

Abstract
Recent reports have demonstrated that lipopolysaccharide (LPS)-induced depressive-like behaviour is mediated via NMDA receptor. In this study, we further investigated the role of GluN2A subunit of NMDA receptor in synaptic processes in the prefrontal cortex (PFC) and hippocampus of GluN2A knockout (KO) mice in LPS-induced depressive-like behavior. Our data suggest that LPS-treated mice, lacking GluN2A subunit, did not exhibit depressive-like behaviour. This was accompanied by unaltered levels of IL-6 and significant changes in neuroplasticity markers and glutamate receptor subunits composition in PFC and hippocampus. In particular, an immune challenge in GluN2A KO mice resulted in unchanged PSA-NCAM levels and proBDNF increase in both brain structures as well as in increase in BDNF levels in hippocampus. Furthermore, the absence of GluN2A resulted in increased levels of all NCAM isoforms in PFC upon LPS which was followed with a decrease in GluN1 and GluN2B subunits. The levels of AMPA receptor subunits (GluA1, GluA3, and GluA4) in the hippocampus of GluN2A mice were unaltered upon the treatment and abundantly present in the PFC of KO mice.
These results indicate that the GluN2A subunit is critical in neuroinflammation-related depression, that its absence abolishes LPS-induced depressive phenotype, sustains PSANCAM levels, increases proBDNF signalling in the PFC and hippocampus and potentiates synaptic stabilization through NCAM in the PFC upon an immune challenge.
Keywords: GluN2A knockout mice, lipopolysaccharide, depressive-like behaviour, synaptosomes, neuroplasticity, glutamatergic neurotransmission
1. Introduction
During the last decade, mounting evidence indicated an important role of inflammatory cytokines in the pathogenesis of various psychiatric disorders, such as major depressive disorder (MDD). The altered profile of inflammatory cytokines, such as interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumour necrosis factor α (TNF-α), was observed in some subpopulations of patients with MDD [1] and significant correlation of this abnormal cytokine profile and severity of symptoms of the disease has been established [2].
Using preclinical models of inflammation, it has been recently shown that the lipopolysaccharide (LPS) induces depressive-like behaviour through modulation of N-methyl-D-aspartate (NMDA) receptor functions [3]. Briefly, LPS induces depressive-like behaviour by activating indoleamine 2,3 dioxygenase (IDO) [4] which leads to an increase kynurenine/tryptophan ratio and subsequent predominant formation of quinolinic acid [5] that activates NMDA receptor. The importance of NMDA receptor in inflammation related-depressive phenotype has been further strengthened by the finding that ketamine, a specific NMDA receptor antagonist, blocks LPS-induced depressive-like behaviour by redirecting glutamatergic neurotransmission toward α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor [3]. Despite the fact that pharmacological studies emphasised the role of NMDA receptor in LPS-induced depression, it’s still remains open how specific NMDA receptor subunits contribute to aberrant behaviour upon LPS treatment.
Besides glutamatergic receptors and numerous cytoskeletal proteins, cell adhesion molecules and neurotrophic factors also play an important role [6] in synapse structure and function. A growing body of evidence has implicated neural cell adhesion molecule (NCAM) and its polysialylated form (PSA-NCAM) as risk factors for major neuropsychiatric and neurodegenerative disorders [7]. The NCAM is one of the most abundant adhesive molecules in hippocampal excitatory synapses [8] involved in synapse adhesion, synaptogenesis, recruiting and stabilizing the vesicular pool [9]. On the other hand, PSA-NCAM has been implicated in neurite growth during synaptogenesis, synapse remodelling and plasticity [10] and has been linked to specific population of NMDA receptors [11, 12]. Additionally, control of PSA-NCAM expression by NMDA receptor activation has been described in several systems, suggesting a functional link between these two proteins [13]. Besides synaptogenesis, PSA-NCAM contributes to survival and differentiation of neurons, the processes which is brain-derived neurotrophic factor BDNF-dependant [14]. Considering inflammation-related depression, it remains unclear whether NCAM and PSA-NCAM are affected by LPS, and if so, to what extent they are dependent on BDNF signalling in the context of GluN2A-containing NMDA receptor. Therefore we hypothesise that GluN2A subunit is involved in LPS-induced depressive-like behaviour and that GluN2A context affects PSA-NCAM-BDNF signalling and other components of glutamatergic transmission under the treatment.
2. Materials and methods
2.1. Animals and Treatments
GluN2A knockout (KO) male mice with null mutation for the GluN2A-coding gene Grin2A were generated as previously described [15] and purchased from Jackson Laboratories. GluN2A KO, heterozygous GluN2A (HET), and wild-type (WT) mice were generated from HET x HET mating in our laboratory. Mouse genomic DNA from tail was used for genotyping GluN2A knockouts and wild- types as previously described [16].
All experiments were performed on 8 week old male WT (n=40) and GluN2A KO (n=40) mice. 7 days before the experiment mice were housed individually in standard shoebox cages with access to food (commercial mouse pellets) and water ad libitum. Light was kept on between 7:00 am and 7:00 pm and room temperature was kept at 20 ± 2ºC. All animal procedures were approved by the Ethical Committee of VINCA Institute of Nuclear Sciences, according to the guidelines of the EU registered Serbian Laboratory Animal Science Association (SLASA)(Application No. 4/2015; 323–07-04657/2015–05/3).
2.2. Experimental design
During the experiments, we used two sets of animals. First set of animals was used for non-invasive sucrose preference test (SPT) (performed 24 to 28 h after the LPS treatment) and immediately after testing animals were sacrificed by cervical dislocation. Brain structures, hippocampus and PFC, were extracted only from these animals, frozen in liquid nitrogen and stored at −80°C until sample preparation for further molecular analyses (Fig. 1A). The second set of animals was used only for examination of locomotor activity and forced swim test (FST). Specifically, locomotor activity was examined 6 and 24 h after treatment and FST was performed 26 h after treatment (Fig. 1B). In each set, animals were divided into four groups (10 animals per grope): groups I and II consisted of male WT mice treated with saline or LPS, respectively, and groups III and IV consisted of male GluN2A KO mice treated with saline or LPS, respectively. The vehicle (VEH) groups were treated with mass-adjusted volume of physiological saline, while LPS-groups were treated with LPS (Escherichia coli, Sigma-Aldrich) previously dissolved in sterile, pyrogen free physiological saline at a single dose of 0.83 mg/kg. The dose of LPS (0,83 mg/kg) was chosen for its ability to reliably induce the acute sickness response and subsequent depressive-like behaviours across the time points examined here [4, 17]. All animal groups were treated intraperitoneally at 8:00 am.
