

Abstract
Transforming growth factor (TGF)-α1 (encoded by TGFB1) is the prototypic member of the TGF-β family of 33 proteins that orchestrate embryogenesis, development and tissue homeostasis1,2. Following its discovery3, enormous interest and numerous controversies have emerged about the role of TGF-β in coordinating the balance of pro- and anti-oncogenic properties4,5, pro- and anti-inflammatory effects6, or pro- and anti-fibrinogenic characteristics7. Here we describe three individuals from two pedigrees with biallelic loss-of-function mutations in the TGFB1 gene who presented with severe infantile inflammatory bowel disease (IBD) and central nervous system (CNS) disease associated with epilepsy, brain atrophy and posterior leukoencephalopathy. The proteins encoded by the mutated TGFB1 alleles were characterized by impaired secretion, function or stability of the TGF-β1-LAP complex, which is suggestive of perturbed bioavailability of TGF-β1. Our study shows that TGF-β1 has a critical and nonredundant role in the development and homeostasis of intestinal immunity and the CNS in humans.
TGF-β1 is translated as a precursor protein, which consists of an N-terminal signal peptide, the latency-associated peptide (LAP) and the C-terminal mature growth factor (TGF-β1). After proteolytic cleavage, LAP and TGF-β1 form the noncovalent small latent complex (SLC)8. The stabilization, secretion, deposition in the extracellular matrix and activation of SLCs are regulated by covalent association with latent TGF-β-binding proteins (LTBPs), resulting in formation of large latent complexes (LLCs)9. Multiple factors are known to control the release of active TGF-β1, for example, proteases, reactive oxygen species and integrins. Active TGF-β1 binds to a heterotetrameric transmembrane complex composed of TGF-β receptor type 1 (TGFBR1) and TGFBR2, which results in the phosphorylation of signal-transducing SMAD molecules and transcription of target genes10.
Dysfunction of TGF-β1 signaling has been implicated in several human diseases, including cancer, cardiovascular diseases, fibrosis, atherosclerosis and developmental defects1. Heterozygous gain-of-function mutations in TGFB1 are associated with Camurati-Engelmann disease (CED), which is characterized by osteosclerotic lesions in the long bones and skull11. Increased TGF-β1-mediated signaling due to mutations in TGFBR1 and TGFBR2 has been documented in patients with Loeys–Dietz syndrome, which is characterized by connective tissue disorders and arterial aneurysms12. Here we report that biallelic loss-of-function mutations in TGFB1 result in very early-onset IBD and CNS dysfunction.
Patient 1 (also referred to as P1 or A.II-1), who was born to non-consanguineous parents from Malaysia (Fig. 1a), presented in the first months of life with bloody diarrhea and subsequently developed severe perianal abscesses and fistulae. Colonoscopy confirmed the diagnosis of chronic active pancolitis associated with diffuse erythema, superficial ulcerations and multiple pseudopolyps (Fig. 1b). Histology showed crypt abscesses and inflammatory infiltrations of the epithelium with mucosal ulcerations (Fig. 1b). In addition, P1 showed eosinophilic esophagitis and esophageal candidiasis. He was refractory to nutrition regimens and intensive conventional anti-inflammatory therapy, including mesalazine, steroids, azathioprine, methotrexate, infliximab, adalimumab and tacrolimus. At the age of 4 years, a total colectomy with ileostomy was performed. P1 also showed global developmental delay associated with impaired speech and cognitive dysfunction. Generalized skeletal muscle atrophy and muscular hypotonia were present, but neither pyramidal tract signs nor evidence for movement disorders were detected. Cranial magnetic resonance imaging (MRI) indicated global brain atrophy and posterior leukoencephalopathy (Fig. 1c). Electroencephalography (EEG) analysis showed a moderate global encephalopathic pattern lacking normal background activity and continuous mixed alpha and beta activity. No interictal epileptic discharges were recorded. Oligoclonal IgG bands and increased levels of IL-1β and IL-8 in the cerebrospinal fluid were suggestive of inflammatory processes.
