

Abstract
Despite the significant progress achieved with Combination Antiretroviral Therapy (cART) in the fight against Human Immunodeficiency Virus (HIV) infection, the difficulty to eradicate the virus together with the rapid emergence of multi-drug-resistant strains clearly underline a pressing need for innovative agents, possibly endowed with novel mechanisms of action. In this context, due to its essential role in the HIV genome replication, the reverse transcriptase-associated ribonuclease H (RNase H) has proven to be an appealing target. To identify new RNase H inhibitors, an in-house cycloheptathiophene-3-carboxamide library was screened, leading to compounds endowed with inhibitory activity whose structural optimization led to the catechol derivative 33, with IC50 value on the RNase H activity in the nanomolar range. Mechanistic studies suggested selective inhibition of the RNase H through the binding to an innovative allosteric site, which could be further exploited to enrich this class of inhibitors.
Keywords: Allosteric Inhibitors, Antiviral Agents, Cycloheptathiophene-3-Carboxamides, HIV-1 Ribonuclease H, Medicinal Chemistry
Graphical Abstract
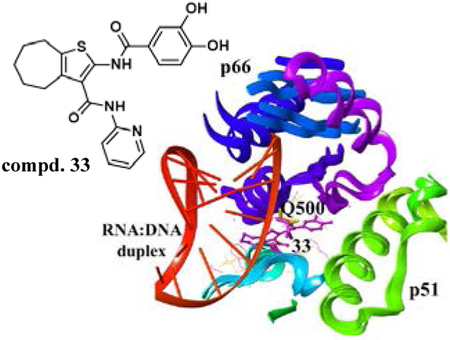
In this paper, by screening an in-house library of cycloheptathiophene-3-carboxamide derivatives and synthesizing a new series of analogues, catechol derivative 33 was identified as a nanomolar inhibitor of the HIV-1 RNase H. Mechanistic studies suggested its interaction with an innovative allosteric site entailing p66 residue Q500, a key residue for the binding of RT to RNA:DNA duplex substrate.
Introduction
Combination Antiretroviral Therapy (cART) for treatment of Human Immunodeficiency Virus (HIV) infection significantly suppresses viral load, preventing the development of AIDS in infected patients, and improving both their quality and expectancy of life. However, a positive cART outcome depends, on the one hand, on the susceptibility of the virus to the drugs and, on the other hand, on the adherence of the patient to the therapy. Lack of compliance often results in the selection of drug resistant variants whose transmission to drug naïve patients is becoming an increasing concern,[1,2] often causing treatment failure and increasing the need for new drugs with alternative mechanisms of action or new binding sites on traditional targets.
In this context, HIV reverse transcriptase (RT)-associated ribonuclease H (RNase H) function provides a promising target[3] since abrogation of this function strongly impairs viral infectivity, ascertaining its essential role in viral replication.[4] RNase H catalyzes both non specific and highly specific hydrolysis of the RNA strand of the RNA:DNA replication intermediate. HIV-1 RT is an heterodimer consisting of two subunits, p66 and p51. p66 Hosts active sites for both RNA- and DNA-dependent DNA synthesis and RNase H activity. The RNase H active site is located at the C-terminus in close contact with the p51 subunit and contains a highly conserved, essential, DEDD motif comprising the carboxylates residues D443, E478, D498, and D549, which coordinate two Mg2+ cations required as cofactors for hydrolysis reaction.[5] While no RNase H inhibitor has reached clinical trials, a limited number of compounds have been reported, distinguished by two classes: metal chelating active site inhibitors, which bind and coordinate the two Mg2+ ion cofactors, and allosteric inhibitors, which induce a conformational change of the active site disabling the RNA:DNA hybrid substrate binding.[6–10] Allosteric inhibitors could be attractive both to counteract the resistant strains development and to avoid the inhibition of related host enzymes, such as the human RNase H1.[11]
Only a small number of allosteric HIV RNase H inhibitors have been reported so far, including structurally different compounds such as vinylogous ureas, thienopyrimidinones, anthraquinones, hydrazones, and isatine derivatives.[6,12–15] These classes of compounds have been hypothesized to bind at different allosteric pockets. In particular, vinylogous ureas[16–18] and the closely related thienopyrimidinones[19] are the most promising class of potent and selective allosteric RNase H inhibitors. The vinylogous urea NSC727447 (1)[16] was initially identified through a high throughput screening of NCI libraries as modestly potent HIV-1 and HIV-2 RNase H inhibitor. A subsequent SAR study identified the cyclized thienopyrimidinone DNTP (2)[17] as a lead candidate for further structural optimization that identified more active thienopyrimidinones, such as the 3’,4’-dihydroxyphenyl derivative GZ552 (3)[19] (Figure 1).
Figure 1.

Known RNase H inhibitors: Vinylogous urea 1 and thienopyrimidinones 2 and 3. Cycloheptathiophene-3-carboxamide derivatives (cHTCs), previously reported as anti-influenza agents.
In the absence of crystallographic data, mass spectrometric protein footprinting and mutagenesis studies implicated p51 thumb residues (C280 and K281) in inhibitor binding, suggesting that these compounds, which are effective against the enzyme and the enzyme/substrate complex, inhibited RNase H activity by occupying a site at the p51 and p66 subunit interface. Indeed, further molecular modeling studies, performed on 1 and 2, suggested that these inhibitors bind to an allosteric site located at the p66 RNase H domain/p51 thumb interface, most probably hampering subunit flexibility, which is essential for RNA:DNA hybrid binding and catalysis.[20]
Considering our continuous interest in the anti-HIV field,[7,9,15,21–26] we noted the strict structural similarity with the vinylogous ureas[16–18] of a series of cycloheptathiophene-3-carboxamide derivatives (cHTCs, Figure 1) recently reported by us as influenza virus inhibitors based on their ability to disrupt the PA-PB1 subunits interaction of the viral RNA polymerase.[27,28] Thus, we decided to assay a set of these compounds for the anti-RNase H activity. The promising results led to synthesis of further analogues, with the aim of improving the anti-RNase H activity and perform an in-depth investigation on their mechanism of action.
Results and Discussion
Exploiting the cHTC derivatives as RNase H inhibitors
Based on their analogy with vinylogous ureas, a set of 20 cHTCs (compounds 4–22, Table 1)[27] were initially tested for anti-HIV RNase H activity. The majority of compounds were characterized by a 2-pyridine ring at the C-3 position of the cycloheptathiophene-3-carboxamide scaffold coupled with a substituted phenyl ring at the C-2 position. Compounds 19–22, carrying a smaller substituent at the C-3 position (i.e. amide, ethyl carboxylate and carboxylic acid), were included. Moreover, to gain further structural insight on this class of RNase H inhibitors, we synthesized additional derivatives (compounds 23–37, Table 1). In particular, maintaining the 2-pyridine ring at the C-3 position, the C-2 position was further explored by varying the nature and position of the substituents on the phenyl ring. In this way, compounds 23–29 were prepared. Additional derivatives bearing poly-substituted phenyl or pyridine rings on C-2 position, such as compounds 30–33, were also synthesized. The phenyl ring was replaced by its bioisoster thiophene in compound 34 as well as by a smaller methyl group in compound 35. The o-fluorophenyl moiety was spaced by the cycloheptathiophene ring by inserting a methylenic unit as in compound 36 while was interchanged with the C-3 pyridine ring in compound 37.
Table 1.