Fig. 1.
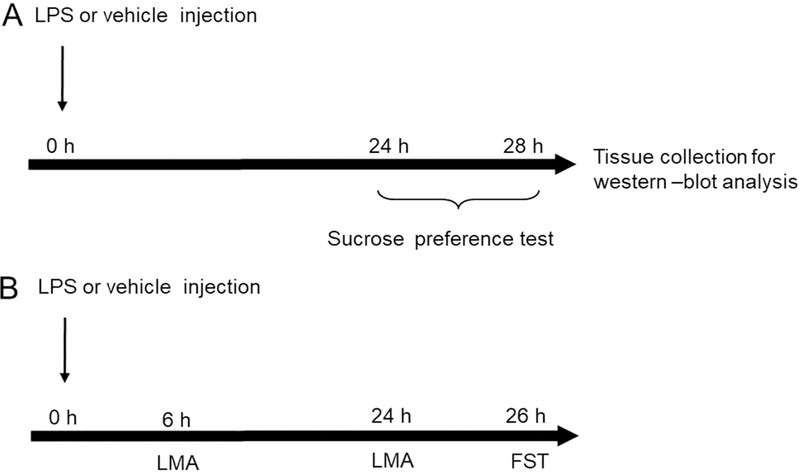
Experimental design. The schedule of treatment and behavioural evaluations (A, B). (A) Sucrose preference test (SPT) was performed 24 to 28 h after LPS (0,83 mg/kg) or vehicle injection. Brain samples were collected 28 h after LPS administration (immediately after SPT). (B) Locomotor activity (LMA) test was performed 6 and 24h after LPS (0,83 mg/kg) or vehicle injection. After LMA test animals were subjected to forced swim test (FST) (26h after LPS or vehicle treatment). Brain samples were not collected from this set of animals.
2.3. Behaviour test and locomotor activity
Locomotor activity.
To confirm that decrease in locomotor activity, as a consequence of sickness behaviour induced by LPS, is recovered at the time of behavioural tests we examined motor activity of animals 6 and 24 h after treatment. Mouse activity was recorded in new home cages without bedding or litter. The ground area of the cage was divided into four equal areas. Mice were placed in the centre of the cage, and their motor performance was recorded and analysed during the 5 min test period using the TSEVIDEOMOT 2 software (version 5.75; TSE Systems, Bad Homburg, Germany). Locomotor activity of animals was expressed as number of quadrant entries during the test period.
Forced Swim Test.
FST previously described by Porsolt, 2000 was used [18]. 26 h after treatment mice were placed individually for 6 min in a Plexiglas cylinder (diameter: 23 cm; height: 31 cm) filled with 24 ± 1°C water to a depth of 30 cm. The water was changed between two testing sessions. During the test, the mice were video recorded from above, and the duration of immobility was automatically measured over the last five minutes of the test period using the mobility function of the TSEVIDEOMOT 2 software (version 5.75; TSE Systems, Bad Homburg, Germany).
Sucrose preference test (SPT).
In order to verify that LPS treatment induced depressive-like behaviour, 24 h after the LPS/vehicle injection animals were subjected to 4 hours period sucrose preference testing to estimate anhedonia. One week prior to testing mice were habituated to drink 1% sucrose solution by daily exposing animals to 2 ml syringe containing a freshly prepared 1% sucrose solution and an identical syringe containing tap water placed one beside the other for 4 hours. Training and testing occurred between 8:00 am and 12:00 pm. Fluid consumption (grams) was measured by weighing syringes before and after session on testing day. The sucrose preference (%) was calculated as sucrose intake (g)/total fluid consumption (g) ratio [3].
2.4. Synaptosome and whole cell preparation for Western-blot detection of proteins
After decapitation, brains were rapidly removed from the skulls and hippocampi and PFC were dissected on ice. Crude hippocampal and PFC synaptosomal fractions were prepared as previously described [19]. Briefly, tissue was homogenized in 0.32 M sucrose, 20 mM HEPES (pH 7.3), 1 mM EDTA, 1 protease inhibitor cocktail, 5 mM NaF and 1 mM NaVO3 and subsequently centrifuged at 2800 rpm for 10 min. Supernatants were further processed by centrifugation at 12000 rpm for 10 min to obtain a pellet with crude synaptosomes. In the next step, pellets were sonicated in RIPA lysis buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 1 mM NaVO3, 5 mM NaF and 1 protease inhibitor cocktail.
Whole cell fraction for cytokine detection was also prepared from hippocampus and PFC. For tissue homogenisation, we used buffer containing 100 mM Tris pH 7, 100 mM NaCl, 10 mM EDTA, 0.5% NP-40, and 0.5% sodium deoxycolate plus protease and phosphatase inhibitors. After centrifugation at 14000 rpm for 20 min at 4°C supernatant was collected.
Protein concentration was determined by the method of Markwell [20] and samples were incubated 5 min at 100ºC in appropriate amount of denaturing buffer according to Laemmli [21]. 60 μg of proteins were subjected to SDS-PAGE electrophoresis using 7.5 %, 10% or 12% gels and subsequently transferred onto PVDF membrane (Immobilon-P membrane, Millipore) using a blot system (Transblot, Bio-Rad). After protein transfer, the blots were blocked with 5% milk in PBS for 1 hour at room temperature (RT) and incubated in appropriate primary and secondary antibodies.
Primary antibodies used in this study were anti-GluA1(1:250), anti-GluA3 (1:500), anti-BDNF and anti-pro-BDNF (1:500) (Santa Cruz Biotechnology); anti-GluN2A (1:1000), anti-GluN2B (1:1000), anti-NCAM (1:2000) and anti-PSA-NCAM (1:500) (Millipore); antiGluA4 (1:500) (Cell Signalling); anti-IL-6 (1:250) and anti-TNF alpha (1:250) (eBioscience). As loading control we used β-actin primary antibody (1:5000) (Abcam). After overnight incubation in primary antibodies membranes were washed three times in PBS-T and incubated 2 hours in appropriate peroxidase-labelled secondary, donkey anti-mouse or goat anti-rabbit antibodies (Santa Cruz Biotechnology). Bands were detected using enhanced chemiluminescent reagent (Pierce), SuperSignal Pico Chemiluminescent Substrate or SuperSignal Femto Maximum Sensitivity Substrate (Thermo Scientific) and exposed to X-ray Film for Western Blot Detection (Thermo Scientific). Image J analysis PC software (NIH, Bethesda, MD) was used for quantification densitometry of protein bands on X-ray film. Amounts of all analysed proteins were normalized to β-actin levels.
