

Abstract
High-risk skin cancer is a rare, but severe, complication associated with discoid lupus erythematosus (DLE). Chronic scar, inflammation, ultraviolet radiation and immunosuppressive medications are proposed explanations for this heightened skin cancer risk; however, the exact mechanism driving skin carcinogenesis in DLE is unknown. The distinct co-localization of multiple independent skin cancers with areas of active inflammation in two DLE patients followed over eight years strongly suggested that lupus inflammation promotes skin carcinogenesis in DLE. To investigate this clinical observation, we subjected lupus-prone MRL/lpr and control (MRL/n) mice to a skin carcinogenesis protocol. Skin tumors developed preferentially within the cutaneous lupus inflammation without scarring in MRL/lpr mice (p < 0.01). The inflammation in MRL/lpr skin was characterized by the accumulation of T regulatory cells, mast cells, M2 macrophages, and markedly elevated TGF-β1 and interleukin (IL)-6 levels, which have been linked to tumor promotion. Importantly, tacrolimus treatment reduced skin inflammation and blocked cancer development in MRL/lpr mice (p = 0.0195). A similar tumor- promoting immune environment was detected in SCCs and the perilesional skin of cancer-prone DLE patients. Therefore, discoid lupus inflammation promotes skin cancer in high-risk DLE patients and blocking the inflammation may be critical for preventing this life-threatening complication of DLE.
Keywords: discoid lupus, chronic inflammation, skin cancer, T regulatory cells, tacrolimus
INTRODUCTION
Discoid lupus erythematosus (DLE) is the most common type of cutaneous lupus and is characterized by scarring lesions with pigmentary changes on the sun-exposed skin (Walling and Sontheimer, 2009). Squamous cell carcinoma (SCC) has been reported in 2.3–3.3% of DLE patients, with substantially higher rates of recurrence and metastasis compared to sporadic SCC (Tao et al., 2012). A four-fold increase in the risk of SCC has been reported in patients with DLE (Grönhagen et al., 2012), which is associated with a 20-fold increase in mortality compared to sporadic SCC (Tao et al., 2012). The poor prognosis of SCC in DLE patients highlights the need to better understand the etiology of skin cancer development in this disease.
Several factors may predispose DLE patients to skin cancer. Ultraviolet (UV) radiation is an established risk factor for SCC and UV exposure triggers lupus flares and DLE lesions occur preferentially on UV-exposed areas of the body (Chen et al., 2013, Fernandez and Kirou, 2016). Immunosuppressive medications used to treat cutaneous lupus, like mycophenolate mofetil, tacrolimus and azathioprine, may promote SCC development by suppressing the antitumor immune response in the skin (Walling and Sontheimer, 2009). However, a similar prevalence of SCC is reported among DLE patients who never received systemic immunosuppressive medications (Tao et al., 2012). Inflammation and scarring are other potential risk factors for skin cancer. Inflammation in DLE lesions is characterized as predominantly T helper 1 (Th1) mediated (Jabbari et al., 2013). Therefore, cancer development within such a tumor-suppressive immune environment, albeit in a small subset of DLE patients, raises the need to determine the nature of inflammation specifically in DLE lesions susceptible to SCC development.
Although there are several potential tumor-inducing factors affecting DLE, the pathophysiology underlying the development of DLE-associated SCC has not been defined. Long-term follow-up of SCC-prone DLE lesions in African American patients suggested the role of inflammation as the promoter of SCC in DLE. We demonstrated DLE inflammation in the absence of scarring and systemic immunosuppression significantly enhanced skin cancer development in mice homozygous for the lymphoproliferation spontaneous mutation (Faslpr) in susceptible MRL/Mp strain (MRL/lpr) (Horiguchi et al., 1987, Kanauchi et al., 1991). Furthermore, we identified an abundance of tumor-promoting immune cells and cytokines in lupus inflammation. We showed that blocking cutaneous lupus inflammation with tacrolimus prevented skin cancer development in MRL/lpr mice accompanied by significant reduction of T regulatory cells (Tregs) in the skin. We conclude that the presence of pro-tumorigenic immune cells and cytokines in a subset of DLE patients can overcome the tumor-suppressing properties of a Th1-mediated immune environment and drive cancer development in DLE.
RESULTS
Skin cancer development and the localization of neoplastic lesions in DLE
A 47-year-old African American female with discoid lupus treated with prednisone, mycophenolate mofetil and acitretin presented with three painful lesions along the peripheral aspects of a depigmented DLE plaque on her right cheek (Figure 1A). Biopsy of the lesions showed SCC in situ (SCCIS) (Figure 1A, 1 and 3) and invasive SCC (Figure 1A, 2). The patient’s facial DLE plaque developed several years prior to her initial presentation and maintained its shape and size during the eight-year follow-up period with widespread depigmentation, active inflammation toward the periphery (depicted in red in Figure 1B), and depressed atrophic scar with telangiectasia most prominent in the center of the lesion (depicted in white in Figure 1B). During eight years of follow-up, patient developed several lesions on this stable DLE plaque that were biopsied to rule out skin cancer. 10 of 11 biopsies that showed neoplastic skin lesions developed at the margins of the DLE plaque where the lupus inflammation was most active (Figure 1B and Supplementary Figure S1). All other biopsies that were diagnosed as active lupus inflammation without scarring (LE) were also located at the margins of the DLE plaque (Figure 1B). Interestingly, skin cancer development exclusively occurred on the depigmented side of the DLE plaque margins on the face, highlighting UV as the main driver of skin cancer initiation. In both skin cancer and perilesional skin of the DLE plaque, no scarring was found, which suggested that chronic scarring did not play a major role in the development of SCC in the DLE lesion (Figure 1C). Instead, we observed marked accumulation of CD45+ leukocytes surrounding the skin cancer compared to normal skin (Figure 1C). Meanwhile, we encountered another African American patient with DLE who developed a skin cancer at the margin of a depigmented DLE plaque on her left cheek (Supplementary Figure S2). Her cancer also developed in active inflammation without scarring (Supplementary Figure S2). These findings point to inflammation as the main driver of cancer development in DLE plaques.
