striatum.
Keywords: Neurodevelopmental Disorders, Autism, de novo Mutation, Genetics of Developmental Delay, KCNQ2, PACS1, PACS2, MAP2K1, KCNH1, GRIN2B, PPP2R5D, CTCF, BCL11A, SETBP1, ARID1B, DNMT3A, ZMYND11, SMARCD1, MECP2, KAT6B, WDR26, PTPN11, CDK13, TBL1XR1, TCF4, SNX5, HNRNPU, TNPO2, STXBP1, ITPR1, DYRK1A, CHD2, SOX5, KCNQ3, TRIP12, TAF1, KAT6A, CSNK2A1, PUF60, CUL3, FOXP1, GNAO1, SMARCA2, MYT1L, GNAI1, ADAP1, FOXG1, SMC1A, PPP2R1A, GABRB2, EEF1A2, NAA10, HDAC8, ADNP, KMT2A, PIK3CA, PTEN, IQSEC2, MEF2C, SMARCA4, COL4A3BP, CAPN15, PURA, CHD3, WDR45, DLX3, TMEM178A, MED13L, ZBTB18, TLK2, DYNC1H1, POGZ, SLC6A1, EP300, SYNGAP1, DDX3X, SATB2, AGO4, SCN2A, BRAF, EHMT1, NSD1, NFIX, CREBBP, SRCAP, TRIO, PPP1CB, USP9X, CASK, SCN8A, CHD8, EFTUD2, SNAPC5, KDM5B, HECW2, KIF1A, SETD5, RAC1, SUV420H1, ANP32A, KANSL1, MEIS2, NONO, HIVEP3, ANKRD11, ASXL3, CTNNB1, AHDC1, GATAD2B, TCF20, SHANK3, ENO3, ASH1L, CHAMP1, PPM1D, KIAA2022, LEO1, CAPRIN1, CNKSR2, MSL3, MBD5, NAA15, SYNCRIP, QRICH1, DLG4, UPF3B, SET, WAC
INTRODUCTION
The importance of de novo mutations (DNMs) underlying neurodevelopmental disorders (NDDs) has been recognized for many years. Some of the strongest genome-wide evidence came from early copy number variation (CNV) studies, which consistently showed an excess of de novo as well as large private CNVs in patients with autism, developmental delay (DD) and epilepsy1-4. Significance based on CNV recurrence was more readily achieved from smaller sample sizes because of elevated mutation rates in regions flanked by segmental duplications5 or hotspots of recurrent rearrangement near telomeres6. In many cases the individual genes underlying the genomic disorders remain unknown.
The advent of next-generation sequencing and exome sequencing rapidly accelerated our ability to specify genes associated with potentially pathogenic de novo single-nucleotide variants (SNVs) for both DD and autism7,8, although recurrent mutations occurred more rarely9,10. Different statistical models for discovery of genes based on recurrent SNV mutation have been developed, including those based on chimpanzee-human divergence11, trinucleotide mutation context12, and clustering of DNMs13-15. Despite extensive CNV analyses of nearly 45,000 patients with autism and DD16-18, few attempts have been made18,19 to integrate the wealth of CNV data with recent exome sequencing results despite a common mutational model of dosage imbalance.
In this study, we perform an integrated meta-analysis combining DNM exome sequence data from individuals with autism spectrum disorder (ASD), intellectual disability (ID) and/or DD20 with CNV morbidity data. Because of the significant comorbidity between ID and ASD21,22 and the fact that autism cases with a severe DNM are enriched in DD23, we overlay these data with known genomic disorders. The goals of this study are threefold: 1) provide an integrated list of candidate NDD genes based on multiple lines of DNM and CNV evidence, 2) compare different models of recurrent mutation, and 3) identify the most likely genes underlying pathogenic microdeletion and microduplication CNVs associated with DD.
RESULTS
Genes enriched for de novo SNV mutation and model comparisons:
We compile de novo variation identified from exome sequencing of 10,927 cases with NDDs from the denovo-db v.1.5 database release20. This includes 5,624 cases with a primary diagnosis of ASD and 5,303 cases with a diagnosis of ID/DD collected from 17 studies11,23-38 (Supplementary Table 1). We consider all protein-altering and likely gene-disruptive (LGD) mutations, including frameshifts, splice donor/acceptor mutations, start losses and stop gains. The combined set of 12,172 DNMs includes 2,357 LGD and 9,815 missense mutations.
We initially applied two statistical models. The first incorporates locus-specific transition/transversion/indel rates and chimpanzee-human coding sequence divergence11 to estimate the number of expected DNMs, hereafter referred to as the chimpanzee-human divergence model or the CH model. The second model, denovolyzeR12, estimates mutation rates based using trinucleotide context and incorporates exome depth and divergence adjustments based on macaque-human comparisons over a ±1 Mbp window and accommodates known mutational biases, such as CpG hotspots. Both models apply their underlying mutation rate estimates to generate prior probabilities for observing a specific number and class of mutations for a given gene. While both models incorporate LGD and missense probabilities, we recently modified the CH model to incorporate CADD scores39,40 allowing us to also specifically test for enrichment for the missense subset of the predicted most severe 0.1% of mutations (i.e., CADD scores over 30 or MIS30). Such missense mutations are more likely to be functionally equivalent to an LGD mutation and have been shown to be significantly enriched in NDD cases compared to controls14. To account for the sensitivity biases, we applied an upper bound baseline mutation rate assumption of 1.8 DNMs (derived from high-coverage genome sequencing data) to the CH model, which exceeds the overall DNM rate of this cohort.
Combined, the two models (union set) identify 253 candidate NDD genes with evidence of excess DNM at a false discovery rate (FDR) <5% and at least two mutations for at least one mutational category (Table 1, Table 2). This includes 145 genes with excess LGD mutations and 123 genes with excess missense mutations. Among these, 29 genes demonstrate evidence of both LGD and missense mutations (Figure 1, Table 1, and Supplementary Table 2). In general, both models highlight similar genes (Figure 1, Supplementary Note), particularly for LGD events where 73.1% (106/145) of genes are shared. This stands in contrast to recurrent missense DNMs where only 51.2% (63/123) of the genes overlap between the models suggesting that additional model refinement is required to more accurately predict pathogenic missense mutations. A more stringent application of the exome-wide Bonferroni family-wise error rate (FWER) identifies a union of 124 genes (Supplementary Table 2, Supplementary Note).
Table 1:
Recurrent DNM gene summary and model comparison.
| ID/DD Only | ASD Only | |||||
|---|---|---|---|---|---|---|
| Variant Category | CH Model Count | denovolyzeR Count | Union Count (FDR (FWER)) | Intersection Count (FDR (FWER)) | Union Count (FDR (FWER)) | Union Count (FDR (FWER)) |
| LGD & MIS30 & Missense | 14 | NA | 14 (5) | NA | 4 (1) | 0 (0) |
| LGD & MIS30 | 1 | NA | 1 (3) | NA | 0 (0) | 0 (0) |
| LGD & Missense | 13 | 22 | 15 (6) | 21 (8) | 19 (10) | 3 (1) |
| LGD | 92 | 108 | 115 (62) | 85 (49) | 95 (55) | 31 (14) |
| MIS30 & Missense | 28 | NA | 31 (18) | NA | 9 (7) | 0 (0) |
| MIS30 | 16 | NA | 14 (10) | NA | 0 (0) | 0 (0) |
| Missense | 46 | 63 | 63 (20) | 42 (22) | 56 (25) | 7 (3) |
| total | 210 | 193 | 253 (124) | 148 (79) | 183 (98) | 41 (18) |
The number of genes reaching statistical significance for DNM enrichment in n = 10,927 independent samples by the CH model and denovolyzeR as well as the unions and intersections of these gene sets are shown for each mutation category (LGD, Missense, MIS30). Also shown are the union counts for the ASD (n = 5,624 independent samples) and ID/DD (n = 5,303) only analyses. Counts represent genes passing an FDR q-value threshold of 0.05. Bracketed numbers represent genes passing a Bonferroni FWER correction (p < 5e-7).
