

Abstract
Rationale:
Pulmonary arterial hypertension (PAH) is a deadly disease of the pulmonary vasculature for which no disease modifying therapies exist. Small vessel stiffening and remodeling are fundamental pathologic features of PAH that occur early and drive further endovascular cell dysfunction. Bone marrow (BM)-derived proangiogenic cells (PACs), a specialized heterogeneous subpopulation of myeloid lineage cells, are thought to play an important role in pathogenesis.
Objective:
To determine if BM-derived PACs directly contributed to experimental pulmonary hypertension (PH) by promoting small vessel stiffening through serotonin 2B receptor (5-HT2B) mediated signaling.
Methods and Results:
We performed BM transplants using transgenic donor animals expressing diphtheria toxin secondary to activation of an endothelial-specific tamoxifen-inducible Cre and induced experimental PH using hypoxia with SU5416 to enhance endovascular injury and ablated BM-derived PACs, after which we measured right ventricular systolic pressures in a closed-chest procedure. BM-derived PAC lineage tracing was accomplished by transplanting BM from transgenic donor animals with fluorescently labeled hematopoietic cells and treating mice with a 5-HT2B antagonist. BM-derived PAC ablation both prevented and reversed experimental PH with SU5416-enhanced endovascular injury, reducing the number of muscularized pulmonary arterioles and normalizing arteriole stiffness as measured by atomic force microscopy. Similarly, treatment with a pharmacologic antagonist of 5-HT2B also prevented experimental PH, reducing the number and stiffness of muscularized pulmonary arterioles. PACs accelerated pulmonary microvascular endothelial cell injury response in vitro, and the presence of BM-derived PACs significantly correlated with stiffer pulmonary arterioles in PAH patients and mice with experimental PH. RNA sequencing of BM-derived PACs showed that 5-HT2B antagonism significantly altered biologic pathways regulating cell proliferation, locomotion and migration, and cytokine production and response to cytokine stimulus.
Conclusions:
Together, our findings illustrate that BM-derived PACs directly contribute to experimental PH with SU5416-enhanced endovascular injury by mediating small vessel stiffening and remodeling in a 5-HT2B signaling-dependent manner.
Keywords: Pulmonary hypertension, serotonin 5-HT2B, bone marrow cells, proangiogenic, hematopoietic stem cells, stiffness, vascular remodeling
INTRODUCTION
Pulmonary arterial hypertension (PAH) is an insidious, deadly illness of the pulmonary vasculature. Despite the existing need for disease-modifying therapeutics, the rational selection of targets is significantly hampered by the still poor understanding of the cellular and molecular mechanisms that mediate PAH pathogenesis.1 Small vessel remodeling, an active pathologic process that results in thickened, muscularized arteriole walls and obliteration of the vessel lumen, is ultimately responsible for the clinical symptoms associated with PAH.2 Small vessel remodeling is hypothesized to occur in response to enhanced endothelial cell death and damage intrinsic to PAH, occurring before the onset of elevated right ventricular pressures3 and potentiating further endovascular cell dysfunction through mechanotransduced alterations in cell metabolism.4 While intrinsic genetic defects in endovascular cell function undoubtedly contribute to arteriole stiffening and remodeling, recent studies also suggest this process is driven in part by myeloid-derived cells.5,6 Bone marrow (BM)-derived proangiogenic cells (PACs) are a subtype of myeloid cells believed to contribute directly to small vessel remodeling. Phenotypically heterogeneous and poorly characterized, PACs are generally described as expressing some combination of endothelial, hematopoietic, or stem cell surface markers (such as VEGFR2, Tie2, CD31, CXCR4, CD34, CD133, and c-Kit).7,8 Their presence in peripheral blood has been well-correlated with PAH in a number of studies,9–12 and BM-derived cells with endothelial or progenitor cell markers have been identified in the walls of remodeled vessels from PAH patients.10,13,14 While PACs are not believed to proliferate and occlude pulmonary vessels themselves, they are hypothesized to promote pathologic vasculogenic-like processes in neighboring endovascular cells.15 However their exact function remains obscure, and to date no study has definitively established their role in promoting (or abrogating) disease.
The molecular mechanisms governing PAC function in disease are equally opaque. Signaling through the serotonin 2B receptor (5-HT2B), responsible for mediating serotonergic diet drug-induced PAH in humans,16 is also necessary for the myeloid contribution to experimental pulmonary hypertension (PH) in hypoxic mice.17 Mice lacking 5-HT2B have similar metrics of hematopoiesis to wild type animals with a notable exception of fewer CD31+CD11b- cells in the BM and peripheral circulation, identified as immature proangiogenic or endothelial-like cells. It is possible that intact 5-HT2B signaling may be necessary for PAC recruitment or function.
We hypothesized that PACs contribute directly to small vessel stiffening and remodeling through a 5-HT2B dependent mechanism. By selectively ablating PACs utilizing a transgenic mouse model we successfully prevent and reverse the development of elevated pulmonary pressures following hypoxia exposure and enhanced endovascular injury with SU5416. PAC ablation also reduced markers of small vessel remodeling and restored vessel wall compliance to normal levels. We then illustrate the effectiveness of 5-HT2B antagonism in preventing experimental PH in the same disease model and, through lineage tracing of hematopoietic cells, show reduced recruitment and altered gene expression profiles of PACs in animals treated with a pharmacologic inhibitor of 5-HT2B. Finally, we show that PACs enhance pulmonary microvascular endothelial injury response in vitro and that stiffened arterioles in human lungs with PAH correlate with the presence of PACs. Our data indicate that BM-derived PACs necessary for the development of experimental PH in the setting of endovascular injury and likely contribute to small artery stiffening and remodeling.
METHODS
The authors declare that all supporting data are available within the article [and its online supplementary files].
Bone marrow transplantation and transgenic mouse models.
All mouse experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee prior to their initiation. 6–12 week old C57BL/6 mice were given a split 12 Gy dose of radiation from a Cs137 source followed by retro-orbital administration of 5×105 BM cells isolated from an age- and sex-matched cogenic donor. Male mice were used to control for the well-documented effects of endogenous estrogen on the hypoxia-induced PH mouse model.18 For the ablation of PACs, C57BL/6 donor mice were bred to express a tamoxifen-inducible Cre under the control of the 5’ endothelial specific stem cell leukemia (SCL) promoter and diphtheria toxin under control of the ROSA26 promoter, preceded by a floxed-STOP codon to prevent transcription in the absence of Cre expression19 (endothelial SCL-CreERT2/DTafl/-, obtained as a generous gift from the laboratory of Dr. H. Scott Baldwin at Vanderbilt University). For lineage tracing, we used C57BL/6 donor mice expressing a Tie2-promoter driven Cre and ROSA26-promoter driven YFP preceded by a floxed STOP codon (Tie2-Cre/YFPfl/-, labeling all hematopoietic cells regardless of lineage20). Donor cells were given 10 weeks for engraftment prior to the initiation of experiments. For in vitro studies, C57BL/6 donor mice were bred to express both endothelial SCL-CreERT2 and tdTomato under control of the ROSA26 promoter, preceded by a floxed STOP codon (endothelial SCL-CreERT2/tdTomatofl/). Transplants were again performed as described.
Induction of experimental PH and hemodynamic phenotyping.
To induce experimental PH with enhanced endovascular injury, we administered the vascular endothelial growth factor receptor 2 (VEGFR2) inhibitor SU5416 (Tocris Biosciences) at 20 mg/kg/week intraperitoneally (IP) while mice were maintained in hypoxia (10% O2) for 3 weeks. Also referred to as the “Sugen-hypoxia” model of experimental PH, inhibition of VEGFR2 in combination with hypoxia results in hypoxic vascular injury and a proliferative vascular remodeling response mimicking human PAH pathology21 that persists up to 10 weeks following the secession of hypoxia and SU5416.22 Control animals were maintained on either room air or in 10% O2 while receiving SU5416 or vehicle injections (0.5% carboxymethylcellulose, 0.4% polysorbate, 0.9% benzyl alcohol (Sigma-Aldrich) in 0.9% sterile saline). For randomization, mice were given an identification number with no relation to experimental group assignment prior to disease induction. In order to ablate BM-derived PACs, SCL-CreERT2/DTafl/- recipient mice were simultaneously given 2 mg of tamoxifen (Sigma-Aldrich) IP every other day or vehicle injections (10% ethanol in sunflower oil, Sigma-Aldrich). For lineage tracing of PACs, Tie2-Cre/YFPfl/- recipient mice were implanted with subcutaneous Alzet pumps delivering the 5-HT2B antagonist SB204741 (Tocris Biosciences) (1 mg/kg/day) or vehicle (50% dimethylsulfoxide (Sigma-Aldrich) and polyethyleneglycol-400 (Fisher Chemical)) 24 hours prior to disease induction. After 3 weeks, mice were placed under surgical anesthesia (Avertin) and a catheter was inserted into the right heart via the right jugular vein in a closed-chested procedure to measure right ventricular systolic pressures (RVSP), as previously described.23 All mice that were alive at the conclusion of the procedure were included in data analysis. Mice were euthanized with phenobarbital prior to collection of biologic samples.