Figure 1. Skin cancer development is associated with areas of active skin inflammation in DLE.
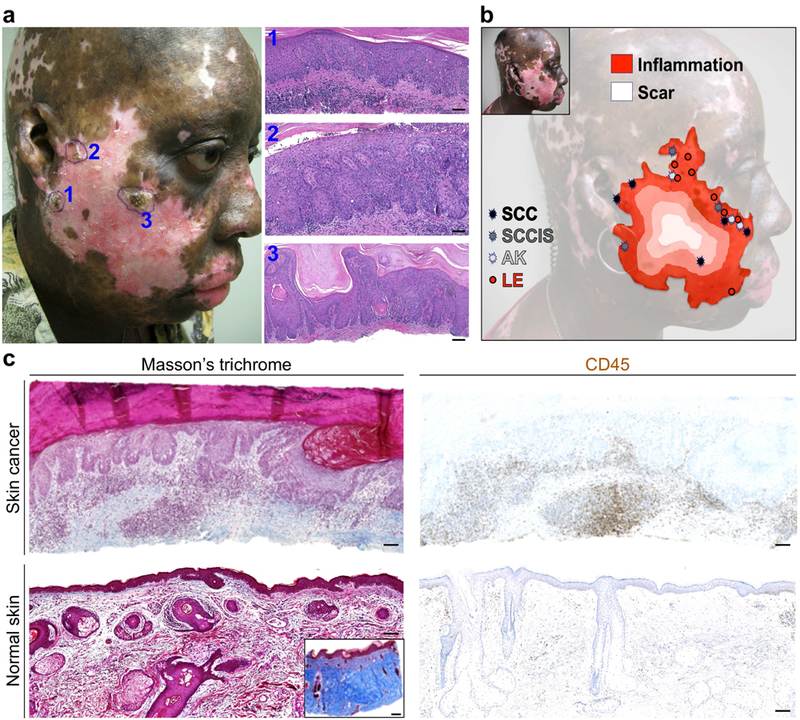
(a) Clinical and pathological images of three independent skin cancers, [1] SCC in situ (SCCIS), [2] SCC and [3] SCCIS, that were diagnosed concurrently at the margins of a depigmented DLE plaque on a patient’s sun-exposed skin are shown. (b) The review of clinical images and pathology reports reveals the locations of neoplastic skin lesions (SCC, SCCIS and AK: actinic keratosis) and active lupus inflammation (LE) that were biopsied to rule out skin cancer during the eight-year follow-up period on the stable DLE plaque. Each lesion is mapped onto a topographic outline of the DLE plaque with the opposite gradients of scarring and inflammation in DLE highlighted in the background. (c) The extent of dermal scarring (Masson’s trichrome stain) and inflammation (CD45 stain) are shown in patient’s skin cancer compared to aged-matched normal facial skin. Note the insert shows positive control for Masson’s trichrome staining in a skin sample with scarring. Cytoplasm and muscle fibers stain red, whereas collagen displays blue coloration; scale bars: 100 µm.
Cutaneous lupus inflammation is associated with skin cancer development
We next examined the tumor-promoting potential of cutaneous lupus inflammation in in lupus-prone MRL/lpr mice compared to their haplotype-matched MRL/n controls (Figure 2). Animals were treated with a single dose of a carcinogen, 7,12-dimethylbenz(a)anthracene (DMBA), on the back skin (Abel et al., 2009, Cipolat et al., 2014, Lopez-Pajares et al., 2013) followed by two treatments with 6 µg of 12-O-tetradecanoylphorbol-13-acetate (TPA). Additionally, mice received UV on their back skin twice weekly for 16 weeks at doses up to 300 mJ/cm2 to induce lupus rash on the upper back skin of MRL/lpr mice (Figure 2A) (Supplementary Figure S3) (Bobe et al., 2006, Furukawa et al., 1996). 9 out of 10 MRL/lpr mice developed one or more skin tumors, whereas 5 out of 10 MRL/n control mice developed tumors (Figure 2B; p = 0.0176, Student’s t test). Most skin tumors in MRL/lpr animals localized to the areas of skin rash and spared the lower back without rash (Figure 2, B and C; p = 0.001, Student’s t test). Marked epidermal hyperplasia, dermal thickening and dense dermal infiltrate was observed in both the MRL/lpr skin tumor and skin rash (Figure 2D). Epidermal hyperplasia observed in MRL/lpr rash was accompanied by increased proliferation of basal and suprabasal keratinocytes, significant accumulation of CD45+ leukocytes, increased CD31+ blood vessels and no scarring compared to MRL/lpr no rash and MRL/n back skin (Supplementary Figure S4). Therefore, cutaneous lupus inflammation without scarring in MRL/lpr mice maintained by chronic UV exposure resulted in a tumor-promoting immune environment in the skin.
Figure 2. Cutaneous lupus inflammation promotes skin cancer development.