Table 2:
Genes enriched for de novo variation in 10,927 ASD/ID/DD patients in denovo-db v.1.5.
| Significance Category | Genes |
|---|---|
| LGD | ADNP*†, AHDC1*†, ANK2†, ANKRD11*†, ANP32A*, ARID1B*†, ARID2†, ASH1L*†, ASXL1†, ASXL3*†, AUTS2†, BCL11A*†, BRPF1†, CAPRIN1*†, CASZ1†, CDC42BPB†, CDKL5†, CHAMP1*†, CHD7†, CHD8*†, CLTC†, CNKSR2*†, CNOT3†, CTNNB1*†, CUL3*†, DLG4*†, DSCAM†, DVL3†, EBF3†, EHMT1*†, ENO3*†, EP300*†, FAM200A†, FAM200B†, FOSL2, FOXP2†, GATAD2B*†, HIST1H1E†, HIVEP2†, HIVEP3*†, HNRNPD†, IRF2BPL†, KANSL1*†, KAT6A*†, KAT6B*†, KCNS3†, KDM5B*†, KDM6A†, KDM6B†, KIAA2022*†, KIF11†, KMT2A*†, KMT2C†, LARP4B†, LEO1*†, MBD5*†, MEIS2*†, MSL3*†, NAA15*†, NFE2L3†, NONO*†, NSD1*†, ODC1†, PDHA1†, PHF12†, PHF21A†, PHF3†, PHIP†, POU3F3†, PPM1D*†, PRR12†, PTCHD1†, QRICH1*†, RAI1†, RPL26†, SET*†, SETBP1*†, SETD2†, SETD5*†, SHANK3*†, SIN3A†, SKIDA1†, SMC1A*†, SON†, SOX5*†, SPAST†, SPEN†, SPRY2†, SRCAP*†, SRRM2, SRSF11†, STARD9†, SUV420H1*†, SYNCRIP*†, SYNGAP1*†, TAB2†, TBR1†, TCF12†, TCF20*†, TNRC6B, TRA2B†, TRIP12*†, UPF3B*†, USP9X*†, VEZF1, WAC*†, WDFY3†, WDR45*†, WDR87†, WHSC1†, YTHDF3†, ZBTB18*†, ZBTB7A†, ZC4H2†, ZNF292† |
| LGD & Missense | CHD2*†, CREBBP*†, DYRK1A*†, FBXO11†, FOXG1*†, FOXP1*†, HNRNPU*†, MEF2C*†, MYT1L*†, NFIX*†, POGZ*†, PTEN*†, PURA*†, TLK2*†, WDR26*† |
| LGD & MIS30 | TCF7L2† |
| LGD & Missense & MIS30 | CASK*†, DDX3X*†, HDAC8*†, IQSEC2*†, MECP2*†, MED13L*†, PPP2R5D*†, PUF60*†, SATB2*†, SCN2A*†, SLC6A1*†, STXBP1*†, TBL1XR1*†, TCF4*† |
| Missense | ABI2†, ACHE†, ADAP1*†, AGAP2†, AGO1†, AGO4*†, AQP10†, BRAF*†, BTF3†, C2orf42†, CABP7†, CAPN15*†, CBL†, CHD4†, CLASP1†, DEAF1†, DLX3*†, DNM1†, EGLN2†, GABRB2*†, GABRB3†, GLRA2†, GNAI1*†, HMGXB3†, HUWE1†, ITPR1*†, KCNC1†, KCNJ6†, MAPK3†, MTF2†, MYO1E†, PBX1†, PLAC8L1†, PLK5†, PPP1CB*†, PRKCA†, PRKD1†, PRPF18†, PSMG4†, PTPN11*†, RAC1*†, RFX8†, RRP8†, RYR2†, SETD1B†, SF3B1†, SHISA6†, SMAD4†, SMARCD1*†, SMC3†, SNAPC5*†, SNX5*†, SUSD4†, SYT1†, TAOK1†, TMEM178A*†, TMEM42†, TNPO3†, TRAF7†, TRRAP†, UNC80†, VAMP2†, WDFY4†, YWHAG† |
| MIS30 | ACTC1†, AGO3†, CACNA1E†, FAM104A†, HIST1H2AC†, KIF5C†, PACS2*†, PAPOLG†, PDK2†, SEPT10†, STC1†, TAF1*†, TNPO2*†, U2AF2† |
| Missense & MIS30 | CDK13*†, CHD3*†, COL4A3BP*†, CSNK2A1*†, CTCF*†, DNMT3A*†, DYNC1H1*†, EEF1A2*†, EFTUD2*†, GNAO1*†, GRIN2B*†, HECW2*†, KCND3†, KCNH1*†, KCNQ2*†, KCNQ3*†, KIF1A*†, MAP2K1*†, NAA10*†, NR2F1†, NR4A2†, PACS1*†, PIK3CA*†, PPP2R1A*†, RAB11A†, SCN8A*†, SMARCA2*†, SMARCA4*†, TRIO*†, ZMYND11*† |
Listing of genes reaching significance for excess of DNM in n = 10,927 independent samples at an FDR of 5% by either the denovolyzeR or CH model (union) for each mutational category (LGD, Missense, MIS30).
Gene also in the intersection set.
Gene in FWER exome-wide significance (p<5e-7) set.
Figure 1: de novo enriched genes and their characteristics.
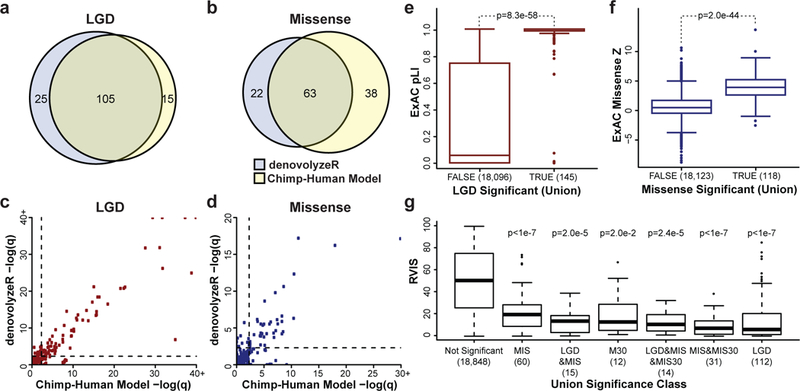
Shown are the results of applying both the chimpanzee-human (CH) divergence model and denovolyzeR to de novo variation in n = 10,927 independent individuals with ASD/ID/DD. The two models show considerable gene overlap (A,B) with correlated significance values (LGD Pearson r2 = 0.94, missense r2 = 0.74) (C,D). CH model LGD outliers include NONO, MEIS2, LEO1, WDR26, and CAPRIN1, and denovolyzeR LGD outliers include ZBTB18 and FAM200B (C). CH model missense (MIS) outliers include CAPN15, SNAPC5, DLX3, TMEM178A, ADAP1, SNX5, SMARCD1, WDR26, and AGO4, and denovolyzeR missense outliers include ITPR1, RAC1, SETD1B, WDFY4, and UNC80 (D). Recurrent mutated LGD genes (TRUE, n = 145 with pLI scores) are highly enriched for genes intolerant to mutation as defined by ExAC pLI score (two-tailed Wilcoxon rank-sum test) (E). Genes significantly enriched for missense DNMs (n = 118 with missense Z scores) are outliers by the ExAC missense depletion Z scores (two-tailed Wilcoxon rank-sum test) (F). Similarly, all subcategories of significant genes (n below each category name) are intolerant to mutation (RVIS percentile) when compared to non-significant genes (Tukey HSD test, p-values are corrected for all possible group comparisons) (G). Boxplots represent Quartiles 1 to 3 with the median indicated. Whiskers span from Q1 - 1.5 IQR to Q3 + 1.5 IQR.
We identified additional evidence of disease association, from a comprehensive database and PubMed literature search, for 204/253 genes (Methods) indicating that 49/253 union genes (10/124 FWER union genes) presented here are novel associations (Supplementary Table 2, Supplementary Table 3). Of these novel genes, 61% (30/49) demonstrate DNM in both ASD and ID/DD patients. We wish to note that neither FDR nor FWER, however, are metrics of pathogenicity but rather simply thresholds of significance to identify genes for further investigation.
Since it has been well established that NDD genes are less tolerant to mutation, we categorize the 253 genes into different functional groups and compare their tolerance to mutation in the general population using three metrics: residual variance to intolerance score (RVIS); probability of loss-of-function intolerance (pLI); and missense constraint scores (missense Z scores). For both pLI and missense Z scores, we utilized the ExAC subset with known neuropsychiatric cohorts removed (45,376 individuals)41. For genes with an enrichment of LGD variants, we observe a significant increase in pLI scores compared to all other genes (p = 8.3 × 10−58 two-tailed Wilcoxon rank-sum test, ROC AUC = 0.90) (Figure 1E). We also observe a significant increase in missense constraint (missense Z scores) among genes with enrichment for missense variation (p = 1.9 × 10−44 two-tailed Wilcoxon rank-sum test, ROC AUC = 0.87) (Figure 1F). Similarly, we observe a significant RVIS depletion for all categories where at least two genes were identified (Figure 1G).
Examination of a combination of constraint metrics is particularly valuable as a small number of genes demonstrate conflicting results, such as the LGD- and missense-enriched gene MEF2C, which is involved in severe ID when disrupted by deletions or mutations42 and demonstrates constraint by RVIS, but not by pLI (RVIS = 18.97, pLI = 2.4 × 10−3, missense Z score = 4.47). Interestingly, among genes without pLI or RVIS support, we identified established ASD/ID genes in addition to the expected potential false positives. Among the union genes 82.6% (209/253) have no detected LGD or missense DNM in controls (n = 2,278 controls; Supplementary Note). The detection of control events may represent incomplete penetrance, variable expressivity, undiagnosed/subclinical controls, or benign variation (primarily in the case of missense variation). None of the recurrent control DNM genes reach exome-wide FDR significance. While some of these are plausible candidates, disease significance should be considered with caution until additional functional and clinical data establish their role.
ASD versus ID/DD genes:
We investigated the distribution of LGD and missense mutations between the ASD and ID/DD cohorts. The majority of this NDD gene set (68.4% (173/253) of the union and 72.3% (107/148) of the intersection) show evidence of DNM in both ASD and ID/DD cohorts, highlighting the utility of joint analyses. Although a small number of genes are specific or enriched for a diagnosis of ID/DD (q < 0.01, one-tailed Fisher’s exact test), none are yet statistically enriched for ASD (Supplementary Figure 1). A few genes (WDFY3, DSCAM and CHD8) trend toward ASD diagnosis. To eliminate potential ascertainment bias in discovery, we repeated the overlap analysis considering gene discovery independently in each cohort (Supplementary Note). Considering all 253 genes and the full set of NDD patients, we calculate that 17.7% (1,932/10,927) of the samples have at least one de novo event in this gene set.
