

Abstract
Photoacoustic (PA) imaging allows visualization of physiology and pathology of tissues with good spatial resolution and relatively deep tissue penetration. The method converts NIR laser excitation into thermal expansion, generating pressure transients that are detected with an acoustic transducer. Here we find that the response of the PA contrast agent indocyanine green (ICG) can be enhanced 17-fold when it is sealed within a rigid nanoparticle. ICG encapsulated in particles composed of porous silicon (pSiNP), porous silica, or calcium silicate all show greater PA contrast relative to equivalent quantities of free ICG, with the pSiNPs showing the strongest enhancement. A liposomal formulation of ICG performs similar to free ICG, suggesting that a rigid host nanostructure is necessary to enhance ICG performance. The improved response of the nanoparticle formulations is attributed to the low thermal conductivity of the porous inorganic hosts and their ability to protect the ICG payload from photolytic and/or thermal degradation. The translational potential of ICG-loaded pSiNPs as photoacoustic probes is demonstrated via whole mouse brain imaging.
Keywords: Indocyanine green, contrast agent, in vivo imaging, ultrasound, brain imaging
Graphical Abstract
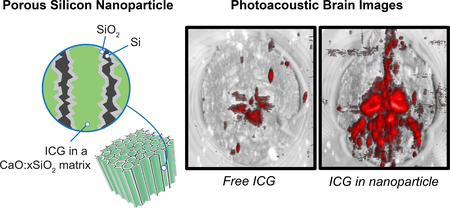
The photoacoustic response of indocyanine green (ICG) is substantially enhanced when it is sealed within rigid porous nanoparticles such as porous silicon, porous silica, or calcium silicate. The enhanced photoacoustic contrast is attributed to the low thermal conductivity of the porous hosts and their ability to protect the ICG payload against photochemical degradation.
Photoacoustic (PA) imaging is a promising biomedical tool that addresses the resolution and depth limits of optical imaging.[1-3] In PA imaging, laser pulses are directed at tissues containing a molecular or nanoparticle chromophore with a strong optical absorbance at the laser wavelength, typically in the near infrared (NIR) region of the spectrum. Non-radiative relaxation converts the optical excitation into a thermoacoustic wave[1, 4, 5] which is detected with audio transducers and reconstructed into a three-dimensional image.[6] Since light is used only to excite the target molecules and the generated acoustic waves are less efficiently scattered than photons in biological tissues, images can be obtained at greater depths and with higher spatial resolution than can be achieved with more conventional fluorescence imaging.
Materials exhibiting a high extinction coefficient at near infrared (NIR) wavelengths and efficient conversion of optical excitation into heat are used as contrast agents for PA imaging.[7-9] One of the more common PA imaging agents is indocyanine green (ICG), an FDA-approved fluorophore with a strong NIR absorbance. One of the challenges of organic imaging agents such as ICG is photochemical degradation, which irreversibly reduces the PA response and limits the quality of images that can be obtained. Metallic nanoparticles with strong plasmon bands in the NIR have been proposed to address this challenge because they are substantially more stable toward photolytic degradation. Another potential advantage of using nanoparticles as PA contrast agents is that they can incorporate affinity agents such as peptides, aptamers, or antibodies[10, 11] to better target the tissues of interest. The PA response of a nanoparticle contrast agent has been found to increase substantially if it is thermally isolated from the surrounding aqueous matrix, because this generates a larger temperature differential immediately after the laser pulse. Thus core-shell nanoparticles composed of gold nanorod cores and silica shells show increased image contrast relative to “bare” gold nanorods.[12] In this study, we reasoned that the same approach may be applicable to molecular contrast agents, and sought to improve the PA response of ICG by trapping it in a nanoparticle of relatively low thermal conductivity. We further reasoned that ICG would be less susceptible to photolytic degradation when trapped in a nanoparticle host. Porous silicon, porous silica, and calcium silicate materials were tested as hosts for ICG. All three materials have low thermal conductivity and have been studied extensively as in vivo biomaterials due to their low toxicity and superior biocompatibility[13-15] relative to metallic nanoparticles--an important consideration in translation to human diagnostic applications.[16-18]