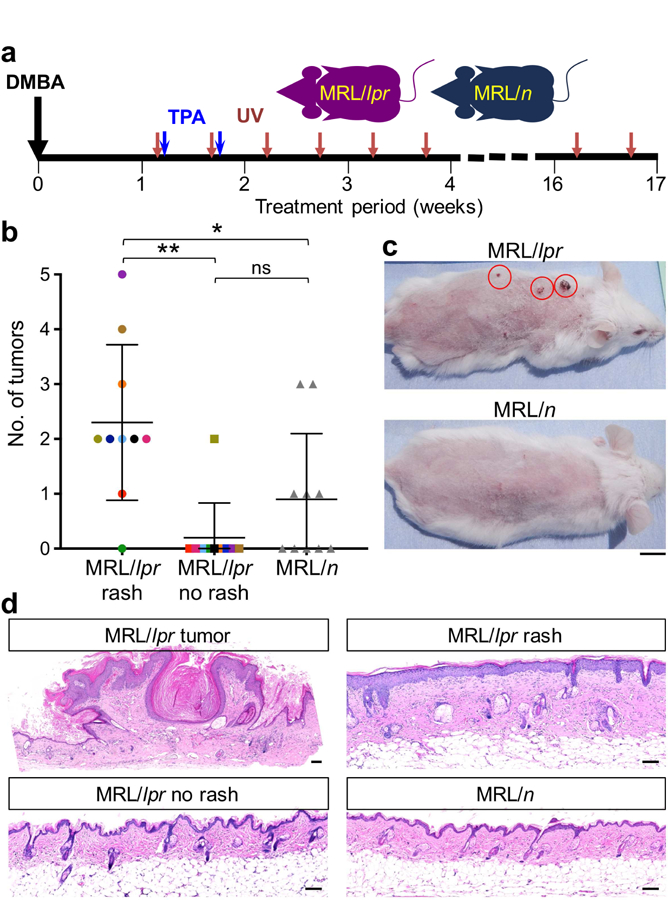
(a) Schematic diagram of the protocol designed to test the role of cutaneous lupus inflammation in skin cancer promotion. All animals received DMBA/TPA to initiate skin carcinogenesis followed by chronic UV exposure in order to induce and maintain skin rash in MRL/lpr mice. (b) Number of tumors developed in MRL/lpr mice on the back skin with rash (upper back), back skin with no rash (lower back) and MRL/n control mice are shown. Colored points highlight the areas of rash and no rash in the same MRL/lpr mouse (*p < 0.05; **p < 0.01 by Student’s t test). (c) Representative images of MRL/lpr and MRL/n mice following skin carcinogenesis protocol are shown. Red circles highlight skin tumors on MRL/lpr skin with rash; scale bar: 1 cm. (d) Representative histological images of skin tumor, skin rash and skin with no rash of MRL/lpr and back skin of MRL/n mice after the completion of skin carcinogenesis protocol are shown; scale bars: 100 µm. Age-matched female mice were used in this study; n = 10 per group.
Cutaneous lupus inflammation is marked by tumor-promoting immune cells and cytokines
Examination of the immune environment of MRL/lpr and MRL/n control mice revealed more activated T cells, as shown by CD44 and PD1 expression, and higher numbers of T-bet+ T helper 1 (Th1), Foxp3+ Tregs and CD8+ T cells in MRL/lpr lymph nodes (Supplementary Figure S5). MRL/lpr skin rash away from the skin tumor sites showed higher number of T cells in close contact with epidermis compared to MRL/lpr no rash and MRL/n control skin (Figure 3A). Most T cells in MRL/lpr rash were CD4+ T cells (Figure 3B). Among CD4+T cell subsets, Foxp3+ Tregs were increased significantly in MRL/lpr skin rash (Figure 3, C and D). In contrast to lymph node, T-bet+ Th1 cells were markedly reduced in MRL/lpr skin rash compared to MRL/lpr no rash and MRL/n control skin (Figure 3, C and D and Supplementary Figure S5). No difference in GATA3+ Th2 cell infiltrate was observed among MRL/lpr rash, MRL/lpr no rash and MRL/n control skin (Figure 3D). These findings suggest that high Treg infiltrate in MRL/lpr skin rash and reduced Th1 cell infiltrate and CD8+ T/Treg ratio play critical roles in tumor promotion by suppressing the antitumor immunosurveillance in cutaneous lupus. In addition, we examined mast cells and M2 macrophages, which also have tumor-promoting potential (Mantovani et al., 2008). Mast cells were significantly increased in the MRL/lpr skin rash compared to both MRL/lpr no rash (p = 0.0227) and MRL/n control skin (Figure 3E, p = 0.014, Student’s t test). Similarly, the number of M2 macrophages was significantly increased in MRL/lpr rash, in comparison to MRL/lpr no rash (p = 0.0077) and MRL/n control skin (Figure 3F, p = 0.0061, Student’s t test). Analysis of spontaneous and UV-induced rash in MRL/lpr mice revealed a similar pattern of CD4+ T cell, mast cell and M2 macrophage induction in these animals as noted in MRL/lpr mice treated with skin carcinogenesis protocol (Supplementary Figure S6). Examination of cytokines associated with tumor-promoting inflammation revealed a significant upregulation of interleukin 6 (IL-6; Supplementary Figure S7, A and B, p < 0.01, Student’s t test) and its downstream STAT3 activation within the keratinocytes of MRL/lpr skin rash compared to MRL/lpr no rash and MRL/n control skin (Supplementary Figure S7C). In addition, cytokines associated with Treg activity, TGF-β1 and IL-10, were markedly upregulated in MRL/lpr rash compared to MRL/lpr no rash and MRL/n control skin (Supplementary Figure S7, p < 0.05, Student’s t test). These findings point to pro-tumorigenic immune environment in MRL/lpr cutaneous lupus.
Figure 3. Tumor-promoting immune cells dominate the immune environment of MRL/lpr skin rash.