Immunohistochemistry.
Lungs were isolated, flushed with PBS, inflated and embedded with optimal cutting temperature compound, flash frozen in liquid nitrogen, and sectioned. Prior to staining lung sections were fixed in a solution of 4% paraformaldehyde (Electron Microscopy Sciences) and 0.01% Triton in PBS. Sections were stained with FITC conjugated rat anti-mouse CD31 (CD31-FITC, BD Biosciences, catalog #558738), Cy3 conjugated mouse monoclonal anti- α smooth muscle actin (αSMA-Cy3, Sigma-Aldrich, SKU C6198), and DAPI (Thermo Fisher Scientific). Lung sections from Tie2-Cre/YFPfl/- transplanted animals were also stained with AF647 conjugated anti-GFP (GFP-AF647, Thermo Fisher Scientific, catalog #A-31852). Control specimens included unstained slides. Slides were labeled only with randomized animal identification number and data acquisition was performed without knowledge of the treatment group. The number of fully (>75% of vessel circumference) and partially muscularized αSMA-positive pulmonary arterioles (<100 μm diameter) were quantified from images taken using an Olympus microscope. In lung sections taken from Tie2-Cre/YFPfl/- transplanted animals, pulmonary arterioles were further evaluated for the presence of cells expressing both YFP and CD31, identified as PACs (negative controls shown in Online Figure I).
Measurement of pulmonary arteriole wall elastic modulus.
Atomic force microscopy (AFM) of whole tissue sections was performed based on previously described methods.24,25 Fresh-frozen lung sections 10 μm thick were stained with anti-mouse CD31 (BD Biosciences, catalog #557355), FITC- conjugated anti-rat IgG secondary (BD Biosciences, catalog #554016), αSMA-Cy3, and DAPI. Lung sections from Tie2-Cre/YFPfl/- transplanted animals were also stained with GFP-AF647. Control specimens included unstained and FITC-conjugated anti-rat secondary only. After staining sections were immersed in PBS and αSMA-positive vessels less than 100 μm in diameter were identified using a Nikon Eclipse Ti microscope. Vessels were then scanned using a Bioscope Catalyst AFM at a scanning frequency of 0.25 Hz and a scan window size of 5–8 μm. Slides were labeled only with randomized animal identification number and data acquisition was performed without knowledge of the treatment group. A total of 12–32 vessels were scanned for each animal, across two or more sections of lung per animal. Wall modulus values were calculated as the average value of each scan, which consisted of 16,384 individual measurements (128×128) spanning an approximately 25–50 um2 area along the vessel wall. Human lung sections were obtained from autopsies or transplant patients with a previous diagnosis of PAH. Fresh-frozen lung sections were stained for anti-human CD31 (BioLegend, catalog #303102), CD45 (Thermo Fisher, catalog #PA5–11671), αSMA-Alexa Fluor 647 (Novus Biologics, catalog #NBP2–34522AF647), and DAPI.
Flow cytometric characterization.
Lung cells were obtained from digesting masticated lungs in a solution of 5% fetal bovine serum, 1 mg/mL collagenase type IV, and 0.02 mg/mL DNase I (Sigma-Aldrich) in RPMI 1640 medium for 45 minutes at 37oC. Peripheral blood mononucleocytes (PBMCs) were isolated from blood drawn from the right jugular. Red blood cells (RBCs) were lysed using RBC lysis buffer (BioLegend). For quantifying the number of PACs in the lungs and peripheral blood, both PBMCs and lung cells were stained with anti- Ter119-Pacific Blue, CD11b-redFuor (Tonbo Biosciences, catalog #10141–234 and #80–0112), CD31-PECy7 (BioLegend, catalog #102524), and DAPI. To quantify engraftment, PBMCs were stained with anti-Ter119-Pacific Blue, CD45.1-PE (BD Biosciences, catalog #561872), CD45.2-PerCPCy5.5 (Tonbo Biosciences, catalog #65–0454), and DAPI. Flow cytometry was performed using a BD LSRFortessa and the data analyzed using FlowJo. Antibody-conjugated beads (Thermo Fisher) were used for single stain compensation controls. Unstained cells and fluorescence-minus-one controls were used for gating populations in all flow cytometry experiments.
Electric cell-substrate impedance sensing (ECIS) in microvascular endothelial cells.
For in vitro ECIS experiments, mouse pulmonary microvascular endothelial cells (PMVECs) were seeded in ECIS well plates (8W1E for electroporation wounding, Applied Biophysics) at a density of 150,000 per well. Following 2 hours of baseline measurements, a wound was applied to the entire plate surface (250 μm diameter) by administering a 60 kHz pulse for 20 seconds and the treatments were added to wells in duplicate. Treatments included: (1) blank controls or 10,000 PACs isolated from the lungs of mice with experimental PH, as described; (2) DMSO or 1 μg/mL SB204741; or (3) 10,000 PACs with DMSO or 1 μg/mL SB204741. Measurements were taken on a single channel at 4 kHz for 24 hours using the ECIS ZTheta (Applied Biophysics). Data was normalized to values at the time the wound was applied and treatment added.
RNA sequencing and analysis.
Adult male C57BL/6 mice were maintained on vehicle for SB204741 in normoxia for 3 weeks as described after which BM cells and PBMCs were collected. BM cells and PBMCs were stained with anti-Ter119-, Gr1-, B220-, and CD3-PacificBlue (Tonbo Biosciences, catalog #75–5931, #75–0452, #75–0032), as well as anti-CD45-FITC (eBioscience, catalog #11–0451-82), CD31-PECy7, CD11b-redFluor, and DAPI. Live (Ter119-Gr1-B220-CD3-) CD31+CD11b- cells were sorted using a BD FACSAria III and collected in PBS (>95% purity). A Qiagen RNeasy Micro kit was used for RNA isolation. Messenger RNA enrichment, cDNA library preparation, and sequencing were performed by the Vanderbilt Technologies for Advanced Genomics center (VANTAGE).
Analysis of RNA-seq data was performed using the Amazon Elastic Compute Cloud. Standard quality control analyses on raw reads were done using FastQC v 0.11.5, with quality trimming and adaptor removal performed using Trimmomatic v 0.36 and read alignment performed using STAR v 2.5.2b. Raw read counts were normalized using the TMM method of library size normalization in edgeR, and differential expression was performed in limma (Biocondutor v3.3). RNA sequencing data will be made publicly available through the National Center for Biotechnology Information’s Gene Expression Omnibus repository.
Differentially expressed genes (p<0.01) were selected for inclusion in the gene ontology (GO) enrichment analysis. A GO slim analysis was performed using the BinGO app in Cytoscape, with GO annotation files downloaded from the GO consortium.26–28 GO enrichment analysis for GO biologic processes was performed using the Web-based GEne SeT AnaLysis Toolkit (WebGestalt).29 Significantly enriched GO terms are defined as having a p<0.05 following a false discovery rate correction.
Statistical analysis.
Statistical analysis was performed using SigmaPlot version 11 and the R programming language. The Shapiro-Wilks test was used to test for data normality. Data was subsequently analyzed using either a 2-way ANOVA with Holm-Sidak post-hoc test or a Kruskal-Wallis rank sum test with Conover-Iman post-hoc test for non-parametric data. A two-tailed student’s t-test was used for single comparisons of two normally distributed groups. Data were considered significantly different for p < 0.05.
RESULTS
Ablation of PACs prevents experimental pulmonary hypertension and normalizes metrics of pulmonary vascular remodeling.