For the porous silicon nanoparticles (pSiNPs), ICG was trapped within the porous matrix using a calcium silicate capping chemistry previously developed for oligonucleotide payloads.[19] The pSiNPs were prepared by electrochemical anodization of highly doped p-type single-crystal silicon wafers in an aqueous ethanolic electrolyte containing hydrofluoric acid and ultrasonic fracture of the resulting porous silicon layer (see Experimental Section).[20] ICG was then loaded and sealed into the pSiNPs by means of calcium silicate precipitation chemistry to generate the Ca-pSiNP-ICG composites (Figure 1a).[19] TEM images of the pSiNPs prior to loading displayed the open pore structure characteristic of this material, with pore diameters on the order of 20 nm (Figure 1b). After the loading and sealing process, the pores in the Ca-pSiNP-ICG composite appeared to be filled (Figure 1c), consistent with prior results.[19] Residual nanocrystalline Si was evident in the Raman spectrum from the observation of a lattice mode[21] at 510 cm−1, although this band was somewhat broadened on the low energy side relative to the pSiNP starting material (Figure S1a, Supporting Information), indicative of some loss of crystallinity. The presence of crystalline silicon was confirmed by powder X-ray diffraction (XRD) measurements (Figure S1b, Supporting Information). The Ca-pSiNP-ICG nanoparticles had an average particle size of (170 nm by dynamic light scattering, Table 1) that was somewhat larger than the pSiNP starting material, and nitrogen adsorption measurements indicated a decrease in surface area associated with the pore filling and sealing process (Figure S2, Supporting Information). The Ca-pSiNP-ICG nanoparticles readily dispersed in water or ethanol and displayed the distinctive green color of ICG compared to the brownish appearance of pSiNPs (Figure 1d). Bands characteristic of ICG were observed in the optical absorbance spectrum (λabs = 790 nm) and also in the Fourier transform infrared (FTIR) spectrum (Figure S2, Supporting Information). The mass loading of ICG was determined by thermogravimetric analysis (TGA) to be 11%, (Figure S2, Supporting Information). The fluorescence from ICG (λem = 820 nm) was quenched by 60% in the Ca-pSiNP-ICG formulation. This can be attributed to energy transfer quenching by the silicon skeleton of the pSiNPs (Figure S3, Supporting Information), although we cannot rule out a contribution from self-quenching due to aggregation of the dye within the pSiNPs.[22, 23] The electrospray ionization mass spectrum of the material released from Ca-pSiNP-ICG into aqueous solution displayed a parent ion at m/z = 751 (Figure S4, Supporting Information), confirming the presence of ICG in the nanoparticles.
Figure 1. Preparation and properties of ICG-containing porous Si nanoparticles.

(a) Procedure used to simultaneously load and seal ICG into the pores of porous silicon nanoparticles (pSiNPs). When pSiNP is incubated in a solution containing ICG and a high concentration of aqueous calcium ion, silicon from the dissolving nanoparticle is locally converted to calcium silicate, sealing the pores and trapping ICG in the nanostructure. The resulting composite of calcium silicate, porous silicon, and indocyanine green is designated Ca-pSiNP-ICG. (b) Transmission electron microscope (TEM) image of the pSiNP starting material, prior to loading. (c) TEM image of Ca-pSiNP-ICG. Scale bars are 100 nm. (d) Photograph of pSiNPs (left) and Ca-pSiNP-ICG (right) dispersed in ethanol (1 mg/mL, based on particle mass). (e) Comparative photoacoustic spectra of ethanol solutions of Ca-pSiNP-ICG, free ICG at concentrations of 150, 100, and 50 μg/mL as indicated, control Ca-pSiNPs that did not contain any loaded ICG, and pure ethanol. The traces represent intensity of the photoacoustic signal as a function of probe laser wavelength. (f) Photoacoustic image overlaid on the ultrasound image with a maximum intensity projection. Tubes contain (from left to right): pure ethanol solvent, Ca-pSiNP, Ca-pSiNP-ICG, and various concentrations of ICG as indicated. All solutions made from ethanol. The black to white and red to white intensity bars correspond to ultrasound and photoacoustic intensity, respectively. Scale bar is 3 mm. The concentration of ICG in the Ca-pSiNP-ICG samples of (e) and (f), quantified by the characteristic optical absorbance of ICG at λabs = 790 nm, corresponds to 15 ± 5 μg/mL.
Table 1.
Summary of properties and relative photoacoustic efficiency of ICG contrast agents studied.
| Absorbancea | Size, nmb | Mass loading of ICG (%)c | PA efficiencyd | |
|---|---|---|---|---|
| ICG 20 μg/mL | 0.7416 | N/A | N/A | 1 |
| Ca-pSiNP-ICG | 0.3518 | 168 | 11.0 | 17 |
| Ca-Silica-ICG | 0.3753 | 146 | 2.6 | 15 |
| CaS-ICG | 0.4524 | 194 | 16.5 | 5 |
| Lip-ICG | 0.6563 | 121 | 57.7 | 1 |
Optical absorbance measured at λ = 790 nm in ethanol (DI water for Lip-ICG). ICG content in each nanoparticle was determined from the optical absorbance at 790 nm. Empty nanoparticles displayed negligible absorbance at this wavelength.
Average nanoparticle diameter, obtained from DLS measurement. Errors ± 5%
Mass loadings determined from TGA analysis (Ca-pSiNP-ICG and Ca-Silica-ICG) or by optical absorbance (CaS-ICG and Lip-ICG).
Relative photoacoustic generation efficiency defined as ratio of SNR to absorbance at λ = 790 nm, normalized to the measured value for free ICG in 20 μg/mL ethanol solution.