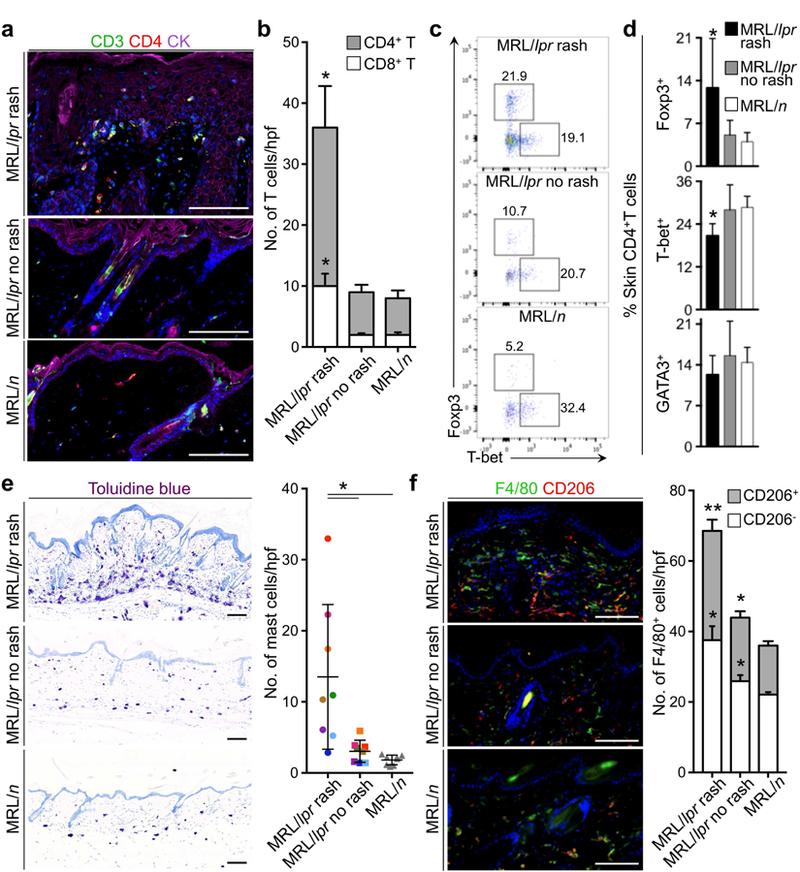
(a) Representative images demonstrate CD4+ T cells in MRL/lpr and MRL/n skin (CK: cytokeratin). (b) CD4+ and CD8+ T cell infiltrates in MRL/lpr rash, no rash and MRL/n control skin are quantified in 10 random high power fields (hpf) per skin section and averaged across the animals in each group. (c) Flow plots show the percentage of Foxp3+ (Treg) and T- bet+ (Th1) CD4+ T cells isolated from MRL/lpr rash, no rash and MRL/n skin. (d) Average % Foxp3+ Tregs, T-bet+ Th1 and GATA3+ Th2 cells in MRL/lpr rash, no rash and MRL/n skin are shown. (e) Representative images of toluidine blue-stained MRL/lpr rash, no rash and MRL/n skin and quantification of Mast cells in 10 random hpf per skin section across the animals in each group are shown. Color-coded points highlight the number of mast cells in rash and no rash skin samples from the same MRL/lpr mouse. (f) Representative images show CD206+ F4/80+ M2 macrophages in MRL/lpr rash, no rash and MRL/n skin and the bar graph demonstrates the average number of macrophages in 10 random hpf per skin section across the animals in the study groups. n = 8 per group; stained cells were counted blindly; *p < 0.05; **p < 0.01 compared to MRL/n control skin by Student’s t test; scale bars: 100 µm.
Tacrolimus treatment reduces lupus inflammation and blocks skin cancer development
To determine whether lupus inflammation is responsible for skin cancer promotion, we examined the effect of blocking inflammation with tacrolimus on skin tumorigenesis in MRL/lpr skin rash (Mok, 2017). Animals were subjected to the skin carcinogenesis protocol and randomized to receive intraperitoneal injection of 1mg/kg tacrolimus or saline (carrier control) three times a week for 9 weeks. During this period, all mice received UV at doses up to 300 mJ/cm2 to induce and maintain lupus rash in saline-treated MRL/lpr control mice (Figure 4A). Tacrolimus treatment markedly reduced lupus rash induced by UV radiation (Supplementary Figure S8) (Furukawa et al., 1995). Six out of eight control mice developed one or more skin tumors, whereas one out of eight tacrolimus-treated mice developed one tumor (Figure 4B; p = 0.0195, Student’s t test). Most tumors in saline-treated MRL/lpr animals localized to skin rash, sparing the lower back skin without rash (Figure 4C). Epidermal hyperplasia and keratinocyte proliferation were reduced in tacrolimus-treated MRL/lpr skin (Figure 4, D and E). Tacrolimus treatment markedly reduced CD4+ T cell infiltrate in MRL/lpr skin rash with no impact on skin- infiltrating CD8+ T cells compared to saline-treated MRL/lpr control skin (p < 0.01, Student’s t test; Figure 4, F and G). Furthermore, quantification of the CD4+ T cell subtypes in MRL/lpr skin using flow cytometry (Supplementary Figure S9) revealed a significant reduction in Foxp3+ Tregs in tacrolimus-treated MRL/lpr skin compared to saline-treated MRL/lpr skin (p < 0.01, Student’s t test; Figure 4H). These findings demonstrate that cutaneous lupus inflammation is required for skin cancer promotion in MRL/lpr mice.
Figure 4. Tacrolimus inhibits tumor development in the context of lupus-associated inflammation.