The proportion of patients with a DNM is significantly higher for ID/DD (26.8% or 1,421/5,303 patients) when compared to ASD (9.1% or 511/5,624 patients) (OR = 3.66, p = 1.62 × 10−133, two-tailed Fisher’s exact test). While this may be partially biased by the differences in DNM sensitivity between exome studies, this observation exceeds the baseline 1.58-fold excess of LGD DNM in ID/DD cohorts and thus reflects differences in heterogeneity between the disorders. Further supporting this bias of LGD events to ID/DD, Simons Simplex Collection (SSC) autism probands who carried an LGD DNM in one of the LGD genes were less likely to be high-functioning. Instead, there was an overrepresentation of ID (≤70 IQ, expected 753/2,445, observed 42/95) and low-to-normal IQ ranges (70 < IQ < 100, expected 1,018/2,445, observed 45/95) compared to the high IQ probands (≥100 IQ, expected 674/2,445, observed 8/95) (p = 4.0 × 10−5, two-tailed likelihood ratio test) (Supplementary Table 4). Among the autism mutation carriers, we observe a nominal enrichment for increased severity of repetitive behavior (RBS-R [t(18) = 3.12, corrected p = 0.048], two-tailed independent samples t-test) but were overall underpowered to further disentangle associated phenotypic features.
Network enrichment and patterns of brain expression:
Examination of the 253 genes identified by the union of both statistical approaches suggests that our set is strongly enriched for functionally related networks of genes. The STRING database, for example, identifies a highly significant 1.8-fold (1,067 edges vs. 573 expected) enrichment in interactions among the 253 union genes (p < 1.0 × 10−16, one-tailed hypergeometric test). Given this high level of interconnectivity, we applied MAGI43, a gene network discovery tool, to identify potential gene clusters, functional enrichments, and additional candidate interactions and highlight the top four protein-protein interaction (PPI) and co-expression networks (each at p < 0.01, one-tailed permutation test) and their associated PANTHER functional enrichments (Figure 2A-D, Supplementary Tables 5-7).
Figure 2: Gene expression and protein-interaction networks.
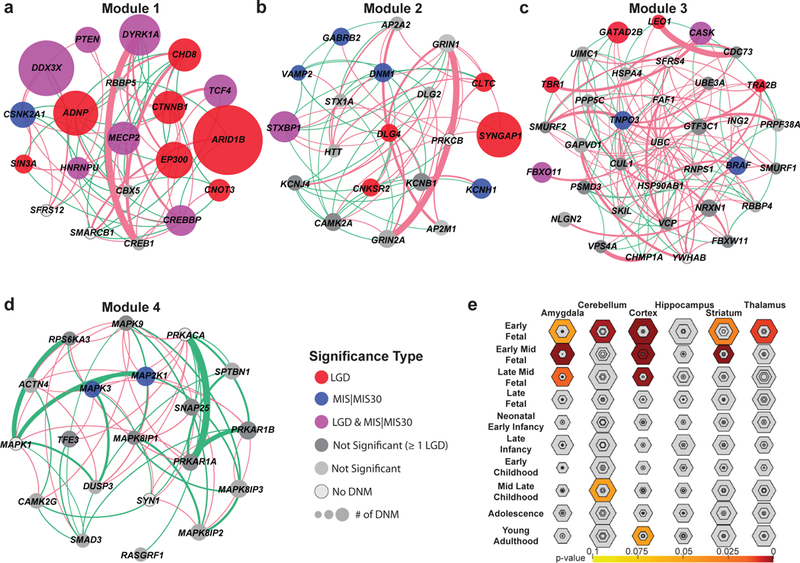
(A-D) MAGI43 analysis of the union set (n = 253 independent genes) highlights the top four modules of co-expression and protein-protein interaction (PPI), including genes significant for DNM enrichment by denovolyzeR (FDR-adjusted Poisson test) or the CH model (FDR-adjusted binomial test) (colored circles) and new candidate genes with DNM that do not yet reach significance (dark gray). The size of the circle represents the relative number of patients with DNMs within this cohort. Edges depict PPIs (pink arcs) and co-expression (green arcs) scaled by their scores from geneMANIA100. (E) Tissue-specific enrichment analyses (TSEA) of the union set (n = 253 independent genes) highlight a strong bias to various developing parts of the brain with the strongest signal early to mid-fetal development (color corresponds to FDR-adjusted one-tailed Fisher’s exact test p-values, shaded regions closer to the center of each hexagon indicate increasing tissue specificity).
Module 1 (20 genes) delineates “regulation of transcription from RNA polymerase II promoter” (p = 0.0269, one-tailed Bonferroni adjusted binomial test) (Figure 2A, Supplementary Tables 5-7) and contains 15 significant genes in addition to three candidates that do not yet reach significance (CREB1, RBBP5, CBX5) and two genes (SREK1, SMARCB1) with no DNM in our current data set. Module 2 highlights multiple functions relating to neurotransmitter signaling (p = 0.0358; one-tailed Bonferroni adjusted binomial test) and synaptic signaling (p = 8.91 × 10−5, one-tailed Bonferroni adjusted binomial test) (Figure 2B, Supplementary Tables 5-7) and contains nine significant genes in addition to ten genes that do not reach significance (DLG2, HTT, AP2A2, AP2M1, KCNJ4, KCNB1, STX1A, GRIN2A, GRIN1, CAMK2A) and one gene with no DNM (PRKCB). Module 3 highlights the “transmembrane receptor protein serine/threonine kinase signaling pathway” (p = 0.002; one-tailed Bonferroni adjusted binomial test) (Figure 2C, Supplementary Tables 5-7) and contains eight significant genes in addition to 21 genes that do not reach significance (SMURF2, SMURF1, CDC73, RNPS1, RBBP4, UBE3A, CUL1, CHMP1A, FBXW11, VCP, VPS4A, PPP5C, PRPF38A, SKIL, HSPA4, PSMD3, UIMC1, GAPVD1, NLGN2, GTF3C1, NRXN1) and six genes with no DNM (ING2, SRSF4, FAF1, UBC, HSP90AB1, YWHAB). Finally, Module 4 highlights c-Jun N-terminal kinase (JNK) (p = 4.65 × 10−5, one-tailed Bonferroni adjusted binomial test) and mitogen-activated protein kinase (MAPK) (p = 3.13 × 10−6, one-tailed Bonferroni adjusted binomial test) cascades (Figure 2D, Supplementary Tables 5-7) and contains two significant genes in addition to 15 genes that do not reach significance (RPS6KA3, RASGRF1, MAPK8IP1, SMAD3, DUSP3, MAPK9, SPTBN1, ACTN4, CAMK2G, TFE3, PRKAR1A, SNAP25, MAPK8IP2, MAPK8IP3, PRKAR1B) and three genes with no DNM (SYN1, MAPK1, PRKACA). Among these nonsignificant genes are many previously identified NDD candidate genes (e.g., NRXN1, GRIN2A, CAMK2A)44-46 suggesting this group as a potential target for future screening and disease gene discovery.
In addition to enrichment in the PPI context, we also note functional cell-specific and tissue-specific enrichment analyses (CSEA and TSEA)47,48 obtained primarily from mouse expression data sets. As expected, the 253 gene set is enriched for the brain expression with a bias toward early to mid-fetal gene expression in the cortex, striatum and amygdala (Figure 2E). Among these, the greatest specificity is observed for early to early mid-fetal cortical development. By CSEA, the de novo gene set shows enrichment in both classes of medium spiny neurons within the striatum (striatum D1+ and D2+ medium spiny neurons Benjamini-Hochberg (BH)-corrected p = 0.013 and p = 0.011, one-tailed Fisher’s exact test) at a pSI (specificity index p-value) threshold of 0.05. Additionally, we observe nominal significance for D1+ and D2+ spiny neurons (uncorrected p = 0.027 and p = 0.023, one-tailed Fisher’s exact test) at a pSI of 0.01 (Supplementary Figure 2A), and through a further analysis of available single-cell sequencing data, two pyramidal neuron subtypes (S1PyrL5, BH p = 0.008; one-tailed Fisher’s exact test) and the hippocampus (CA1Pyr1, BH p = 0.046; one-tailed Fisher’s exact test) (Supplementary Figure 2B, Supplementary Note).
As a final analysis, we also assess the expression patterns of the union gene set using human RNA-seq data sets. The gene set, irrespective of class of mutation, is significantly enriched for a pan-neuronal pattern of expression when compared to control sets selected based on synonymous DNM in cases and genes with recurrent DNM in controls samples (Figure 3, Supplementary Figure 3). This analysis does not highlight specific cell types when compared to control genes (p = 0.52, two-tailed corrected Wilcoxon rank-sum test) but does show slightly higher expression across neuronal cell types (p = 0.0001; two-tailed corrected Wilcoxon rank-sum test) even after controlling for gene length (Supplementary Figure 3). The 253 union gene set shows a strikingly broad expression profile across adult human cortical neuron types, including GABAergic (inhibitory) and glutamatergic (excitatory) neurons, compared to control genes (Figure 3A, Supplementary Figure 3, Supplementary Note).