In order to determine if BM-derived PACs contributed to the development of experimental PH, we employed a chimeric mouse model that would allow us to selectively ablate hematopoietic cells with a uniquely endothelial genetic signature. These mice, developed previously to study the contribution of hematopoietic proangiogenic cells to tumor endothelium, display little to no expression of Cre in hematopoietic stem cells (as opposed to their 3’-SCL counterparts), making them an ideal lineage-restricted driver strain.19 After transplant and reconstitution (Online Figure II) mice were placed in either hypoxia with weekly SU5416 injections to induce experimental PH or normoxia and received either vehicle or tamoxifen injections for three weeks (Figure 1A). Following right heart catheterization mice receiving tamoxifen for PAC ablation were found to have significantly lower pressures than their vehicle-treated counterparts (Figure 1B), though no effect on RV hypertrophy was observed (Figure 1C). The number of muscularized small arteries was increased in vehicle-treated mice following hypoxic vascular injury, with PAC ablation significantly reducing the number of fully, but not partially, muscularized arteries (Figure 1D-E). As stiffening of the distal pulmonary vasculature is an early event in experimental PH and important in the potentiation of further vascular remodeling,3,4,30 we next assessed whether PAC ablation altered pulmonary arteriole stiffness. Using AFM, we found that vehicle treated animals exposed to hypoxia and SU5416 had significantly stiffer pulmonary arterioles, as we and others have previously reported for other mouse models of PAH using this same technique,3,25 and that ablation of BM-derived PACs normalized vessel stiffness (Figure 1F-H). In mice exposed to hypoxia alone, tamoxifen treatment failed to reduce RVSP, vessel muscularization, or arteriole stiffness. Furthermore, significantly fewer muscularized arterioles were observed in these mice compared to hypoxia and SU5416 exposed animals. (Figure 1B-F), suggesting that PAC-mediated effects occur only in the setting of SU5416-mediated endovascular injury. Tamoxifen treatment also failed to reduce elevated RVSP in mice transplanted with BM from mice expressing SCL-CreERT2 but not the inducible diphtheria toxin gene, illustrating that diphtheria toxin expression and subsequent ablation of PACs is necessary PH prevention (Online Figure III). The observed effects of PAC ablation are unlikely due to alterations in inflammatory cell populations, as flow cytometric characterization of PBMCs performed on mice with BM from endothelial SCL-CreERT2/tdTomatofl/- donors show that <1% of donor T cells, B cells, and monocytic cells express tdTomato (Online Figure IV).
Figure 1: Ablation of BM-derived PACs reduces elevated RVSP and inhibits the muscularization and stiffening of pulmonary arterioles in experimental PH with SU5416-enhanced endovascular injury.
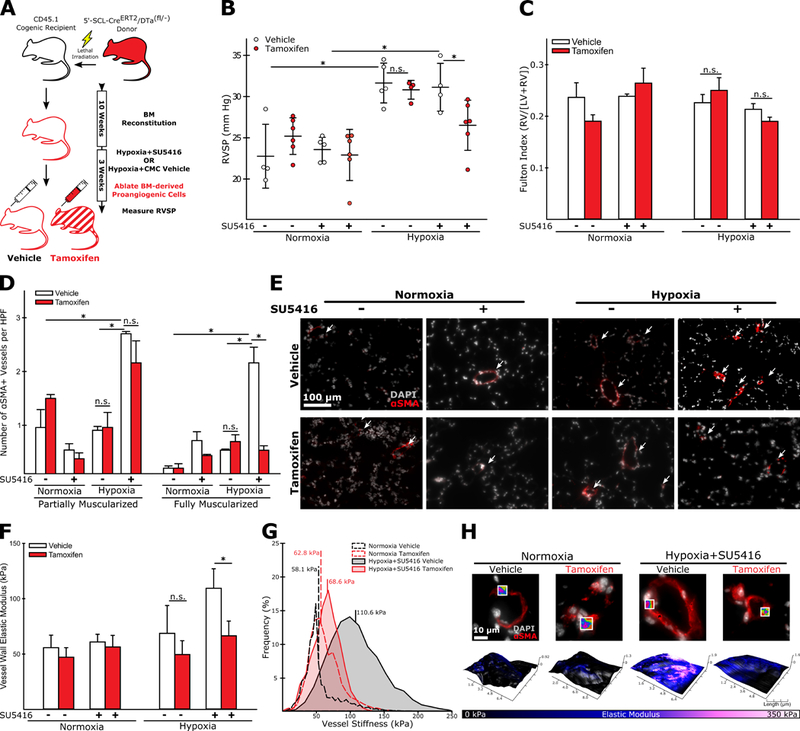
(A) Experimental approach. (B) Ablation of BM-derived PACs normalizes RVSP in hypoxia+SU5416 treated animals. This effect is not observed in animals exposed to hypoxia alone. (C) PAC ablation has no effect on RV remodeling in either animal model (n=4–6). (D-E) Animals exposed to hypoxia+SU5416 have significantly more muscularized arterioles (< 100 μm diameter) than animals exposed to hypoxia alone, and PAC ablation significantly reduces the number of fully muscularized arterioles in hypoxia+SU5416 exposed but not in hypoxia alone exposed animals (n=3). (F) Similarly, PAC ablation normalizes arteriole stiffness in animals exposed to hypoxia+SU5416, but not in animals exposed to hypoxia alone (n=3–6). (G) Representative modulus distributions and (H) scan windows for individual vessels. (B-D, F) Mean +/−S.E., n.s. = not significant, *p<0.05 following (B,C,F) 2-way ANOVA and Holm-Sidak post-hoc test or (D) Kruskal-Wallis rank sum test with Conover-Iman post-hoc test.
Pharmacologic inhibition of 5-HT2B prevents experimental pulmonary hypertension and vascular remodeling.
We next tested the hypothesis that 5-HT2B signaling mediated the recruitment or function of BM-derived PACs to the lungs during experimental PH. To perform lineage tracing on PACs, we used mice expressing both a Tie2-promoter driven Cre and a ROSA26-promoter driven fluorescent reporter (YFP) proceeded by a loxP-flanked STOP codon as BM donors. As Tie2 is expressed on hematopoietic stem cells, this model allowed us to positively identify all BM cells regardless of lineage.20 We combined this approach with cell surface-labeling of CD31+CD11b- negative BM-derived cells, a cell population containing PACs and reportedly reduced in number in 5-HT2B deficient mice.17 This strategy allowed us to determine how 5-HT2B antagonism alters the recruitment of multiple inflammatory cell lineages, including BM-derived PACs, during experimental PH (Online Figure V). Both engraftment efficiency and the efficiency of Cre-mediated recombination was comparable regardless of treatment, with the majority (>80%) of donor CD45.2+ lymphocytes expressing YFP (Online Figure VI). Following transplantation and reconstitution, the mice were placed in either normoxia or hypoxia and subcutaneously implanted with an osmotic pump delivering the 5-HT2B antagonist SB204741 or vehicle for three weeks (Figure 2A). Pharmacologic inhibition of 5-HT2B with SB204741 was sufficient to normalize RVSP as measured by right heart catheterization compared to vehicle treated controls (Figure 2B), though it had no effect on RV remodeling (Figure C). Treatment with SB204741 also reduced the number of fully (but not partially) muscularized arterioles (Figure 2D). Measurements of pulmonary arteriole wall elastic modulus with AFM show a significant reduction in vascular stiffness following SB204741 treatment in mice exposed to hypoxia and SU5416 (Figure 2F-H), recapitulating the results we observed when ablating PACs. Also similar to PAC ablation, SB204741 treatment failed to normalize RVSP, vessel muscularization, and arteriole stiffness in mice exposed to hypoxia in the absence of SU5416 (Figure 2B-F).
Figure 2: Antagonism of the 5-HT2B receptor normalizes elevated RVSP and reduces the muscularization and stiffening of pulmonary arterioles in experimental PH with SU5416-enhanced endovascular injury.
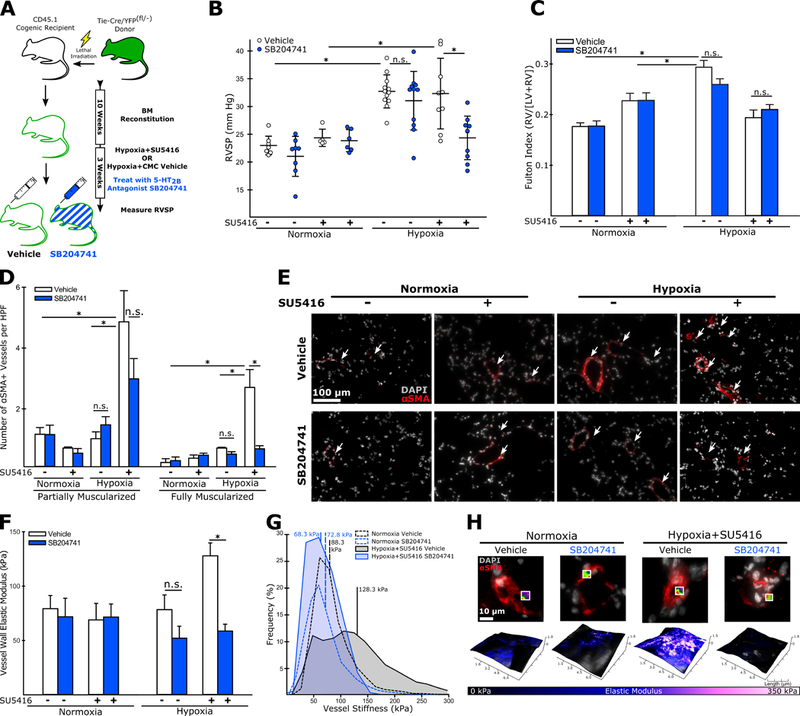
(A) Experimental approach. (B) 5-HT2B antagonist, SB204741, normalizes RVSP in mice exposed to hypoxia+SU5416. This effect is not observed in animals exposed to hypoxia alone. (C) SB204741 has no effect on RV remodeling in either animal model (n=7–8). (D-E) Animals exposed to hypoxia+SU5416 have significantly more muscularized arterioles (< 100 μm diameter) than animals exposed to hypoxia alone, and SB204741 significantly reduces the number of fully muscularized arterioles in hypoxia+SU5416 exposed but not in hypoxia alone exposed animals (n=3–5). (F) SB204741 normalizes arteriole stiffness in animals exposed to hypoxia+SU5416, but not in animals exposed to hypoxia alone (n=3–6). (G) Representative modulus distributions and (H) scan windows for individual vessels. (B-D, F) Mean +/−S.E., n.s. = not significant, *p<0.05 following (B,C,F) 2-way ANOVA and Holm-Sidak post-hoc test or (D) Kruskal-Wallis rank sum test with Conover-Iman post-hoc test.