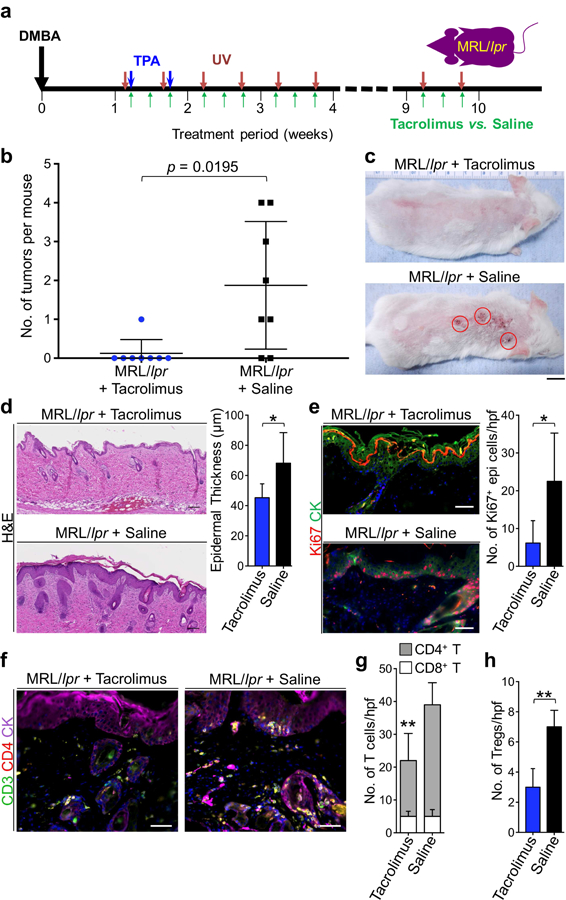
(a) Schematic diagram of the protocol used to test the effect of tacrolimus on skin cancer promotion in the setting of chronic cutaneous lupus inflammation. All mice received DMBA/TPA to initiate skin carcinogenesis followed by UV to induce and maintain skin rash. (b) Number of tumors developed in MRL/lpr mice receiving tacrolimus is compared to the number of tumors developed in MRL/lpr control mice receiving saline (p = 0.0195 by Student’s t test). (c) Representative images of tacrolimus- and saline-treated MRL/lpr mice that completed carcinogenesis protocol are shown. (d) Representative H&E stains of skin rash in MRL/lpr mice treated with tacrolimus or saline are shown. Epidermal thickness is determined based on measurements obtained from 10 random areas of skin per mouse. (e) Representative Ki67 and cytokeratin immunofluorescent staining of tacrolimus- and saline-treated MRL/lpr mice are shown. Bar graph demonstrates the average number of Ki67+ epidermal keratinocytes per hpf in skin rash from tacrolimus-treated MRL/lpr mice compared to that of saline-treated MRL/lpr mice. (f) Representative CD3, CD4 and cytokeratin stainings of skin rash from tacrolimus- and saline- treated MRL/lpr mice are shown. (g) The average number of CD4+ and CD8+ T cell infiltrates is quantified by counting the number of cells in 10 random hpf per skin rash section and averaging the counts across the tacrolimus-treated compared to saline-treated MRL/lpr mice. (h) Bar graph shows the average number of Foxp3+ Tregs in the skin of tacrolimus-treated and saline- treated MRL/lpr mice as calculated using the histological counts of CD4+ T cells in the skin and the percentage of these cells that are Foxp3+ on flow cytometry (Supplementary Figure S9). n = 8 per group; stained cells were counted blindly; *p < 0.05; **p < 0.01 compared to saline-treated MRL/lpr control skin rash by Student’s t test; scale bars: 100 µm.
Tumor-promoting immune environment is present in cancer-prone DLE lesions
We examined the immune environment surrounding the skin cancers that developed in our clinical cases as well as perilesional skin (away from the cancer sites) of the same patients, DLE lesions from three African American patients with no skin cancer history (control DLE, Supplementary Figure S10) and normal facial skin from age-matched individuals. Massive infiltration of CD3+ T cells was detected in skin cancer, perilesional skin in cancer-prone DLE and control DLE lesions, which primarily consisted of CD4+ T cells (Figure 5A and Supplementary Figure S11). Although skin cancer, perilesional skin in cancer-prone DLE and control DLE lesions contained a higher number of CD8+ T cells compared to normal skin, we discovered a significant reduction in CD8+ T cell infiltrate in skin cancer and perilesional skin of cancer-prone DLE compared to control DLE lesions (Figure 5, A and B and Supplementary Figure S11). A substantial number of CD4+ T cells in skin cancer and perilesional skin in cancer- prone DLE consisted of Foxp3+ Tregs (Figure 5, A and B and Supplementary Figure S11). Interestingly, Treg infiltrates were significantly lower in control DLE lesions compared to skin cancer and perilesional skin in cancer-prone DLE (Figure 5, A and B). M2 macrophages were significantly increased in skin cancer, perilesional skin in cancer-prone DLE and control DLE lesions compared to normal skin (Figure 5, A and B and Supplementary Figure S11). In addition, mast cells were increased in skin cancer of DLE patients (Supplementary Figure S11 and S12). Consistent with the role of pro-tumorigenic cytokine signaling in cutaneous lupus- associated tumor promotion in mice, the patients’ skin cancers and perilesional skin showed an increase in nuclear phospho-STAT3 staining in keratinocytes, which validated STAT3 signaling upregulation in cancer-prone DLE skin (Supplementary Figure S11 and S12). Together, the infiltration of Tregs, markedly reduced CD8+ T/Treg ratio (1.5 in perilesional skin of cancer- prone DLE compared to 7.3 in control DLE lesions), increased M2 macrophages, mast cells and, STAT3 pathway activation in keratinocytes of skin cancer and perilesional skin of our DLE patients constituted a bonafide tumor-promoting immune environment in cancer-prone DLE plaques.
Figure 5. Patient’s DLE plaque margins are infiltrated by tumor-promoting immune cells surrounding the sites of skin cancer development.
