

Abstract
Background
Previous studies have demonstrated that CF epithelial cells exhibit increased cholesterol content at the plasma membrane compared to wild type controls as measured by electrochemical methods. Microtubule dysregulation that impacts intracellular transport has also been identified in CF cells and is reversible with histone deacetylase 6 (HDAC6) inhibition, a regulator of tubulin acetylation. The hypothesis of this study is that increased membrane cholesterol content in CF cells is dependent on HDAC6 regulation.
Methods
Electrochemical measurement of membrane cholesterol in mouse trachea and in primary human CF bronchial epithelial cells is used to monitor CFTR correction and manipulation of cholesterol processing by HDAC6 inhibition.
Results
Data demonstrate that induction of Cftr expression in an inducible CF mouse model restores tubulin acetylation levels and normalizes membrane cholesterol content. To test the relationship between tubulin acetylation, membrane cholesterol levels were measured in a CF mouse model depleted of Hdac6 expression (CF/HDA). CF/HDA mouse trachea have WT membrane cholesterol levels while CF mice have approximately two-fold increase in membrane cholesterol compared to WT consistent with previous studies. Pharmacological inhibition of HDAC6 in primary human CF bronchial epithelial cells also reduces membrane cholesterol levels.
Conclusions
This study demonstrates that elevated membrane cholesterol in CF epithelium is regulated by HDAC6 function and that the electrochemical measure of membrane cholesterol correlates with both genetic and pharmacological CFTR correction.
Keywords: Cystic fibrosis, microtubule, cholesterol
1. Introduction
Cystic fibrosis (CF) is caused by a loss of function of the cystic fibrosis transmembrane conductance regulator (CFTR) (1, 2). Previous work has identified that loss of CFTR function impacts several aspects of cholesterol processing resulting in perinuclear cholesterol accumulation, increased rate of de novo cholesterol synthesis, and increased cholesterol content located at the plasma membrane (3, 4). Subsequent studies have identified that perinuclear cholesterol accumulation characteristic of CF epithelial cells is due to reduced acetylation and reduced stability of microtubules impairing endosomal transport (5, 6). Histone deacetylase 6 (HDAC6) is a cytosolic deacetylase that contributes to the regulation of microtubule acetylation (7). Inhibition of HDAC6 increases tubulin acetylation in CF cells and restores intracellular transport and redistribution of perinuclear cholesterol (5). It is hypothesized that HDAC6 inhibition will restore the elevated membrane cholesterol content characteristic of CF cells to wild type levels. These data will establish that the membrane cholesterol measurement can be used as a biomarker to monitor the correction of intracellular events characteristic of CF in response to novel therapeutic approaches.
Membrane cholesterol is measured by electrochemical detection methods using an electrode containing cholesterol oxidase. This method has been used previously to detect elevated cholesterol at the membrane in both cultured CF cell models and CF mouse trachea tissue (4, 8). The electrochemical measurement determines the amount of cholesterol at the surface of the plasma membrane accessible to the cholesterol oxidase bound to the electrode. Therefore, this method does not distinguish between excess cholesterol stored within the outer leaflet of the plasma membrane or increased rates of cholesterol efflux. The use of cavity electrodes where there is a gap between the plasma membrane and the electrode surface suggests that increased rates of efflux account for the elevated cholesterol measurements characteristic of CF cells. Our previous studies demonstrate that elevated cholesterol measurements in CF cells correlate with CFTR genotype, are dependent on de novo cholesterol synthesis, and likely develop over time as an adaptive response to the loss of CFTR function (8–10).
Since this measurement correlates with CFTR genotype and appears to develop as a result of cellular changes brought about by CFTR dysfunction, electrochemical measurements of membrane cholesterol can serve as a measure of cellular responsiveness to CFTR targeted therapies. New therapies are being developed that directly target the potentiation of mutant CFTR function or correction of its trafficking (11, 12). This approach has achieved significant success particular with CFTR mutants that traffic correctly (13–15). As new therapies are developed, a specific need in the CF field is the development of a non-invasive marker of disease that correlates with CFTR function and also gives insight into the cellular biology of disease mechanisms.
Current measures of CFTR correction in response to CFTR-targeted therapies are the nasal potential difference (NPD) assay and the sweat chloride test (11, 16–18). The NPD and sweat chloride tests are effective assays of CFTR function, but provide no information as to cellular responses to CFTR function. The sweat chloride test has the advantage of being a sensitive and reproducible assay of CFTR function. However, there is a lack of correlation on an individual basis between reduced sweat chloride and improvements in lung function in the original VX-770 study, only a correlation when responses are averaged (19). These data may indicate that how cells are responding to corrected CFTR function differentially impacting efficacy on an individual basis. Having a method to assess cell biological changes associated with CFTR correction that can complement CFTR functional assays may help evaluate efficacy of CFTR-targeted therapies. In this study we demonstrate that inducible expression of CFTR in a CF mouse model restores microtubule acetylation in primary nasal epithelium and completely reverses elevated membrane cholesterol content characteristic of CF tissues from excised trachea. Mechanistically, we demonstrate that ablation of Hdac6 expression in a CF mouse model achieves the same outcome and normalizes membrane cholesterol content. These data demonstrate that membrane cholesterol can be used to monitor the intracellular impact of CFTR function correction and to determine the impact on intracellular regulation of microtubule function.