5-HT2B antagonism reduces the fraction and alters the activity of BM-derived PACs during experimental pulmonary hypertension.
We next quantified the fraction of PACs in the lungs and peripheral blood following induction of experimental PH with SU5416 enhanced endovascular injury and vehicle or SB204741 treatment. Mice exposed to hypoxia and SU5416 and treated with SB204741 had a significantly reduced fraction of BM-derived (YFP+) PBMCs expressing the endothelial marker CD31 and absent for the immature myeloid marker CD11b (CD31+CD11b-) as assessed by flow cytometry (Figure 3A). We also observed a numerically small but statistically significant increase in the fraction of YFP+CD31+CD11b- cells in the lungs of vehicle treated, hypoxia and SU5416 exposed animals compared to vehicle treated controls. This fraction was reduced in mice treated with SB204741 (Figure 3B). Importantly, neither hypoxic vascular injury nor SB204741 treatment altered the total number of lung cells present, or the fraction of lung cells that expressed YFP, suggesting the absence of large scale inflammatory infiltrate. There was also no significant effect on macrophage recruitment to the lung, another important inflammatory cell type in PAH pathogenesis31 (Online Figure VIIC-E). While BM-derived c-Kit+ cells have previously been reported to accumulate in the walls of remodeled vessels and potentially mediate vessel remodeling in a 5-HT2B dependent manner,12,13,17 we observed a non-significant increase in the fraction of cKit+ cells following hypoxia with SB204741 exerting no measurable effects on the fraction present in the lungs. However, c-Kit expression was consistently enriched in our BM-derived PAC population regardless of treatment conditions, with 10–15% of YFP+CD31+CD11b- cells expressing c-Kit, compared to 0.5–0.8% of all BM-derived cells (Online Figure VIIF-G).
Figure 3: 5-HT2B antagonism reduces the number of BM-derived PACs in the lungs following hypoxia+SU5416.
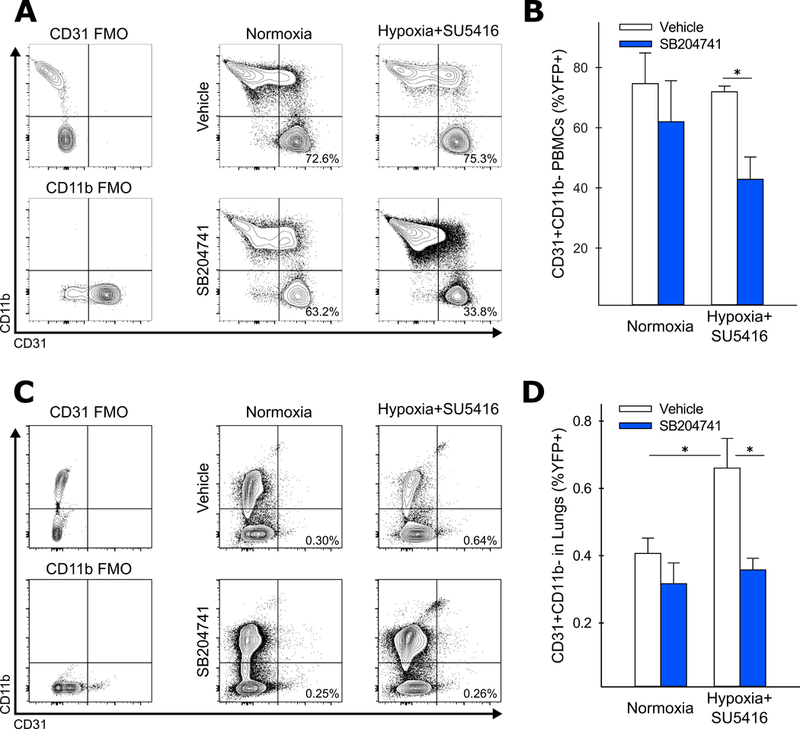
(A-B) Antagonism of 5-HT2B does not alter the fraction of BM-derived CD31+CD11b- cells in the peripheral circulation during normoxia (expressed as the percentage of total YFP labeled cells), but significantly reduces the fraction of these cells present in animals exposed to hypoxia+SU5416 (n=6–8). (C-D) The fraction of BM-derived CD31+CD11b- cells is significantly elevated in the lungs of mice exposed to hypoxia+SU5416, and reduced in mice treated with SB204741 (n=3–4). (A, C) Representative contour plots, gates, and percentages for each treatment condition (FMO = fluorescence minus one control). (B, D) Mean +/− S.E., *p<0.05 following 2-way ANOVA and Holm-Sidak post-hoc test.
In order to determine if BM-derived PACs localized to the walls of remodeling arterioles, we performed immunostaining on fixed frozen lung sections. Quantification of lung immunostaining showed an increased number of YFP+CD31+ cells in the walls of remodeling (α-SMA positive) arterioles, as well as a larger fraction of pulmonary arterioles containing at least one positively identified YFP+CD31+ cell embedded in the vessel wall (Figure 4A-B). Interestingly, pharmacologic antagonism of 5-HT2B in the absence of hypoxic vascular injury significantly increased the localization of PACs to the walls of pulmonary arterioles, while leaving the total fraction of YFP+CD31+CD11b- cells unchanged (as evidenced by flow cytometry data).
Figure 4: BM-derived PACs accumulate in the walls of remodeled pulmonary arterioles and are associated with arteriole wall stiffening.
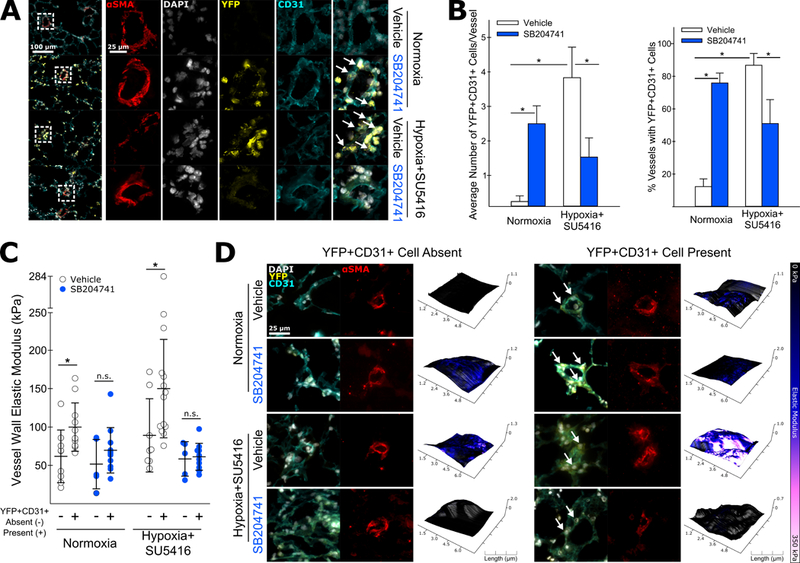
(A) Lung sections were incubated with fluorescently labeled antibodies specific αSMA to identify pulmonary arterioles. Co-staining with DAPI, anti-GFP, and anti-CD31 allowed for the identification of CD31+ BM-derived cells. BM-derived PACs (YFP+CD31+) were observed to accumulate in the walls of pulmonary arterioles in mice exposed to hypoxia+SU5416. (B) Both an increased number of BM-derived CD31+ cells adjacent to remodeled vessels and an increased frequency of vessels with at least one BM-derived CD31+ cell were observed in vehicle treated mice exposed to hypoxia+SU5416. SB204741 treatment reduced both the number of YFP+CD31+ cells per vessel as well as the percentage of vessels with at least one adjacent YFP+CD31+ cell. Conversely, SB204741 treatment significantly increased both of these metrics in the absence of hypoxic vascular injury (n=3–4, mean +/− S.E. *p<0.05 following 2-way ANOVA with Holm-Sidak post-hoc test). (C-D) In normoxic and SU5416+hypoxia treated animals, arterioles with at least one adjacent BM-derived CD31+ cell were significantly stiffer than those without adjacent BM-derived CD31+ cells. In either case, SB204741 treatment normalized the stiffness of the vessel walls (n=5–14, mean +/− S.E. *p<0.05 following two-tailed student’s t-test).