Effect of cHTCs on the HIV-1 RT-associated RNase H function.
| Compd | 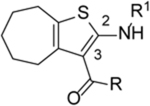 |
HIV-1 RNase H IC50 [μM][a] |
|
|---|---|---|---|
| R | R1 | ||
| 4 |  |
 |
21.89 ± 0.44 |
| 5 | “ | 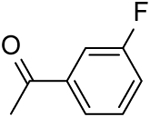 |
20.47 ± 0.90 |
| 6 | “ | 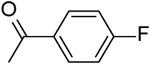 |
4.72 ± 0.04 |
| 7 | “ | 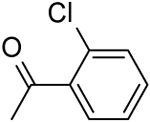 |
23.90 ± 0.80 |
| 8 | “ | 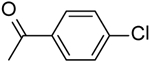 |
17.78 ± 2.44 |
| 9 | “ | 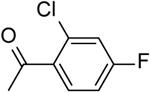 |
7.50 ± 0.10 |
| 10 | “ | 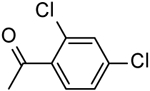 |
12.24 ± 0.70 |
| 11 | “ | 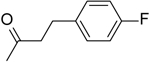 |
14.50 ± 0.90 |
| 12 | “ | 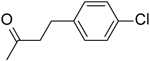 |
9.90 ± 3.10 |
| 13 | “ | 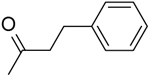 |
14.60 ± 1.40 |
| 14 | “ | 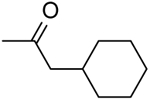 |
67.10 ± 2.50 |
| 15 | “ | -H | 20.10 ± 2.81 |
| 16 | “ | 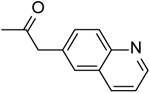 |
16.44 ± 1.44 |
| 17 | “ | 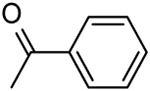 |
14.80 ± 0.80 |
| 18 | “ | 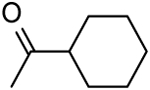 |
16.60 ± 0.50 |
| 19 | -NH2 | 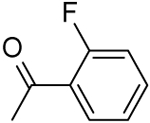 |
13.73 ± 2.16 |
| 20 | -OEt | “ | 25.20 ± 1.40 |
| 21 | “ | 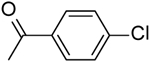 |
72.58 ± 2.81 |
| 22 | -OH | “ | >100 |
| 23 | 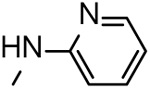 |
 |
4.57 ± 0.01 |
| 24 | “ | 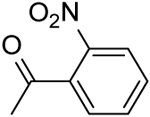 |
53.48 ± 1.70 |
| 25 | “ | 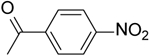 |
20.47 ± 0.90 |
| 26 | “ | 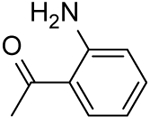 |
14.88 ± 4.10 |
| 27 | “ | 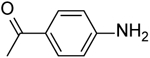 |
21.43 ± 4.40 |
| 28 | “ | 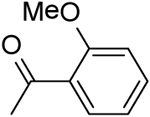 |
14.25 ± 0.20 |
| 29 | “ | 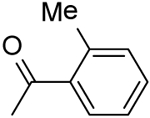 |
18.30 ± 1.80 |
| 30 | “ | 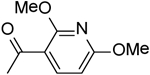 |
21.50 ± 1.00 |
| 31 | “ | 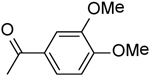 |
4.01 ± 1.00 |
| 32 | “ | 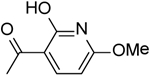 |
15.07 ± 1.30 |
| 33 | “ | 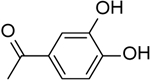 |
0.84 ± 0.10 |
| 34 | “ | 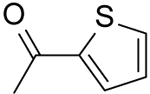 |
10.36 ± 1.96 |
| 35 | “ | -COCH3 | >100 |
| 36 | “ | 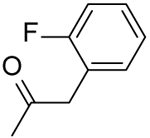 |
9.90 ± 3.10 |
| 37 | 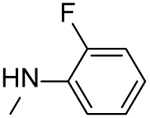 |
 |
13.70 ± 1.60 |
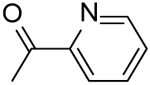
| NSC727447 |  |
4.00 ± 0.50 | |
IC50: compound concentration required to inhibit the HIV-1 RT-associated RNase H activity by 50%. The table reports the average and standard deviation of three independent experiments, made in duplicate.
Synthesis of cHTC derivatives
The synthesis of the various 2-pyridinylcycloheptathiophene-3-carboxamide derivatives 23–36 and 2-fluorophenylthiophene-3-carboxamide derivative 37 was accomplished as reported in Scheme 1, applying the two-steps Gewald synthesis.[29,30] In particular, a first Knoevenagel condensation of the cycloheptanone with 2-cyano-N-pyridin-2-ylacetamide[31] or 2-cyano-N-(2-fluorophenyl)acetamide,[31] followed by cyclization performed in the presence of sulphur and N,N-diethylamine in ethanol, gave compounds 38[27] and 39, respectively. Compounds 23–25, 28–31, and 34–36 were then obtained by coupling reaction of intermediate 38 with the appropriate acyl chlorides in dry pyridine. Analogously, the target compound 37 was obtained by intermediate 39. The o-amino and p-amino derivatives 26 and 27 were obtained by reduction of the nitro precursors 24 and 25 in the presence of 10% Pd/C, while O-demethylation of dimethoxy derivatives 30 and 31, performed with BBr3, provided the dihydroxy derivatives 32 and 33, respectively.
Scheme 1. Synthetic route to cHTCs.

Reagents and conditions: a) ammonium acetate, glacial acetic acid, benzene, reflux, 16 h; b) sulfur, N,N-diethylamine, 40–50 °C, 1.5 h, the two steps 25–59%; c) R1COCl, dry pyridine, 2–72 h, 7–49%; d) 10% Pd/C, ammonium formate, MeOH/DMF, rt, 1–120 h, 30–80%; e) BBr3, dry CH2Cl2, rt, 2 h, 19–60%.
Biological evaluation
A total of 35 functionalized cHTC derivatives were assayed against HIV-1 RNase H (Table 1). Results confirmed that the cycloheptathiophene scaffold is particularly suitable to achieve inhibition. Indeed all compounds, with the exception of 22 and 35, were inhibitory. In particular compounds 6, 9, 12, 23, 31, 33, 34, and 36, carrying aromatic rings at both the C-2 and C-3 positions, showed IC50 values in the low micromolar range, differing from the few examples of compounds bearing two aromatic rings on the cycloheptathiophene nucleus which were previously reported to be inactive.[17] The pyridine ring seems a particularly suitable C-3 substituent, since its replacement by smaller groups, such as ester or acid, caused a sharp decrease or even abrogation of inhibitory potency (compounds 20–22). Several substituted phenyl rings were suitable for the C-2 position. In agreement with previously reported for thienopyrimidinone derivatives,[19] the best substituent was the catechol moiety, represented by the most potent compound in this study, compound 33, displaying an IC50 value of 0.84 μM. When the hydroxyl groups were O-methylated, as in compound 31, a 5-fold decrease in potency was observed. The bioisosteric substitution of the 2-phenyl ring with the thiophene one preserved the inhibitory activity (compound 34), while its replacement with a smaller methyl group, as in acetyl derivative 35, completely abolished activity. Addition of a methylene or ethylene spacer between the C-2 amide linkage and the phenyl moiety did not significantly affect activity (compare 36, 11, 12, and 13 vs 4, 6, 8, and 17). The two aromatic rings at the C-2 and C-3 position can be interchanged; indeed, compound 37 showed even better activity of the analog 4 (IC50 values of 13.70 and 21.89 μM, respectively).
Mode of action studies
To better understand their mechanism of action, the most potent derivatives 6, 9, 12, 23, 31, 33, 34, and 36 were selected for further analysis. Firstly, to exclude any interaction with the RNase H catalytic center, the selected compounds were tested for their ability to coordinate Mg2+ ions. Based on the UV spectra recorded in the absence and in the presence of MgCl2, all derivatives were unable to chelate the divalent ions, since no shift was observed in the maximum of absorbance (Figure S1). Hence, these results confirmed that they act with a mechanism different from RNase H active site inhibitors.
Secondly, to examine their specificity of action, the same compounds were tested against the RT-associated RNA-dependent DNA polymerase (RDDP) function, using as a positive control the non nucleoside RT inhibitor efavirenz. Compounds 1 and 3 were also included to comparative purposes (Table 2).
Table 2.
Effects of selected cHTC derivatives and reference compounds on the HIV-1 RT-associated RNA dependent DNA polymerase function.
| Compd | HIV-1 RDDP IC50 [μM][a] |
|---|---|
| 6 | > 50 |
| 9 | > 50 |
| 12 | > 50 |
| 23 | > 50 |
| 31 | > 50 |
| 33 | 20.90 ± 1.70 |
| 34 | > 50 |
| 36 | > 50 |
| 1 | > 50 |
|
3 efavirenz |
25.40 ± 3.70 0.023 ± 0.0016 |
IC50: compound concentration required to inhibit the HIV-1 RT-associated RNA dependent DNA polymerase activity by 50%. The table reports the average and standard deviation of three independent experiments, made in duplicate.