2. Materials and Methods
2.1. Mice
HDAC6 mice: Four genotypes of mice were used in this experiment: wild type mice (WT), ΔF508 mutated mice (CF), HDAC6 knockout mice (HDA), and ΔF508 and HDAC6 double-knockout mice (CF/HDA). All mice were provided by the CF Center animal core facility at Case Western Reserve University. CFTR wild-type mice were siblings of CF mice. All mice were used between six and eight weeks of age and are back-crossed over ten generations onto a C57Bl/6 background. All mice were fed a standard chow. To decrease the incidence of intestinal obstruction that is common in CF mice, mice were allowed access to sterile water with osmotic laxative, PEG-3350 with electrolytes (Kremers Urban). All mice were maintained on a 12 h light, 12 h dark cycle at a mean ambient temperature of 22 °C. Mice were cared for in accordance with the Case Western Reserve University IACUC guidelines by the CF Animal Core Facility.
2.2. Inducible Cftr-on mice
The creation of a conditional Cftr mouse allele that can restore CFTR function in a CF mouse has been previously described (20). This allele Cftr-on, or Cftrinvfl10, was combined with a tamoxifen inducible ubiquitously expressed Cre recombinase transgene which provides the ability to restore CFTR function throughout the body of a CF mouse at a specific time (21). The particular inducible system used contains Cre recombinase that is expressed through a ubiquitous ROSA promoter so that Cre is always present in every cell in the mouse (Jackson laboratories, #008463). However, the Cre is fused to a mutated estrogen receptor ligand binding domain that selectively binds to tamoxifen. This fusion makes Cre recombinase restricted to the cytoplasm of the cell and can only gain access to the nucleus to mediate conditional activation of Cftr after exposure to tamoxifen. Tamoxifen is used to induce Cre recombinase function, and thus recombination between existing loxP sites, by intraperitoneal injection of mice. Mice homozygous for Cftr-on allele and containing the tamoxifen cre transgene were given intraperitoneal injections for 3 consecutive days with 3mg/40gram body weight of tamoxifen at 4–6 weeks of age. After a minimum of two weeks post treatment, nasal potential difference was assessed (22–24) as well as the electrochemical analysis of membrane cholesterol.
2.3. Electrochemical measurements of cholesterol
The trachea of mouse was excised, pinned on a gelatin Petri plate and was submerged in pH 7.4 HBR (Hepes-buffered Ringer) solution at room temperature. Platinum disk (10 μm diameter) microelectrodes were polished and ultrasonic cleaned. The electrodes were rinsed and immersed in 1) 4 mM of 3,3′-dithiodipropionic acid di(N-hydroxysuccinimide ester) (DTSP) in dimethyl sulfoxide (DMSO) solution for 45 minutes; 2) 33 unit/mL recombinant cholesterol oxidase (Sigma-Aldrich, Co., 40 unit/mg) solution for 2 hours; 3) ~0.56 mol/L glutaraldehyde (TCI America, ~5.6 mol/L) solution for 30 minutes; 4) cholesterol oxidase solution (same as above) at 4°C overnight. The control experiments were conducted with a bare platinum disk microelectrode (same as above), which was polished and ultrasonic cleaned before each use.
2.4. Data Acquisitions
Amperometric measurements were conducted using a two-electrode cell and a voltammeter-amperometer (Chem-Clamp, Dagan corp.). The three-pole Bessel filter was set to 100 Hz. The output current was further filtered with a 50/60 Hz noise eliminator (Hum Bug, Quest Scientific). An Ag/AgCl (saturated KCl) reference electrode was used for all experiments, and the applied potential was 800 mV versus normal hydrogen electrode (NHE) for all experiments. All experiments were performed in HBR solution at room temperature. The electrode was initially immersed in electrolyte away from the tissue sample for acquisition of baseline data. The electrode was repositioned to touch the inner surface of trachea tissue for acquisition of electrode response. The electrode was then positioned away from tissue to ascertain the baseline. Data was collected by Lab View Signal Express software and analyzed in Origin 8.0.