2.5. Statistical analysis
Analysis was performed using STATISTICA 7 software. The results regarding the expression levels of GluN2A were analysed by student t-test. All other results were processed by two-way ANOVA and post hoc Tukey test was used to determine differences between the experimental groups in order to assess the influence of either genotype, treatment or their interaction on measured proteins levels or behaviour parameters. Data are presented as a mean ± S.E.M. and statistical significance was accepted at p<0.05.
3. Results
3.1. Effects of LPS on locomotor activity and behaviour
Locomotor activity -
Changes in locomotor activity, as effect of LPS-induced sickness response, were observed at 6 and 24 h after treatment. We showed that LPS treatment decreased locomotor activity in WT as well as in GluN2A KO mice 6 h after treatment(LPS: F(1,26)=29.15, p<0.05) (Fig. 2A) but 24 hours after treatment there were no changes in locomotor activity between LPS treated mice and untreated controls (Fig. 2B).
Fig. 2.
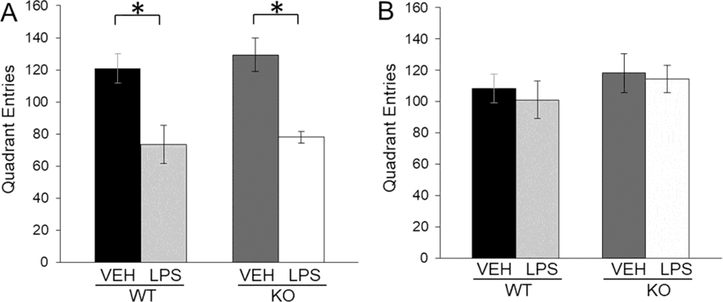
Quadrant entries in locomotor activity test (± SEM) of male wild-type (WT) and GluN2A knockout (KO) mice 6h (A) and 24h (B) after LPS or VEH treatment (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
Sucrose preference test -
LPS treatment induced a decrease in the consumption of sucrose solution only in WT mice (LPS x genotype interaction: F(1,34)=7.20, p<0.05) (Fig. 3A). LPS treatment did not affect depressive like behaviour in GluN2A KO mice.
Fig. 3.
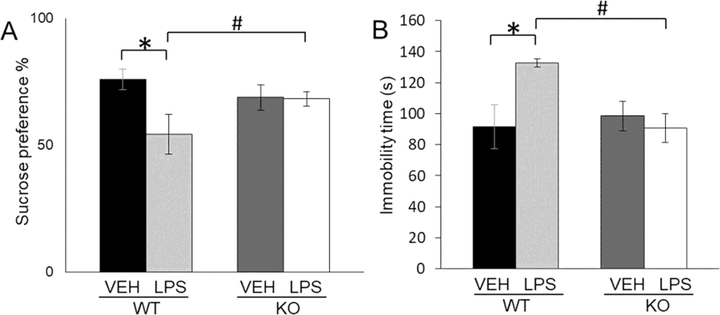
Sucrose preference (%) (± SEM) of male wild-type (WT) and GluN2Aknockout (KO) mice upon LPS or VEH treatment (n=10) (A). Immobility time in the forced swim test (± SEM) of male wild-type (WT) and GluN2A knockout (KO) mice upon LPS or VEH treatment (n=10) (B). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
Forced Swim test -
LPS treatment increased immobility only in WT mice (LPS x genotype interaction: F(1,23)=5.65 p<0.05) (Fig. 3B) and as well as sucrose preference test did not affect developing of depressive like behaviour in KO mice.
3.2. Cytokine levels in GluN2A KO and WT mice upon LPS treatment.
Our results revealed that in both analysed tissues, LPS increased levels of IL-6 in WT but not in GluN2A KO mice (LPS: F(1,32)=16,75, p<0.05; LPS x genotype interaction: F(1,32)=4.15, p<0.05 for hippocampus; LPS: F(1,32)=37,63 p<0.05; LPS x genotype interaction: F(1,32)=182.92, p<0.05 for PFC) (Figs. 4A and B).
Fig. 4.

The levels of IL-6 in the hippocampal (A) and PFC (B) whole cell fraction of male wild-type (WT) and GluN2A knockout (KO) mice upon LPS treatment compared to controls (VEH). Values are presented as mean ± SEM % of control values (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
3.3. Effects of LPS on neuroplasticity measured by BDNF and proBDNF levels
In WT mice, LPS treatment significantly decreased BDNF levels in both examined brain structures and increased its levels only in hippocampus of GluN2A KO mice (LPS x genotype interaction: F(1,29)=35.23, p<0.05 for hippocampus; LPS: F(1,36)=45,87, p<0.05 for PFC) (Fig. 5A and C).
Fig. 5.
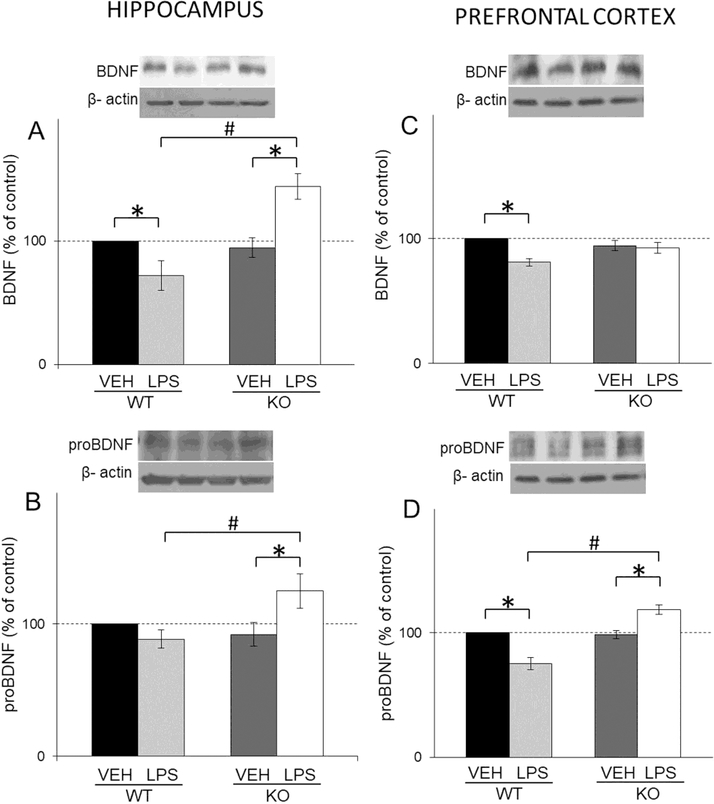
The protein levels of BDNF (A, C) and proBDNF (B, D) in the hippocampal and PFC synaptosomal fraction of male wild-type (WT) an GluN2Aknockout (KO) mice upon LPS treatment compared to controls (VEH). Values are presented as mean ± SEM % of control values (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
As for pro-BDNF, in GluN2A KO mice, LPS treatment significantly increased the levels of this protein in hippocampus and PFC and decreased its levels in PFC of WT (LPS x genotype interaction: F(1,49)=7,39, p<0.05 for hippocampus; LPS x genotype interaction F(1,45)=49,22, p<0.05 for PFC ) (Fig. 5B and D).