The photoacoustic response of Ca-pSiNP-ICG was compared to free ICG (15−150 μg/mL) by scanning tubes containing solutions of the relevant contrast agent with 790 nm pulsed laser irradiation. The effective concentration of ICG in the Ca-pSiNP-ICG samples was 15 ± 5 μg/mL (Figure 1 and Figure S5, Supporting Information). The Ca-pSiNP-ICG formulation produced a photoacoustic signal that was significantly (p < 0.01) greater than for samples containing comparable or even much larger concentrations of free ICG. The signal was 8-fold larger than that from a solution of free ICG at a concentration of 10 μg/mL (Figure S6, Supporting Information), and the signal was also significantly (p < 0.01) stronger than that observed from the highest concentration of free ICG studied (150 μg/mL). The PA measurements were replicated on four independent Ca-pSiNP-ICG preparations (Figure S7, Supporting Information).
To evaluate the factors involved in enhancing the PA signal, we prepared and tested three other nanoparticle types containing encapsulated ICG: a porous calcium silicate nanoparticle (CaS-ICG), a microporous silica nanoparticle sealed with calcium silicate (Ca-Silica-ICG), and a liposomal nanoparticle (Lip-ICG) (Table 1). The ICG was incorporated into each of the nanoparticles during their synthesis. The CaS-ICG nanoparticles were prepared by mixing ICG and CaCl2 solution (2 M) with silicic acid (see Experimental Section and Figure S8, Supporting Information). The Ca-Silica-ICG samples were prepared by mixing commercial microporous silica nanoparticles (pore size < 2 nm) with ICG and CaCl2 solution (Figure S9, Supporting Information). These nanoparticles displayed different pore dimensions and ICG loading efficiency, but they had average diameters similar to the pSiNPs used in this work (between 120 and 200 nm, Table 1). For this comparison, the concentration of each nano-formulation was adjusted to display a similar optical absorbance at 790 nm (the absorbance band of ICG), which was approximately equal to the absorbance displayed by free ICG at a concentration of 10 μg/mL (Figure S10a, Supporting Information). All three of the inorganic nanoparticle samples (Ca-pSiNP-ICG, CaS-ICG, and Ca-Silica-ICG) displayed substantially larger photoacoustic signals relative to free ICG (Figure S10bc, Supporting Information). On the basis of SNR values (normalized to the absorbance value at 790 nm), the photoacoustic generation efficiencies were between 5- to 17-fold greater than for free ICG, with the Ca-pSiNP-ICG material showing the largest efficiency (Table 1).
In contrast to the behavior of the relatively rigid inorganic nanoparticles, a “softer” nanoparticle based on a liposome (Lip-ICG) showed no enhancement of the photoacoustic signal from encapsulated ICG. The Lip-ICG formulation was prepared by extrusion through polycarbonate membranes containing 100 nm pores as described previously.[24] In order to make a direct comparison, Lip-ICG and Ca-pSiNP-ICG nanoparticles were prepared of comparable size (Table 1 and Figure S10a, Supporting Information), and their concentrations were adjusted to yield comparable optical absorbance at λ = 788 nm (Figure S10b, Supporting Information). Photoacoustic signals recorded using λ = 788 nm pulsed laser irradiation (Figure S11, Supporting Information) showed 6-fold lower response from the Lip-ICG sample relative to Ca-pSiNP-ICG. The response of the liposomal formulation was thus quite similar to the response of a comparable concentration of free ICG.
The poor PA imaging performance of the liposomal formulation relative to the inorganic nanoparticles was consistent with steady-state laser heating measurements. In these experiments, the samples were continuously irradiated with a high power laser (power density 1.1 W/cm2) emitting at λ = 808 nm and the temperature was recorded as a function of time (Figure 2). The Ca-pSiNP-ICG and Lip-ICG samples were dispersed in 20% ethanol (in DI water) and the concentrations adjusted such that the optical absorbance at λ = 808 nm was comparable for each sample (Figure 2c). Irradiated Ca-pSiNP-ICG rapidly attained a steady state temperature that was 47 °C greater than the sample in the dark (ambient temperature was ~25 °C), it was stable under irradiation, and it maintained a constant elevated temperature for the duration of the experiment (Figure 2b). By contrast, Lip-ICG displayed a lower level of steady-state heat generation, and the initial elevation in temperature decreased rapidly with increasing irradiation time. The behavior of the Lip-ICG formulation is consistent with a photobleaching process, and it was mirrored by the behavior of free ICG (Figure 2c).[25, 26] Free ICG is known to be photolytically unstable under laser irradiation,[27, 28] and oxidized porous Si has previously been shown to protect a trapped fluorescent molecular payload from the photobleaching process.[29] In order to quantify this in the present system, we monitored fluorescence intensity from free ICG and from Ca-pSiNP-ICG during irradiation with λ = 790 nm pulsed laser light as a function of time. Fluorescence from free ICG lost >10% of its intensity during 5 min of irradiation, while the fluorescence spectrum from nanoparticle-encapsulated ICG did not change substantially under these conditions (Figure S12, Supporting Information). We conclude that the more rigid, sealed pSiNPs effectively protect loaded ICG from photobleaching, and postulate that this results in a more constant response and a higher steady-state temperature elevation upon sustained laser irradiation. While the greater photostability that ICG exhibits when it is loaded into Ca-pSiNP-ICG might contribute to the large photoacoustic signal enhancement seen with this nanoparticle host, it is likely that other factors are involved as well. For example, the relative rigidity of a solid porous host is expected to inhibit rotations, vibrations, and Brownian motion of dye molecules trapped within, and this could influence PA performance.