2.5. Double potential pulse coulometry measurements of cholesterol
Human CF bronchial epithelial cells (CFBE) were cultured on filters. They were treated with 10 μM tubastatin or vehicle for 24 hours before measurements. To reduce the background of measurements, 10 um diameter carbon disk microelectrodes were used in this experiment. Solution of nano platinum was prepared by adding 600 ul of 0.01 M NaBH4 dropwise into 5 ml aqueous solution containing 1.25 umol H2PtCl6 and 1.22 umol sodium citrate. Carbon microelectrodes were immersed in the solution for 20 minutes and was baked at 80°C for 10 minutes. The electrodes were then modified with cholesterol oxidase by the same method mentioned above. Double pulse coulometry measurements were conducted by a two-electrode system: carbon microelectrodes as working electrodes and Ag/AgCl (saturated KCl) as the reference electrode. The voltammeter-amperometer circularly provided the double potential pulse waveform of 5 seconds of quiet time potential of 200 mV followed by two 0.25 second of 800 mV potential pulses with a short 200 mV interval of 0.5 second (versus NHE). The three-pole Bessel filter was set to 100 Hz. The output current was further filtered with a 50/60 Hz noise eliminator (Hum Bug, Quest Scientific). To keep the cells alive, measurements were made in culture medium. After gently pointing the electrodes to CFBE cell membrane, the potential and responding current were recorded by Lab View Signal Express software, and then processed by Python and Origin. (Double pulse method: The charge collected by the second pulse is background signal and was calculated and subtracted from the charge produced by the first pulse which measures the signal of oxidized cholesterol plus background. Therefore, the difference between the two charges represented the amount of cholesterol.) (10).
2.6. Nasal TEPD assay
Nasal potential difference (NPD) measurements were obtained as previously described with modifications (25). Mice were anesthetized using a ketamine/xylazine rodent cocktail given intraperitoneally, dosed at 0.012 ml/g mouse weight until plane of anesthesia is reached approximately 10 minutes later.
3.0. Results
3.1. Restoration of CFTR function in CF mice reduces membrane cholesterol content
To test the ability of the membrane cholesterol measurement to monitor in vivo CFTR correction, we utilized an inducible CFTR mouse model that employs the tamoxifen dependent Cre-lox system (20). In order to confirm CFTR correction in these mice, transepithelial nasal potential difference (NPD) assay was performed to monitor CFTR function. Forskolin stimulated chloride transport in WT, in CF mice not corrected with tamoxifen treatment, and in CF mice with CFTR expression corrected is compared. Uncorrected CF mice have zero response to Chloride-free Ringers and forskolin treatment, while corrected CFTR expression results in stimulated chloride transport identical to WT transport (figure 1A–D). These data demonstrate CFTR expression and function are corrected in this mouse model and represent a model to examine potential biomarkers of CFTR function.
Figure 1.
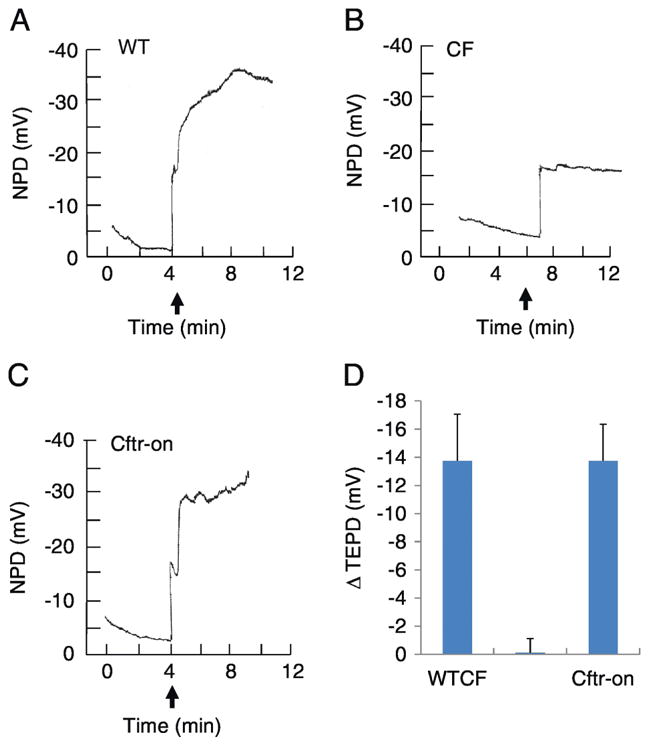
Chloride transport as measured by nasal potential difference (NPD) assay is restored in Cftr-corrected (CF-on) mice compared to control (WT) and Cftr −/− (CF) mice. A, B, and C) Representative NPD traces of WT CF, and Cftr-on mice. D) Composite data of the change in transepithelial potential difference (ΔTEPD) (Cftr −/−) (n= 4).
To determine the effect of CFTR correction on membrane cholesterol content, trachea are excised from mice and examined for membrane cholesterol content by electrochemical methods utilizing an electrode with cholesterol oxidase as described in Methods. In previous studies, we have demonstrated that CF nasal and tracheal epithelium have an approximately two-fold increase in membrane cholesterol content compared to control WT mice (4, 26). To determine the impact of CFTR correction on membrane cholesterol regulation, the Cftr-on mice are used. Membrane cholesterol values between WT and Cftr-on mice are identical (Figure 2A). Compared to Cftr −/− mice, correction of CFTR expression results in an identical two-fold decrease in membrane cholesterol content observed previously between WT and Cftr −/− mice (figure 2B). These data demonstrate that membrane cholesterol regulation is dependent on CFTR activity and that this method can potentially be used to evaluate the cellular impact of CFTR correction.