3.4. Effects of LPS on PSA-NCAM and NCAM levels
In the hippocampus, LPS treatment decreased PSA-NCAM levels only in WT mice (LPS: F(1,16)=13,36, p<0.05; LPS X genotype interaction: F(1,16)=5.46, p<0.05) (Fig. 6A) while in the PFC treatment decreased PSA-NCAM levels in WT and increased its levels in KO mice (LPS X genotype interaction: F(1,87)=68.27, p<0.05) (Fig. 6B).
Fig. 6.
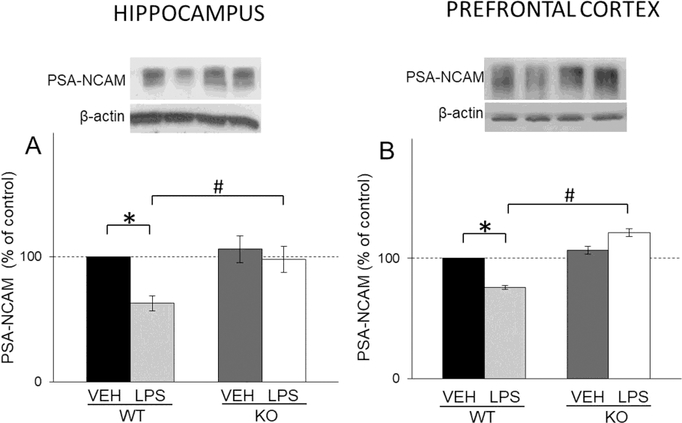
The levels of PSA-NCAM in the hippocampal (A) and PFC (B) synaptosomal fraction of male wild-type (WT) and GluN2A knockout (KO) mice upon LPS treatment compared to controls (VEH). Values are presented as mean ± SEM % of control values (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
Neither treatment nor genotype had effects on the levels of hippocampal NCAM isoforms (120/140/180 kDa) (Figs. 7A, B and C). On the other hand, levels of PFC 180 and 140 kDa NCAM isoforms increased in WT comparing to KO (genotype: F(1,44)=65,10 p<0.05 for 180kDa NCAM isoform; F(1,44)=616,88, p<0.05 for 140 kDa NCAM isoform (Figs. 7E and F). LPS treatment increased all NCAM isoforms in KO mice and decreased NCAM 140 kDa isoforms in WT mice (LPS X genotype interaction: F(1,44)=17.38 p<0.05 for 180 kDa NCAM isoform; F(1,44)=355.92, p<0.05 for 140 kDa NCAM isoform; F(1,28)=22.85, p<0.05 for 120kDa NCAM isoform) (Figs. 7D, E and F).
Fig. 7.
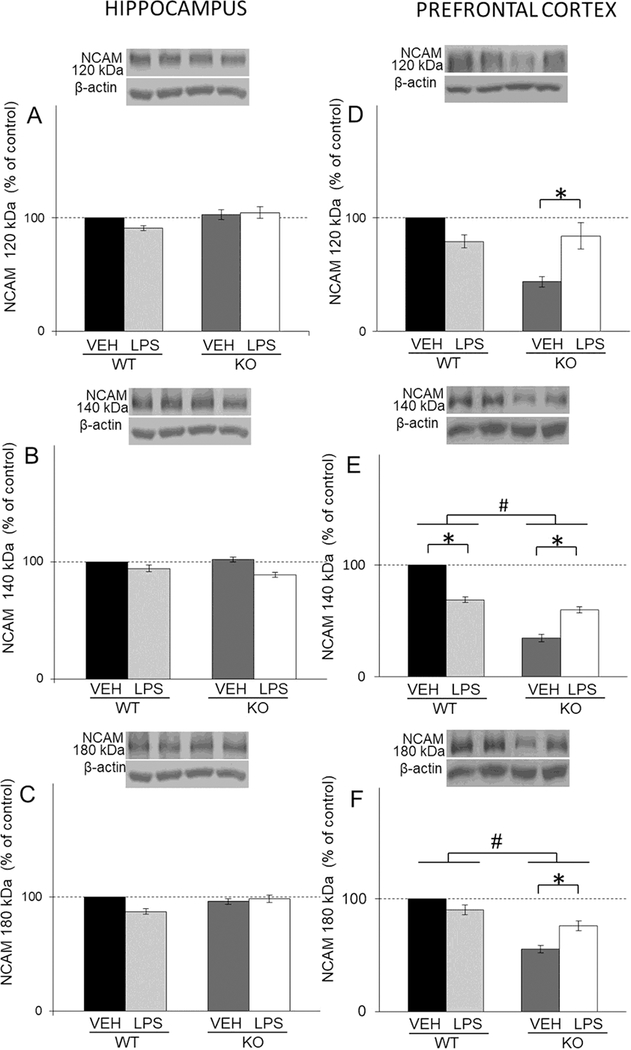
The levels of NCAM isoforms (120/140/180 kDa) in the hippocampal (A, B, C) and PFC (D, E, F) synaptosomal fraction of male wild-type (WT) an GluN2A knockout (KO) mice upon LPS treatment compared to controls (VEH). Values are presented as mean ± SEM % of control values (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
3.5. Effects of LPS on NMDA and AMPA receptor subunits levels
Our results revealed that LPS treatment did not affect NMDA receptor subunits in hippocampus (Fig. 8A, B and C). On the other hand, in PFC, LPS increased all NMDA receptor subunits in WT mice, and decreased levels of GluN1 and GluN2B subunits in KO mice (LPS: F(1,31)=4,24, p<0.05; LPS X genotype: F(1,31)=86.72, p<0.05 for GluN1; t(18)=7,78, p<0.05for GluN2A; LPS: F(1,44)=11,25, p<0.05; LPS X genotype :F(1,44)=58.39, p<0.05 for GluN2B respectively) (Figs. 8D, E and F).
Fig. 8.