Figure 3: Expression in human cortical neurons.
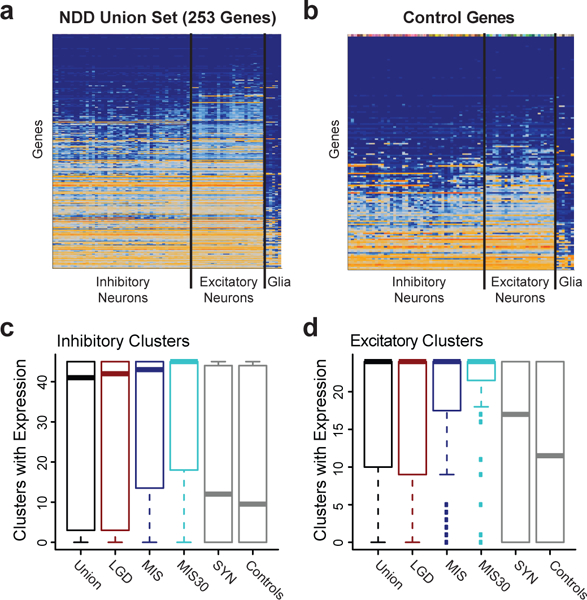
(A-B) Heatmaps demonstrating a broad pattern of inhibitory and excitatory neuronal expression (median log2 (CPM+1)) in the union gene set (n = 253 independent genes) compared to control genes (n = 156 independent genes). Expression level is indicated by a color gradient from low expression (dark blue) to high (orange). Rows represent individual genes and are ordered by the number of clusters (transcriptomic defined cell types) with expression (median CPM > 1), and columns represent 41 inhibitory neuronal, 24 excitatory neuronal, and 6 glial transcriptional clusters, each representing a distinct cell type. (C-D) The number of inhibitory and excitatory clusters with expression in NDD genes (Union n = 253, LGD n = 145, MIS n = 123, MIS30 n = 59) compared to controls (synonymous (SYN) n = 101, Control n = 156 independent genes). The signal is strongest for NDD genes with the most severe missense mutations (MIS30). Boxplots represent Quartiles 1 to 3 with the median indicated. Whiskers span from Q1 - 1.5 IQR to Q3 + 1.5 IQR.
Projected rates of gene discovery:
Based on the number of genes that reach significance for DNM in our cohort of 10,927 cases, we estimate the potential yield by mutational class and the CH model. To this end, we subsampled smaller populations from our set 10,000 times each and tested for how many genes would reach significance using the CH model in a resampled cohort. We assess logistic growth models for each mutation class and select the best fitting model by Bayesian information criteria (BIC) to predict future performance. For genes with excess LGD DNMs, we observe what appears to be a rapid upcoming plateau in gene discovery with an asymptote at 216 genes (95% CI 208-225) (ΔBIC linear model - asymptotic regression model = 259) (Figure 4). Similarly, for genes with an excess of MIS30 DNMs, the model predicts an asymptote of only 65 genes (95% CI 63-67) (ΔBIC linear model - Weibull model = 250) (Figure 4). By contrast, genes with an excess of recurrent missense mutations cannot yet be projected (Supplementary Note).
Figure 4: Estimation of gene discovery rates in future cohorts.
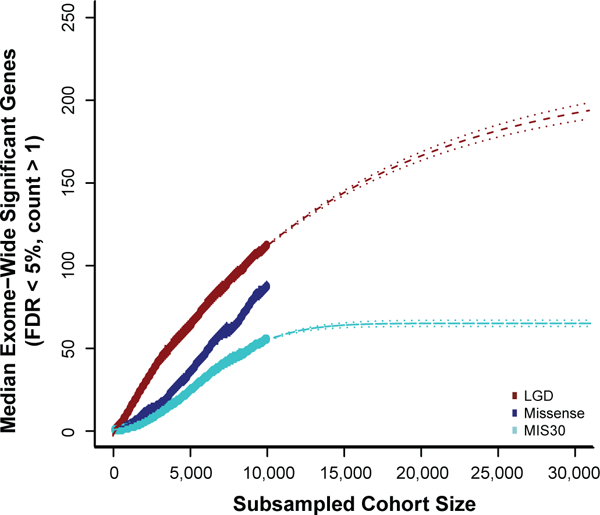
We estimate the number of genes reaching significance under the CH model at varying population sizes subsampled from the total cohort of 10,927 individuals. Both the number of significant genes with recurrent LGD and MIS30 DNMs appear to be saturating with limited new gene discovery as sample sizes grow. De novo missense variants (including MIS30), however, as a more general class demonstrate a more complex growth pattern with no best-fit line and, thus, likely represent the most important reservoir for new gene discovery as sequence data are generated from additional ASD and DD cohorts.
CNV intersection:
In order to identify potentially dosage-sensitive genes underlying pathogenic CNVs, we intersect the 253 candidate gene set with a list of 58 genomic disorders based on previous CNV morbidity maps and the DECIPHER database (Table 3, Supplementary Table 2, Figure 5). Considering all genes with a de novo variant (n = 6,886), we find that 30 of 253 significant genes intersect a genomic disorder region, compared to 426 of 6,633 non-significant genes intersecting a disorder. This represents a significant (p = 0.0017, two-tailed Fisher’s exact test) enrichment (LR+ 1.85 [95% CI 1.38 - 2.43]) compared to expectations supporting the notion that neurodevelopmental CNVs and DNMs converge on a common genetic etiology of gene-dosage imbalance. While we are underpowered to detect enrichment for any specific mutational and CNV class interactions by post-hoc testing, as expected LGD-significant genes and deletion disorders demonstrated the strongest enrichment among the four combinations (p = 0.1, two-tailed Fisher’s exact test, LR+ = 1.77 [95% CI 1.2 - 2.54]). Given the known complexity of gene regulation in CNVs49, we highlight candidates representing all interaction types (Table 3, Supplementary Table 2) with the expectation that the strongest candidates will correspond to a simple model of haploinsufficiency.
Table 3:
Intersection between pathogenic CNVs and recurrently mutated genes.
| denovo-db v.1.5
Counts |
Union CH Model and denovolyzeR | Genomic Disorders | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LGD de novo Variants | Missense de novo Variants | MIS30 de novo Variants | |||||||||||||
| Gene Symbol* | All (n=10,927) | ASD (n=5,624) | ID/DD (n=5,303) | All (n=10,927) | ASD (n=5,624) | ID/DD (n=5,303) | All (n=10,927) | ASD (n=5,624) | ID/DD (n=5,303) | Significance (FDR ≤ 5%, count > 1) | Deletion Syndrome | Duplication Syndrome | CNV Significance Type18 | Decipher† Classification | |
| Significant in Morbidity Map18 (37 Disorders) | MAPK3 | 0 | 0 | 0 | 3 | 2 | 1 | 1 | 1 | 0 | MIS | 16p11.2-deletion | 16p11.2-duplication | DEL AND DUP | |
| KANSL1 | 8 | 0 | 8 | 1 | 1 | 0 | 0 | 0 | 0 | LGD | 17q21.31-deletion | 17q21.31-duplication | DEL | Category 1 | |
| KIF1A | 0 | 0 | 0 | 11 | 1 | 10 | 9 | 0 | 9 | MIS | 2q37-deletion | None | DEL | Category 1 | |
| EHMT1 | 9 | 0 | 9 | 3 | 0 | 3 | 2 | 0 | 2 | LGD | 9q34-deletion | 9q34-duplication | DEL AND DUP | Category 1 | |
| SHANK3 | 10 | 6 | 4 | 1 | 1 | 0 | 0 | 0 | 0 | LGD | Phelan-McDermid-syndrome-deletion | None | DEL AND DUP | Category 1 | |
| PHF21A | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | LGD | Potocki-Shaffer-syndrome | None | DEL | Category 1 | |
| GABRB3 | 1 | 1 | 0 | 6 | 2 | 4 | 0 | 0 | 0 | MIS | Prader-Willi/Angelman | PWS-duplication | DEL AND DUP | Category 1 | |
| RAI1 | 3 | 1 | 2 | 3 | 2 | 1 | 0 | 0 | 0 | LGD | Smith-Magenis-syndrome-deletion | Potocki-Lupski-syndrome-duplication | DEL AND DUP | Category 1 | |
| NSD1 | 8 | 1 | 7 | 5 | 2 | 3 | 1 | 0 | 1 | LGD | Sotos-syndrome-deletion | None | DEL | Category 1 | |
| WHSC1 | 4 | 1 | 3 | 2 | 1 | 1 | 1 | 0 | 1 | LGD | Wolf-Hirschhorn-deletion | None | DEL AND DUP | Category 1 | |
| 21 Additional Genomic Disorders | SIN3A | 3 | 0 | 3 | 3 | 2 | 1 | 1 | 0 | 1 | LGD | 15q24 deletion (A to E Inclusive) | None | Other SD Pairs | |
| CLTC | 4 | 0 | 4 | 2 | 0 | 2 | 1 | 0 | 1 | LGD | 17q23 deletion | None | |||
| PPM1D | 8 | 1 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | LGD | 17q23.1q23.2 deletion | None | |||
| BCL11A | 6 | 2 | 4 | 3 | 0 | 3 | 0 | 0 | 0 | LGD | 2p15-16.1 microdeletion syndrome | None | |||
| PAPOLG | 0 | 0 | 0 | 3 | 3 | 0 | 3 | 3 | 0 | MIS30 | 2p15-16.1 microdeletion syndrome | None | |||
| POU3F3 | 2 | 0 | 2 | 2 | 0 | 2 | 2 | 0 | 2 | LGD | 2q11.2q13 deletion | None | |||
| RFX8 | 0 | 0 | 0 | 3 | 2 | 1 | 0 | 0 | 0 | MIS | 2q11.2q13 deletion | None | |||
| HECW2 | 1 | 0 | 1 | 9 | 2 | 7 | 7 | 2 | 5 | MIS30 & MIS | 2q33.1 | None | Category 1 | ||