Figure 2.
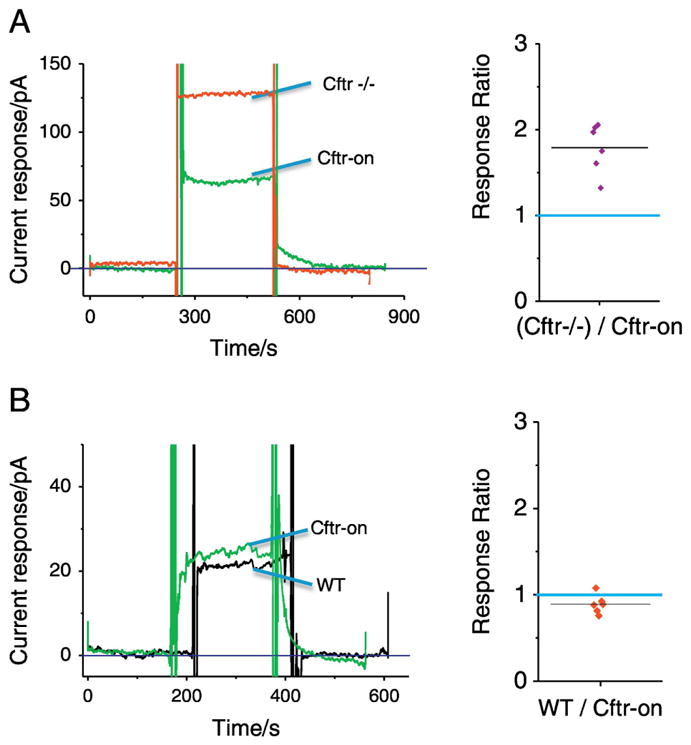
Induction of Cftr expression in CF mouse model restores membrane cholesterol content. A) Left panel shows representative current responses of plasma membrane cholesterol in mouse trachea of Cftr −/− (red) and Cftr-corrected (Cftr-on; green) mice. The right panel is the summary of response ratios of (Cftr −/−)/Cftr-on. CF mice have a 1.9 ± 0.1 fold increase response compared to Cftr-on mice (p < 0.01; n=5). Blue line shows WT/WT ration of 1.0. B) Comparison of Cftr-on and WT mouse membrane cholesterol content. Left panel shows representative current responses of plasma membrane cholesterol in mouse trachea of WT (black) and Cftr-corrected (Cftr-on; green) mice. The right panel is the summary of response ratios of WT/Cftr-on. There is no significant difference in responses between WT and Cftr-on mice.
3.2. Effect of HDAC6 depletion on membrane cholesterol content in CF mouse trachea
To test the hypothesis that microtubule regulation is the mechanistic link between CFTR function and membrane cholesterol content, we knocked out expression of the tubulin deacetylase HDAC6 from F508del (CF) mice as previously described (27). We have previously shown that inhibition of HDAC6 in cell models of CF restores intracellular cholesterol transport (5). There are four experimental groups of mice for this study: WT, Hdac6 −/− (HDA), F508del (CF), and CF/HDA. Our technique only allows for the side-by-side comparison of two experimental groups at a time. The first comparison was between WT and CF mice to ensure that our results are consistent with previous studies. Like earlier findings, CF mouse trachea has a 2.2 ± 0.1 fold increase in membrane cholesterol compared to WT mouse trachea (figure 3A).
Figure 3.

Effect of Hdac6 depletion on membrane cholesterol content in a CF mouse model. A) Representative trace of cholesterol measurement in F508del (CF) (red) and in control WT (black) mouse tracheal tissue. B) Representative trace of cholesterol measurement in tracheal tissue from F508del (CF) mice (red) and in F508del mice lacking HDAC6 expression (CF/HDA) (magenta). C) Representative trace of cholesterol measurement in tracheal tissue from CF/HDA mice (magenta) and in WT mice (black). D) Representative trace of cholesterol measurement in tracheal tissue from Hdac6 −/− (HDA) mice (orange) and in WT mice (black). E) Summary data showing ratios of responses between experimental pairs. Average ratio is shown by bar in each column. Replicates are at least 5 for each comparison. Significance determined by ANOVA using the Newman-Keuls post-hoc test for multiple comparisons (*p < 0.05 compared to WT/WT value of 1.0 shown be blue line).
To determine the impact of HDAC6 depletion on membrane cholesterol in CF mice, we next compared CF and CF/HDA mouse trachea. Membrane cholesterol content is increased 2.7 ± 0.4 fold in F508del mouse trachea compared to F508del/Hdac6 mouse trachea (figure 3B). This response indicates that CF/HDA trachea tissue is responding like WT mice and suggests that Hdac6 depletion is able to correct cholesterol processing independently of CFTR function.