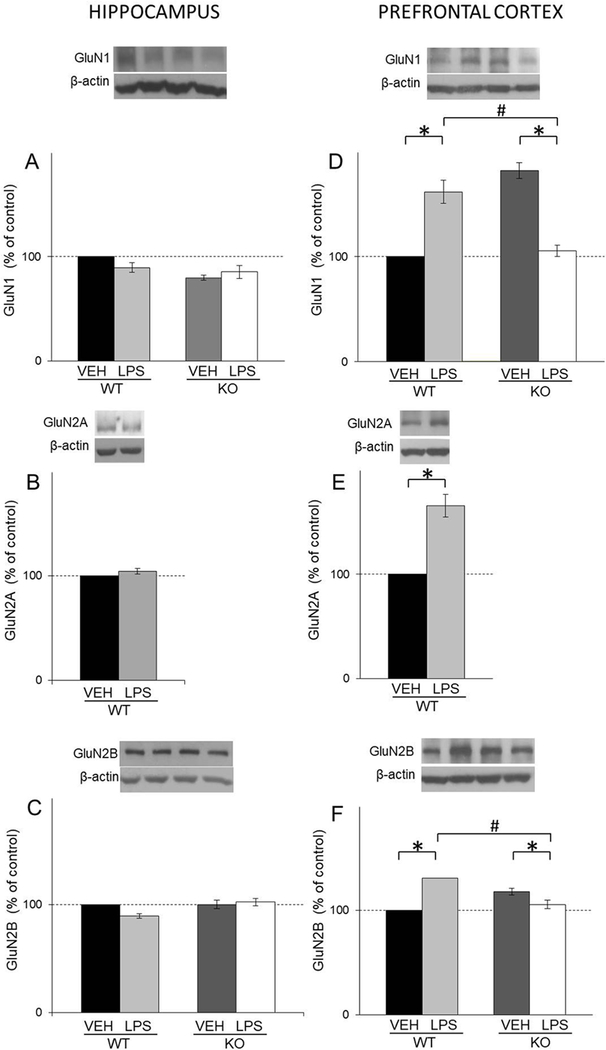
The levels of GluN1, GluN2A and GluN2B in the hippocampal (A,B,C) and PFC (D, E, F) synaptosomal fraction of male wild-type (WT) an GluN2A knockout (KO) mice upon LPS treatment compared to controls (VEH). Values are presented as mean ± SEM % of control values (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
As for AMPA receptor subunits GluA1, GluA3 and GluA4, the treatment reduced the levels of GluA1 and GluA4 in hippocampus of WT mice (LPS: F(1,12)=25,25, p<0.05 for GluA1; F(1,16)=24,7, p<0.05 for GluA4 respectively) (Figs. 9A and C). Additionally, statistical analysis revealed increased levels of GluA4 AMPA receptor subunits in treated GluN2A KO mice compared to treated WT (LPS X genotype: F(1,16)=112.85, p<0.05) (Fig. 9C). In PFC, we observed increased levels of GluA1, GluA3 and GluA4 of GluN2A KO compared to WT mice (genotype: F(1,42)=251.37, p<0.05 for GluA1; F(1,45)=354.09, p<0.05for GluA3; F(1,39)=225.6, p<0.05 for GluA4 respectively) (Figs. 9D, E and F). Additionally, in this tissue LPS treatment increased levels of GluA3 and deceased levels of GluA1 in GluN2A KO mice (LPS X genotype: F(1,42)=12.55 p<0.05 for GluA1; F(1,45)=64.06, p<0.05 for GluA3, respectively) (Figs. 9D and E).
Fig. 9.
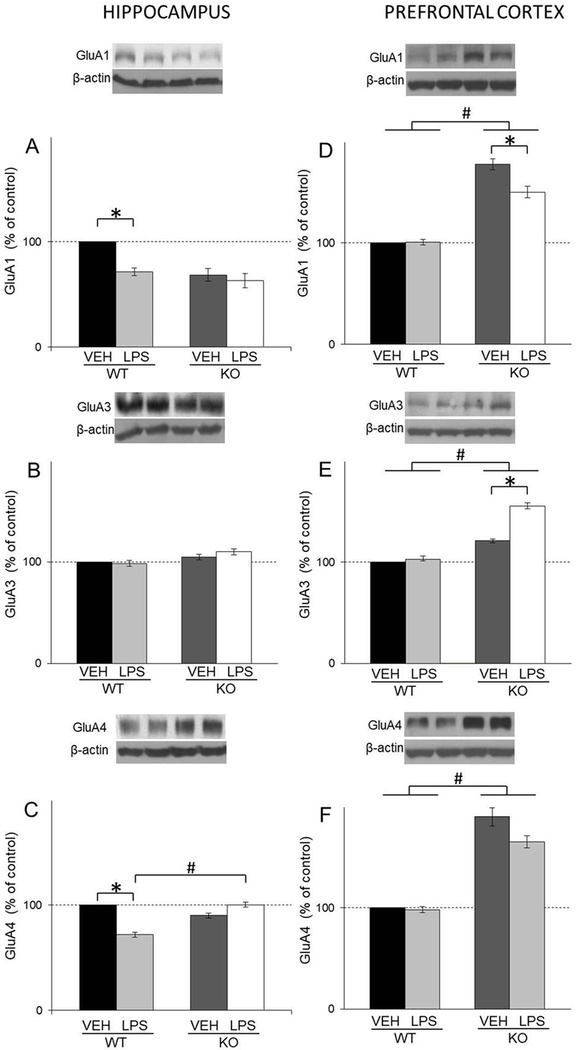
The levels of GluA1, GluA3 and GluA4 in the hippocampal (A, B, C) and PFC (D, E, F) synaptosomal fraction of male wild-type (WT) an GluN2A knockout (KO) mice upon LPS treatment compared to controls (VEH). Values are presented as mean ± SEM % of control values (n=10). Statistically significant differences are given as p < 0.05. *VEH vs. LPS, #WT vs. KO.
4. Discussion
LPS-induced depressive-like behaviour is, in a part, mediated by the activation of NMDA receptor by quinolinic acid [3]. However, little is currently known about the specific roles of NMDA receptor subunits in the mediation of LPS-altered behaviour. In this study, we used GluN2A KO mice to determine whether genetic inactivation of this NMDA receptor subunit is involved in LPS-induced alterations in behaviour. We observed that the absence of the GluN2A subunit fully abrogates LPS-induced depressive phenotype in mice. In accordance with the existing literature [22–24] our results confirmed that LPS treatment induced depressive-like behaviour in WT rodents 24 hours after the injection. In contrast, in GluN2A KO mice LPS did not induced depressive-like behaviour, which was clearly shown by higher consumption of sucrose and decreased immobility time in FST in respect to WT counterparts. Additionally, our results demonstrate that at the time of assessing the depressive phenotype, 24 hours after the LPS injection, locomotor activity was back to normal meaning that sickness behaviour subsided and that obtained behavioural changes can be ascribed solely to depressive-like behaviour.