We next assessed whether or not the presence or absence of PACs was correlated with increased vessel wall stiffness. After determining the vessel wall elastic modulus using AFM, we subdivided vessels into either positive or negative for YFP+CD31+ cells in the vessel wall. On average, the measured vessel wall elastic modulus was significantly higher for vessels with at least one positively identified YFP+CD31+ cell in vehicle treated animals exposed to hypoxia and SU5416 (Figure 4C-D). While a significant fraction of vessels in SB204741 treated animals were identified as having greater than one YFP+CD31+ vessel, the presence or absence of these cells was not correlated with a significant difference in vessel stiffness in either hypoxic or normoxic conditions.
In order to determine how 5-HT2B antagonism might alter the function or phenotype of PACs, we performed RNA sequencing on PACs isolated from mice treated with either vehicle or SB204741 in normoxic conditions (Online Figure VIII). The results of both a GO slim analysis and full GO Biologic Process analysis for PACs isolated from both BM and peripheral blood was notable for a significant enrichment of genes broadly regulating immunologic processes, cytoskeletal regulation and cell motility, and cell differentiation. A more detailed GO enrichment analysis for all biological process categories indicated that, in PACs isolated from BM, 5-HT2B antagonism altered phosphorylation events associated with cellular metabolism, processes involved in cell development and differentiation, and cell motility and migration. Contrastingly in peripheral blood isolated PACs, genes regulating cytokine production and response, cell motility and migration, and cell division were significantly enriched (Online Figure IX).
BM-derived PACs are necessary for maintenance of the experimental PH phenotype in mice.
We next sought to determine if BM-derived PAC presence was necessary for maintaining the PH phenotype. Previous studies employing the same hypoxic vascular injury model of PH illustrate that the combination of SU5416 and hypoxia is sufficient to maintain elevated RVSP in normoxic conditions for up to 10 weeks after the initial induction,22 and has been used previously to study the efficacy of interventions to “reverse” or hasten the remission of established experimental PH.32 Adapting our protocol from these previous studies, we induced experimental PH in mice transplanted with BM from endothelial-SCL-CreERT2/DTafl/- donors (Figure 5A, Online Figure X). After 3 weeks we moved the mice to room air for an additional 3 weeks and administered vehicle or tamoxifen to ablate BM-derived PACs. We found that BM-derived PAC ablation significantly reduced RVSP in mice exposed to hypoxia+SU5416 (Figure 5B), though no effect on RV remodeling was observed (Figure 5C). PAC ablation also reduced the number of muscularized arterioles, though this effect was not statistically significant (Figure 5D). Arteriole stiffness remained elevated in vehicle-treated mice following their return to room air but was significantly reduced in mice following BM-derived PAC ablation (Figure 5E-G).
Figure 5: Ablation of BM-derived PACs reverses experimental pulmonary hypertension and normalizes vessel muscularization and stiffness.
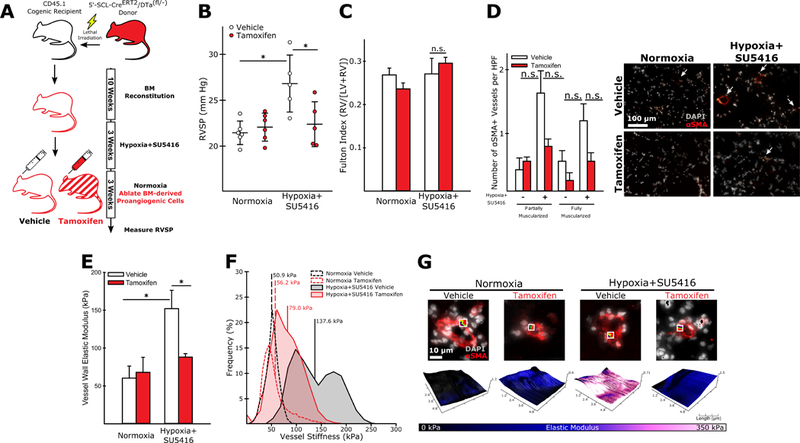
(A) Experimental approach. (B) Mice exposed to hypoxia+SU5416 with intact PACs (vehicle treated) had significantly elevated pressures compared to controls, while PAC ablation significantly reduced RVSP (n=5–6). (D) BM-derived PAC ablation caused a non-significant reduction in the number of muscularized arterioles in the lungs of animals exposed to hypoxia+SU5416 (n=3). (E) AFM measurements demonstrate normalization in pulmonary arteriole wall stiffness following BM-derived PAC ablation in animals exposed to hypoxia+SU5416 (n=3). (F) Representative modulus distributions and (G) scan windows for individual vessels. (B-D, E) Mean+/− S.E. *p<0.05 following (B,C,E) 2-way ANOVA and Holm-Sidak post-hoc test or (D) Kruskal-Wallis rank sum test with Conover-Iman post-hoc test.
BM-derived PACs isolated from the lungs of mice with experimental pulmonary hypertension promote endothelial cell proliferation, migration, and spreading in vitro.
To determine the effect of PACs on PMVECs, we used Electric Cell-Substrate Impedance Sensing (ECIS) to monitor the recovery of endothelial cell monolayer confluence following injury in the presence or absence of PACs. After induction of experimental PH, BM-derived (CD45+) CD31+CD11b- cells (PACs) were isolated from the lungs (further flow cytometric characterization in Online Figure XI shows that this cell population excludes major inflammatory cell groups, including T and B cells). Following injury, PMVECs co-cultured with PACs demonstrated enhanced resistance at t1/2 = 11 h and enhanced impedance and reduced capacitance at both 11 h and 22 h (Figure 6A). These measurements correspond directly to cell barrier quality (resistance) and cell spreading and migration (capacitance) or both (impedance). PAC co-culture also accelerated the rate of PMVEC spreading and migration (Figure 6A). We repeated this experiment on PMVECs cultured in the presence of SB204741 or vehicle and found that the drug did not alter PMVEC barrier quality, spreading, or migration (Figure 6B). Interestingly the drug also failed to appreciably alter these same quantities in PMVECs cultured with PACs (Figure 6C), suggesting 5-HT2B inhibition may be working further upstream to inhibit the pathogenic effect of PACs, perhaps at the point of cell differentiation or proliferation in the myeloid compartment.
Figure 6: BM-derived PACs promote pulmonary endothelial cell migration, spreading, and recovery of barrier integrity following injury.