Figure 2. Comparison of photostability of ICG and nanoparticle formulations of ICG.

(a) Optical absorbance spectra of Ca-pSiNP-ICG, Ca-pSiNP and free ICG at concentrations of 20 and 30 μg/mL, in ethanol (100 μL total volume). The wavelength of excitation by laser irradiation (λ = 808 nm) used in (b) is indicated with the gray dotted line. (b) Temporal temperature profiles measured under steady-state irradiation of the indicated formulations from (a) with (λ = 808 nm) laser light. All samples are in 1:1 (v:v) ethanol:DI water. (c) Optical absorbance spectra of Ca-pSiNP-ICG and Lip-ICG 30 in 20% ethanol (1:4 ethanol:DI water, by volume, (200 μL total volume), and the laser wavelength used in (d) is indicated with the gray dotted line. (d) Temporal temperature profiles measured under steady-state laser irradiation (λ = 808 nm) comparing Lip-ICG with Ca-pSiNP-ICG samples from (c). A pure DI water control is also shown. Nanoparticle samples are in 20% ethanol (1:4 ethanol:DI water, by volume).
Control experiments were performed to test if trapping of the ICG payload within the porous nanoparticle was a necessary condition for the high PA response observed. For these experiments we blocked the empty pore openings of the pSiNPs via thermally induced silane dehydrocoupling of octadecylsilane (H3Si-C18H37).[30] Spectroscopic measurements combined with centrifugation indicated that the resulting particle (C18-pSiNP) effectively excluded ICG, and photoacoustic measurements on a mixture of C18-pSiNP and free ICG showed no signal enhancement relative to free ICG alone (Figure S13, Supporting Information), establishing that PA signal enhancement is not caused simply by proximity of the pSiNPs to ICG. A second control experiment was performed in which the empty pSiNP surface was modified with primary amines using the (3-aminopropyl)-dimethylmethoxysilane coupling agent, and ICG was loaded onto the resulting positively charged surface via electrostatic interaction (pSiNP-NH2-ICG). This experiment was performed to test if the dye payload must be sealed within a rigid host in order to exhibit PA signal enhancement. The rationale for these experiments was that a surface-adsorbed dye would be in contact with the liquid phase and so would experience better thermal coupling to the solution. Thus the pSiNP-NH2-ICG construct received no calcium silicate sealing chemistry. These samples showed no enhancement in the PA signal relative to a comparable concentration of free ICG (Figure S13, Supporting Information), indicating that simple adsorbtion of ICG onto the surface of the nanoparticle is not sufficient to achieve PA contrast enhancement.
The porous silicon-based host contained an additional feature that may contribute to heating that is not seen with the simpler oxide-based hosts: it contains elemental silicon. This material displayed pronounced quenching of fluorescence from the ICG payload (Figure S3, Supporting Information) that was not observed with the other nanoparticle hosts. Prior studies of porous silicon have shown that it can act as an efficient quencher of fluorescence from a proximal organic dye.[23, 31] Silica and calcium silicate, being insulators, do not have a high density of electronic states available to quench fluorescence in a similar manner. For the silicon-containing nanoparticle, fluorescence quenching provides an additional means to channel the excited state of an emissive molecular guest into a non-radiative (thermal) pathway, and this non-radiative quenching process may be responsible for the slightly greater signals generated by the pSiNP constructs relative to the porous silica-based nanoparticles. In addition, the efficient quenching of fluorescence by the silicon host may limit photooxidation of the dye by intercepting its excited state before it has time to generate singlet oxygen or participate in other photochemical degradation pathways.
Taken together, the above results suggest that photoacoustic signal enhancement of a molecular dye contrast agent is a general characteristic of the rigid porous inorganic host, which we postulate to arise from two complementary factors: First, the relatively low thermal conductivity of the porous silicon, silica and calcium silicate host materials[32-35] allows the particles to better retain heat generated during the laser pulse, yielding a larger temperature differential and resulting in a stronger photoacoustic signal. A similar effect has been invoked to explain the improved PA signals seen from gold nanorods that have been encapsulated in silica shells.[12] Second, as seen previously with thermally oxidized porous Si,[29] encapsulation of the dye in a relatively oxygen-impermeable structure shuts down photobleaching processes that degrade the free dye.