Test compounds were unable to inhibit DNA polymerase function at the highest tested concentration of 50 μM, analogous to 1, with the exception of compound 33. However, the IC50 value of 20.90 μM was 20-fold weaker than that for RNase H activity. A similar behavior was previously reported for other dihydroxythienopyrimidinones[19] and also confirmed by us for compound 3. A possible explanation for the anti-polymerase activity displayed by 33 could arise from its potency. In particular, cHCT derivatives most likely bind at an allosteric site and induce long-range conformational alterations in the RT heterodimer, affecting both RNase H and polymerase activities, but this effect was observed only for the most potent compound 33. Alternatively, 33 might have an allosteric site and/or a binding mode in the same site different from that recognized by the other cHTC compounds.
We therefore explored the possible binding of compound 33 to the previously reported RNase H allosteric site close to helix αI of p51 subunit,[18] by studying the effect on an RT bearing the C280A mutation (p66wt/p51C280A RT), the most significant substitution reported for the first generation vinylogous ureas inhibitors[18] (Table 3). Indeed, vinylogous ureas have been postulated to bind at the p66/p51 interface and protein footprinting, mutagenesis, and docking studies suggested that residue C280 in helix αI of p51 subunit was particularly important for ligand binding.[16–19] Derivative 31 was also studied in parallel along with thienopyrimidinone 3. As previously reported,[19] compound 3 fully retained its inhibitory activity on the p66wt/p51C280A mutated RT, and, the same behavior was observed for compound 33 that showed an IC50 value of 0.52 μM. In contrast, compound 31 failed to inhibit the mutant enzyme, strengthening the hypothesis the two related cHTC derivatives might have a different binding site and/or binding modes in the same site.
Table 3.
Effects of selected cHTC derivatives and reference 3 on the RT-associated RNase H function of p66wt/p51C280A HIV-1 RT.
| Compd | HIV-1 RNase H IC50 [μM][a] |
|---|---|
| 31 | > 50 |
| 33 | 0.52 ± 0.01 |
| 3 | 0.24 ± 0.002 |
IC50: compound concentration required to inhibit the HIV-1 RT-associated RNase H activity by 50%. The table reports the average and standard deviation of three independent experiments, made in duplicate.
To further explore the binding site of cHTC derivatives, differential scanning fluorimetry (ThermoFluor) assay was performed for some active compounds (6, 12, 31, and 33) to determine their effect on the stability of the RT heterodimer. In contrast to thienopyrimidinones, which reduced the Tm by 0.5–5.5 °C,[19] no decreased Tm was measured for cHTC derivatives (data not shown). The inability of these compounds to destabilize the RT heterodimer suggests either that they do not bind to the p66/p51 interface or that they bind to the subunit interface but differently from most of the thienopyrimidinones such that they do not affect dimer stability. Thus, from this assay, a different behavior emerged between our cHTCs and compound 3.
To achieve further insights on the derivatives allosteric binding to RNase H domain and gain a deeper understanding of the key structural aspects of their interaction, we performed docking studies for compounds 31 and 33 along with 3. Since no crystal structure with RT co-crystallized with vinylogous ureas is available, we used the crystal structure of unbounded RT[32] and performed Induced Fit Docking (IFD) to take into account side-chain conformational changes induced by the ligand. The procedure comprises a combination of the docking program Glide,[33] followed by Prime for side-chain rearrangements and minimization of residues within the binding pocket.[34] The binding box was centred on p66 residue Q500 and occupied a volume of 20×20×20 Å, allowing exploration of the entire RNase H domain.
Although the putative binding pocket of some vinylogous ureas was previously found located in the α helix containing residues C280 and K281,[17,18] docking experiments placed compounds 31 and 33, as well as the reference 3, close to the near p51 helix α79, which includes residues 254–267 (Figure 2a,c, and e). Probably this difference in binding site is due to the different geometry and/or greater hindrance of these derivatives compared to other vinylogous ureas as 1 and 2. In details, the cycloheptathiophene ring of 31 occupies a small hydrophobic pocket formed by residues W535, P537, P421, and L425, the C-2 carbonyl group establishes a hydrogen bridge with Q500, and the carbonyl group of the C-3 amide linker forms a hydrogen bond with W266 (p51) (Figure 2b). However, 31 cannot accommodate its dimethoxyphenyl group in the pocket due to steric hindrance, as a result this group is exposed to the solvent. Compound 33 adopts a slight different orientation (Figure 2 d) with the C-2 carbonyl group that, similar to 31, forms hydrogen bonds with the p66 Q500, while the C-3 carbonyl group interacts with p51 K424. The RT-33 complex appears particularly stabilized by a favourable hydrogen bond between the hydroxyl group of the catechol moiety and p66 L533. The same stabilizing effect was observed for 3 (Figure 2f), which adopted a slightly different binding site from compound 33 with a small overlapping of the catechol moiety. The difference in complex stability is also shown by the different Glide Score and variation of interaction energy reported (Table 4).
Figure 2.

Putative binding mode of a) 31, c) 33, e) reference compound 3; 2D representation of binding pocket interacting residues for compounds b) 31, d) 33, and f) 3.
Table 4.
Docking score and ΔTotal Interaction Energy (ΔTot E) of [Compound • RT] complexes expressed in Kcal/mol.
| Compd | Glide docking score | ΔTot E |
|---|---|---|
| 31 | −7.36 | −21.95 |
| 33 | −9.5 | −27.12 |
| 3 | −10.03 | −28.91 |
Hence, the lower activity of compound 31 with respect to 33 and 3, when C280 is mutated in alanine, could be due to the weaker 31-RT complex stability: in fact, mutation of C280 could have a destabilizing effect inhibitor binding, leading to its complete loss of activity.[18]
Interestingly, docking simulations showed that, even if the binding of compounds 31 and 33 was slightly different, residue Q500 emerged a key residue in the cycloheptathiophene-3-carboxamides binding. Q500 has been reported as one of the key residues responsible for the RNA:DNA hybrid binding[35] and it has recently been proposed as a possible druggable binding site for allosteric RNase H inhibitors.[36–38] Theoretically, compounds interacting with residue Q500 (Figure 3), or residues in its vicinity that are important for RNA:DNA substrate positioning, could potentially interfere with duplex accommodation in the RNase H active site.[36]
Figure 3.

Alignment of RT-33 complex with RNA:DNA-RT complex (pdb code 4B3P[20]). Compound 33 is shown in ball and stick, residue Q500 in stick. Binding pocket residues are represented in magenta lines for RT-33 complex and yellow for RNA:DNA-RT complex.
Based on these results, we hypothesized that the cHTCs binding to the RNase H allosteric site could be affected by competition with the RNA:DNA duplex, especially for compounds that are weaker inhibitors. To test this hypothesis, RNase H activity of compounds 31 and 33 was re-assayed by modifying the order of addition of the RNA:DNA duplex substrate. Indeed, similar to reports for the N-[3-(aminocarbonyl)-4,5-dimethyl-2-thienyl]-2-furancarboxamide,[17] when the inhibitors were added after the duplex substrate, weaker inhibitory activity was measured (IC50 values of 20.0 ± 7.0 μM and 5.3 ± 0.2 μM, for 31 and 33, respectively), 5-fold higher than the one obtained on the regular assay, further suggesting cHTC derivatives interfere with the dsRNA:DNA accommodation.
To further compare the binding mode of compound 33 and 3, we performed a Yonetani-Theorell analysis[39] on the combined effects of these two compounds on the HIV-1 RNase H function (Figure 4). Such an analysis reveals whether simultaneous binding (or inhibition) of two compounds is possible or not. Results showed that 33 and 3 inhibitors are kinetically mutually exclusive, demonstrating that they either bind to the same site or that, in any case, the binding of the first prevents the binding of the second. Possibly, as suggested by the docking study, the binding sites are slightly different, with an overlapping of the catechol moiety (Figure 2).
Figure 4.

Yonetani–Theorell plot of the combination of 33 and 3 on HIV-1 RT RNase H activity. HIV-1 RT was incubated in the presence of 33 (3.3 μM, 1.1 μM, 0.55 μM, 0.27 μM, and 0.13 μM) combined with increasing concentrations of 3: 0.27 μM (●), 0.55 μM (○), 1.1 μM (▼), and 3.3 μM (Δ). Data were obtained by a single experiment, made in duplicate.