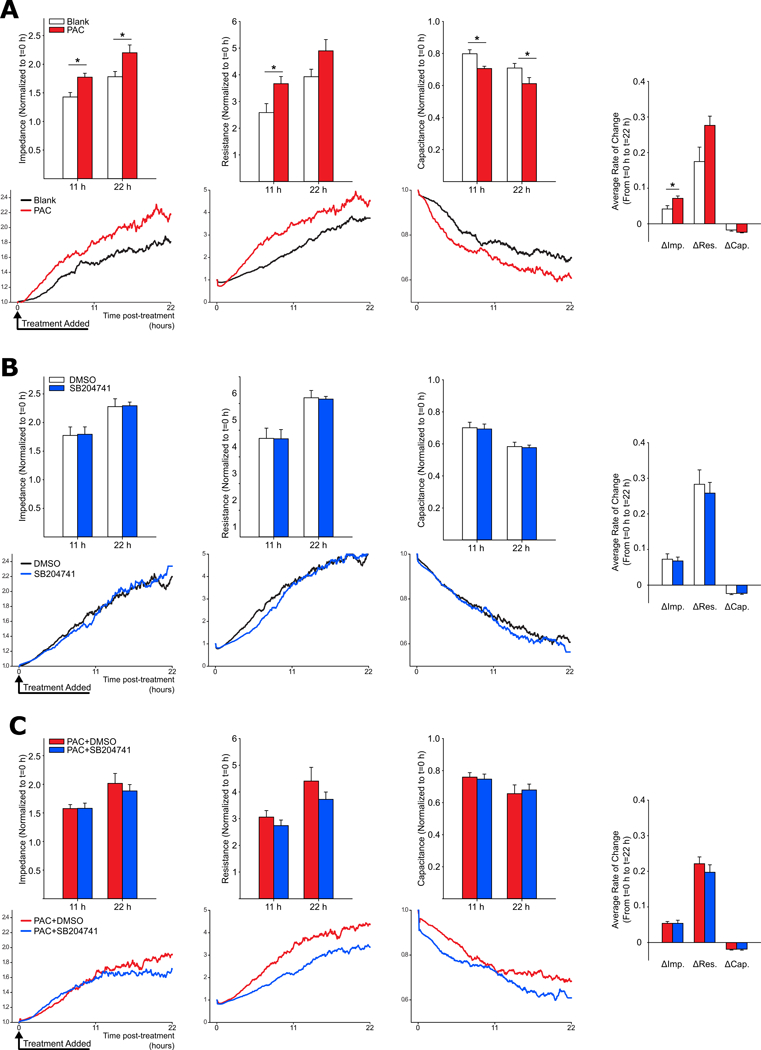
Average values for impedance, resistance, and capacitance measured at 11h and 22h post-injury using ECIS are shown in the top graph of A, B, and C, with corresponding representative tracings of each value over time shown immediately below. (A) PMVECs co-cultured with PACs demonstrate rapid recovery of barrier function and enhanced cell spreading and migration following injury. Both the relative amount and rate of change in measured electrical impedance (indicative of endothelial cell spreading, migration, and barrier integrity) are increased in PMVECs cultured with PACs. (B) The 5-HT2B antagonist SB204741 has no effect on PMVEC injury response. (C) SB204741 treatment also fails to normalize PMVEC response to injury in the presence of PACs, suggesting 5-HT2B signaling may be an important mediator of disease pathology prior to PAC recruitment to sites of endovascular injury (n=4, mean+/−S.E. *p<0.05 following 2-way ANOVA with Holm-Sidak post-hoc test or two-tailed student’s t-test for average rate-of-change comparisons).
BM-Derived CD31+ cell presence correlates with stiffer arterioles in the lungs of human PAH patients.
After establishing that BM-derived PACs are necessary for both initiation and maintenance of the experimental PH phenotype in mice, we next sought to establish whether the presence of an analogous cell in humans might also correlate with remodeled and stiffened pulmonary arterioles. We performed AFM on pulmonary arterioles identified from lung section taken from a small cohort of patients with either idiopathic or hereditary PAH (Supplemental Table 1). Co-staining with αSMA, CD31, and the hematopoietic marker CD45 allowed us to identify pulmonary arterioles with at least one adjacent BM-derived CD31 expressing cell (CD45+CD31+). Upon scanning these vessels we found them to be significantly stiffer on average than vessels lacking an adjacent CD45+CD31+ cell, recapitulating our results from the animal experiments (Figure 7).
Figure 7: The presence of hematopoietic (CD45+) cells expressing the endothelial marker CD31 predicts pulmonary arteriole stiffening in lungs from a cohort of PAH patients.
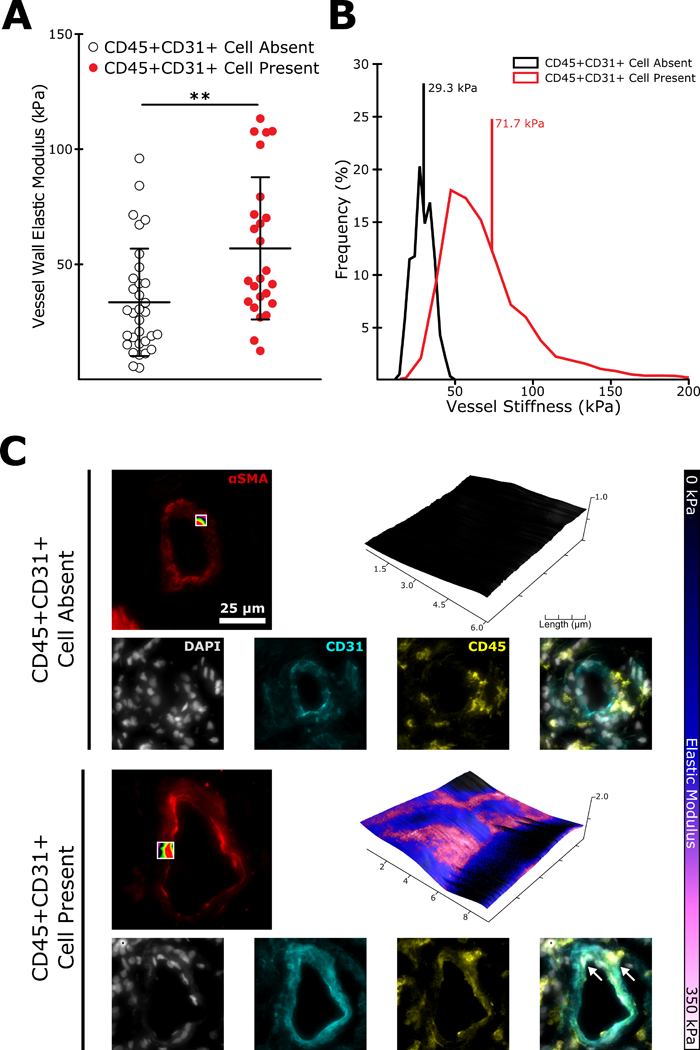
(A) Sections of human lung taken from PAH patients were co-stained for αSMA to identify pulmonary arterioles (< 100 μm in diameter) and CD45 and CD31 to identify BM-derived PACs. Pulmonary arterioles with at least one adjacent CD45+CD31+ cell were on average significantly stiffer than those with no identified CD45+CD31+ cell (n=25–32, mean+/− S.E. **p<0.01 following two-tailed student’s t-test). (B) Representative modulus distributions and (C) scan windows for individual vessels.
DISCUSSION
By utilizing a combination of genetically-targeted ablation and pharmacologic inhibition, we have illustrated that BM-derived PACs contribute directly to experimental PH in the setting of enhanced endovascular injury and that the 5-HT2B receptor is a critical mediator of this contribution. Prior evidence for PAC involvement in vascular remodeling is primarily derived from observations that BM-derived cells expressing endothelial or progenitor surface markers accumulate in the walls of remodeled arterioles,13,14,33 and that increased or decreased numbers of these cells are found in the peripheral blood of PAH patients (depending on the surface markers employed for identification or the patient cohort studied).34–36 Pharmacologic blockade of chemokine receptors such as CXCR4 and CXCR712,37 and the progenitor cell marker c-Kit17 has been successful in preventing experimental PH presumably by targeting subsets of this population, but the ubiquity of chemokine receptor expression among hematopoietic cell types (as well as native vascular endothelium)38 and lack of specificity among tyrosine kinase receptor antagonists makes interpretation of these results difficult.
Employing an inducible endothelial-specific Cre developed for studying hematopoietic proangiogenic cells19 allowed us to circumvent the problem of surface marker heterogeneity. By specifically targeting all BM-derived cells with this endothelial phenotype for destruction, we both prevented the development of experimental PH and reversed established illness. Notably, both PAC ablation and 5-HT2B antagonism were only efficacious in animals exposed to both hypoxia and SU5416, suggesting that PAC function is closely related to the endothelial cell injury response. RV remodeling is reportedly modest for both the hypoxia-alone and hypoxia+SU5416 mouse models39, and the success of both interventions in reducing RVSP and while failing to impact RV hypertrophy suggests that they were primarily targeted to the pulmonary microvasculature.
The normalization of arteriole compliance following PAC ablation is particularly notable, especially considering the important role arteriole stiffening plays in experimental PH and PAH pathogenesis. Recent evidence from mouse models and human patients suggests that small vessel stiffening is an early and perhaps initiating event in experimental PH and PAH.3 In hypoxia-driven experimental PH, lysyl oxidase induced crosslinking of collagen occurring within minutes to hours, and inhibition of lysyl oxidases is sufficient to prevent disease onset.40 Stiffer substrates induce metabolic reprogramming in native endothelial and smooth muscle cells, facilitating their transition to the proliferative and synthetic phenotype that predominates during active vascular remodeling.4,30 It is possible that PACs may be the cell type mediating this transition by helping to directly modulate vascular stiffness.