Finally, we demonstrated the feasibility of the use of the nanoparticle-based photoacoustic imaging agent in tissues using ex vivo mouse brains (fixed in 1% agarose gel, n = 3 per each group). We injected aliquots of Ca-pSiNP-ICG and ICG (at equivalent concentrations, based on the ICG absorbance at λ = 790 nm) at the lambda point of each brain, which is the location of some of the largest blood vessels in this organ (Figure S14, Supporting Information). Ca-pSiNP-ICG, ICG, and PBS-injected brains were positioned in a water tank approximately 1 cm beneath the acoustic transducer and imaged. Strong photoacoustic signals were detected in the Ca-pSiNP-ICG-injected brains in the region emanating from the injection point, and signals from deeper in the tissues were also visible (Figure 3). By contrast, only weak signals were detected in brains injected with free ICG. The presence of ICG in both brain samples was verified by its characteristic photoacoustic spectrum, obtained from brain coronal cross sections (Figure S15, Supporting Information). The corresponding photoacoustic spectrum from the control (PBS-injected) brain was featureless. The ex vivo photoacoustic data validate that the Ca-pSiNP-ICG construct is an effective PA contrast agent for imaging of tissues, and that the substantial improvement in contrast afforded by the nanoparticle host in vitro is also apparent in animal tissue imaging.
Figure 3.

Photoacoustic images of fixed mouse brain comparing contrast agents: free ICG vs a comparable quantity of ICG encapsulated in a nanoparticle. Images at the far left are photoacoustic image (red) overlaid on ultrasound image (grayscale) after injection of control (PBS), free ICG, and Ca-pSiNP-ICG, as indicated. Each set of 4 images contain: brain (i) top view, (ii) location of sectioning, (iii) coronal cross-section, and (iv) axial cross-section. Scale bar is 2 mm.
The calcium silicate sealing chemistry used in this study to trap ICG in the Ca-pSiNP-ICG formulation has been used previously to load siRNA as a potential drug payload, and this chemistry allows conjugation of polyethylene glycol linkers and peptide-based targeting groups to a pSiNP host, which have shown selective homing and delivery of the payload to injured brain tissues from systemic circulation.[19] The results of the present work showing that this nanoparticle can stabilize a photolytically unstable dye, maintain a large photothermal temperature differential, and significantly enhance the performance of a photoacoustic imaging agent in animal tissues establishes the feasibility of expanding this selective tissue targeting system into photoacoustic imaging and photothermal therapy applications.
Experimental Section
Preparation of Porous Silicon Nanoparticles:
The pSiNPs were prepared following the published “perforation etching” procedure with slight modification.[20] A highly boron-doped p++-type single-crystal silicon wafer (1 mΩ cm resistivity, 100 mm diameter, Virginia Semiconductor, Inc.) was anodically etched in an electrolyte consisting of 3:1 (v:v) of 48% aqueous hydrofluoric acid (HF): absolute ethanol. Prior to preparation of the pSi layer, the silicon wafer was etched a thin porous layer called “sacrificial layer” with 3:1 (v:v) 48% aqueous HF:ethanol and removed with aqueous potassium hydroxide (KOH, 2 M). The etching waveform consisted of a lower current density 50 mA cm−2 of 1.2 sec, followed by a higher current density 400 mA cm−2 of 0.363 sec. This waveform was repeated for 500 cycles, generating a pSi film with “perforations” as cleavage points during ultrasonication repeating approximately every 120 nm. The etched pSi layer was collected from the silicon substrate by applying 4 mA cm−2 current density for 250 sec in a solution containing 1:20 (v:v) aqueous HF:ethanol. The collected pSi films were fractured into nanoparticles in deionized water (DI H2O, 6 mL) for 24 hr. The pSiNPs were purified three times by centrifugation (14,000 rpm, 20 min).
Preparation of ICG-loaded inorganic nanoparticles (Ca-pSiNP-ICG, Ca-Silica-ICG, CaS-ICG, and pSiNP-NH2-ICG):
ICG was loaded into the pSiNPs following the published “self-sealing chemistry” procedure with slight modification.[19] The pSiNPs (1 mg in 100 μL ethanol) were mixed with ICG solution (2 mg/mL in DI water, 150 μL), DI water (250 μL) and aqueous CaCl2 solution (2 M, 500 μL). The mixture was subjected to ultrasonication (50T ultrasonic bath, VWR International) in an ice water bath for 30 min. The ICG-loaded pSiNPs (Ca-pSiNP-ICG) were purified by three sequential centrifugation steps (14,000 rpm, 20 min), where the supernatant was discarded and the pellet was redispersed in DI water, 70% ethanol, and finally 100% ethanol. The control calcium silicate-sealed pSiNPs (Ca-pSiNP) were prepared without ICG following the above procedure but omitting ICG from the reaction. The Ca-Silica-ICG and CaS-ICG samples were prepared in the same manner, but commercial silica nanoparticles (1 mg in 100 μL ethanol) or silicic acid was added to the reaction mixture instead of pSiNPs. The silicic acid used in this preparation was synthesized by dissolution of 1 mg pSiNP in aqueous KOH (50 μL, 2 M) and filtration of the solution through an Amicon centrifugal filter unit (100 kDa cutoff) prior to use. The aminated pSiNPs (pSiNP-NH2-ICG) were prepared by mixing pSiNPs (1 mg in 1 mL ethanol) with 12 μL of (3-aminopropyl)-dimethylmethoxysilane for ~3 h. The mixture was purified three times by centrifugation (14,000 rpm, 20 min) from ethanol to remove excess reagents. The aminated pSiNPs (1 mg in 850 μL ethanol) were mixed with ICG solution (2 mg/mL in ethanol, 150 μL) and agitated for 2 hrs. The resulting pSiNP-NH2-ICG construct was purified three times by centrifugation (14,000 rpm, 20 min) from ethanol to eliminate excess ICG.