Compounds 31 and 33 were also tested for their ability to inhibit HIV-1 (IIIB strain) and HIV-2 (ROD strain) replication in acutely infected MT-4 cells evaluating in parallel the cytotoxicity. Compounds were unable to affect viral replication at concentrations less than those that were cytotoxic (CC50 = 6.24 and 30.69 μM for 31 and 33, respectively).
The lack of antiviral activity might be explained by the fact that the compounds in vivo are not potent enough to compete with the RNA:DNA duplex at the RNase H allosteric site or that their binding site is hampered by the duplex.
Conclusions
New anti-HIV-1 RNase H inhibitor derivatives have been developed based on the cycloheptathiophene-3-carboxamide scaffold that, with one exception, do not inhibit the RDDP function. Since they are unable to chelate Mg2+ ions, we hypothesized an allosteric mechanism of inhibition. Evaluation of the anti-RNase H activity of the C280A RT mutant, a key residue of the vinylogous urea binding, and ThermoFluor analysis showed that cHTCs have a different binding site from compounds 1–3 previously reported. Docking studies suggested that the new derivatives recognize an allosteric binding site located below the catalytic site, entailing p66 residue Q500 as key residue for the binding. This residue, known to interact with the RNA strand within the RNA:DNA duplex substrate, has already captured the attention in the search for novel druggable allosteric site to inhibit the RNase H. Despite being unable to correlate RNase H inhibitory activity with inhibition of HIV replication, this study should promote synthesis of more potent allosteric RNase H inhibitors endowed with anti-HIV activity in vivo.
Experimental Section
Chemistry
All reactions were routinely checked by TLC on silica gel 60F254 (Merck) and visualized using UV or iodine. Flash column chromatography separations were carried out on Merck silica gel 60 (mesh 230–400). Melting points were determined in capillary tubes (Büchi Electrothermal Mod. 9100) and are uncorrected. HRMS spectra were registered on Agilent Technologies 6540 UHD Accurate Mass Q-TOF LC/MS, HPLC 1290 Infinity. Purity of the target compounds was determined by LC/MS on Agilent Technologies 6550 iFUNNEL Q-TOF equipped with HPLC 1290 Infinity with DAD detector and evaluated to be higher than 95%. HPLC conditions to assess the purity of final compounds were as follows: column, Phenomenex AERIS Widepore C4, 4.6mm × 100 mm (6.6 μm); flow rate, 0.85 mL/min; acquisition time, 10 min; DAD 190−650 nm; oven temperature, 30 °C; gradient of acetonitrile in water containing 0.1% of formic acid (0−100% in 10 min). 1H NMR and 13C NMR spectra were recorded at 200 MHz (Bruker Avance DPX-200) and 400 MHz (Bruker Avance DRX-400) using residual solvents such as chloroform (δ = 7.26) or dimethylsulfoxide (δ = 2.48) as an internal standard. Chemical shifts are given in ppm () and the spectral data are consistent with the assigned structures. Reagents and solvents were purchased from common commercial suppliers and used as such. After extraction, organic solutions were dried over anhydrous Na2SO4, filtered, and concentrated with a Büchi rotary evaporator at reduced pressure. Yields are of purified product and were not optimized. All starting materials were commercially available unless otherwise indicated.
General procedure for C-2 amide bond formation.
To a solution of the appropriate 2-aminocycloheptathiophene derivative (1.0 equiv) in pyridine, the suitable acyl chloride (2.0 equiv) was added. The reaction mixture was maintained at room temperature until no starting material was detected by TLC (2–72 h) and worked up and purified as defined in the description of the compounds.
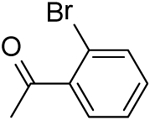
2-(2-Bromobenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (23).
Compound 23 was prepared starting from 38[27] by the general procedure, using 2-bromobenzoyl chloride. After 19 h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (15%), to give 23 in 16% yield: mp 166–168 °C; 1H NMR (DMSO-d6): δ = 1.40–1.60 (m, 4H, cycloheptane CH2), 1.65–1.80 (m, 2H, cycloheptane CH2), 2.55–2.75 (m, 4H, cycloheptane CH2), 7.00–7.10 (m, 1H, pyridine CH), 7.25–7.40 (m, 2H, aromatic CH), 7.45–7.75 (m, 3H, aromatic CH and pyridine CH), 8.10 (d, J = 8.2 Hz, 1H, pyridine CH), 8.25 (d, J = 4.0 Hz, 1H, pyridine CH), 10.40 and 11.00 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 27.2, 27.5, 28.6, 29.0, 31.6, 114.3, 118.5, 119.9, 127.6, 129.9, 131.8, 132.9, 133.0, 133.8136.1, 138.3, 141.7, 151.0, 163.9, 164.5; HRMS m/z [M+H]+ calcd for C22H20BrN3O2S: 470.0539, found: 470.0541.
2-(2-Nitrobenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (24).
Compound 24 was prepared starting from 38[27] by the general procedure, using 2-nitrobenzoyl chloride. After 20 h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (20%) and then crystallized by EtOH, to give 24 in 7% yield: mp 204–207 °C; 1H NMR (DMSO d6): δ = 1.45–1.65 (m, 4H, cycloheptane CH2), 1.75–1.85 (m, 2H, cycloheptane CH2), 2.65–2.80 (m, 4H, cycloheptane CH2), 7.05–7.10 (m, 1H, pyridine CH), 7.70–7.85 (m, 4H, pyridine CH and aromatic CH), 8.10 (d, J = 8.0 Hz, 1H, pyridine CH), 8.15 (d, J = 8.3 Hz, 1H, aromatic CH), 8.25 (d, J = 3.7 Hz, 1H, pyridine CH) 10.60 and 11.30 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 27.1, 27.4, 28.6, 29.0, 31.5, 114.2, 118.4, 120.0, 124.7, 128.6, 131.1, 131.3, 132.8, 133.3, 133.5, 138.2, 141.9, 147.0, 148.1, 150.9, 162.3, 164.6 ppm. HRMS m/z [M+H]+ calcd for C22H20N4O4S: 437.1239, found: 437.1288.
2-(4-Nitrobenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (25).
Compound 25 was prepared starting from 38[27] by the general procedure, using 4-nitrobenzoyl chloride. After 24h, the reaction mixture was poured into ice/water and extracted with EtOAc. The organic layers were evaporated to dryness, yielding a solid that was purified by flash chromatography eluting with EtOAc/cyclohexane (40%), to give 25 in 46% yield: mp 230–234 °C; 1H NMR (DMSO-d6): δ = 1.50–1.90 (m, 6H, cycloheptane CH2), 2.70–2.80, (m, 4H, cycloheptane CH2), 7.10–7.20 and 7.75–7.95 (m, each 1H, pyridine CH), 8.10 (d, J = 8.8 Hz, 2H, aromatic CH), 8.20 (d, J = 7.8 Hz, 1H, pyridine CH), 8.30–8.40 (m, 3H, aromatic CH and pyridine CH), 10.50 and 11.25 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 27.1, 27.4, 28.6, 29.1, 31.4, 114.4, 118.1, 120.2, 124.0, 128.5, 133.0, 133.4, 138.1, 138.3, 142.6, 148.1, 150.0, 150.8, 161.1, 165.0 ppm. HRMS m/z [M+H]+ calcd for C22H20N4O4S: 437.1239, found: 437.1293.
2-(2-Aminobenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (26).
To a solution of 24 (0.1 g, 0.24 mmol) in a MeOH/DMF (1:2) mixture (10 mL) Pd/C 10% (0.12 g) and ammonium formate (0.07 g, 1.21 mmol) were added. The reaction mixture was maintained at room temperature for 120 h and then filtered over celite. The filtrate was evaporated to dryness affording to a crude product that was purified by flash chromatography eluting with EtOAc/cyclohexane (15%), to give 26 (0.03g, 30%): mp 217–219 °C; 1H NMR (DMSO d6): δ = 1.50–1.70 (m, 4H, cycloheptane CH2), 1.75–1.85 (m, 2H, cycloheptane CH2), 2.65–2.80 (m, 4H, cycloheptane CH2), 6.50 (t, J = 7.4 Hz, 1H, aromatic CH), 6.75 (d, J = 8.21 Hz, 1H, pyridine CH), 7.10 (t, J = 5.9 Hz, 1H, aromatic CH), 7.15 (t, J = 7.5 Hz, 1H, aromatic CH), 7.50 (d, J = 7.9 Hz, 1H, aromatic CH), 7.75 (d, J = 7.3 Hz, 1H, pyridine CH), 8.10 (d, J = 8.3 Hz, 1H, pyridine CH), 8.25 (d, J = 4.3 Hz, 1H, pyridine CH), 10.60 and 11.30 ppm (s, each 1H, NH); HRMS m/z [M+H]+ calcd for C22H22N4O2S: 407.1541, found: 407.1561.