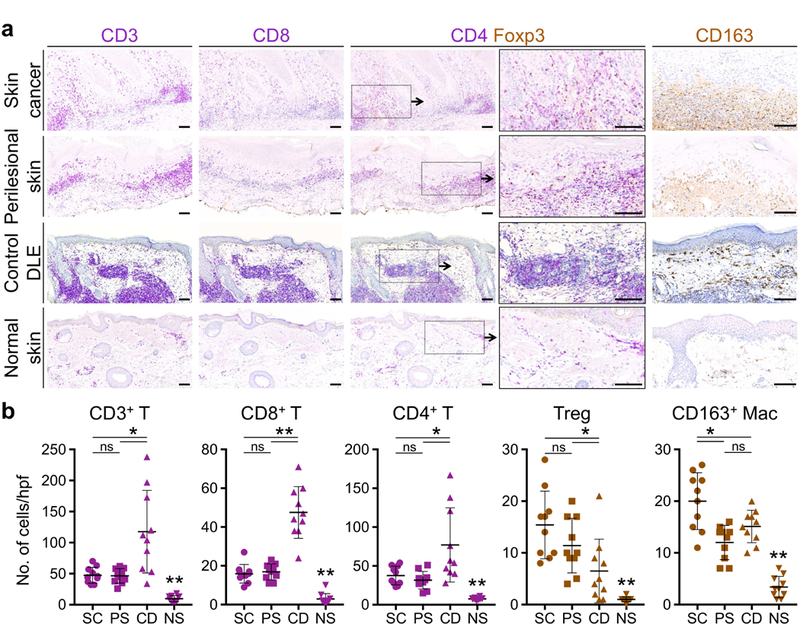
(a) T cell and macrophage accumulation in the skin cancer and perilesional skin of cancer-prone DLE lesion is compared to DLE lesions from three cancer-free African American patients (control DLE) and an age-matched normal facial skin as shown by representative images of CD3, CD8, CD4/Foxp3 and CD163-stained tissue sections. (b) Average number of CD3+ T, CD8+ T, CD4+ T, Foxp3+ Tregs and CD163+ M2 macrophage are determined by counting the cells in 10 random hpf of the skin cancer (SC), perilesional skin of DLE (PS), normal skin (NS) excision sections, and 10 random hpf across punch biopsy specimen from three independent control DLE lesions (CD; Supplementary Figure 10). Stained cells were counted blindly; ns: not significant, *p < 0.05, **p < 0.0001 by Student’s t test; scale bars: 100 µm.
DISCUSSION
Our clinical and experimental findings provide critical insights into the etiology of SCC development in susceptible DLE patients. Skin cancer develops through a stepwise process involving cancer initiation and cancer promotion (Abel et al., 2009). UV exposure likely initiates skin cancer by inducing oncogenic mutations as all neoplastic lesions in our patients developed in depigmented sites. However, UV radiation does not appear to play a major role in the promotion of DLE-associated skin cancers since depigmentation spreads from the center of DLE lesions as they grow out from papules to plaques over time. If UV radiation also acted as a tumor promoter, neoplastic lesions would have been expected to develop mostly in the center of the depigmented plaque, which had the longest exposure to UV radiation. This central pattern of SCC development would have also been expected if chronic scar played a significant role in cancer promotion because the center of DLE plaques has the highest degree of scarring (Walling and Sontheimer, 2009). In contrast, if immunosuppressants were the cause of tumor promotion in DLE, an even distribution of cancerous lesions across the DLE plaques would be predicted. However, most skin cancers developed at the periphery of the depigmented DLE plaques co-localizing with the areas of active inflammation. In addition, the majority of the murine tumors co-localized with skin rash, suggesting chronic inflammation is the main promoter of skin carcinogenesis in cutaneous lupus.
The role of inflammation as the dominant promoter of skin cancer in DLE is supported by increased pro-tumorigenic immune infiltrates consisting of Tregs, mast cells and M2 macrophages in MRL/lpr skin rash and perilesional skin of the DLE plaques. MRL/lpr mice suffer from systemic autoimmunity and lymphoproliferative disease and their cutaneous lupus lesions lacks the scaring seen in DLE (Cohen and Eisenberg, 1991). Nonetheless, MRL/lpr cutaneous lupus inflammation that resembled cancer-prone DLE immune environment in our patients promoted skin tumor development. Among the immune cells that infiltrated the perilesional skin of the DLE plaques, CD8+ T cell are commonly associated with antitumor immunity (Vesely and Schreiber, 2013). However, CD8+ T cell infiltrate detected in the skin cancer and perilesional skin of the DLE plaques may function to augment the tumor-promoting effect of lupus inflammation (Kwong et al., 2010, Roberts et al., 2007). In addition to immune cells, highly elevated pro-tumorigenic cytokines like IL-6 and its downstream STAT3 signaling activation within the keratinocytes in MRL/lpr skin rash and perilesional skin in DLE plaques establish a strong tumor-promoting immune axis, which is also implicated in several other types of solid cancers (Bollrath et al., 2009, Bromberg and Wang, 2009, Sansone et al., 2007, Schafer and Brugge, 2007, Zhang et al., 2012). Tacrolimus is an immunosuppressive mediation and a calcineurin inhibitor, which can block antitumor immunity and directly promote skin carcinogenesis (Wu et al., 2010). Therefore, the significant reduction in skin cancer susceptibility with tacrolimus treatment in MRL/lpr mice strongly supports the dominant role of lupus inflammation in driving skin cancer development in cutaneous lupus. The immune environments of MRL/lpr skin rash and the DLE plaques of cancer-prone patients are immunosuppressive against cytotoxic immune cells including Th1 and CD8+ T cells. These observations stand in contrast to a Th1 dominant immune environment described in common DLE lesions (Jabbari et al., 2013). No patient in this published study, which included African American and Caucasian patients, had any history of skin cancer ((Jabbari et al., 2013) and personal communication). Considering that only 2.3–3.3% of DLE patients develop skin cancer and the susceptible patients continue to develop multiple new skin cancers over time (Tao et al., 2012), the tumor-promoting immune environment we find in cancer-prone DLE patients may be distinct from the skin inflammation detected in the majority of DLE patients who are not at increased risk of developing skin cancer. Interestingly, the comparison of skin cancer and perilesional skin in our cancer-prone DLE patients to control DLE lesions in African American patients with no history of skin cancer demonstrated a significant increase in Tregs and marked reduction in CD8+ T cells in cancer-prone DLE lesions. These differences support a switch from Th1-dominant immunity in common DLE lesions to a tumor-promoting immune environment in the high-risk subset of DLE.