5-HT2B has long been recognized as a significant contributor to PAH pathogenesis, and more recently as an indispensable mediator of the hematopoietic contribution to disease.16,17 We have previously shown that pharmacologic blockade of 5-HT2B is effective in preventing familial experimental PH using a genetic mouse model and that 5-HT2B antagonism significantly normalizes the expression of genes regulating cytoskeletal maintenance and contractility in the lung.25 Our results in the current study suggest that this effect may be secondary to the recruitment of PACs following hypoxic vascular injury. 5-HT2B antagonism exerted similar effects on the PAH phenotype to PAC ablation, effectively normalizing RVSP and metrics of pulmonary vessel remodeling (including vascular stiffness). 5-HT2B antagonism also altered the fraction of CD31+CD11b- BM-derived cells in circulation and in lung tissue during experimental PH, a cell population with enriched proangiogenic potential and previously reported to be reduced in the peripheral blood of 5-HT2B knockout mice.17,41,42 This dual effect suggests that the 5-HT2B antagonist may be exerting its effects in the BM compartment by preventing the proliferation or differentiation of PACs from a precursor population. The results from our GO enrichment analysis complement these findings, indicating a significant alteration in genes regulating both cell differentiation and proliferation in BM- and peripheral blood-isolated PACs. This hypothesis is consistent with the previously published data showing 5-HT2B promotes myeloerythroid differentiation potential in mouse and human hematopoietic stem cells.17 Our in vitro data further support this hypothesis by showing PACs enhance PMVEC injury response, while simultaneous antagonism of 5-HT2B fails to inhibit this effect, suggesting that in vivo observations are secondary to alterations in PAC number or function prior to their recruitment to the lungs.
Also consistent with previous studies was our observation that CD31+ BM-derived cells accumulate in the walls of muscularized arterioles during experimental PH.10,14,43 We observed that vessels with at least one associated BM-derived CD31+ cell were significantly stiffer in both normoxic and hypertensive animals. We observed a similar correlation in PAH patient lungs, suggesting that BM-derived PACs may also mediate small vessel stiffening and remodeling in human illness. However, these results should be interpreted cautiously, as the limited number of cell surface markers used for characterization and inherent heterogeneity of the cell population in question may confound the analysis, and it is unclear if these cells are the same population as those ablated in our mouse model.
In mice 5-HT2B antagonism reduced both the localization of CD31+ BM-derived cells to the walls of remodeling arterioles and the stiffness of vessels with associated CD31+ BM-derived cells. Surprisingly, in the absence of hypoxic vascular injury we found increased localization of these cells secondary to 5-HT2B antagonist treatment, despite the absence of observable changes in the total fraction of these cells as measured by flow cytometry. Despite this apparent increase in localization during normoxia, the normalization of vessel wall stiffness suggests that 5-HT2B antagonism also exerts direct, functional effects on this cell population, impairing their ability to induce vascular remodeling. Our GO enrichment results support this conclusion by showing alterations in cytokine production and signaling pathways secondary to 5-HT2B antagonism, the hypothesized mechanism whereby PACs contribute to vessel remodeling.15
There are several important limitations to our study that must be addressed. Firstly, our utilization of transgenic tools necessitated a reliance on the mouse model of endovascular injury, rather than the rat model (which produces a more robust phenotype). The modest phenotype observed in mice makes subtler changes more difficult to detect and does not reproduce all aspects of human disease, and findings must be cautiously interpreted. Additionally, our reliance on systemic inhibition of the 5-HT2B receptor precludes a more precise understanding of its role in mediating BM-derived PAC recruitment and function. 5-HT2B is expressed in both BM and native pulmonary vascular endothelium,16,17 and it is unclear if the observed effects on PAC recruitment and function were downstream of some other signaling event. Development of an animal model allowing targeted genetic ablation of the 5-HT2B receptor from PACs would more precisely elucidate its role in their biology and contribution to illness. This being said, our in vitro data suggest that 5-HT2B antagonism does not directly alter the interaction between PACs and injured pulmonary microvascular endothelium, nor does it promote alterations in endothelium itself in response to vascular injury. Therefore, it is reasonable to conclude that 5-HT2B signaling inhibits PAC function or reduces PAC number prior to their recruitment to the lungs.
The stiffening and muscularization of peripheral arterioles is a complex process involving numerous cell types. Exactly how BM-derived PACs promote and maintain this process remains unclear. While it is unlikely that these cells themselves proliferate and contribute to vessel muscularization, it is possible that they may in some fashion signal to native endovascular cells promoting the process of vessel remodeling. Our in vitro data show that PACs directly enhance endothelial cell spreading, migration, and barrier quality, the latter of which is also a function of cell proliferation. These effects are observed in the context of vascular injury (consistent with our animal model) and could represent a pathologically-enhanced injury response mechanism, whereby endothelial cells respond inappropriately to noxious stimuli.
In the present study we have illustrated that PACs are indispensable for both the development and maintenance of the experimental PH phenotype and strongly correlate with the presence of stiffened and muscularized pulmonary arterioles. By implicating the 5-HT2B receptor as a critical mediator of the recruitment and function of these cells during hypoxic vascular injury, we have further defined the function of 5-HT2B signaling in experimental PH. This discovery provides additional impetus to pursue pharmacologic targeting of 5-HT2B as a potential therapy for PAH, and encourages further exploration of PAC function in vascular remodeling in the hopes of identifying novel molecular mediators of illness.
Supplementary Material
Novelty and Significance
What Is Known?
Pulmonary hypertension (PH) is a deadly, incurable disease in which the pulmonary vasculature is slowly obliterated through a process of active endovascular remodeling.
Bone marrow-derived proangiogenic cells (PAC) are a heterogeneous subpopulation of myeloid derived cells previously observed in the walls of remodeling vessels in human and experimental PH.
Intact signaling from the serotonin 2B receptor (5-HT2B) in bone marrow cells is necessary for the development of experimental PH, and pharmacologic inhibition of 5-HT2B reduces the number of PACs in the peripheral blood of mice.
What New Information Does This Article Contribute?
Using a transgenic chimeric animal model in which we selectively destroy PACs, we show that PACs directly contribute to experimental PH in mice.
We also show that 5-HT2B inhibition prevents experimental PH, reduces the number of PACs in the lungs of mice with experimental PH, and alters the function of PACs in vivo.
PH is a deadly illness with no cure and a poorly understood pathogenesis. Bone marrow-derived PACs are correlated with disease severity and arteriole remodeling in PH, but their exact function (for good or ill) and the signaling mechanisms regulating their contribution to illness remains poorly understood. By selectively destroying PACs from the bone marrow of mice, we prevent and reverse existing experimental PH induced by a combination of endovascular injury and hypoxia. We also identify the 5-HT2B receptor as a mediator of PAC function, and illustrate that endovascular injury is necessary for PAC contributions to disease in vivo and in vitro. This study is the first to provide a causal (not correlative) link between PACs and PH, and further delineates a role for 5-HT2B in PH pathogenesis. In doing so, we provide further impetus to pursue 5-HT2B as a target for PH treatment and further clarify the mechanisms governing PH pathogenesis.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Matthew Bersi for his contributions to ECIS data analysis and Dr. Muthian Gladson for assistance with immunohistochemistry data acquisition.
SOURCES OF FUNDING
This work was supported by the National Institutes of Health (R35-HL135790, R01-HL115103, F30-HL126280) and American Heart Association (15PRE23260021).