2-(4-Aminobenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (27).
Compound 27 was prepared starting from 25 by using the same procedure as used for the synthesis of compound 26 (1h), with the exception that it was purified by crystallization by EtOH (80% yield): mp 210–212 °C; 1H NMR (DMSO-d6): δ = 1.40–1.60 (m, 4H, cycloheptane CH2), 1.70–1.85 (m, 2H, cycloheptane CH2), 2.55–2.70 and 3.30–3.40 (m, each 2H, cycloheptane CH2), 6.95–7.05 (m, 1H, pyridine CH), 7.70–7.85 (m, 1H, pyridine CH), 8.20–8.35 (m, 3H, aromatic CH and pyridine CH), 8.50 (d, J = 3.7 Hz, 1H, pyridine CH), 8.70 (d, J = 8.8 Hz, 2H, aromatic CH), 10.60 and 11.30 ppm (s, each 1H, NH); HRMS: m/z [M+H]+ calcd for C22H22N4O2S: 407.1541, found: 407.1563.
2-(2-Methoxybenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (28).
Compound 28 was prepared starting from 38[27] by the general procedure, using 2-methoxybenzoyl chloride. After 72 h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (20%), to give 28 in 47% yield: mp 226–228 °C; 1H NMR (DMSO-d6): δ = 1.50–1.70 (m, 4H, cycloheptane CH2), 1.75–1.85 (m, 2H, cycloheptane CH2), 2.70–2.85 (m, 4H, cycloheptane CH2), 3.95 (s, 3H, CH3), 7.10–7.20 (m, 2H, pyridine CH and aromatic CH), 7.25 (d, J = 8.4 Hz, 1H, aromatic CH), 7.60 (t, J = 7.3 Hz, 1H, aromatic CH), 7.85 (t, J = 7.1 Hz, 1H, pyridine CH), 8.05 (d, J = 6.9 Hz, 1H, aromatic CH), 8.25 (d, J = 8.3 Hz, 1H, pyridine CH), 8.35 (d, J = 4.3 Hz, 1H, pyridine CH), 10.40 and 12.00 ppm (s, each 1H, NH); HRMS m/z [M+H]+ calcd for C22H20BrN3O2S: 422.1494, found: 422.1552.
2-(2-Methylbenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (29).
Compound 29 was prepared starting from 38[27] by the general procedure, using 2-methylbenzoyl chloride. After 72 h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (20%), to give 29 in 26% yield: mp 258–261 °C; 1H NMR (DMSO-d6): δ = 1.40–1.60 (m, 4H, cycloheptane CH2), 1.60–1.80 (m, 2H, cycloheptane CH2), 2.25 (s, 3H, CH3), 2.50–2.70 (m, 4H, cycloheptane CH2), 7.05 (dd, J = 2.1 and 7.0 Hz, 1H, aromatic CH), 7.10–7.25 (m, 2H, pyridine CH and aromatic CH), 7.25–7.35 (m, 1H, aromatic CH), 7.45 (d, J = 7.4 Hz, 1H, aromatic CH), 7.60–7.75 (m, 1H, pyridine CH), 8.10 (d, J = 8.3 Hz, 1H, pyridine CH), 8.25 (d, J = 3.5 Hz, 1H, pyridine CH), 10.40 and 10.80 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ =20.4, 27.2, 27.5, 28.6, 29.0, 31.6, 114.2, 117.9, 119.9, 126.1, 127.4, 130.8, 131.5, 132.4, 132.8, 133.9, 137.6, 138.2, 142.6, 148.0, 151.1, 164.8, 165.9; HRMS m/z [M+H]+ calcd for C23H23N3O2S: 406.1545, found: 406.1594.
2,6-Dimethoxy-N-(3-(pyridin-2-ylcarbamoyl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)nicotinamide (30).
Compound 30 was prepared starting from 38[27] by the general procedure, using 2,6-dimethoxynicotinoyl chloride. After 16 h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (15%), to give 30 in 48% yield: mp 224–226 °C; 1H NMR (DMSO-d6): δ = 1.40–1.80 (m, 6H, cycloheptane CH2), 2.60–2.80 (m, 4H, cycloheptane CH2), 3.85 and 3.95 (s, each 3H, CH3), 6.50 (d, J = 8.5 Hz, 1H, aromatic CH), 7.05–7.10 (m, 1H, pyridine CH), 7.75–7.85 (m, 1H, pyridine CH), 8.10–8.40 (m, 3H, pyridine CH and aromatic CH), 10.40 and 11.60 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 27.4, 27.6, 28.7, 29.0, 31.8, 53.9, 103.1, 106.5, 113.8, 118.8, 119.6, 132.5, 133.0, 138.2, 141.5, 144.1, 148.0, 151.6, 160.4, 160.9, 164.5, 165.2; HRMS m/z [M+H]+ calcd for C23H24N4O4S: 453.1552, found: 453.1601.
2-(3,4-Dimethoxybenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (31).
The title compound was prepared starting from 38[27] by the general procedure, using 3,4-dimethoxybenzoyl chloride. After 3 h, the reaction mixture was poured into ice/water yielding a precipitate that was filtered and crystallized by EtOH, to give 31 in 42% yield: mp 194–196 °C; 1H NMR (DMSO-d6): δ = 1.50–1.70 (m, 4H, cycloheptane CH2), 1.75–1.85 (m, 2H, cycloheptane CH2), 2.60–2.75 (m, 4H, cycloheptane CH2), 3.70 and 3.80 (s, each 3H, CH3), 7.00–7.20 (m, 2H, pyridine CH and aromatic CH), 7.30–7.50 (m, 2H, aromatic CH), 7.80 (t, J = 7.7 Hz 1H, pyridine CH), 8.15 (d, J = 8.6 Hz, 1H, pyridine CH), 8.25–8.35 (m, 1H, pyridine CH), 10.30 and 10.80 ppm (s, each 1H, NH); HRMS m/z [M+H]+ calcd for C24H25N3O4S: 452.1599, found: 452.1652.
2-Hydroxy-6-methoxy-N-(3-(pyridin-2-ylcarbamoyl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)nicotinamide (32).
To a solution of 30 (0.29 g, 0.79 mmol) in CH2Cl2 dry (3 mL), a 1M solution of BBr3 in CH2Cl2 (3.9 ml, 3.91 mmol) was added dropwise maintaining the temperature at 0 °C. The reaction mixture was stirred at room temperature for 2 h and then poured into ice/water obtaining a precipitate, which was filtered and purified by crystallization by EtOH, to give 32 (0.17 g, 60%): mp 191–193 °C; 1H NMR (CDCl3): δ = 1.45–1.60 (m, 4H, cycloheptane CH2), 1.60–1.80 (m, 2H, cycloheptane CH2), 2.55–2.75 (m, 4H, cycloheptane CH2), 4.85 (s, 3H, CH3), 6.10 (d, J = 8.4 Hz, 1H, pyridine CH), 7.20–7.30 (m, 1H, pyridine CH), 7.80–8.00 (m, 2H, pyridine CH and aromatic CH), 8.30–8.40 (m, 2H, pyridine CH and aromatic CH), 11.00 and 12.80 ppm (s, each 1H, NH); HRMS m/z [M+H]+ calcd for C22H22N4O4S: 439.1395, found: 439.1445.
2-(3,4-Dihydroxybenzamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (33).
Compound 33 was prepared starting from 31, by using the same procedure as used for the synthesis of compound 32 in 19% yield: mp 213–215 °C; 1H NMR (DMSO-d6): δ = 1.40–1.75 (m, 6H, cycloheptane CH2), 2.55–2.75 (m, 4H, cycloheptane CH2), 6.75 (d, J = 8.2 Hz, 1H, aromatic CH), 7.05–7.40 (m, 3H, pyridine CH and aromatic CH), 7.75 (t, J = 7.9 Hz, 1H, pyridine CH), 8.10 (d, J = 8.2 Hz, 1H, pyridine CH), 8.25 (d, J = 3.9 Hz, 1H, pyridine CH), 9.25, 9.70, 10.25, and 10.75 ppm (s, each 1H, OH and NH); HRMS m/z [M+H]+ calcd for C22H21N3O4S: 424.1286, found: 424.1342.