| SATB2‡ | 9 | 0 | 9 | 6 | 0 | 6 | 5 | 0 | 5 | LGD & MIS30 & MIS | 2q33.1 | None | Category 1 | ||
| ABI2 | 0 | 0 | 0 | 3 | 2 | 1 | 1 | 1 | 0 | MIS | 2q33.1 | None | Category 1 | ||
| SF3B1 | 0 | 0 | 0 | 5 | 3 | 2 | 1 | 0 | 1 | MIS | 2q33.1 | None | Category 1 | ||
| CAPN15 | 0 | 0 | 0 | 3 | 0 | 3 | 1 | 0 | 1 | MIS | ATR-16 | None | Category 1 | ||
| SMC1A | 8 | 0 | 8 | 2 | 0 | 2 | 1 | 0 | 1 | LGD | None | Xp11.22-linked ID | |||
| HUWE1 | 0 | 0 | 0 | 9 | 0 | 9 | 3 | 0 | 3 | MIS | None | Xp11.22-linked ID | |||
| WDR45 | 8 | 0 | 8 | 2 | 1 | 1 | 0 | 0 | 0 | LGD | None | Xp11.22-p11.23 microduplication | |||
| MECP2 | 11 | 4 | 7 | 7 | 0 | 7 | 4 | 0 | 4 | LGD & MIS30 & MIS | None | Xq28 (MECP2) duplication | |||
| CREBBP | 3 | 0 | 3 | 13 | 3 | 10 | 1 | 1 | 0 | LGD & MIS | Rubinstein-Taybi syndrome | None | Category 1 | ||
| SUV420H1 | 7 | 4 | 3 | 3 | 3 | 0 | 1 | 1 | 0 | LGD | SHANK2 FGFs deletion | None | |||
| YWHAG | 0 | 0 | 0 | 3 | 1 | 2 | 1 | 0 | 1 | MIS | Wms-distal deletion | Wms-distal duplication | |||
| AUTS2 | 4 | 0 | 4 | 1 | 1 | 0 | 0 | 0 | 0 | LGD | Wms-prox deletion | Wms-prox duplication | |||
| 14 Significant Regions18 | SATB2‡ | 9 | 0 | 9 | 6 | 0 | 6 | 5 | 0 | 5 | LGD & MIS30 & MIS | 2q33.1 (SATB2) deletion | |||
| MEF2C | 4 | 0 | 4 | 5 | 1 | 4 | 0 | 0 | 0 | LGD & MIS | 5q14 (MEF2C) deletion | ||||
| CHD4 | 1 | 0 | 1 | 8 | 1 | 7 | 2 | 0 | 2 | MIS | 12p13 (SCNN1A to PIANP) | ||||
| WDFY4 | 0 | 0 | 0 | 5 | 4 | 1 | 1 | 0 | 1 | MIS | 1q11.23 duplication | ||||
DNM counts and significance categories from n = 10,927 independent samples are shown for genes from the union significance set (n = 253 genes denovolyzeR or CH model FDR < 5%) that intersect a previously established genomic disorder region. Gene symbols with underlines represent confirmation of known CNV associations. SD: segmental duplication
DECIPHER syndrome classification system; see URLs.
SATB2 is significant both by focal deletions and as part of the 2q33.1 region.
Figure 5: Integration of de novo SNVs and CNV morbidity map.
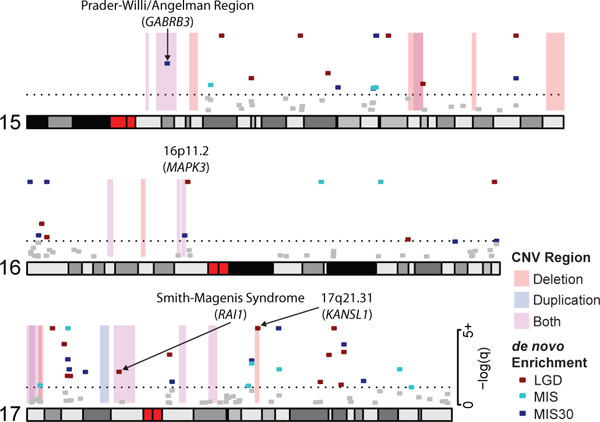
Shown are examples of pathogenic CNVs (blue, red and purple shading) associated with genomic disorders from chromosomes 15, 16, and 17, which intersect with genes that show a significant excess of DNM in n = 10,927 independent patients (red, turquoise and blue points representing the minimum q-value from either denovolyzeR or CH model, the dashed line represents a q-value of 0.05). The analysis confirms known associations, such as RAI1, and KANSL1 and candidate association for MAPK3. Recurrent severe missense mutations of GABRB3 have been associated with autism and may be relevant to the recurrent 15q11 duplication. We note that mutations and deletions of the imprinted genes SNRPN (no DNM in our data set) and UBE3A (1 LGD and 1 missense DNM in our data set) are known to cause the core phenotype of Prader-Willi and Angelman syndromes, respectively, but do not reach significance in this analysis.
Many genomic disorders intersect with a single DNM-enriched gene, confirming a known CNV gene association, including KANSL1 (Koolen-de Vries)50, SHANK3 (Phelan-McDermid)51, RAI1 (Smith-Magenis)52, NSD1 (Sotos)53, WHSC1 (Wolf-Hirschhorn)54, BCL11A (2p15-16.1 microdeletion)55, EHMT1 (9q34 deletions / Kleefstra syndrome)56, and CREBBP (Rubinstein-Taybi)57 (Table 3, Supplementary Table 2). In addition, this analysis also highlights genes that have been implicated as candidates by case reports, functional studies, or smaller CNVs (Table 3, Supplementary Table 2). Among these, we identify an excess of recurrent missense mutation in MAPK3 mapping to the 16p11.2 microdeletion/microduplication region associated with ASD and ID58. Recurrent LGD mutations in PHF21A, a gene previously implicated by translocations and focal CNVs59,60, map to the Potocki-Shaffer deletion region, while an excess of missense mutations in KIF1A correspond to the 2q37 deletion syndrome region61,62. Recurrent LGD DNMs in SIN3A, a REST and MECP2 interactor, map to the 15q24 deletion region63-65. PPM1D has been linked to ID66 and is located in the 17q23.1q23.2 deletion region. CLTC has been linked to multiple malformations and DD and is located in the 17q23 deletion region67. Genes enriched for recurrent missense DNM, YWHAG and GABRB3, co-localize to the Williams-Beuren distal and Prader-Willi deletion/duplication regions, respectively68-70.
Finally, we also consider as part of this analysis the 14 regions identified as significant for CNV burden18 and identify five candidate intersections (Table 3). These include SATB2 in the 2q33.1 region 18,71; MEF2C, which demonstrated focal deletions, functions at cortical synapses and has been independently linked to hyperkinesis and epilepsy72,73; CHD4 in the 12p13 duplication region, which has been linked by both a genome-wide association study (GWAS) and DNM to an ID syndrome74,75; and WDFY4 in the 10q11.23 duplication region, which appears to be a novel finding at this time.
A second category of CNVs are those that intersect more than a single gene (Table 3, Supplementary Table 2). The 2q33.1 region contains several potentially high-impact candidates, including HECW2, which has been linked to neurodevelopmental delay, ID and epilepsy by missense mutations27,76; SATB2, which has been independently identified by focal CNVs18,71; ABI2, which is a candidate for autosomal recessive ID77; and SF3B1, which interacts directly with the ID gene PQBP178. Similarly, in the 2p15-p16.1 deletion region we identify both the primary gene BCL11A55 as well as a second candidate gene in the minimal critical region, PAPOLG79, which is enriched for severe missense DNM. Finally, in the 2q11.2q13 deletion region we identify both POU3F3, which has been linked to ID and dysmorphic features by focal deletions80, and RFX8, which has limited functional information in the literature.
DISCUSSION
Exome sequencing of parent-child trios is a particularly powerful tool for the identification of genes, which when disrupted lead to pediatric NDD. The use of two DNM models, CH model and denovolyzeR, identifies a high-confidence intersection (n = 148 genes) and a comprehensive union set (253 genes), which reach significance by one or both models. An advantage of using both is that we identify high-risk candidate genes unique to each model (n = 75 additional genes), including several where DNMs have already been associated with neurodevelopmental disease. Examination of this gene set in the context of general population (i.e., ExAC) confirms that this set list is enriched for genes that are constrained in the general population (LGD pLI p = 8.3 × 10−58, missense Z score p = 1.9 × 10−44, two-tailed Wilcoxon rank-sum test, RVIS p < 1 × 10−7 to 2.0 × 10−2, Bonferroni adjusted Tukey HSD test) (Figure 1).