It is worth mentioning that GluN2A KO mice exhibited antidepressant-like profiles in the FST and tail suspension test, as compared to WT controls [25]. However, our behavioural data are not in line with this study and they emphasise that GluN2A KO antagonize the effect of LPS on behaviour.
Previous study demonstrated that LPS altered the cytokine levels in various brain structures. For instance, LPS increased levels of IL-1β, TNF-α and IL-6 in hypothalamus, hippocampus and brainstem in mice [26]. Furthermore, single dose of 0.83 mg/kg LPS increased the expression of IL-1β, TNF-α and IFN-γ mRNA in the brain [4] and plasma level of IL-6 [3] even 28 h after the treatment. LPS also launches inflammatory cascade and IDO activity in the brain contributing to depressive-like behaviour, which is in a part mediated by the activation of NMDA receptor by quinolinic acid [3]. Our results regarding cytokine expression are in line with these studies, since we have demonstrated that the level of IL- 6 is increased in both brain structures in LPS-treated WT mice. In our study, elevated marker of inflammation in LPS treated WT may lead to IDO activation and quinolinic acid production which activate NMDA receptor and cause depressive phenotype in these animals [3]. However, in KO mice the levels of IL-6 were unchanged upon LPS, which could be a consequence of involvement of NMDA receptor in microglial activation. In particular, previous study suggested that stress-induced microglia proliferation was a consequence of corticosterone-induced activation of the NMDA receptor within the CNS, while pharmacological blockade of NMDA receptor prevented stress-induced microglial activation [27].
The observed depressive-like behaviour in LPS treated WT mice appears to be linked to the decrease in BDNF signalling and PSA-NCAM in the hippocampus and PFC. Namely, we have found significant decrease of mature BDNF protein level in synaptosomal fraction of these two brain structures upon the treatment. This is in accordance with already published data indicating that LPS decreases BDNF in PFC and hippocampus (CA3, dentate gyrus) [28, 30], which is considered to be important step in developing depressive-like behaviour [28, 29, 31]. Decrease in BDNF levels could be associated with IL-6 increase in LPS treated WT mice since preclinical studies have suggested that pro-inflammatory cytokines could directly reduce BDNF gene transcription via pathways such as cyclic AMP-response element-binding protein phosphorylation or nuclear factor-κB activation (NF-kB) [32–34]. On the other hand, in LPS treated KO mice we detected increased levels of BDNF in hippocampus and its unaltered levels in PFC which will be discussed below in the text.
The alterations in NMDA receptor activation have been shown to modulate the expression of NCAM [11], as well as PSA-NCAM in vivo and in vitro [11, 13], the proteins that play an important role in the regulation of plasticity processes in different brain structures. Moreover, it has been reported that the changes in NCAM function are inhibited by the use of the NMDA receptor antagonist, MK-801 [35], while on the other hand, there is evidence that NMDA receptor can downregulate the expression of PSA-NCAM in adult brain stem synapses [11]. Our results showed a decrease in PSA-NCAM levels at the hippocampal and PFC synaptosomes in WT mice upon LPS treatment that matched with previous data showing relation between decreased PSA-NCAM levels and stress-induced cognition deficits [36] and behavioural alterations in experimental model of depression [10, 37]. Further, the regulation of NCAM polysialylation is additionally linked to BDNF signaling, since PSANCAM-deficient hippocampus, characterised with defective long-term potentiation, is associated with a reduced activation of BDNF signaling and can be selectively rescued by BDNF [38]. In agreement with this, decreased BDNF levels in the hippocampal and PFC synaptosomes of LPS treated WT mice, suggest a potential mechanistic link between PSANCAM and BDNF-associated signaling in LPS-induced depressive-like behaviour. On the other hand, unaltered PSA-NCAM levels in GluN2A KO mice upon the treatment could be preserved by elevated levels of proBDNF signaling.
Regarding NCAM levels, our results are in accordance with published data showing that mice deficient in all NCAM isoforms display depressive-like behaviour [39]. However, a decrease of 140 kDa NCAM isoform that we observed in the PFC of LPS-treated WT indicated that effects of neuroinflammation on NCAM are isoform and brain structure specific. Interestingly, LPS-treated GluN2A KO exhibited increased levels of all three isoforms of NCAM proteins in the PFC, implying that their elevation in GluN2A KO upon an immune challenge may be involved in preventing the development of depressive-like behaviour. Overall, above results suggested that the absence of inflammation-induced depressive phenotype in GluN2A KO could be attributed to the increased proBDNF levels in both brain structures, elevated NCAM in the PFC and preserved PSA-NCAM in the PFC and hippocampus.
Considering the role of glutamate receptors in neuroinflammation–related depression and regulation of neural plasticity, we further analysed the effects of LPS on the levels of NMDA and AMPA receptor synaptic subunits. The most striking results were that hippocampal NMDA and AMPA receptor subunits were less affected by LPS then in the PFC. LPS induced a decrease of hippocampal GluA1 and GluA4 subunits of AMPA receptor of WT. Additionally, in the PFC, an immune challenge increased all NMDA receptor subunits in WT and decreased them in KO mice. Moreover, abundantly present AMPA receptor subunits in the PFC of KO mice were more affected by LPS in comparison to WT. This data suggested that effects of neuroinflammation on glutamate receptors composition were more pronounced in mice PFC and that they are affected by LPS in an opposite manner in WT and GluN2A mice.
Furthermore, the alteration in NMDA receptor subunit composition that was not followed by the changes in PSA NCAM levels, particularly in KO mice, indicate that LPS-induced changes were not responsible for sustaining PSA levels upon an immune challenge in this mice genotype. This is in agreement with previous study indicating that PSA-NCAM synthesis is NMDA receptor independent [40]. In contrast, levels of all NCAM isoforms in the PFC of both WT and GluN2A KO mice were oppositely related to levels of NMDA receptor subunits suggesting that their expression is negatively regulated by NMDA receptor.
Regarding proBDNF in both brain structures of GluN2A KO mice, its increase in the PFC by LPS could result from higher levels of AMPA receptor subunits in this genotype, particularly from GluA3. Indeed, it has been shown that AMPA receptor activation regulated BDNF expression in brain [41]. In contrast, an increase of BDNF in hippocampus of LPS-treated GluN2A KO could, in a part, be a result of AMPA receptor regulation since the levels of GluA4 subunit were higher in this genotype then in their WT counterparts. In addition, BDNF increase could result from the activation of PI3/AKT/mTOR signalling pathway which promotes translation of BDNF [42]. Indeed, we observed increase in mTOR signaling only in the hippocampus of LPS-treated GluN2A KO mice (data not shown). It is worth mentioning that previous study also documented that inflammation, particularly chronic LPS treatment, increased BDNF via an epigenetic mechanism [43].