Preparation of ICG-loaded liposome nanoparticle (Lip-ICG):
ICG was loaded into liposomes following the published procedure with slight modification.[24] Lipid stock solution was prepared with concentration of 10 mg/mL in chloroform and ICG was dissolved in methanol with 1 mg/mL. The lipid film was prepared with a mixture of 30.8 mg DMPC, 6.7 mg DSPE-PEG (2000) Methoxy, and 148.6 μg ICG (molar ratio DMPC:DSPE-PEG:ICG = 250:12.5:1) and the solvents were evaporated in a vacuum desiccator. The lipid film was hydrated with DI water (20 mL) and subsequently extruded through a 100 nm pore-size polycarbonate membrane at 40 °C. Lip-ICG was purified using Sephadex G-50 beads in a 100 kDa dialysis tube overnight under continuous water elution to remove free ICG not encapsulated by liposome. The purified Lip-ICG was concentrated with a 100 kDa centrifugal filter to prepare the final concentration (40 – 80 μg/mL). Loaded ICG content was measured by UV-Vis absorbance and calculated based on a standard curve.
Characterization:
Transmission electron microscope (TEM) images were obtained with a JEOL-1200 EX II instrument. The hydrodynamic size and zeta potential was measured using dynamic light scattering (DLS, Zetasizer ZS90, Malvern Instruments). Fourier transform infrared (FTIR) spectra were obtained with a Thermo Scientific Nicolet 6700 FTIR instrument. Raman spectra were recorded using a Renishaw inVia Raman microscope with 532 nm laser excitation source. Powder x-ray diffraction (XRD) spectra were obtained at ambient temperature on a Bruker D8 Advance diffractometer using Cu Kα (λ = 1.5418 Å) radiation (40 kV, 40 mA), a scan speed of 0.1 sec/step, a step size of 0.02° in 2θ, and a 2θ range of 10–80°. Thermogravimetric analysis (TGA) was carried out using a STA 6000 Simultaneous Thermal Analyzer (PerkinElmer) in a nitrogen ambient. Electrospray ionization mass spectrometry (ESI-MS) was performed using a nanoACQUITY UPLC-TripleTOF 5600. Optical absorbance spectra were acquired using a Molecular Devices Spectra Max spectrophotometer or a Nanodrop2000 (Thermo Fisher Scientific) instrument. Fluorescence spectra were obtained using a Molecular Devices Spectra Max GEMINI XPS.
Photoacoustic imaging:
The photoacoustic instrument was a Vevo 2100 commercial photoacoustic scanner (Visualsonics) described previously.[36] The system consisted of a flashlamp-pumped Q-switched Nd:YAG laser with optical parametric oscillator and second harmonic generator operating at 20 Hz between 680 and 970 nm with a 1 nm step size and a pulse of 4 to 6 nm. The peak energy was 45 ± 5 mJ at 20 Hz at the source. The laser was coupled into 21 to 30 MHz-centered transducers (LZ250 and LZ400). The full field of view of the transducer was 14–23 mm wide. The acquisition rate was 5 frames per second.
A total volume of 17 μL of each sample was placed in polyethylene tubing (Harvard apparatus) with an outer diameter of 1.27 mm and an inner diameter of 0.85 mm, and the tubing was fixed in a custom-built phantom holder.[37] The phantom holder incorporated 16 equally spaced holes with diameters ranging from 1.7 to 2.5 mm. The samples were positioned 1 cm beneath the acoustic transducer for optimal signal collection. The laser energy was calibrated and optimized using a built-in energy meter prior to measurements. The samples were typically scanned with 100% laser energy and the signal was amplified between 10 to 40 dB for optimal visualization. We also performed 3D scans to image all parts of the tubing using 790 or 788 nm excitation; photoacoustic spectra were collected in the wavelength range from 680 to 970 nm.
For ex vivo experiments, mice were sacrificed after blood perfusion with PBS, and fixed in 4 % paraformaldehyde (PFA) for 24 h. Then, 15 μL of PBS, free ICG or Ca-pSiNP-ICG solution was injected twice at the lambda point of the brain at an interval of 15 min. 25 mL of 1% agarose gel was poured into a 100 mm well plate and allowed to cool to make a bottom layer. Each brain was placed on top of the cooled agarose gel, and another 25 mL of 1% agarose gel was poured over the tissue carefully, to avoid bubble formation. The prepared agarose-fixed tissues were positioned 1 cm from the transducer in a water tank to obtain the photoacoustic images.