Fig. 1|. Identification of a biallelic TGFB1 mutation in patient 1 with very early-onset inflammatory bowel disease and global neurological defects.
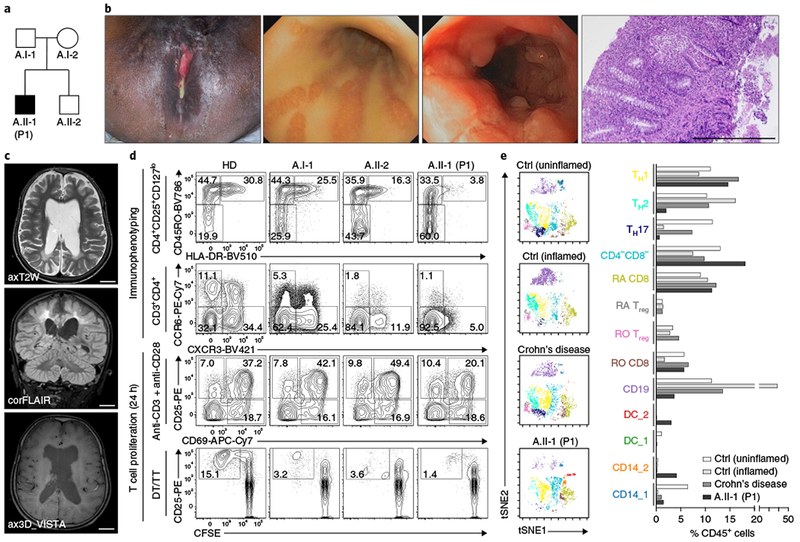
a, Pedigree of patient 1 (P1, A.II-1) born to a nonconsanguineous Malaysian family (family A). b, Images showing severe perianal disease with purulent discharge (leftmost image) in P1, massive suppuration (second image from left) and superficial ulcerations and multiple pseudopolyps (third image from left) in the colon, as revealed by endoscopy, and crypt abscesses and inflammatory infiltrations of the epithelium with mucosal ulceration, as documented by histology (rightmost image; scale bar, 500 μm). c, MRI images of the brain indicating global brain atrophy and posterior periventricular leukencephalopathy in P1. Scale bars, 2 cm. d, Representative immunophenotypic analysis of CD4+CD25+CD127lo Treg cells, CCR6−CXCR3+ TH1 and CCR6+CXCR3− TH 17 T cells and assessment of CD3+CD4+ T cell activation and proliferation in response to anti-CD3/anti-CD28 or specific antigens (DT/TT, diphtheria and tetanus toxoid). Immunophenotyping was performed in two independent experiments. e, CyTOF analysis of the composition of lamina propria mononuclear cells derived from two control (Ctrl) patients without IBD (uninflamed and inflamed), a patient with Crohn’s disease and P1. Left, clusters of CD45+ viSNE27 plots were manually gated and color-coded for various populations on the basis of similar marker expression. Right, graphical representations depicting percentages of the indicated immune cell populations. CyTOF analysis was performed once owing to limited availability of patient material.
P1 had a history of recurrent upper and lower respiratory tract infections and chronic cytomegalovirus (CMV) retinitis. Laboratory studies showed leukocytosis, thrombocytosis and hypochromic anemia. Serum levels of IgG (4,044 mg/dl; normal: 576–1,507) and IgE (2,665 IU/ml; normal: <90) were high, whereas IgA and IgM levels were within normal ranges. Immunophenotypic analysis of peripheral blood mononuclear cells (PBMCs) showed decreased proportions of activated memory regulatory T (Treg) cells, as well as CCR6−CXCR3+ T helper 1 (TH1) and CCR6+CXCR3− TH17 T cells (Fig. 1d). T cell activation following stimulation with anti-CD3 and anti-CD28, and T cell proliferation in response to specific antigens (diphtheria and tetanus toxoid), were reduced as compared to healthy donors and first-degree relatives (Fig. 1d). Mass cytometry (CyTOF) analysis of colonic lamina propria mononuclear cells showed a decreased frequency of CD45RO+ and CD45RA+FOXP3+, CCR6−CXCR3−, CCR6+CXCR3− and CD103+ T cells as compared to patients without IBD (control patients without (uninflamed) or with (inflamed) inflammation) and a patient with Crohn’s disease (Fig. 1e and Supplementary Fig. 1). TGF-β1 exerts both stimulatory and inhibitory immunomodulatory effects6; however, we cannot exclude the possibility that some of the clinical and immunological features we observed could have been influenced by infections or drug-associated immunosuppression. P1 is currently in stable clinical condition at the age of 11 years.