Our results should be interpreted considering several limitations. Pharmacological experiments using specific NMDA receptor subunit antagonist or KO mice for a different NMDA receptor subunit could be used to define a specific involvement of other NMDA receptor subunit in LPS-induced depressive-like behaviour. Also, since clinical data demonstrate that there are sex-specific differences in the frequency and treatment response of patients suffering from MDD, it would be useful to perform the similar experimental approach in female WT and GluN2A mice and to compare them with males.
Overall, the results of this study suggest that the GluN2A subunit is critical in neuroinflammation-related depression. The absence of this NMDA receptor subunit may abolish the depressive outcome when challenged with LPS by sustaining PSA-NCAM levels and increasing proBDNF signalling in the PFC and hippocampus and potentiating synaptic stabilization through NCAM in the PFC. These results also point out that specific inhibition of GluN2A NMDA receptor subunit could present a new avenue in the treatment of neuroinflammation-related depression.
Highlights:
GluN2A KO mice displayed resiliance to depressive-like behaviour upon LPS
GluN2A KO mice abolishes LPS induced inflammation
LPS treated GluN2A KO mice showed elevated proBDNF in the PFC & hippocampus and increased NCAM in PFC
GluN2A KO mice exhibited sustained PSA-NCAM levels in the PFC and hippocampus
GluN2A KO potentiated synaptic stabilization through NCAM in the PFC upon LPS
Acknowledgements
This study was supported by a grant from the Ministry of Education and Sciences of Serbia (Grant III41029) and NIH grant R21MH098793.
Footnotes
Conflict of interest
The other authors declare no conflict of interest.
Compliance with ethical standards
All animal procedures were approved by the Ethical Committee of VINCA Institute of Nuclear Sciences, according to the guidelines of the EU registered Serbian Laboratory Animal Science Association (SLASA).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL, A meta-analysis of cytokines in major depression, Biological psychiatry 67(5) (2010) 446–57. [DOI] [PubMed] [Google Scholar]
- [2].Martinez JM, Garakani A, Yehuda R, Gorman JM, Proinflammatory and “resiliency” proteins in the CSF of patients with major depression, Depression and anxiety 29(1) (2012) 32–8. [DOI] [PubMed] [Google Scholar]
- [3].Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, Kelley KW, Dantzer R, NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice, Neuropsychopharmacology 38(9) (2013) 1609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R, Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3dioxygenase activation in mice, Molecular psychiatry 14(5) (2009) 511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Saito K, Markey SP, Heyes MP, Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse, Neuroscience 51(1) (1992) 25–39. [DOI] [PubMed] [Google Scholar]
- [6].Yamagata M, Sanes JR, Weiner JA, Synaptic adhesion molecules, Current opinion in cell biology 15(5) (2003) 621–32. [DOI] [PubMed] [Google Scholar]
- [7].Brennaman LH, Maness PF, NCAM in neuropsychiatric and neurodegenerative disorders, Advances in experimental medicine and biology 663 (2010) 299–317. [DOI] [PubMed] [Google Scholar]
- [8].Shin MH, Lee EG, Lee SH, Lee YS, Son H, Neural cell adhesion molecule (NCAM) promotes the differentiation of hippocampal precursor cells to a neuronal lineage, especially to a glutamatergic neural cell type, Experimental & molecular medicine 34(6) (2002) 401–10. [DOI] [PubMed] [Google Scholar]
- [9].Polo-Parada L, Bose CM, Plattner F, Landmesser LT, Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice, The Journal of neuroscience : the official journal of the Society for Neuroscience 24(8) (2004) 1852–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dityatev A, Dityateva G, Sytnyk V, Delling M, Toni N, Nikonenko I, Muller D, Schachner M, Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses, The Journal of neuroscience : the official journal of the Society for Neuroscience 24(42) (2004) 9372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bouzioukh F, Tell F, Jean A, Rougon G, NMDA receptor and nitric oxide synthase activation regulate polysialylated neural cell adhesion molecule expression in adult brainstem synapses, The Journal of neuroscience : the official journal of the Society for Neuroscience 21(13) (2001) 4721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kochlamazashvili G, Senkov O, Grebenyuk S, Robinson C, Xiao MF, Stummeyer K, GerardySchahn R, Engel AK, Feig L, Semyanov A, Suppiramaniam V, Schachner M, Dityatev A, Neural cell adhesion molecule-associated polysialic acid regulates synaptic plasticity and learning by restraining the signaling through GluN2B-containing NMDA receptors, The Journal of neuroscience : the official journal of the Society for Neuroscience 30(11) (2010) 4171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Singh J, Kaur G, Transcriptional regulation of PSA-NCAM expression by NMDA receptor activation in RA-differentiated C6 glioma cultures, Brain research bulletin 79(3–4) (2009) 157–68. [DOI] [PubMed] [Google Scholar]
- [14].Vutskits L, Djebbara-Hannas Z, Zhang H, Paccaud JP, Durbec P, Rougon G, Muller D, Kiss JZ, PSA-NCAM modulates BDNF-dependent survival and differentiation of cortical neurons, The European journal of neuroscience 13(7) (2001) 1391–402. [DOI] [PubMed] [Google Scholar]
- [15].Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, et al. , Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit, Nature 373(6510) (1995) 151–5. [DOI] [PubMed] [Google Scholar]
- [16].Gao C, Frausto SF, Guedea AL, Tronson NC, Jovasevic V, Leaderbrand K, Corcoran KA, Guzman YF, Swanson GT, Radulovic J, IQGAP1 regulates NR2A signaling, spine density, and cognitive processes, The Journal of neuroscience : the official journal of the Society for Neuroscience 31(23) (2011) 8533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mormede C, Palin K, Kelley KW, Castanon N, Dantzer R, Conditioned taste aversion with lipopolysaccharide and peptidoglycan does not activate cytokine gene expression in the spleen and hypothalamus of mice, Brain, behavior, and immunity 18(2) (2004) 186–200. [DOI] [PubMed] [Google Scholar]
- [18].Porsolt RD, Animal models of depression: utility for transgenic research, Reviews in the neurosciences 11(1) (2000) 53–8. [DOI] [PubMed] [Google Scholar]