Nonstandard Abbreviations and Acronyms:
- αSMA
Alpha smooth muscle actin
- AFM
Atomic force microscopy
- BM
Bone marrow
- DMSO
Dimethyl sulfoxide
- ECIS
Electric cell-substrate impedance sensing
- GO
Gene ontology
- IP
Intraperitoneal
- PBMC
Peripheral blood mononucleocytes
- PAC
Proangiogenic cell
- PAH
Pulmonary arterial hypertension
- PH
Pulmonary hypertension
- PMVEC
Pulmonary microvascular endothelial cell
- RBC
Red blood cell
- RVSP
Right ventricular systolic pressure
- 5-HT2B
Serotonin 2B receptor
- SCL
Stem cell leukemia
- VEGFR2
Vascular endothelial growth factor receptor 2
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Sutendra G, Michelakis ED. Pulmonary arterial hypertension: challenges in translational research and a vision for change. Science Translational Medicine. 2013;5:208sr5. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. The Journal of Clinical Investigation. 2012;122:4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu F, Haeger CM, Dieffenbach PB, Sicard D, Chrobak I, Coronata AMF, Velandia MMS, Vitali S, Colas RA, Norris PC, Marinković A, Liu X, Ma J, Rose CD, Lee S-J, Comhair SAA, Erzurum SC, McDonald JD, Serhan CN, Walsh SR, Tschumperlin DJ, Fredenburgh LE. Distal vessel stiffening is an early and pivotal mechanobiological regulator of vascular remodeling and pulmonary hypertension. JCI Insight. 2016;1:e86987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang Y-Y, Rehman S, Sugahara M, Qi Z, Gorcsan J, Vargas SO, Saggar RR, Saggar RR, Wallace WD, Ross DJ, Haley KJ, Waxman AB, Parikh VN, De Marco T, Hsue PY, Morris A, Simon MA, Norris KA, Gaggioli C, Loscalzo J, Fessel J, Chan SY. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. Journal of Clinical Investigation. 2016;126:3313–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan L, Chen X, Talati M, Nunley BW, Gladson S, Blackwell T, Cogan J, Austin E, Wheeler F, Loyd J, West J, Hamid R. Bone Marrow–derived Cells Contribute to the Pathogenesis of Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine. 2016;193:898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asosingh K, Farha S, Lichtin A, Graham B, George D, Aldred M, Hazen SL, Loyd J, Tuder R, Erzurum SC. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood. 2012;120:1218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rose JA, Erzurum S, Asosingh K. Biology and flow cytometry of proangiogenic hematopoietic progenitors cells. Cytometry. Part A : the Journal of the International Society for Analytical Cytology. 2015;87:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, Tang WHW, Loyd JE, Theil K, Tubbs R, Hsi E, Lichtin A, Erzurum SC. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei L, Zhang B, Cao W, Xing H, Yu X, Zhu D. Inhibition of CXCL12/CXCR4 suppresses pulmonary arterial smooth muscle cell proliferation and cell cycle progression via PI3K/Akt pathway under hypoxia. Journal of Receptor and Signal Transduction Research. 2014:1–11. [DOI] [PubMed] [Google Scholar]
- 10.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LSG, Marchesan D, Yang J, Suntharalingam J, Soon E, Exley A, Stewart S, Hecker M, Zhu Z, Gehling U, Seeger W, Pepke-Zaba J, Morrell NW. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine. 2009;180:780–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foris V, Kovacs G, Marsh LM, Bálint Z, Tötsch M, Avian A, Douschan P, Ghanim B, Klepetko W, Olschewski A, Olschewski H. CD133+ cells in pulmonary arterial hypertension. European Respiratory Journal. 2016;48:459–469. [DOI] [PubMed] [Google Scholar]
- 12.Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian M, Renaud JF, Simonneau G, Lombet A, Humbert M. Targeting of c-kit+ haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. European Respiratory Journal. 2011;37:1392–1399. [DOI] [PubMed] [Google Scholar]
- 13.Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, Huertas A, Hammad H, Lambrecht B, Simonneau G, Launay J-M, Cohen-Kaminsky S, Humbert M. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine. 2011;184:116–23. [DOI] [PubMed] [Google Scholar]
- 14.Satoh K, Kagaya Y, Nakano M, Ito Y, Ohta J, Tada H, Karibe A, Minegishi N, Suzuki N, Yamamoto M, Ono M, Watanabe J, Shirato K, Ishii N, Sugamura K, Shimokawa H. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation. 2006;113:1442–50. [DOI] [PubMed] [Google Scholar]
- 15.Lanzola E, Farha S, Erzurum SC, Asosingh K. Bone marrow-derived vascular modulatory cells in pulmonary arterial hypertension. Pulmonary Circulation. 2013;3:781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Launay J-M, Hervé P, Peoc’h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, Maroteaux L. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nature Medicine. 2002;8:1129–1135. [DOI] [PubMed] [Google Scholar]
- 17.Launay J-M, Hervé P, Callebert J, Mallat Z, Collet C, Doly S, Belmer A, Diaz SL, Hatia S, Côté F, Humbert M, Maroteaux L. Serotonin 5-HT2B receptors are required for bone-marrow contribution to pulmonary arterial hypertension. Blood. 2012;119:1772–1780. [DOI] [PubMed] [Google Scholar]
- 18.Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, Nilsen M, Loughlin L, Thomas M, MacLean MR. Sex-Dependent Influence of Endogenous Estrogen in Pulmonary Hypertension. American Journal of Respiratory and Critical Care Medicine. 2014;190:456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Göthert JR, Gustin SE, van Eekelen JAM, Schmidt U, Hall MA, Jane SM, Green AR, Göttgens B, Izon DJ, Begley CG. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–77. [DOI] [PubMed] [Google Scholar]
- 20.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez-Arroyo JG, Voelkel NF, Bogaard HJ. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulmonary Circulation. 2012;2:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, Kourembanas S. The Sugen 5416/Hypoxia Mouse Model of Pulmonary Hypertension Revisited: Long-Term Follow-Up. Pulmonary Circulation. 2014;4:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W-C, Park S- H, Hoffman C, Philip C, Robinson L, West J, Grunig G. Right ventricular systolic pressure measurements in combination with harvest of lung and immune tissue samples in mice. Journal of Visualized Experiments : JoVE. 2013:e50023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sewell-Loftin M-K, Brown CB, Baldwin HS, Merryman WD. A novel technique for quantifying mouse heart valve leaflet stiffness with atomic force microscopy. The Journal of Heart Valve Disease. 2012;21:513–20. [PMC free article] [PubMed] [Google Scholar]
- 25.West JD, Carrier EJ, Bloodworth NC, Schroer AK, Chen P, Ryzhova LM, Gladson S, Shay S, Hutcheson JD, Merryman WD. Serotonin 2B Receptor Antagonism Prevents Heritable Pulmonary Arterial Hypertension. PloS One. 2016;11:e0148657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. [DOI] [PubMed] [Google Scholar]
- 28.Gene Ontology Consortium: going forward. Nucleic Acids Research. 2015;43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Research. 2013;41:W77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang Y-Y, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Black SM, Fratz S, Fineman JR, Vargas SO, Haley KJ, Waxman AB, Chau BN, Fredenburgh LE, Chan SY. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Reports. 2015;13:1016–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123:1986–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long L, Ormiston ML, Yang X, Southwood M, Gräf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nature Medicine. 2015;21:777–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majka SM, Skokan M, Wheeler L, Harral J, Gladson S, Burnham E, Loyd JE, Stenmark KR, Varella-Garcia M, West J. Evidence for cell fusion is absent in vascular lesions associated with pulmonary arterial hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2008;295:L1028–L1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smadja DM, Mauge L, Sanchez O, Silvestre J-S, Guerin C, Godier A, Henno P, Gaussem P, Israel-Biet D. Distinct patterns of circulating endothelial cells in pulmonary hypertension. European Respiratory Journal. 2010;36:1284–1293. [DOI] [PubMed] [Google Scholar]
- 35.JunHui Z, XingXiang W, GuoSheng F, YunPeng S, FuRong Z, JunZhu C. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respiratory Medicine. 2008;102:1073–1079. [DOI] [PubMed] [Google Scholar]
- 36.Smadja DM, Gaussem P, Mauge L, Israel-Biet D, Dignat-George F, Peyrard S, Agnoletti G, Vouhe PR, Bonnet D, Levy M. Circulating Endothelial Cells: A New Candidate Biomarker of Irreversible Pulmonary Hypertension Secondary to Congenital Heart Disease. Circulation. 2009;119:374–381. [DOI] [PubMed] [Google Scholar]
- 37.Young KC, Torres E, Hatzistergos KE, Hehre D, Suguihara C, Hare JM. Inhibition of the SDF-1/CXCR4 Axis Attenuates Neonatal Hypoxia-Induced Pulmonary Hypertension. Circulation Research. 2009;104:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold MET, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. Journal of Experimental Medicine. 2006;203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed A a, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D, Farkas D, Conrad DH, Nicolls MR, Voelkel NF. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nave AH, Mi ikova I, Niess G, Steenbock H, Reichenberger F, Talavera ML, Veit F, Herold S, Mayer K, Vadasz I, Weissmann N, Seeger W, Brinckmann J, Morty RE. Lysyl Oxidases Play a Causal Role in Vascular Remodeling in Clinical and Experimental Pulmonary Arterial Hypertension. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34:1446–1458. [DOI] [PubMed] [Google Scholar]
- 41.Wara AK, Croce K, Foo S, Sun X, Icli B, Tesmenitsky Y, Esen F, Rosenzweig A, Feinberg MW. Bone marrow-derived CMPs and GMPs represent highly functional proangiogenic cells: implications for ischemic cardiovascular disease. Blood. 2011;118:6461–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Cho H-J, Kim S-W, Liu B, Choi YJ, Lee J, Sohn Y-D, Lee M-Y, Houge MA, Yoon Y. CD31+ cells represent highly angiogenic and vasculogenic cells in bone marrow: novel role of nonendothelial CD31+ cells in neovascularization and their therapeutic effects on ischemic vascular disease. Circulation Research. 2010;107:602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, Farver C, Comhair SAA, Xu W, Licina L, Huang L, Anand-Apte B, Yoder MC, Tuder RM, Erzurum SC. Circulating Angiogenic Precursors in Idiopathic Pulmonary Arterial Hypertension. The American Journal of Pathology. 2008;172:615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.