In order to verify that membrane cholesterol content in CF/HDA mice is indeed restored to a WT level, a direct comparison between the CF/HDA and WT mouse trachea was performed with the prediction that CF/HDA and WT membrane cholesterol levels should be similar. Membrane cholesterol levels in WT trachea are 1.5 ± 0.1 fold higher (n=6, p < 0.001) than CF/HDA (figure 3C). The observation that membrane cholesterol is lower in CF/HDA tissue compared to even WT suggests that HDAC6 function is a key regulator of cholesterol movement in CF tissues to the plasma membrane.
Finally, to address the role of HDAC6 in regulating plasma membrane cholesterol content in mice with WT Cftr, trachea membrane cholesterol content was compared between WT and HDA mice. Interestingly, WT mice have reduced membrane cholesterol content than trachea from HDA mice (WT/HDA: 0.79 ± .02 fold (n=6, p < 0.001)) (figure 3D). The mechanism of this small but significant change is unclear.
As stated, only side-by-side comparisons can be made with a single electrode resulting in multiple comparisons. Therefore, summarized data showing the relative effect of Hdac6 depletion on membrane cholesterol content is provided in figure 3E. These data demonstrate that CF samples have an approximately two-fold increase in membrane cholesterol compared to either WT or the CF/HDA tracheal tissue. These data support the hypothesis that membrane cholesterol content in CF is regulated by Hdac6 function.
3.3. Reduction of membrane cholesterol content in human CF bronchial epithelial cells (CFBE) by pharmacological HDAC6 inhibition
We have recently published that electrochemical measurement of membrane cholesterol can be done in vivo on human buccal tissue using a double-pulse technique to act as an internal reference and reduce background noise (28). This technique was used on CFBE cells grown on filters in air-liquid interface to measure the impact of HDAC6 inhibition on membrane cholesterol. Only CF cells were examined as only two side-by-side measurements can be made with a single electrode and mouse studies above demonstrate limited impact of HDAC6 inhibition in WT tissues. Therefore, studies were focused on CFBE cells treated with tubastatin (10 μM) for 24 h or carrier to determine if HDAC6 inhibition could influence membrane cholesterol content. Tubastatin treatment reduces the double-pulse measure by 12.1 ± 3.1% compared to non-treated controls (figure 4). These data demonstrate that membrane cholesterol content in primary CF human epithelial cells is responsive to HDAC6 inhibition.
Figure 4.

HDAC6 inhibition reduces membrane cholesterol content in primary human CF bronchial epithelial (CFBE) cells. A) Representative traces of difference charge measured on carrier treat CFBE (NT, red) and 10uM tubastatin treated CFBE (tub, cyan) during 0.25 s of potential pulses. B) Representative traces of difference charge measured on two carrier treated samples as a control for CFBE measurement variability. C) Summary of difference charge ratios comparing tubastatin (Tub) treated CFBE cells over carrier treated CFBE cells (Tub/NT) and side-by-side carrier treated CFBE cell pairs (NT/NT) as a control for variability. Significance determined by t-test; (n= 10 electrodes tested on 5 cell pairs for Tub/NT; n= 6 electrodes tested on 3 cells pairs for NT/NT); p < 0.03).
3.4. VX-809 treatment reduces membrane cholesterol in CFBE cells
In order to determine if the electrochemical measurement of membrane cholesterol could be used to monitor the efficacy of the CFTR corrector VX-809 on cellular phenotypes, WT and F508del CFBE cells were treated with VX-809 (3.3 μM, 24 h). Representative traces showing relative levels of CFTR function in WT and F508del CFBE cells with and without VX-809 treatment are shown (Figure 5A). As mentioned above, we can only do side-by-side cholesterol measurement comparisons currently so data are presented as ratios of untreated/treated values. VX-809 treated CFBE cells do exhibit a reduction in the membrane cholesterol measurement resulting in an approximately 20% increase CF un-treated/treated ratio (Figure 5E). As a control, WT cells were also treated with VX-809 under the same conditions. VX-809 treatment had no effect on membrane cholesterol in WT cells (Figure 5E). For comparison, WT and CFBE cells were examined side-by-side with either cholesterol oxidase modified electrodes or BSA modified electrodes as a control. The membrane cholesterol is elevated in CFBE cells consistent with other model systems (Figure 5B,C, D). However, BSA modified electrodes show no difference between CF and WT measurements. These data demonstrate directly that pharmacological F508del CFTR correction impacts CF cell biology and that impact can be monitored by measuring membrane cholesterol levels.
Figure 5.