- [19].Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS, mTORdependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists, Science 329(5994) (2010) 959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Markwell MA, Haas SM, Bieber LL, Tolbert NE, A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples, Analytical biochemistry 87(1) (1978) 206–10. [DOI] [PubMed] [Google Scholar]
- [21].Laemmli UK, Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Nature 227(5259) (1970) 680–5. [DOI] [PubMed] [Google Scholar]
- [22].Adzic M, Djordjevic J, Mitic M, Brkic Z, Lukic I, Radojcic M, The contribution of hypothalamic neuroendocrine, neuroplastic and neuroinflammatory processes to lipopolysaccharide-induced depressive-like behaviour in female and male rats: Involvement of glucocorticoid receptor and C/EBP-beta, Behavioural brain research 291 (2015) 130–9. [DOI] [PubMed] [Google Scholar]
- [23].Brkic Z, Petrovic Z, Franic D, Mitic M, Adzic M, Male-specific effects of lipopolysaccharide on glucocorticoid receptor nuclear translocation in the prefrontal cortex of depressive rats, Psychopharmacology 233(18) (2016) 3315–30. [DOI] [PubMed] [Google Scholar]
- [24].Bay-Richter C, Janelidze S, Hallberg L, Brundin L, Changes in behaviour and cytokine expression upon a peripheral immune challenge, Behavioural brain research 222(1) (2011) 193–9. [DOI] [PubMed] [Google Scholar]
- [25].Boyce-Rustay JM, Holmes A, Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice, Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 31(11) (2006) 2405–14. [DOI] [PubMed] [Google Scholar]
- [26].Datta SC, Opp MR, Lipopolysaccharide-induced increases in cytokines in discrete mouse brain regions are detectable using Luminex xMAP technology, Journal of neuroscience methods 175(1) (2008) 119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nair A, Bonneau RH, Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation, Journal of neuroimmunology 171(1–2) (2006) 72–85. [DOI] [PubMed] [Google Scholar]
- [28].Zhang JC, Wu J, Fujita Y, Yao W, Ren Q, Yang C, Li SX, Shirayama Y, Hashimoto K, Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation, The international journal of neuropsychopharmacology 18(4) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, Han M, Wu J, Ushida Y, Suganuma H, Hashimoto K, Prophylactic effects of sulforaphane on depression-like behavior and dendritic changes in mice after inflammation, The Journal of nutritional biochemistry 39 (2017) 134–144. [DOI] [PubMed] [Google Scholar]
- [30].Nowacka MM, Paul-Samojedny M, Bielecka AM, Plewka D, Czekaj P, Obuchowicz E, LPS reduces BDNF and VEGF expression in the structures of the HPA axis of chronic social stressed female rats, Neuropeptides 54 (2015) 17–27. [DOI] [PubMed] [Google Scholar]
- [31].Bai M, Zhu X, Zhang Y, Zhang S, Zhang L, Xue L, Yi J, Yao S, Zhang X, Abnormal hippocampal BDNF and miR-16 expression is associated with depression-like behaviors induced by stress during early life, PloS one 7(10) (2012) e46921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lahiri T, Moore PE, Baraldo S, Whitehead TR, McKenna MD, Panettieri RA Jr., Shore SA, Effect of IL-1beta on CRE-dependent gene expression in human airway smooth muscle cells, American journal of physiology. Lung cellular and molecular physiology 283(6) (2002) L1239–46. [DOI] [PubMed] [Google Scholar]
- [33].Parry GC, Mackman N, Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription, Journal of immunology 159(11) (1997) 5450–6. [PubMed] [Google Scholar]
- [34].Murphy PG, Borthwick LA, Altares M, Gauldie J, Kaplan D, Richardson PM, Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons, The European journal of neuroscience 12(6) (2000) 1891-9. [DOI] [PubMed] [Google Scholar]
- [35].Schuster T, Krug M, Hassan H, Schachner M, Increase in proportion of hippocampal spine synapses expressing neural cell adhesion molecule NCAM180 following long-term potentiation, Journal of neurobiology 37(3) (1998) 359–72. [DOI] [PubMed] [Google Scholar]
- [36].Pham K, Nacher J, Hof PR, McEwen BS, Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus, The European journal of neuroscience 17(4) (2003) 879–86. [DOI] [PubMed] [Google Scholar]
- [37].Wainwright SR, Galea LA, The neural plasticity theory of depression: assessing the roles of adult neurogenesis and PSA-NCAM within the hippocampus, Neural plasticity 2013 (2013) 805497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Muller D, Djebbara-Hannas Z, Jourdain P, Vutskits L, Durbec P, Rougon G, Kiss JZ, Brain-derived neurotrophic factor restores long-term potentiation in polysialic acid-neural cell adhesion molecule-deficient hippocampus, Proceedings of the National Academy of Sciences of the United States of America 97(8) (2000) 4315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Aonurm-Helm A, Jurgenson M, Zharkovsky T, Sonn K, Berezin V, Bock E, Zharkovsky A, Depression-like behaviour in neural cell adhesion molecule (NCAM)-deficient mice and its reversal by an NCAM-derived peptide, FGL, The European journal of neuroscience 28(8) (2008) 1618–28. [DOI] [PubMed] [Google Scholar]
- [40].Rodriguez JJ, Dallerac GM, Tabuchi M, Davies HA, Colyer FM, Stewart MG, Doyere V, Nmethyl-D-aspartate receptor independent changes in expression of polysialic acid-neural cell adhesion molecule despite blockade of homosynaptic long-term potentiation and heterosynaptic long-term depression in the awake freely behaving rat dentate gyrus, Neuron glia biology 4(3) (2008) 169–78. [DOI] [PubMed] [Google Scholar]
- [41].Mackowiak M, O’Neill MJ, Hicks CA, Bleakman D, Skolnick P, An AMPA receptor potentiator modulates hippocampal expression of BDNF: an in vivo study, Neuropharmacology 43(1) (2002) 1–10. [DOI] [PubMed] [Google Scholar]
- [42].Dijkhuizen PA, Ghosh A, BDNF regulates primary dendrite formation in cortical neurons via the PI3-kinase and MAP kinase signaling pathways, Journal of neurobiology 62(2) (2005) 278–88. [DOI] [PubMed] [Google Scholar]
- [43].Tao W, Chen Q, Zhou W, Wang Y, Wang L, Zhang Z, Persistent inflammation-induced upregulation of brain-derived neurotrophic factor (BDNF) promotes synaptic delivery of alpha-amino-3hydroxy-5-methyl-4-isoxazolepropionic acid receptor GluA1 subunits in descending pain modulatory circuits, The Journal of biological chemistry 289(32) (2014) 22196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]