Immunosuppressive medications increase the risk of skin cancer in solid organ transplant recipients (Chockalingam et al., 2015). However, the efficacy of tacrolimus in blocking skin cancer in lupus-prone mice demonstrates the dominant role of cutaneous DLE inflammation as the driver of skin cancer. The effect of tacrolimus on reducing skin-infiltrating CD4+ T cells and Tregs highlights the critical role of these T cells in inducing tumor development while suppressing the antitumor immune response in DLE. Tregs can enhance tumorigenesis by producing growth factors (Zaiss et al., 2013) and stimulating other tumor-promoting immune cell types (Takeuchi and Nishikawa, 2016). For example, Tregs stimulate mast cells to produce IL-6 via TGF-β1 (Ganeshan and Bryce, 2012), which in turn induces keratinocyte proliferation through the activation of JAK-STAT3 pathway. DLE lesions contain significantly higher numbers of infiltrating mast cells as compared to acute forms of cutaneous lupus (Kaczmarczyk-Sekula et al., 2015). In addition, Tregs suppress antitumor immune responses through the release of cytokines like IL-10 and TGF-β. The role that each of these cytokines play in DLE-associated skin cancer will be elucidated in future studies.
Although only a small percentage of the DLE patients develop skin cancer, SCCs that arise within DLE lesions are associated with high risk of recurrence, early metastasis and mortality in contrast to common forms of SCC, which usually have a much better prognosis (Shapera and Kim, 2016). Furthermore, cancer development in the context of chronic inflammation is an important complication seen in other, more prevalent inflammatory conditions, such as hypertrophic lichen planus (Kutlubay et al., 2009, Singh et al., 2006), inflammatory bowel disease (IBD), viral hepatitis and human papilloma virus (HPV)-induced cervicitis (Multhoff et al., 2011). This association between cancer development and chronic inflammation underscores the importance of developing innovative immune-targeting therapies against cancer in DLE and other pro-tumorigenic inflammatory diseases. For instance, the induction of a Treg-dominant infiltrate in the inflamed skin as described in our DLE studies plays a crucial role in other cancer- prone chronic inflammatory diseases (Kryczek et al., 2011). Therefore, blocking Tregs and signaling pathways that recruit them to the site of chronic inflammation may prove effective in the prevention and treatment of cancer in high-risk DLE and other chronic inflammatory diseases.
MATERIALS AND METHODS
Human samples
The biopsies of the lesions and adjacent skin were fixed in formalin and embedded in paraffin. Formalin-fixed paraffin-embedded tissue sections (4 µm) were stained with hematoxylin and eosin (H&E) and Masson’s trichrome stains (Sigma-Aldrich) according to the manufacturer’s protocol. Immunostainings were performed using a Ventana Ultra automated immunostainer (Ventana Medical Systems, Tucson, AZ). Stained cells were counted blindly. Refer to supplemental material for the list of antibodies (Supplementary Table 1).
Mice
All mice were housed under pathogen-free conditions in an animal facility at Massachusetts General Hospital in accordance with animal care regulations. MRL/lpr and MRL/n mice were purchased from the Jackson Laboratory (Bar Harbor, ME).
Statistical analysis
Two-tailed Student’s t-test was used as the test of significance for tumor counts, RNA and protein expression levels and other quantitative measurements. p value < 0.05 is considered significant. All the bar graphs show mean + SD.
Study approval
Washington University School of Medicine IRB approved the clinical studies. Written informed consent was obtained from patients including the permission to obtain and publish their photographs and study their skin cancers. Massachusetts General Hospital IACUC approved the animal studies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Barbara Gilchrest and Dr. Ethan Lerner for critically reading the manuscript. We thank Dr. Lynn Cornelius and Mary Tabacchi for assistance with the clinical study. Shadmehr Demehri, M.D., Ph.D., holds a Career Award for Medical Scientists award from the Burroughs Wellcome Fund. AHA is supported by Howard Hughes Medical Institute. AZ, SMT, AHA, TJC, KHN, SD were supported by grants from the Burroughs Wellcome Fund, Sidney Kimmel Foundation, Cancer Research Institute and NIH (5K08AR068619 and DP5OD021353091).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTERESTS
MJA is a consultant for Biogen and Adgero and study investigator for Vanda, Biogen and Veloce. Other authors declare no competing financial interests.