Measurement of thermal profile under continuous laser exposure:
Temporal heating profiles were obtained under continuous laser exposure, on samples consisting of 200 μL of free ICG or Ca-pSiNP-ICG in 1:1 (by volume) ethanol:DI water. For comparison with the liposomal formulations, Lip-ICG or Ca-pSiNP-ICG samples were prepared in solutions composed of 1:4 (by volume) ethanol:DI water. Samples were placed in 96 well plates and exposed for 300 sec to a near-infrared light source (1.1 W/cm2, BWF2 808 nm diode laser, B & W TEK Inc.). The temperature during laser irradiation was recorded using an infrared (IR) camera (FLIR SC305, FLIR Systems, Inc.).
Measurement of stability of fluorescence from ICG formulations under laser irradiation:
Ethanol mixtures of free ICG (20 μg/mL), or Ca-pSiNP-ICG containing an equivalent quantity of ICG, were diluted with deionized water to form a 1:1 (by volume) ethanol:DI mixture. The resulting samples were placed in cuvettes and excited with λ = 790 nm light from a tripled YAG-pumped optical parametric oscillator (Opolette 355, Opotek Inc.), operating at 20 Hz with nominal pulse width 8 ns and an average power density of 0.3 mW (~0.5 cm diameter expanded beam). Fluorescence spectra were obtained every 10 sec for a period of 5 min using a cooled CCD (charge coupled device) spectrometer (QE Pro, Ocean Optics) fitted with a 800 nm long-pass emission filter.
Supplementary Material
Acknowledgements
J. Kang and D. Kim contributed equally to this work. This work was supported by the Defense Advanced Research Projects Agency (DARPA) under Cooperative Agreement HR0011–13-2–0017, by the National Science Foundation under grant No. CBET-1603177, and by the National Institutes of Health, through grant No. R01 AI132413–01. J. Kang acknowledges financial support from the UCSD Frontiers of Innovation Scholars Program (FISP) fellowship. D. Kim acknowledges the Basic Science Research Program of the Korea National Research Foundation (NRF) funded by the Ministry of Education (Grant No. 2016R1A6A3A03006343). Y. Han and J.-H. Park acknowledge the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (Grant No. NRF-2017R1E1A1A01074847).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jinyoung Kang, Department of Nanoengineering, University of California San Diego, 9500 Gilman Drive, La Jolla, California, 92093, USA.
Junxin Wang, Department of Nanoengineering, University of California San Diego, 9500 Gilman Drive, La Jolla, California, 92093, USA.
Ali Hariri, Department of Nanoengineering, University of California San Diego, 9500 Gilman Drive, La Jolla, California, 92093, USA.
Dokyoung Kim, Department of Anatomy and Neurobiology, College of Medicine, Center for Converging Humanities, Kyung Hee University, 26 Kyungheedae-Ro, Dongdaemun-Gu, Seoul 02447, Republic of Korea
Yunho Han, Department of Bio and Brain Engineering & KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea.
Ji-Ho Park, Department of Bio and Brain Engineering & KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea
Dr. Jonathan M. Zuidema, Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, California, 92093, USA
Jesse V. Jokerst, Department of Nanoengineering, Materials Science and Engineering Program, University of California San Diego, 9500 Gilman Drive, La Jolla, California, 92093, USA
Michael J. Sailor, Department of Chemistry and Biochemistry, Department of Nanoengineering, Department of Bioengineering, Materials Science and Engineering Program, University of California San Diego, La Jolla, California, 92093, USA, msailor@ucsd.edu
References
- [1].Beard P, Interface Focus 2011, 1, 602–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhang EZ, Laufer JG, Pedley RB, Beard PC, Phys. Med. Biol. 2009, 54, 1035–1046. [DOI] [PubMed] [Google Scholar]
- [3].Ntziachristos V, Razansky D, Chem. Rev. 2010, 110, 2783–2794. [DOI] [PubMed] [Google Scholar]
- [4].Kim C, Favazza C, Wang LHV, Chem. Rev. 2010, 110, 2756–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lu HD, Wilson BK, Heinmiller A, Faenza B, Hejazi S, Prud’homme RK, ACS Appl. Mater. Inter. 2016, 8, 14379–14388. [DOI] [PubMed] [Google Scholar]