While intolerance metrics are useful to enrich for pathogenic genes, our analysis suggests caution in strict application of a specific cutoff or even a single metric. Several known pathogenic genes are borderline by only one intolerance score, while several are poorly constrained by both metrics (e.g., MECP2 and Rett syndrome; RVIS = 32.4, pLI = 0.66). For example, we identify 20 genes that are intolerant to mutation by pLI but not RVIS (RVIS > 20). Some of these are well-established genes (e.g., KANSL1 and the Koolen-de Vries syndrome)50 and the basis for this discrepancy is unknown but may relate to the fact that part of the gene is duplicated complicating genome-wide analyses of intolerance. Among targets poorly constrained by both metrics, the Bohring-Opitz syndrome gene, ASXL1, was recently highlighted for the presence of somatic mosaic variants in the ExAC population (from which both the RVIS and pLI score are derived)81.
Our projection estimates indicate that gene discovery based on recurrent LGD or severe missense mutations (MIS30) will soon plateau (Figure 3). Clinical interpretation of patients with the only DNM in a previously unobserved gene will remain a challenge. Partitioning patients based on additional phenotypic criteria, sub-selecting genes based on functional pathway enrichment43, integration of inherited variation (e.g., TADA)82, or targeting a small number of genes in much larger cohorts10,11 are all potential strategies for associating specific genes with a phenotype under these conditions. For example, analysis of this set of DNMs in the context of MAGI modules further identified key functional categories, including neurotransmitter/synaptic signaling and JNK/MAPK cascades. This analysis identifies 46 genes with DNMs among the four modules that do not yet reach significance but likely represent functionally important targets for future screens.
In contrast to LGD and MIS30 DNM, the number of genes that will be identified by missense DNM generally has not yet begun to approach an asymptote. Samples sizes are just now beginning to reach the level where signatures of recurrence and missense clustering are being detected for a relatively modest number of genes—most of which are only nominally significant14,15. We propose that this class of mutation (less severe or clustered missense DNMs) represents the most promising reservoir for future gene discovery but will require much larger whole-exome and whole-genome data sets to tease apart. As the number of exomes grows for both ASD and DD, the maintenance and curation of de novo databases will be especially important in this regard20.
Previous studies have implicated larger CNVs and increased mutation burden with more severe phenotypic outcomes and here we observe that the majority of DNM in the 253 genes originates from ID/DD cases (>3:1, Supplementary Table 8). Importantly, DNM-enriched genes significantly overlap known pathogenic CNV regions (p = 0.0017, two-tailed Fisher’s exact test, LR+ 1.85 [95% CI 1.38 - 2.43]) supporting a common genetic etiology. These specific targets have offered both independent confirmation of existing single-gene associations (e.g., KANSL1 in the 17q21.31 region), additional support for candidate genes (MAPK3 in the 16p11.2 region), and further support for potentially oligogenic effects with multiple compelling candidate genes (HECW2, SATB2, ABI2, and SF3B1 in the 2q33.1 deletion region). This is consistent with findings suggesting a role for multiple hits in ASD83,84. Among the genes with recurrent mutation and CNV intersection, MAPK3 is particularly interesting with respect to the chromosome 16p11.2 microduplication. Several functional studies on 16p11.2 deletion and duplication mice as well as Drosophila models have suggested that MAPK3 is a key regulator of the syndrome: being downstream of other ASD target genes; involved in axon targeting and regulation of cortical cytoarchitecture; and being the most topologically important gene in the region by PPIs85-87. Our analysis builds on these studies by providing evidence of recurrent missense mutation enrichment in human NDDs.
Finally, it is interesting that the 253 genes we highlight in this meta-analysis demonstrate a pan-neuronal expression pattern with the majority of genes being expressed in all GABAergic (inhibitory) and glutamatergic (excitatory) neuron types (Figure 3), suggesting these genes have the potential to alter many paths in the adult cortical circuit. While the majority of genes are broadly expressed across neuronal cell types, a subset demonstrates evidence of specific expression. More specifically, we observed enrichment for genes specifically expressed in the D1+ (19 genes) and D2+ (18 genes) medium spiny neurons of the striatum (13 genes are shared in the D1+ and D2+ lists) in mouse brain (Supplementary Figure 2A). Previously, Dougherty and colleagues highlighted this particular brain region based on a survey of genes reported as autism candidate risk genes48. Using a CSEA, we now extend this observation to NDD genes enriched for recurrent DNM. Remarkably, a similar enrichment was recently reported in autistic individuals with multiple DNMs in coding and putative noncoding regulatory DNA84. We also observe a similar signature for genes where nominal significance has been observed for clustered DNMs. While many of the genes enriched for D1+ and D2+ expression are not exclusive to the striatum and are more broadly expressed (as demonstrated by the enrichment signal at the lowest specificity threshold), the striatum has been implicated in ID and autism pathology by numerous studies88-97. The striatum is particularly compelling as it has been linked to repetitive behaviors88 core to the autism phenotype and also to genes known to be involved in DD, including CHD8, SHANK3, FOXP2, and KCNA489,90,93,96,97. While the striatum is most strongly linked to autism core phenotypes, our observation of enrichment in a more general DD cohort suggests that, while the general bias of cortex genes to ID and striatum genes to ASD92 still holds, the diverse expression patterns of genes across the brain at complex developmental time points may have substantial functional overlap among subtypes of NDDs that will require deep phenotyping and imaging to tease apart.
In conclusion, the 253 genes that show evidence for recurrent DNM represent a starting point for further functional and phenotypic investigations. Of these genes, 124 reach a strict GWAS threshold of significance strongly arguing that DNM in these genes contributes significantly to disease. Overall, the genes we highlight demonstrate strong conservation, refine pathogenic CNVs, define distinct functional pathways, and support the role of striatal networks in the pathogenicity of both ASD and ID/DD. Strikingly, the majority of the genes identified in this study present with DNMs in both ASD and ID/DD. While we expect a degree of diagnostic overlap21,22, our results support a common genetic etiology among broad neurodevelopmental phenotypes. These genes are candidates for a genotype-first paradigm98, where downstream follow-up of patients with the same de novo disrupted gene is likely to provide additional insight into unique phenotypic features associated with these different genetic subtypes99 and additional support for their role in NDD.
METHODS
Data set:
We analyzed de novo single-nucleotide and indel variants from whole-exome sequencing (WES) data generated for 10,927 cases with neurodevelopmental diagnoses of ASD or ID/DD compiled in the denovo-db v.1.5 release20 (Supplementary Table 1). The subset of denovo-db v.1.5 cohorts used was specifically chosen to avoid potential sample overlap as described in Geisheker et al.14 and in the release documentation for denovo-db v.1.5 (see URLs). Briefly, we first assumed minimal overlap between studies based in Europe and America and studies with exclusion criteria, including participation in another study (e.g., SSC and the Autism Sequencing Consortium or ASC). This was supported by screening for individuals with shared mutational sites and second events14. We excluded studies from The Autism Simplex Collection or TASC due to known sample overlaps; additionally, we utilized only the more recent MSSNG data set35 to avoid redundant annotations. All variants were annotated to RefSeq transcripts using SnpEff and collapsing to the most severe variant across isoforms. Variants were further binned into LGD (stop loss/gain, splice, and frameshift), missense, or synonymous categories for analysis. While these data are derived from a diverse set of WES platforms with differing sensitivities, our DNM-based analysis assumed samples to have perfect sensitivity. The combined set of 12,172 DNMs includes 2,357 LGD and 9,815 missense mutations, representing the largest such analysis to date. Each source study reports validated sites or validation rates ranging from 88.2% to 100% (with the exception of the DDD study, which opted for a high sensitivity approach) (Supplementary Table 1). Among these events, 1,106 LGD and 2,594 missense DNMs were validated/confirmed in the original studies while the remainder have unknown validation status (invalidated sites are not included in this analysis). CNV region data was obtained from a previous study of 29,085 children with developmental disorders and 19,584 population controls18 as well as a curated list of genomic disorders maintained by DECIPHER (v.9.18) (see URLs).
Statistics:
All standard statistical tests not reported by a described application (see relevant Methods) were performed using the R statistical language (v3.2.4), and non-parametric tests were used wherever possible. Wilcoxon rank-sum tests were used for distribution comparisons, with the exception of the Tukey Honest Significant Difference test for RVIS testing across all categories, and t-test for IQ comparisons. Fisher’s exact test was used for all count comparisons. Likelihood ratio tests for goodness of fit were performed using one million multinomial sampling simulations. Multiple testing correction was applied where appropriate using either the Benjamini-Hochberg FDR or Bonferroni FWER as described in the relevant sections.