REFERENCES
- Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nature protocols 2009;4(9):1350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobe P, Bonardelle D, Benihoud K, Opolon P, Chelbi-Alix MK. Arsenic trioxide: A promising novel therapeutic agent for lymphoproliferative and autoimmune syndromes in MRL/lpr mice. Blood 2006;108(13):3967–75. [DOI] [PubMed] [Google Scholar]
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130- mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009;15(2):91–102. [DOI] [PubMed] [Google Scholar]
- Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009;15(2):79–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, Halliday GM, Damian DL. Non-melanoma skin cancer: carcinogenesis and chemoprevention. Pathology 2013;45(3):331–41. [DOI] [PubMed] [Google Scholar]
- Chockalingam R, Downing C, Tyring SK. Cutaneous Squamous Cell Carcinomas in Organ Transplant Recipients. Journal of clinical medicine 2015;4(6):1229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Hoste E, Natsuga K, Quist SR, Watt FM. Epidermal barrier defects link atopic dermatitis with altered skin cancer susceptibility. eLife 2014;3:e01888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annual review of immunology 1991;9:243–69. [DOI] [PubMed] [Google Scholar]
- Fernandez D, Kirou KA. What Causes Lupus Flares? Curr Rheumatol Rep 2016;18(3):14. [DOI] [PubMed] [Google Scholar]
- Furukawa F, Imamura S, Takigawa M. FK506: therapeutic effects on lupus dermatoses in autoimmune-prone MRL/Mp-lpr/lpr mice. Archives of dermatological research 1995;287(6):558–63. [DOI] [PubMed] [Google Scholar]
- Furukawa F, Kanauchi H, Wakita H, Tokura Y, Tachibana T, Horiguchi Y, et al. Spontaneous autoimmune skin lesions of MRL/n mice: autoimmune disease-prone genetic background in relation to Fas-defect MRL/1pr mice. The Journal of investigative dermatology 1996;107(1):95–100. [DOI] [PubMed] [Google Scholar]
- Ganeshan K, Bryce PJ. Regulatory T cells Enhance Mast Cell Production of IL-6 via Surface- bound TGFβ(). J Immunol 2012;188(2):594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönhagen CM, Fored CM, Granath F, Nyberg F. Increased risk of cancer among 3663 patients with cutaneous lupus erythematosus: a Swedish nationwide cohort study. Br J Dermatol 2012;166(5):1053–9. [DOI] [PubMed] [Google Scholar]
- Horiguchi Y, Furukawa F, Ohshio G, Horio T, Imamura S. Effects of ultraviolet light irradiation on the skin of MRL/l mice. Archives of dermatological research 1987;279(7):478–83. [DOI] [PubMed] [Google Scholar]
- Jabbari A, Suarez-Farinas M, Fuentes-Duculan J, Gonzalez J, Cueto I, Franks AG, Jr., et al. Dominant Th1 and Minimal Th17 Skewing in Discoid Lupus Revealed by Transcriptomic Comparison with Psoriasis. The Journal of investigative dermatology 2013. [DOI] [PMC free article] [PubMed]
- Kaczmarczyk-Sekula K, Dyduch G, Kostanski M, Wielowieyska-Szybinska D, Szpor J, Bialas M, et al. Mast cells in systemic and cutaneous lupus erythematosus. Pol J Pathol 2015;66(4):397–402. [DOI] [PubMed] [Google Scholar]
- Kanauchi H, Furukawa F, Imamura S. Characterization of cutaneous infiltrates in MRL/lpr mice monitored from onset to the full development of lupus erythematosus-like skin lesions. The Journal of investigative dermatology 1991;96(4):478–83. [DOI] [PubMed] [Google Scholar]
- Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol 2011;186(7):4388– 95. [DOI] [PubMed] [Google Scholar]
- Kutlubay Z, Kocaturk E, Demirkesen C, Kavala M, Sarigul S, Zindanci I. Squamous cell carcinoma arising from hypertrophic lichen planus. European journal of dermatology: EJD 2009;19(2):175–6. [DOI] [PubMed] [Google Scholar]
- Kwong BY, Roberts SJ, Silberzahn T, Filler RB, Neustadter JH, Galan A, et al. Molecular analysis of tumor-promoting CD8+ T cells in two-stage cutaneous chemical carcinogenesis. The Journal of investigative dermatology 2010;130(6):1726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pajares V, Yan K, Zarnegar BJ, Jameson KL, Khavari PA. Genetic pathways in disorders of epidermal differentiation. Trends in genetics: TIG 2013;29(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454(7203):436–44. [DOI] [PubMed] [Google Scholar]
- Mok CC. Calcineurin inhibitors in systemic lupus erythematosus. Best Pract Res Clin Rheumatol 2017;31(3):429–38. [DOI] [PubMed] [Google Scholar]
- Multhoff G, Molls M, Radons J. Chronic Inflammation in Cancer Development. Frontiers in Immunology 2011;2. [DOI] [PMC free article] [PubMed]
- Roberts SJ, Ng BY, Filler RB, Lewis J, Glusac EJ, Hayday AC, et al. Characterizing tumor- promoting T cells in chemically induced cutaneous carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America 2007;104(16):6770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. The Journal of Clinical Investigation 2007;117(12):3988–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ZT, Brugge JS. IL-6 involvement in epithelial cancers. The Journal of Clinical Investigation 2007;117(12):3660–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapera EA, Kim PD. Squamous Cell Cancer Arising in an African American Male Cheek from Discoid Lupus: A Rare Case and Review of the Literature. Case Reports in Surgery 2016;2016. [DOI] [PMC free article] [PubMed]
- Singh SK, Saikia UN, Ajith C, Kumar B. Squamous cell carcinoma arising from hypertrophic lichen planus. J Eur Acad Dermatol Venereol 2006;20(6):745–6. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. International immunology 2016;28(8):401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Zhang X, Guo N, Chen S, Huang C, Zheng L, et al. Squamous cell carcinoma complicating discoid lupus erythematosus in Chinese patients: review of the literature, 1964–2010. Journal of the American Academy of Dermatology 2012;66(4):695–6. [DOI] [PubMed] [Google Scholar]
- Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Annals of the New York Academy of Sciences 2013;1284:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus: issues in diagnosis and treatment. Am J Clin Dermatol 2009;10(6):365–81. [DOI] [PubMed] [Google Scholar]
- Wu X, Nguyen BC, Dziunycz P, Chang S, Brooks Y, Lefort K, et al. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010;465(7296):368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Grone A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 2013;38(2):275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yang J, Qian J, Li H, Romaguera JE, Kwak LW, et al. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood 2012;120(18):3783–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.