CFTR function and membrane cholesterol measurements in mock treated WT and CF HBE cells and in CF HBE cells with VX-809 treatment. A) Left panel: Representative traces of Ussing chamber analyses of WT and CF HBE cells treated with either DMSO carrier or VX-809 (3.3 μM, 24h). Additions of forskolin (10 μM) and VX-770 (1 μM) to stimulate CFTR function and the CFTR inhibitor inh172 (10 μM) are indicated by arrows. Right panel: Summarized data showing change in Isc (ΔIsc) elicited by inh172 treatment to identify magnitude of CFTR response. The number of filters assayed are shown in parentheses. B,C) representative traces of WT and CF HBE cells measured with cholesterol-oxidase modified electrodes or BSA-modified electrodes as a control. D) Ratio of CF/WT cholesterol measurement values with either cholesterol oxidase or BSA modified electrodes. Significance determined by t-test; (n= 6 electrodes tested on 3 cell pairs for CF and WT cells measured with either cholesterol oxidase-modified or BSA-modified electrodes. P < 0.001). E) Summary data of untreated/VX-809 treated WT and CF HBE cells. Significance determined by t-test; (n= 6 electrodes tested on 3 cell pairs for CF and WT; p=0.01). Data are reported as a ratio of VX-809 treated over mock-treated cells.
4.0. Conclusions
Previous work demonstrates reduced microtubule acetylation in CF cells and tissues and that inhibition of HDAC6 restores those acetylation levels (5, 6). We have postulated that the reduction of tubulin acetylation impairs microtubulediated intracellular transport in CF cells. This impairment results in perinuclear cholesterol accumulation in CF cells due to inefficient endosomal transport (3–5, 29). HDAC6 inhibition restores intracellular transport and leads to redistribution of accumulated cholesterol in CF models (5). We have also demonstrated that inhibition of HDAC6 leads to reduced inflammatory signaling in epithelial cells suggesting that cholesterol measurements are related to CF inflammatory signaling (5). The anti-inflammatory benefit of HDAC inhibition has also been confirmed by Bodas et al (30). Previous findings show that impaired cholesterol transport leads to inefficient transport of cholesterol to the endoplasmic reticulum (ER) resulting in enhanced de novo cholesterol synthesis from sterol response element activation (4). We have also identified that detection of cholesterol at the plasma membrane using an electrochemical method is also elevated in CF cells and tissues (8, 9, 26, 31). It was found that this increase was likely related to the increased rate of de novo cholesterol synthesis (8). The hypothesis tested in this study was that restoring intracellular transport in CF cells via HDAC6 inhibition would be detectable as a decrease in available cholesterol for electrochemical detection at the plasma membrane. It was also a goal to demonstrate that the membrane cholesterol measurement could serve as a complement to CFTR functional assays to determine how effectively new therapies are in correcting cellular manifestations of CF.
Data demonstrate that restoration of CFTR expression in an inducible CFTR mouse model restores both tubulin acetylation and WT levels of membrane cholesterol. To test whether this correlation was causative, the impact of HDAC6 inhibition on membrane cholesterol in CF models was tested and found to be corrective. These data help establish that microtubule alterations regulated by HDAC6 mediate many cellular effects of impaired CFTR function and suggest that microtubule regulation may be the link between CFTR function and several secondary phenotypes associated with CF.
Broader benefits of HDAC6 inhibition in a CF context are observed in CF mice where Hdac6 expression has been ablated (CF/HDA). These CF/HDA mice exhibit improved growth and weight gain, as well as restored female fertility compared to CF mice (27). These findings indicate that microtubule acetylation influencing intracellular transport is a key process in pathophysiology of multiple CF phenotypes and that HDAC6 inhibition is an effective approach to restoring those pathways to more WT-like patterns. The ability to detect membrane cholesterol normalization in response to CFTR expression induction and assign an HDAC6-dependent mechanism to that correction suggest that the membrane cholesterol measurement may be a biomarker for the correction of CFTR function and an assessment on the degree of cellular regulation restored by that correction.
The advent of new CFTR-targeted therapies has highlighted the need for effective biomarkers to assess potential efficacy of newly developed compounds. The current markers for evaluating CFTR activation are the nasal potential difference (PD) assay and the sweat chloride test (18). These assays are effective in detecting increased CFTR-mediated chloride transport stimulated by CFTR potentiators and correctors, but there is limited information about efficacy in reversing cellular events associated with CFTR dysfunction or potential efficacy. Having an accessible marker that reflects intracellular responses to CFTR correction may complement sweat chloride measurements to better predict how individual subjects will respond to CFTR-targeted therapies. Now that this study has established that the cholesterol measurement can be made in primary CFBE cells and the mechanism linking CFTR function to cholesterol regulation, future studies will focus on determining the impact of CFTR correctors and potentiators on cholesterol measurements in CFBE cells.