N-(Pyridin-2-yl)-2-(thiophene-2-carboxamido)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (34).
Compound 34 was prepared starting from 38[27] by the general procedure, using thiophene-2-carbonyl chloride. After 2h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (20%), to give 34 in 49% yield: mp 221–223 °C; 1H NMR (DMSO-d6): δ = 1.40–1.80 (m, 6H, cycloheptane CH2), 2.60–2.70 (m, 4H, cycloheptane CH2), 7.00–7.15 (m, 2H, pyridine CH and thiophene CH), 7.70–7.85 (m, 3H, pyridine CH and thiophene CH), 8.10 (d, J = 8.6 Hz, 1H, pyridine CH), 8.20–8.30 (m, 1H, pyridine CH), 10.30 and 10.80 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 27.1, 27.5, 28.5, 29.1, 31.4, 114.3, 119.9, 127.9, 129.0, 131.5, 132.7, 135.6, 137.7, 138.3, 143.0, 148.0, 151.0, 158.2, 164.9; HRMS: m/z [M+H]+ calcd for C20H19N3O2S2: 398.0952, found: 398.1007.
2-Acetamido-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (35).
To a solution of 38[27] (0.20 g, 0.69 mmol) in THF dry (5 mL), Et3N (0.14 ml, 1.04 mmol) and acetyl chloride (0.048 ml, 0.69 mmol) were added. After 2 h, the reaction mixture was poured into ice/water, producing a precipitate that was filtered and purified by flash chromatography, eluting with EtOAc/cyclohexane (20%), to give 35 (0.032 g, 14%): mp 250–251°C; 1H NMR (DMSO-d6): δ = 1.40–1.80 (m, 6H, cycloheptane CH2), 2.00 (s, 3H, CH3), 2.50–2.70 (m, 4H, cycloheptane CH2), 7.00–7.10 and 7.70–7.85 (m, each 1H, pyridine CH), 8.10 (d, J = 8.8 Hz, 1H, pyridine CH), 8.20–8.30 (m, 1H, pyridine CH), 10.30 and 10.40 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 23.4, 27.2, 27.5, 28.5, 29.0, 31.5, 114.1, 117.3, 119.9, 132.2, 132.5, 138.2, 142.4, 148.1, 151.1, 164.8, 166.7; HRMS m/z [M+H]+ calcd for C17H19N3O2S: 330.1232, found 330.1284.
2-(2-(2-Fluorophenyl)acetamido)-N-(pyridin-2-yl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (36).
Compound 36 was prepared starting from 38[27] by the general procedure, using 2-(2-fluorophenyl)acetyl chloride. After 18 h, the reaction mixture was poured into ice/water yielding a precipitate, which was filtered and purified by flash chromatography eluting with EtOAc/cyclohexane (15%), to give 36 in 15% yield: mp 164–166 °C; 1H NMR (DMSO-d6): δ = 1.40–1.60 (m, 4H, cycloheptane CH2), 1.60–1.80 (m, 2H, cycloheptane CH2), 2.50–2.65 (m, 4H, cycloheptane CH2), 3.70 (s, 1H, CH2), 7.00–7.30 (m, 5H, pyridine CH and aromatic CH), 7.70–7.80 (m, 1H, pyridine CH), 8.10 (d, J = 8.1 Hz, 1H, pyridine CH), 8.25 (d, J = 4.3 Hz, 1H, pyridine CH), 10.40 and 10.60 ppm (s, each 1H, NH); HRMS m/z [M+H]+ calcd for C23H22FN3O2S: 424.1450, found: 424.1499.
2-Amino-N-(2-fluorophenyl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxamide (39).
A mixture of 2-cyano-N-(2-fluorophenyl)acetamide[31] (0.88 g, 4.49 mmol), cycloheptanone (2.10 ml, 17.96 mmol), ammonium acetate (0.45g, 5.9mmol), and glacial acetic acid (0.89 ml, 15.71mmol), in benzene (45 mL) was refluxed for 16 h in a Dean-Stark apparatus. After cooling, the reaction mixture was diluted with CHCl3, washed with H2O, with 10% Na2CO3 solution and then with H2O. The organic phases was evaporated to dryness, affording to the crude Knoevenagel product, which was used in the next step without further purification. This material was dissolved in EtOH (15mL) and added of sulphur (0.57g, 17.96 mmol) and N,N-diethylamine (1.86 ml, 17.96 mmol). The reaction mixture was maintained at 40–50 °C for 1.5 h and then evaporated to dryness, yielding a residue that after washing with cyclohexane gave 39 (0.35g, 25%); mp 100–104 °C; 1H NMR (DMSO-d6): δ = 1.45–1.95 (m, 6H, cycloheptane CH2), 2.60–2.70 and 2.80–2.90 (m, each 2H, cycloheptane CH2), 6.05 (bs, 2H, NH2), 7.00–7.25 (m, 3H, aromatic CH), 7.80–7.90 (m, 1H, aromatic CH), 9.25 ppm (bs, 1H, NH).
N-(3-((2-Fluorophenyl)carbamoyl)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)picolinamide (37).
Compound 37 was prepared starting from 39 by the general procedure, using picolinoyl chloride. After 16 h, the reaction mixture was poured into ice/water and extracted with EtOAc. The organic layers were evaporated to dryness, yielding a solid that was purified by flash chromatography eluting with EtOAc/cyclohexane (20%), to give 37 in 17% yield: mp 200–202 °C; 1H NMR (DMSO-d6): δ = 1.55–1.70 (m, 4H, cycloheptane CH2), 1.75–1.85, 2.75–2.85, and 2.85–2.95 (m, each 2H, cycloheptane CH2), 7.20–7.35 (m, 3H, pyridine CH and aromatic CH), 7.60–7.70 (m, 2H, pyridine CH and aromatic CH), 8.10 (t, J = 7.7 Hz, 1H, aromatic CH), 8.15 (d, J = 7.6 Hz, 1H, pyridine CH), 8.70 (d, J = 4.3 Hz, 1H, pyridine CH), 9.80 and 11.90 ppm (s, each 1H, NH); 13C NMR (CDCl3): δ = 27.1, 27.6, 28.7, 28.9, 31.8, 114.7, 118.9, 119.4, 121.9, 122.7, 124.3, 124.6, 126.4, 133.0, 133.1, 137.3, 143.5, 148.8, 148.9, 158.9, 161.5, 161.6 ppm. HRMS m/z [M+H]+ calcd for C22H20FN3O2S: 410.1294, found: 410.1342.
Biological Methods
Expression and purification of recombinant HIV-1 RT:
HIV-1 RT group M subtype B. Heterodimeric RT was expressed essentially as described.[40] Briefly, E. coli strain M15 containing the p6HRT-Prot vector was grown to an optical density at 600 nm of 0.7 and induced with 1.7 mM isopropyl β-D-1-thiogalactopyranoside for 4 h. Protein purification was carried out with a BioLogic LP system (BioRad), using a combination of immobilized metal affinity and ion exchange chromatography. Cell pellets were resuspended in lysis buffer (50 mM sodium phosphate buffer pH 7.8, containing 0.5 mg/mL lysozyme), incubated on ice for 20 min, and after adding NaCl to a final concentration of 0.3 M, were sonicated and centrifuged at 30,000×g for 1 h. The supernatant was loaded onto a Ni2+-NTA-Sepharose column pre-equilibrated with loading buffer (50 mM sodium phosphate buffer pH 7.8, containing 0.3M NaCl, 10% glycerol, and 10 mM imidazole) and washed thoroughly with wash buffer (50 mM sodium phosphate buffer pH 6.0, containing 0.3 M NaCl, 10% glycerol, and 80 mM imidazole). RT was eluted with an imidazole gradient in wash buffer (0 – 0.5). Fractions were collected, protein purity was checked by SDS-PAGE and found to exceed 90%. Enzyme-containing fractions were pooled and diluted 1:1 with 50 mM sodium phosphate buffer pH 7.0, containing 10% glycerol and then loaded into a Hi-trap heparin HP GE (Healthcare Lifescience) pre-equilibrated with 10 column volumes of loading buffer (50 mM sodium phosphate buffer pH 7.0, containing 10% glycerol and 150 mM NaCl). The column was washed with loading buffer and the RT was eluted with Elute Buffer 2 (50 mM Sodium Phosphate pH 7.0, 10% glycerol, 1M NaCl). Fractions were collected, and purified protein was dialyzed and stored in buffer containing 50 mM Tris-HCl pH 7.0, 25 mM NaCl, 1mM EDTA, and 50% glycerol. Catalytic activities and protein concentrations were determined. Enzyme-containing fractions were pooled and aliquots were stored at −80 °C.