- [6].Xu MH, Wang LHV, Rev. Sci. Instrum. 2006, 77. [Google Scholar]
- [7].Wu D, Huang L, Jiang MS, Jiang HB, Int. J. Mol. Sci. 2014, 15, 23616–23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang LHV, Hu S, Science 2012, 335, 1458–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pu KY, Shuhendler AJ, Jokerst JV, Mei JG, Gambhir SS, Bao ZN, Rao JH, Nat. Nanotechnol. 2014, 9, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Weber J, Beard PC, Bohndiek SE, Nat. Methods 2016, 13, 639–650. [DOI] [PubMed] [Google Scholar]
- [11].Li JJ, Cheng FF, Huang HP, Li LL, Zhu JJ, Chem. Soc. Rev. 2015, 44, 7855–7880. [DOI] [PubMed] [Google Scholar]
- [12].Chen YS, Frey W, Kim S, Kruizinga P, Homan K, Emelianov S, Nano Lett. 2011, 11, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang H, Dunphy DR, Jiang X, Meng H, Sun B, Tarn D, Xue M, Wang X, Lin S, Ji Z, J. Am. Chem. Soc. 2012, 134, 15790–15804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Canham LT, in Porous Silicon for Biomedical Applications (Ed.: Santos HA), Woodhead Publishing, Cambridge, 2014, pp. 3–20. [Google Scholar]
- [15].Wu C, Chang J, Fan W, J. Mater. Chem. 2012, 22, 16801–16809. [Google Scholar]
- [16].Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV, Nat. Biotechnol. 2007, 25, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Su C-H, Sheu H-S, Lin C-Y, Huang C-C, Lo Y-W, Pu Y-C, Weng J-C, Shieh D-B, Chen J-H, Yeh C-S, J. Am. Chem. Soc. 2007, 129, 2139–2146. [DOI] [PubMed] [Google Scholar]
- [18].Pernodet N, Fang X, Sun Y, Bakhtina A, Ramakrishnan A, Sokolov J, Ulman A, Rafailovich M, Small 2006, 2, 766–773. [DOI] [PubMed] [Google Scholar]
- [19].Kang J, Joo J, Kwon EJ, Skalak M, Hussain S, She ZG, Ruoslahti E, Bhatia SN, Sailor MJ, Adv. Mater. 2016, 28, 7962–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Qin ZT, Joo J, Gu L, Sailor MJ, Part. Part. Syst. Char. 2014, 31, 252–256. [Google Scholar]
- [21].Li B, Yu D, Zhang S-L, Phys. Rev. B 1999, 59, 1645. [Google Scholar]
- [22].Letant S, Vial J, J. Appl. Phys. 1997, 82, 397–401. [Google Scholar]
- [23].Wu EC, Park J-H, Park J, Segal E, Cunin F, Sailor MJ, ACS Nano 2008, 2, 2401–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yoon HJ, Lee HS, Lim JY, Park JH, ACS Appl. Mater. Inter. 2017, 9, 5683–5691. [DOI] [PubMed] [Google Scholar]
- [25].Saxena V, Sadoqi M, Shao J, J. Pharm. Sci. 2003, 92, 2090–2097. [DOI] [PubMed] [Google Scholar]
- [26].Hou L, Fang J, Wang W, Xie Z, Dong D, Zhang N, J. Mater. Chem. B 2017, 5, 3348–3354. [DOI] [PubMed] [Google Scholar]
- [27].Bringley JF, Penner TL, Wang RZ, Harder JF, Harrison WJ, Buonemani L, J. Colloid Interf. Sci 2008, 320, 132–139. [DOI] [PubMed] [Google Scholar]
- [28].Wang YW, Fu YY, Peng QL, Guo SS, Liu G, Li J, Yang HH, Chen GN, J Mater. Chem. B 2013, 1, 5762–5767. [DOI] [PubMed] [Google Scholar]
- [29].Xu WJ; Riikonen J; Nissinen T; Suvanto M; Rilla K; Li BJ; Wang Q; Deng F; Lehto VP, J. Phys. Chem. C 2012, 116, 22307–22314. [Google Scholar]
- [30].Kim D, Joo J, Pan YL, Boarino A, Jun YW, Ahn KH, Arkles B, Sailor MJ, Angew. Chem. Int. Ed. 2016, 55, 6423–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gu L, Orosco M, Sailor MJ, Phys. Status Sol. (a) 2009, 206, 1374–1376 [Google Scholar]
- [32].Ebert H-P, Hemberger F, Int. J. Therm. Sci. 2011, 50, 1838–1844. [Google Scholar]
- [33].Koshida N; Nakajima T; Yoshiyama M; Ueno K; Nakagawa T; Shinoda H, MRS Proceedings 2011, 536, 105. [Google Scholar]
- [34].Boukai AI, Bunimovich Y, Tahir-Kheli J, Yu J-K, Goddard Iii WA, Heath JR, Nature 2008, 451, 168. [DOI] [PubMed] [Google Scholar]
- [35].Koshida N, in Handbook of Porous Silicon (Ed.: Canham LT), Springer, Switzerland, 2014, p. 207. [Google Scholar]
- [36].Wang J, Chen F, Arconada-Alvarez SJ, Hartanto J, Yap L-P, Park R, Wang F, Vorobyova I, Dagliyan G, Conti PS, Nano Lett. 2016, 16, 6265–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Arconada-Alvarez SJ, Lemaster JE, Wang J, Jokerst JV, Photoacoustics 2017, 5, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.