In summary, we have identified that the electrochemical measurement of cholesterol at the plasma membrane can be used to monitor the cellular benefits of Cftr activation using an inducible Cftr mouse model. We have also identified that HDAC6 regulation is the mechanism linking CFTR function to membrane cholesterol regulation and show that inhibition of HDAC6 can normalize CF membrane cholesterol levels in both mouse trachea and primary human nasal epithelial cells. HDAC6 results demonstrate that the plasma membrane cholesterol measurement is related directly to microtubule regulation and the associated intracellular transport issues that impact cholesterol transport as we have previously shown in CF. Data also demonstrate that treatment of F508del CFBE cells with the CFTR corrector VX-809 results in a small but significant decrease in membrane cholesterol. These data are consistent with our previous work that treatment with a CFTR corrector partially restores cholesterol processing in CFBE cells (5). Our previous studies showing that we are able to measure membrane cholesterol levels in human subjects suggest that this technique may be a useful approach to complement direct measures of CFTR function to determine the impact of CFTR-targeted therapies on cellular manifestations of cystic fibrosis. Future studies, however, will have to consider the impact of diet on these measurements. In this study, all mice are on an identical diet limiting variability, but an examination of the effects of fasting and different dietary components on the membrane cholesterol values will need to be completed before clinical studies can be completed.
Highlights.
Membrane cholesterol levels are regulated by CFTR function.
CFTR-mediated control of membrane cholesterol content is dependent on HDAC6 activity.
Activation of F508del CFTR in primary cells restores membrane cholesterol regulation.
Acknowledgments
This work is supported by NIH grants R01EB009481 and 2P30DK027651, and CFF RDP R447-CR11. The authors thank P. Bead for technical assistance.
Footnotes
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Declarations of interest
none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Collins FS, Drumm ML, Cole JL, Lockwood WK, Vande Woude GF, Iannuzzi MC. Construction of a general human chromosome jumping library, with application to cystic fibrosis. Science (New York, NY) 1987;235(4792):1046–9. doi: 10.1126/science.2950591. [DOI] [PubMed] [Google Scholar]
- 2.Drumm ML, Pope HA, Cliff WH, Rommens JM, Marvin SA, Tsui LC, et al. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990;62(6):1227–33. doi: 10.1016/0092-8674(90)90398-x. [DOI] [PubMed] [Google Scholar]
- 3.White NM, Corey DA, Kelley TJ. Mechanistic similarities between cultured cell models of cystic fibrosis and niemann-pick type C. American journal of respiratory cell and molecular biology. 2004;31(5):538–43. doi: 10.1165/rcmb.2004-0117OC. [DOI] [PubMed] [Google Scholar]
- 4.White NM, Jiang D, Burgess JD, Bederman IR, Previs SF, Kelley TJ. Altered cholesterol homeostasis in cultured and in vivo models of cystic fibrosis. American journal of physiology Lung cellular and molecular physiology. 2007;292(2):L476–86. doi: 10.1152/ajplung.00262.2006. [DOI] [PubMed] [Google Scholar]
- 5.Rymut SM, Harker A, Corey DA, Burgess JD, Sun H, Clancy JP, et al. Reduced microtubule acetylation in cystic fibrosis epithelial cells. American journal of physiology Lung cellular and molecular physiology. 2013;305(6):L419–31. doi: 10.1152/ajplung.00411.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rymut SM, Ivy T, Corey DA, Cotton CU, Burgess JD, Kelley TJ. Role of Exchange Protein Activated by cAMP 1 in Regulating Rates of Microtubule Formation in Cystic Fibrosis Epithelial Cells. American journal of respiratory cell and molecular biology. 2015;53(6):853–62. doi: 10.1165/rcmb.2014-0462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 8.Fang D, West RH, Manson ME, Ruddy J, Jiang D, Previs SF, et al. Increased plasma membrane cholesterol in cystic fibrosis cells correlates with CFTR genotype and depends on de novo cholesterol synthesis. Respiratory research. 2010;11:61. doi: 10.1186/1465-9921-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang D, Devadoss A, Palencsar MS, Fang D, White NM, Kelley TJ, et al. Direct electrochemical evaluation of plasma membrane cholesterol in live mammalian cells. Journal of the American Chemical Society. 2007;129(37):11352–3. doi: 10.1021/ja074373c. [DOI] [PubMed] [Google Scholar]
- 10.West RH, Lu H, Shaw K, Chiel HJ, Kelley TJ, Burgess JD. Double Potential Pulse Chronocoulometry for Detection of Plasma Membrane Cholesterol Efflux at Disk Platinum Microelectrodes. Journal of the Electrochemical Society. 2014;161(6):B111–b6. doi: 10.1149/2.005406jes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graeber SY, Dopfer C, Naehrlich L, Gyulumyan L, Scheuermann H, Hirtz S, et al. Effects of Lumacaftor/Ivacaftor Therapy on CFTR Function in Phe508del Homozygous Patients with Cystic Fibrosis. American journal of respiratory and critical care medicine. 2018 doi: 10.1164/rccm.201710-1983OC. [DOI] [PubMed] [Google Scholar]