HIV-1 DNA polymerase-independent RNase H activity determination:
HIV RT-associated RNase H activity was measured as described[41] in 100 μL reaction volume containing 50 mM Tris-HCl buffer pH 7.8, 6 mM MgCl2, 1 mM dithiothreitol (DTT), 80 mM KCl, 0.25 μM hybrid RNA/DNA 5’-GAUCUGAGCCUGGGAGCU-Fluorescin-3’ (HPLC, dry, QC: Mass Check) (available from Metabion) 5’-Dabcyl-AGCTCCCAGGCTCAGATC-3’ (HPLC, dry, QC: Mass Check) and 20 ng of wt RT according to a linear range of dose-response curve. The reaction mixture was incubated for 10 min at 37 °C in a multilabel counter plate reader Victor 3 (Perkin Elmer model 1420–051) and product were quantified with at 490/528 nm (excitation/emission wavelength).
Yonetani-Theorell analysis:
The Yonetani-Theorell analysis was performed as described previously.[13,39]
Evaluation of MgCl2 coordination:
The coordination properties for the compounds was determined as reported.[42] Briefly, compounds were solubilized in 1 mL of 10% ethanol and 10 mM Tris–HCl, pH 7.8. The UV-Vis spectrum was recorded from 250 to 600 nm before and after addition of 6 mM MgCl2.
HIV-1 RNA-dependent DNA polymerase activity determination:
RNA-dependent DNA polymerase (RDDP) activity was measured as described[43] in 25 μL volume containing 60 mM Tris-HCl buffer pH 8.1, 8 mM MgCl2, 60 mM KCl, 13 mM DTT, 2.5 μM poly(A)-oligo(dT),100 μM dTTP, and 6 ng of wt RT, according to a linear range of dose-response curve. After enzyme addition, the reaction mixture was incubated for 30 min at 37 °C and the stopped by addition of EDTA. Reaction products were detected by picogreen addition and measured with a multilabel counter plate reader Victor 3 (Perkin Elmer model 1420–051) equipped with filters for 502/523 nm (excitation/emission wavelength).
Detection of protein inhibitor interactions by Differential Scanning Fluorimetry:
Thermal stability assays were performed according to Nettleship et al.[44] In a LightCycler 480 96-well plate (Roche) we incubated10 μM inhibitor in 50 μl of reaction buffer containing 20 mM HEPES, pH 7.5, 10 mM MgCl2, 100 mM NaCl, 300 nM HIV-1 RT and a 1:1000 dilution of Sypro Orange dye (Invitrogen). The mixture was heated from 30 to 90 °C in increments of 0.2 °C. Fluorescence intensity was measured using excitation and emission wavelengths of 483 and 568 nm, respectively. Changes in protein thermal stability (ΔTm) upon inhibitor binding were analyzed by using the LightCycler 480 software. All assays were performed in triplicate.
In Vitro Antiviral Assays.
Evaluation of the antiviral activity of the compounds against HIV-1 strain IIIB in MT-4 cells was performed using the MTT assay as previously described.[45,46] Stock solutions (10 x final concentration) of test compounds were added in 25 μl volumes to two series of triplicate wells so as to allow simultaneous evaluation of their effects on mock- and HIV-infected cells at the beginning of each experiment. Serial 5-fold dilutions of test compounds were made directly in flat-bottomed 96-well microtiter trays using a Biomek 3000 robot (Beckman instruments, Fullerton, CA). Untreated HIV- and mock-infected cell samples were included as controls. HIV-1(IIIB) stock (50 μl) at 100–300 CCID50 (50 % cell culture infectious doses) or culture medium was added to either the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effects of test compound on uninfected cells in order to assess the cytotoxicity of the test compounds. Exponentially growing MT-4 cells were centrifuged for 5 minutes at 220 g and the supernatant was discarded. The MT-4 cells were resuspended at 6 × 105 cells/ml and 50 μl volumes were transferred to the microtiter tray wells. Five days after infection, the viability of mock-and HIV-infected cells was examined spectrophotometrically using the MTT assay. The MTT assay is based on the reduction of yellow colored 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Acros Organics) by mitochondrial dehydrogenase activity in metabolically active cells to a blue-purple formazan that can be measured spectrophotometrically. The absorbances were read in an eight-channel computer-controlled photometer (Infinite M1000, Tecan), at two wavelengths (540 and 690 nm). All data were calculated using the median absorbance value of three wells. The 50% cytotoxic concentration (CC50) was defined as the concentration of the test compound that reduced the absorbance (OD540) of the mock-infected control sample by 50%. The concentration achieving 50% protection against the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration (EC50).
Docking analysis
Ligand preparation:
Ligands were docked in the global minimum energy conformation as determined by molecular mechanics conformational analysis. Three-dimensional ligand input structures were generated using Maestro GUI.[47] The molecules were then subjected to a conformational search of 1000 steps with an energy window for saving structure of 10 kJ/mol using the MMFFs (Merck molecular force fields)[48] and the GB/SA water implicit solvation model.[49] The Polak-Ribier Conjugate Gradient (PRCG) method was applied for the minimization step, 5000 iterations converging on gradient with a threshold of 0.05 kJ(mol•Å)-1.
Protein preparation:
The atomic coordinates of the unbounded protein with pdb code 1hmv[50] were imported into the Maestro GUI.[47] The protein was further optimized using the Protein Preparation Wizard,[47] missing side chains were added with Prim e.[51]
Docking and post docking:
Molecular docking studies were performed using IFD protocol.[34] The docking grid was defined by centring on Q500 and occupies a volume of 20×20×20 Å. Default settings were applied. Complexes thus obtained were subjected to a post-docking procedure based on energy minimization and subsequent binding energies calculation. Binding energies were obtained applying molecular mechanics and continuum solvation models using the Molecular Mechanics Generalized Born/Surface Area (MM-GBSA) method.[52] The resulting complexes were considered for the binding modes graphical analysis with Maestro.[47]
Supplementary Material
Acknowledgements
This work was carried out with financial supports by PRIN 2011 (grant no. 2010W2KM5L_004). S.F.J. Le Grice and T. Masaoka were supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services. The authors wish to thank Kristien Erven, Kris Uyttersprot, and Roberto Pellegrino for dedicated technical assistance.
Footnotes
Supporting Information
Effect of MgCl2 on the spectrum of absorbance of compounds 6, 9, 12, 23, 31, 33, 34, and 36 (figure S1).