Recurrent variant analysis:
Enrichment of de novo LGD and missense variation per gene was calculated using two statistical models. The CH model as previously described11 was run using the default setting and assuming a baseline rate of 1.8 de novo variants per individual (Supplementary Table 1). We note that ID/DD cohorts tend to have higher DNM rates than ASD cohorts, potentially relating to a combination of both technical (chosen sensitivity, platform differences) and biological biases, with the DDD cohort9 demonstrating the highest DNM rate among large cohorts24. We anticipate that false positives would be randomly distributed and not enriched within specific genes. Observed coding DNM (LGD or missense) rates in the exome studies range from 0.87 to 1.36 among the large cohorts (Supplementary Table 1). While these variances in DNM rate likely influence our results, all rates are below the 1.8 DNM per individual rate used in the CH model; thus, overall our statistics will be conservative. In addition, we ran a recently published modified version40 that separately tests for enrichment of variants with CADD v.1.3 scores39 of 30 and higher, which are predicted to be the most damaging of missense variation. Similarly, denovolyzeR12 (R package version v0.2.0) was run using default settings. Each test (LGD, missense, MIS30) was individually adjusted to a q-value by the Benjamini-Hochberg procedure based on the number of genes in the model (exome wide) and genes with q-value < 0.05 and a DNM count of two or more were considered for the union set. Wherever necessary, gene symbols were adjusted to match those used in the individual models (CH model, denovolyzeR, CSEA). In cases where no model was generated for a gene of interest, “no model” is indicated in the significance column of Supplementary Table 2. Each analysis was corrected genome-wide for the number of genes present in the corresponding models (18,946 for CH model and 19,618 for denovolyzeR).
While the q-value threshold should control the FDR within a single list, the combination of two models has the potential to increase the upper bound of the FDR for both the union and intersection. Although we assume that the FDR should remain at 5% on average, the upper limits to the merged FDRs can be defined as follows:
Application of these upper bounds estimates the maximal error rates in the unions as LGD FDR < 8.59%; Missense FDR < 7.56%. As MIS30 is only examined by one model, its estimated FDR remains at 5%. Similarly, we estimate the maximal error rates in the intersections as LGD FDR < 5.7%; Missense FDR < 6.75%. We wish to stress that we anticipate these to be upper bounds. In addition to the described treatment, to increase the stringency of our union set, we excluded any gene with a single DNM from further consideration. As a result, we discarded q-values for 1,183 LGD, 4,246 missense, and 1,083 MIS30 genes of which 113 reach corrected significance but are primarily small genes. This represents elimination of ~5-15% of the genes further reducing the overall FDR. Despite their likely enrichment for false positives, 11 of 113 single-hit genes have been previously implicated in an NDD (Supplementary Table 2) and are thus potentially of interest for future studies with additional samples.
Identification of novel genes:
To address novelty, we considered statistically significant genes as defined by five recent publications involving large-scale exome, whole-genome, or targeted sequencing (DeRubeis et al.26, Sanders et al.19, an in-press revised version of Stessman et al.10, Yuen et al.35, DDD 201724) and three well-curated databases (OMIM, SFARI Gene, and ID Gene Database Project, see URLs; all database queries made on 02/19/2018). We undertook the following approach to ‘novelty’ when comparing these data sets to our list of 253 significant genes. Any gene listed as significant (regardless of thresholds or alternative methods of significance (CNV integration, private variation, inherited variation)) in any of these data sets was not considered ‘novel’ in our data set. Additionally, we considered SFARI genes of any score category as known. For the 80/253 genes not “known” by this initial screen, we followed up with literature searches for evidence of any link to case reports or human studies of ID, autism, or mental retardation, or DD. For 49/80 genes, no evidence was found through the PubMed search; this represents our most conservative set of ‘novel’ genes (Supplementary Table 3). While this pool is the most likely to contain false positive findings, 10 of these 49 genes were also significant under an FWER correction (SMARCD1, SNX5, TNPO2, ADAP1, CAPN15, CHD3, TMEM178A, AGO4, SNAPC5, and ANP32A). Clinical evidence, even single case reports, adds credence to our gene list as high-confidence candidates for NDD pathology.
Network analysis:
Identification of clustered gene modules was performed using the MAGI (merging affected genes into integrated networks)43 enrichment tool with default settings, followed by visualization incorporating co-expression and physical interaction data from geneMANIA100. Significance for MAGI modules was performed by permuting DNM across genes according to their mutation rates in the CH model 100 times and enumerating the number of random modules with scores greater than or equal to the module of interest.
Expression analysis in mouse:
Functional enrichment was examined using the CSEA and TSEA tools48. CSEA48 was additionally applied to candidate genes using single-cell transcriptomic profiling data from mouse cortex and hippocampus101 using custom R scripts. Raw mRNA count data was downloaded from (see URLs, and only genes with at least 25 total molecules and at least 1 molecule in ≥100 cells were retained. Molecule counts were then incremented by a pseudo-count of 0.125 and RPKM-normalized. RPKMs were averaged over each of the 47 cell subclasses identified by the authors using the BackSPIN clustering algorithm. Significantly enriched transcripts in each subclass were identified using the pSI package47 with a minimum expression value of 3 RPKM and default settings otherwise.
Expression analysis in humans:
15,928 single nuclei were isolated from the middle temporal gyrus of adult post-mortem brains of three human donors and profiled with RNA-sequencing. Raw read (fastq) files were aligned to the GRCh38 human genome sequence (Genome Reference Consortium, 2011) with the RefSeq transcriptome version GRCh38.p2 (current as of 4/13/2015). For alignment, Illumina sequencing adapters were clipped from the reads using the fastqMCF program (ea-utils, see URLs). After clipping, the paired-end reads were mapped using Spliced Transcripts Alignment to a Reference (STAR)102 with default settings. Unsupervised clustering identified 71 distinct transcriptomic clusters, including 41 GABAergic (inhibitory) neuronal, 24 glutamatergic (excitatory) neuronal, and 6 non-neuronal cell types (unpublished). For each gene, the expression pattern was characterized as the number of cell types with appreciable expression (median counts per million [CPM] > 1) in three broad classes: inhibitory and excitatory neurons and glia. Heatmaps were constructed of log-normalized expression (log2CPM + 1) of NDD risk genes and control genes across cell types. The number of inhibitory and excitatory neuronal and glial types that expressed NDD risk genes and control genes were quantified and visualized as empirical cumulative distributions. Distributions were compared with two-sided Wilcoxon rank-sum tests for each broad class of cell types, and p-values were Bonferroni corrected for multiple testing. A cell type specificity or marker score (beta) was defined for all genes to measure how binary expression was among clusters, independent of the number of clusters labeled. First, the proportion (x) of samples in each cluster that expressed a gene above background level (CPM > 1) was calculated. Then, scores were defined as the squared differences in proportions between all pairs (i,j) of n clusters normalized by the sum of absolute differences plus a small constant (ε) to avoid division by zero. Scores ranged from 0 to 1, and a perfectly binary marker had a score equal to 1.
Shapiro-Wilk tests rejected (p < 0.05) the null hypothesis that distributions were normally distributed of cell type counts for each broad class, maximum average expression, and marker scores. Therefore, distributions were compared with two-sided Wilcoxon rank-sum tests, and p-values were Bonferroni corrected for multiple testing.
Projected rates of gene discovery:
Prediction of future LGD and missense variation discovery rates was determined by sampling (with replacement) populations of 100 to 10,900 cases 10,000 times each and calculating DNM statistics using the CH model. The number of genes with two or more mutations and a q-value below 0.05 were then enumerated for each simulation and linear as well as logistic growth models were fit to each curve with the best model being chosen by BIC. Model fits and confidence bounds were performed using the base stats and propagate (see URLs) packages in the R statistical language.
URLS
denovo-db, http://denovo-db.gs.washington.edu/; denovo-db v.1.5 documentation, http://denovo-db.gs.washington.edu/denovo-db.v.1.5.pdf; DECIPHER, https://decipher.sanger.ac.uk; Online Mendelian Inheritance in Man (OMIM), https://www.omim.org; SFARI Gene, https://gene.sfari.org/; ID Gene Database Project, http://gfuncpathdb.ucdenver.edu/iddrc/iddrc/home.php; Linnarsson Lab Single-cell analysis of mouse cortex,http://linnarssonlab.org/cortex; ea-utils fastqMCF program, https://expressionanalysis.github.io/ea-utils/; R package ‘propagate’, https://CRAN.R-project.org/package=propagate; DECIPHER Syndrome Overview https://decipher.sanger.ac.uk/disorders#syndromes/overview; Human MTG single nucleus RNA-seq data, http://celltypes.brain-map.org/download
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank Tychele Turner and Jay Shendure for helpful discussion and Tonia Brown for edits. This research was supported, in part, by the following: the Simons Foundation Autism Research Initiative (SFARI 303241) and US National Institutes of Health (NIH R01MH101221) to E.E.E. The J.D.D. lab is supported by a NARSAD Independent Investigator Award from the Brain and Behavior Research Foundation, as well as NIH (5R01MH107515-03). We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
DATA AVAILIBILITY STATEMENT
All variant data in this study is available to download from denovo-db v.1.5 (see URLs). Human MTG single-nucleus RNA-seq data and clusters can be downloaded from the Allen Institute for Brain Science web site: http://celltypes.brain-map.org/download.
COMPETING FINANCIAL INTERESTS
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc.