- 12.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgeworth D, Keating D, Ellis M, Button B, Williams E, Clark D, et al. Improvement in exercise duration, lung function and well-being in G551D-cystic fibrosis patients: a double-blind, placebo-controlled, randomized, cross-over study with ivacaftor treatment. Clinical science (London, England: 1979) 2017;131(15):2037–45. doi: 10.1042/CS20170995. [DOI] [PubMed] [Google Scholar]
- 14.Flume PA, Wainwright CE, Elizabeth Tullis D, Rodriguez S, Niknian M, Higgins M, et al. Recovery of lung function following a pulmonary exacerbation in patients with cystic fibrosis and the G551D-CFTR mutation treated with ivacaftor. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2018;17(1):83–8. doi: 10.1016/j.jcf.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Ronan NJ, Einarsson GG, Twomey M, Mooney D, Mullane D, NiChroinin M, et al. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores After CFTR Modulation With Ivacaftor. Chest. 2017 doi: 10.1016/j.chest.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 16.De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. 2014;10(11):2053–74. doi: 10.4161/15548627.2014.973737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muhlebach MS, Clancy JP, Heltshe SL, Ziady A, Kelley T, Accurso F, et al. Biomarkers for cystic fibrosis drug development. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2016;15(6):714–23. doi: 10.1016/j.jcf.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahrens RC, Standaert TA, Launspach J, Han SH, Teresi ME, Aitken ML, et al. Use of nasal potential difference and sweat chloride as outcome measures in multicenter clinical trials in subjects with cystic fibrosis. Pediatric pulmonology. 2002;33(2):142–50. doi: 10.1002/ppul.10043. [DOI] [PubMed] [Google Scholar]
- 19.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. The New England journal of medicine. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodges CA, Grady BR, Mishra K, Cotton CU, Drumm ML. Cystic fibrosis growth retardation is not correlated with loss of Cftr in the intestinal epithelium. American journal of physiology Gastrointestinal and liver physiology. 2011;301(3):G528–36. doi: 10.1152/ajpgi.00052.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 22.Brady KG, Kelley TJ, Drumm ML. Examining basal chloride transport using the nasal potential difference response in a murine model. American journal of physiology Lung cellular and molecular physiology. 2001;281(5):L1173–9. doi: 10.1152/ajplung.2001.281.5.L1173. [DOI] [PubMed] [Google Scholar]
- 23.Kelley TJ, Cotton CU, Drumm ML. In vivo activation of CFTR-dependent chloride transport in murine airway epithelium by CNP. Am J Physiol. 1997;273(5 Pt 1):L1065–72. doi: 10.1152/ajplung.1997.273.5.L1065. [DOI] [PubMed] [Google Scholar]
- 24.Kelley TJ, Cotton CU, Drumm ML. Regulation of amiloride-sensitive sodium absorption in murine airway epithelium by C-type natriuretic peptide. The American journal of physiology. 1998;274(6 Pt 1):L990–6. doi: 10.1152/ajplung.1998.274.6.L990. [DOI] [PubMed] [Google Scholar]
- 25.Kelley TJ, Thomas K, Milgram LJ, Drumm ML. In vivo activation of the cystic fibrosis transmembrane conductance regulator mutant deltaF508 in murine nasal epithelium. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(6):2604–8. doi: 10.1073/pnas.94.6.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang D, Fang D, Kelley TJ, Burgess JD. Electrochemical analysis of cell plasma membrane cholesterol at the airway surface of mouse trachea. Analytical chemistry. 2008;80(4):1235–9. doi: 10.1021/ac7019909. [DOI] [PubMed] [Google Scholar]
- 27.Rymut SM, Corey DA, Valerio DM, Erokwu BO, Flask CA, Kelley TJ, et al. Improved Growth Patterns in Cystic Fibrosis Mice after Loss of Histone Deacetylase 6. Scientific reports. 2017;7(1):3676. doi: 10.1038/s41598-017-03931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu X, Kelley TJ, Chiel HJ, Burgess JD. Communication-Microelectrode Detection of Cholesterol Efflux from the Human Buccel Mucosa. Journal of the Electrochemical Society. 2016;163(8):B453–b5. doi: 10.1149/2.1001608jes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manson ME, Corey DA, White NM, Kelley TJ. cAMP-mediated regulation of cholesterol accumulation in cystic fibrosis and Niemann-Pick type C cells. American journal of physiology Lung cellular and molecular physiology. 2008;295(5):L809–19. doi: 10.1152/ajplung.90402.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bodas M, Mazur S, Min T, Vij N. Inhibition of histon-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respiratory research. 2018;19(1):2. doi: 10.1186/s12931-017-0705-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang D, Jiang D, Lu H, Chiel HJ, Kelley TJ, Burgess JD. Observation of cellular cholesterol efflux at microcavity electrodes. Journal of the American Chemical Society. 2009;131(34):12038–9. doi: 10.1021/ja903684f. [DOI] [PMC free article] [PubMed] [Google Scholar]