References
- 1.Frentz D, Van de Vijver DAMC, Abecasis AB, Albert J, Hamouda O, Jørgensen LB, Kücherer C, Struck D, Schmit J-C, Vercauteren J, et al. BMC Infect. Dis 2014, 14, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frentz D, van de Vijver D, Abecasis A, Albert J, Hamouda O, Jørgensen L, Kücherer C, Struck D, Schmit J-C, Vercauteren J, et al. PLoS One 2014, 9, e94495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corona A, Esposito F, Tramontano E. Future Virol. 2014, 9, 445–448. [Google Scholar]
- 4.Schatz O, Cromme FV, Grüninger-Leitch F, Le Grice SFJ. FEBS Lett. 1989, 257, 311–314. [DOI] [PubMed] [Google Scholar]
- 5.Esposito F, Corona A, Tramontano E. Mol. Biol. Int 2012, 2012, 586401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corona A, Masaoka T, Tocco G, Le Grice SFJ, Tramontano E. Futur. Med. Chem 2013, 5, 2127–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corona A, Di Leva FS, Thierry S, Pescatori L, Cuzzucoli Crucitti G, Subra F, Delelis O, Esposito F, Rigogliuso G, Costi R, Cosconati S, Novellino E, Di Santo R, Tramontano E. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuzzucoli Crucitti G, Métifiot M, Pescatori L, Messore A, Madia VN, Pupo G, Saccoliti F, Scipione L, Tortorella S, Esposito F, Corona A, Cadeddu M, Marchand C, Pommier Y, Tramontano E, Costi R, Di Santo R. J. Med. Chem 2015, 58, 1915–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meleddu R, Distinto S, Corona A, Bianco G, Cannas V, Esposito F, Artese A, Alcaro S, Matyus P, Bogdan D, Cottiglia F, Tramontano E, Maccioni E. Eur. J. Med. Chem 2015, 93, 452–460. [DOI] [PubMed] [Google Scholar]
- 10.Chung S, Himmel DM, Jiang J-K, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. J. Med. Chem 2011, 54, 4462–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poongavanam V, Olsen JMH, Kongsted J. Integr. Biol. (Camb) 2014, 6, 1010–1022. [DOI] [PubMed] [Google Scholar]
- 12.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. ACS Chem. Biol 2006, 1, 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esposito F, Kharlamova T, Distinto S, Zinzula L, Cheng Y-C, Dutschman G, Floris G, Markt P, Corona A, Tramontano E. FEBS J. 2011, 278, 1444–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esposito F, Corona A, Zinzula L, Kharlamova T, Tramontano E. Chemotherapy 2012, 58, 299–307. [DOI] [PubMed] [Google Scholar]
- 15.Meleddu R, Cannas V, Distinto S, Sarais G, Del Vecchio C, Esposito F, Bianco G, Corona A, Cottiglia F, Alcaro S, Parolin C, Artese A, Scalise D, Fresta M, Arridu A, Ortuso F, Maccioni E, Tramontano E. Chem. Med. Chem 2014, 9, 1869–1879. [DOI] [PubMed] [Google Scholar]
- 16.Wendeler M, Lee HF, Bermingham A, Miller JT, Chertov O, Bona MK, Baichoo NS, Ehteshami M, Beutler J, O’Keefe BR, Gӧtte M, Kvaratskhelia M, Le Grice S. ACS Chem. Biol 2008, 3, 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung S, Wendeler M, Rausch JW, Beilhartz G, Gotte M, O’Keefe BR, Bermingham A, Beutler JA, Liu S, Zhuang X, Le Grice SF. Antimicrob. Agents Chemother. 2010, 54, 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung S, Miller JT, Johnson BC, Hughes SH, Le Grice SFJ. J. Biol. Chem 2012, 287, 4066–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masaoka T, Chung S, Caboni P, Rausch JW, Wilson JA, Taskent-Sezgin H, Beutler JA, Tocco G, Le Grice SFJ. J. Med. Chem 2013, 56, 5436–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lapkouski M, Tian L, Miller JT, Le Grice SFJ, Yang W. Nat. Struct. Mol. Biol 2013, 20, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabarrini O, Massari S, Cecchetti V. Future Med. Chem 2010, 2, 1161–1180. [DOI] [PubMed] [Google Scholar]
- 22.Tabarrini O, Massari S, Daelemans D, Meschini F, Manfroni G, Bottega L, Gatto B, Palumbo M, Pannecouque C, Cecchetti V. Chem. Med. Chem 2010, 5, 1880–1892. [DOI] [PubMed] [Google Scholar]
- 23.Massari S, Sabatini S, Tabarrini O. Curr. Pharm. Des 2013, 19, 1860–1879. [DOI] [PubMed] [Google Scholar]
- 24.Tabarrini O O, Massari S, Sancineto L, Daelemans D, Sabatini S, Manfroni G, Cecchetti V, Pannecouque C. Chem. Med. Chem 2011, 6, 1249–1257. [DOI] [PubMed] [Google Scholar]
- 25.Sancineto L, Mariotti A, Bagnoli L, Marini F, Desantis J, Iraci N, Santi C, Pannecouque C, Tabarrini O. J. Med. Chem 2015, 58, 9601–9614. [DOI] [PubMed] [Google Scholar]
- 26.Zhan P, Pannecouque C, De Clercq E, Liu X. J. Med. Chem 2015, doi: 10.1021/acs.jmedchem.5b00497 [DOI] [Google Scholar]
- 27.Massari S, Nannetti G, Goracci L, Sancineto L, Muratore G, Sabatini S, Manfroni G, Mercorelli B, Cecchetti V, Facchini M, Palù G, Cruciani G, Loregian A, Tabarrini O. J. Med. Chem 2013, 56, 10118–10131. [DOI] [PubMed] [Google Scholar]
- 28.Massari S, Nannetti G, Desantis J, Muratore G, Sabatini S, Manfroni G, Mercorelli B, Cecchetti V, Palù G, Cruciani G, Loregian A, Goracci L, Tabarrini O. J. Med. Chem 2015, 58, 3830–3842. [DOI] [PubMed] [Google Scholar]
- 29.Gewald K, Schinke E, Böttcher H. Chem. Ber 1966, 99, 94–100. [Google Scholar]
- 30.Sabnis RW, Rangnekar DW, Sonawane ND. J. Heterocycl. Chem 1999, 36, 333–345. [Google Scholar]
- 31.Gorobets NY, Yousefi BH, Belaj F, Kappe CO. Tetrahedron 2004, 60, 8633–8644. [Google Scholar]
- 32.Rodgers DW, Gamblin SJ, Harris BA, Ray S, Culp JS, Hellmig B, Woolf DJ, Debouck C, Harrison SC. Proc. Natl. Acad. Sci. U. S. A 1995, 92, 1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 34.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. J. Med. Chem 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- 35.Sarafianos SG, Das K, Tantillo C, Clark AD Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. EMBO J. 2001, 20, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felts AK, Labarge K, Bauman JD, Patel DV, Himmel DM, Arnold E, Parniak MA, Levy RM. J. Chem. Inf. Model 2011, 51, 1986–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauman JD, Patel D, Dharia C, Fromer MW, Ahmed S, Frenkel Y, Vijayan RS, Eck JT, Ho WC, Das K, Shatkin AJ, Arnold E. J. Med. Chem 2013, 56, 2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Himmel DM, Myshakina NS, Ilina T, Van Ry A, Ho WC, Parniak MA, Arnold E. J. Mol. Biol 2014, 426, 2617–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yonetani T. Methods Enzymol. 1982, 87, 500–509. [DOI] [PubMed] [Google Scholar]
- 40.Le Grice SF, Cameron CE, Benkovic SJ. Methods Enzymol. 1995, 262, 130–144. [DOI] [PubMed] [Google Scholar]
- 41.Costi R, Métifiot M, Chung S, Cuzzucoli Crucitti G, Maddali K, Pescatori L, Messore A, Madia VN, Pupo G, Scipione L, Tortorella S, Di Leva FS, Cosconati S, Marinelli L, Novellino E, Le Grice SF, Corona A, Pommier Y, Marchand C, Di Santo R. J. Med. Chem 2014, 57, 3223–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esposito F, Sanna C, Del Vecchio C, Cannas V, Venditti A, Corona A, Bianco A, Serrilli AM, Guarcini L, Parolin C, Ballero M, Tramontano E. Pathog. Dis 2013, 68, 116–124. [DOI] [PubMed] [Google Scholar]
- 43.Xu L, Grandi N, Del Vecchio C, Mandas D, Corona A, Piano D, Esposito F, Parolin C, Tramontano E. J. Microbiol 2015, 53, 288–293. [DOI] [PubMed] [Google Scholar]
- 44.Nettleship JE, Brown J, Groves MR, Geerlof A. Methods Mol. Biol 2008, 426, 299–318. [DOI] [PubMed] [Google Scholar]
- 45.Pannecouque C, Daelemans D, De Clercq E. Nat. Protoc 2008, 3, 427–434. [DOI] [PubMed] [Google Scholar]
- 46.Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. J. Virol. Methods 1988, 20, 309–321. [DOI] [PubMed] [Google Scholar]
- 47.Suite Schrödinger, in, Schrödinger, LLC, New York, NY, USA. [Google Scholar]
- 48.Halgren TA. J. Comput. Chem 1996, 17, 520–552. [Google Scholar]
- 49.Hasel W, Hendrickson TF, Still WC. Tetrahedron Comput. Methodol 1988, 1, 103–116. [Google Scholar]
- 50.Glide, version 5.9; Schrodinger, LLC, New York, NY, 2013, LLC, New York, NY. [Google Scholar]
- 51.Jacobson MP, Friesner RA, Xiang Z, Honig B. J. Mol. Biol 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
- 52.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srnivasan J, Case DA, Cheatham TE. Acc. Chem. Res 2000, 33, 889–897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.