Reporting summary: Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
REFERENCES
- 1.Sebat J et al. Strong association of de novo copy number mutations with autism. Science 316, 445–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharp AJ et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77, 78–88 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuzun E et al. Fine-scale structural variation of the human genome. Nat Genet 37, 727–32 (2005). [DOI] [PubMed] [Google Scholar]
- 4.de Vries BB et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet 77, 606–16 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey JA, Yavor AM, Massa HF, Trask BJ & Eichler EE Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 11, 1005–17 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vries BB et al. Clinical studies on submicroscopic subtelomeric rearrangements: a checklist. J Med Genet 38, 145–50 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firth HV & Wright CF The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol 53, 702–3 (2011). [DOI] [PubMed] [Google Scholar]
- 8.O’Roak BJ et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43, 585–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stessman HA et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet 49, 515–526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Roak BJ et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46, 944–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner TN et al. Proteins linked to autosomal dominant and autosomal recessive disorders harbor characteristic rare missense mutation distribution patterns. Hum Mol Genet 24, 5995–6002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geisheker MR et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat Neurosci 20, 1043–1051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lelieveld SH et al. Spatial Clustering of de Novo Missense Mutations Identifies Candidate Neurodevelopmental Disorder-Associated Genes. Am J Hum Genet 101, 478–484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper GM et al. A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaminsky EB et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13, 777–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coe BP et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46, 1063–71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanders SJ et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turner TN et al. denovo-db: a compendium of human de novo variants. Nucleic Acids Res 45, D804–D811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matson JL & Shoemaker M Intellectual disability and its relationship to autism spectrum disorders. Res Dev Disabil 30, 1107–14 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Diagnostic and statistical manual of mental disorders. 5. American Psychiatric Association (2013). [Google Scholar]
- 23.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Ligt J et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367, 1921–9 (2012). [DOI] [PubMed] [Google Scholar]
- 26.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halvardson J et al. Mutations in HECW2 are associated with intellectual disability and epilepsy. J Med Genet 53, 697–704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto R et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J Hum Genet 61, 199–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krumm N et al. Excess of rare, inherited truncating mutations in autism. Nat Genet 47, 582–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Lin MC, Kornblum HI, Papazian DM & Nelson SF Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 23, 3481–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lelieveld SH et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci 19, 1194–6 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Michaelson JJ et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreno-Ramos OA, Olivares AM, Haider NB, de Autismo LC & Lattig MC Whole-Exome Sequencing in a South American Cohort Links ALDH1A3, FOXN1 and Retinoic Acid Regulation Pathways to Autism Spectrum Disorders. PLoS One 10, e0135927 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rauch A et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–82 (2012). [DOI] [PubMed] [Google Scholar]
- 35.RK CY et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tavassoli T et al. De novo SCN2A splice site mutation in a boy with Autism spectrum disorder. BMC Med Genet 15, 35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner TN et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am J Hum Genet 98, 58–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuen RK et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med 1, 160271–1602710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang T et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 7, 13316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Meur N et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet 47, 22–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hormozdiari F, Penn O, Borenstein E & Eichler EE The discovery of integrated gene networks for autism and related disorders. Genome Res 25, 142–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Endele S et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42, 1021–6 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Ching MS et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet 153B, 937–47 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephenson JR et al. A Novel Human CAMK2A Mutation Disrupts Dendritic Morphology and Synaptic Transmission, and Causes ASD-Related Behaviors. J Neurosci 37, 2216–2233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dougherty JD, Schmidt EF, Nakajima M & Heintz N Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38, 4218–30 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu X, Wells AB, O’Brien DR, Nehorai A & Dougherty JD Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci 34, 1420–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deshpande A & Weiss LA Recurrent reciprocal copy number variants: Roles and rules in neurodevelopmental disorders. Dev Neurobiol 78, 519–530 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Koolen DA et al. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur J Hum Genet 24, 652–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phelan K & Rogers RC Phelan-McDermid Syndrome in GeneReviews(R) (eds. Adam MP et al.) (Seattle (WA: ), 1993). [Google Scholar]
- 52.Bi W et al. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet 115, 515–24 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Han JY et al. Identification of a novel de novo nonsense mutation of the NSD1 gene in monozygotic twins discordant for Sotos syndrome. Clin Chim Acta 470, 31–35 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Izumi K et al. Interstitial microdeletion of 4p16.3: contribution of WHSC1 haploinsufficiency to the pathogenesis of developmental delay in Wolf-Hirschhorn syndrome. Am J Med Genet A 152A, 1028–32 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Shimbo H et al. Haploinsufficiency of BCL11A associated with cerebellar abnormalities in 2p15p16.1 deletion syndrome. Mol Genet Genomic Med 5, 429–437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleefstra T et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet 46, 598–606 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Fergelot P et al. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am J Med Genet A 170, 3069–3082 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Kumar RA et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 17, 628–38 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Labonne JD et al. A microdeletion encompassing PHF21A in an individual with global developmental delay and craniofacial anomalies. Am J Med Genet A 167A, 3011–8 (2015). [DOI] [PubMed] [Google Scholar]
- 60.McCool C, Spinks-Franklin A, Noroski LM & Potocki L Potocki-Shaffer syndrome in a child without intellectual disability-The role of PHF21A in cognitive function. Am J Med Genet A 173, 716–720 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Leroy C et al. The 2q37-deletion syndrome: an update of the clinical spectrum including overweight, brachydactyly and behavioural features in 14 new patients. Eur J Hum Genet 21, 602–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klebe S et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet 20, 645–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Halder D et al. Suppression of Sin3A activity promotes differentiation of pluripotent cells into functional neurons. Sci Rep 7, 44818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Witteveen JS et al. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat Genet 48, 877–87 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–8 (1999). [DOI] [PubMed] [Google Scholar]
- 66.Jansen S et al. De Novo Truncating Mutations in the Last and Penultimate Exons of PPM1D Cause an Intellectual Disability Syndrome. Am J Hum Genet 100, 650–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeMari J et al. CLTC as a clinically novel gene associated with multiple malformations and developmental delay. Am J Med Genet A 170A, 958–66 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Fusco C et al. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur J Hum Genet 22, 64–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buxbaum JD et al. Association between a GABRB3 polymorphism and autism. Mol Psychiatry 7, 311–6 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Guella I et al. De Novo Mutations in YWHAG Cause Early-Onset Epilepsy. Am J Hum Genet 101, 300–310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Asadollahi R et al. The clinical significance of small copy number variants in neurodevelopmental disorders. J Med Genet 51, 677–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrington AJ et al. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife 5(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paciorkowski AR et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics 14, 99–111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kohannim O et al. Discovery and Replication of Gene Influences on Brain Structure Using LASSO Regression. Front Neurosci 6, 115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weiss K et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am J Hum Genet 99, 934–941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berko ER et al. De novo missense variants in HECW2 are associated with neurodevelopmental delay and hypotonia. J Med Genet 54, 84–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harripaul R et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry (2017). [DOI] [PubMed] [Google Scholar]
- 78.Wang Q, Moore MJ, Adelmant G, Marto JA & Silver PA PQBP1, a factor linked to intellectual disability, affects alternative splicing associated with neurite outgrowth. Genes Dev 27, 615–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levy J et al. Molecular and clinical delineation of 2p15p16.1 microdeletion syndrome. Am J Med Genet A 173, 2081–2087 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Dheedene A, Maes M, Vergult S & Menten B A de novo POU3F3 Deletion in a Boy with Intellectual Disability and Dysmorphic Features. Mol Syndromol 5, 32–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carlston CM et al. Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum Mutat 38, 517–523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He X et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 9, e1003671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Werling DM et al. Limited contribution of rare, noncoding variation to autism spectrum disorder from sequencing of 2,076 genomes in quartet families. BioRxiv (2017). [Google Scholar]
- 84.Turner TN et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park SM, Park HR & Lee JH MAPK3 at the Autism-Linked Human 16p11.2 Locus Influences Precise Synaptic Target Selection at Drosophila Larval Neuromuscular Junctions. Mol Cells 40, 151–161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pucilowska J et al. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci 35, 3190–200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blizinsky KD et al. Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc Natl Acad Sci U S A 113, 8520–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Langen M et al. Changes in the development of striatum are involved in repetitive behavior in autism. Biol Psychiatry 76, 405–11 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Platt RJ et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep 19, 335–350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reim D et al. Proteomic Analysis of Post-synaptic Density Fractions from Shank3 Mutant Mice Reveals Brain Region Specific Changes Relevant to Autism Spectrum Disorder. Front Mol Neurosci 10, 26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Balsters JH, Mantini D & Wenderoth N Connectivity-based parcellation reveals distinct cortico-striatal connectivity fingerprints in Autism Spectrum Disorder. Neuroimage (2017). [DOI] [PubMed] [Google Scholar]
- 92.Shohat S, Ben-David E & Shifman S Varying Intolerance of Gene Pathways to Mutational Classes Explain Genetic Convergence across Neuropsychiatric Disorders. Cell Rep 18, 2217–2227 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Kaya N et al. KCNA4 deficiency leads to a syndrome of abnormal striatum, congenital cataract and intellectual disability. J Med Genet (2016). [DOI] [PubMed] [Google Scholar]
- 94.Flanigan M & LeClair K Shared Motivational Functions of Ventral Striatum D1 and D2 Medium Spiny Neurons. J Neurosci 37, 6177–6179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanders SJ First glimpses of the neurobiology of autism spectrum disorder. Curr Opin Genet Dev 33, 80–92 (2015). [DOI] [PubMed] [Google Scholar]
- 96.Schreiweis C et al. Humanized Foxp2 accelerates learning by enhancing transitions from declarative to procedural performance. Proc Natl Acad Sci U S A 111, 14253–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen YC et al. Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci 19, 1513–1522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stessman HA, Bernier R & Eichler EE A genotype-first approach to defining the subtypes of a complex disease. Cell 156, 872–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bernier R et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS ONLY REFERENCES
- 100.Warde-Farley D et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zeisel A et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–42 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.