To elucidate the genetic etiology, we performed whole-exome sequencing and identified a compound heterozygous mutation in TGFB1 (ENST00000221930.5) (c.[328C>T];[1159T>C], p.[Arg110Cys];[Cys387Arg]). Segregation of the sequence variant with the disease phenotype was confirmed by Sanger sequencing, which indicated that the heterozygous mutation located in the sequence encoding the LAP domain was inherited from the mother, whereas the mutation in the sequence encoding the mature TGF-β1 domain was inherited from the father (Fig. 2a and Supplementary Fig. 2). In contrast to CED11, radiographs of P1 were consistent with osteopenia (data not shown).
Fig. 2|. Effects of TGFB1 mutations on the biosynthesis and bioavailability of TGF-β1.
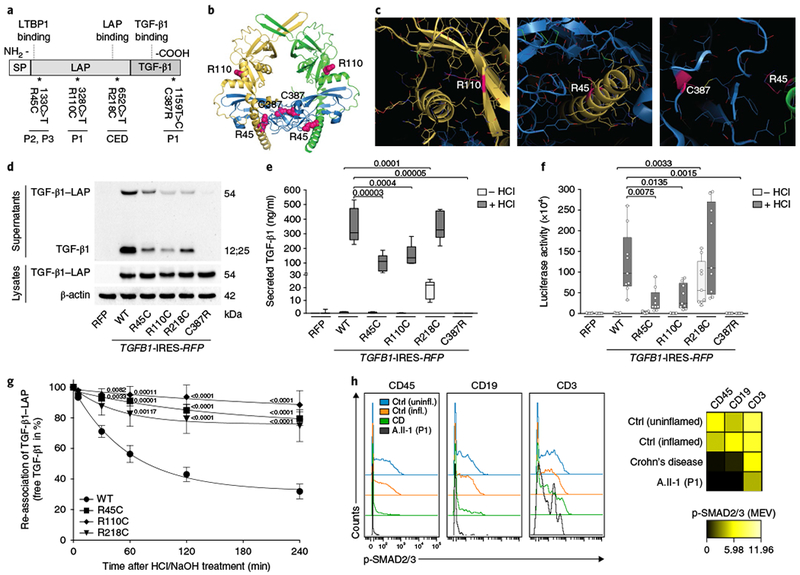
a, Schematic illustration showing the distribution of identified alterations relative to the TGF-β1 structure depicting the N-terminal signal peptide (SP), the latency-associated peptide (LAP) and the C-terminal mature growth factor (TGF-β1). The mutations identified in patients, including a previously described gain-of-function mutation in TGFB1 causing CED11, are depicted for the DNA and protein sequences. b, Structural visualization of the identified TGF-β1 alterations using the crystal structure of latent TGF-β1 (PDB accession 3RJR)13. The structure is depicted as a ribbon model with highlighted secondary structure. Color code: yellow and green, pro-domain dimer; blue, TGF-β1 dimer; magenta spheres, mutation sites. c, Detailed views of the altered sites (stick model, with altered side chains highlighted in magenta). d, Representative immunoblot (n = 3) for TGF-β1 levels in lysates and conditioned medium from HEK293T cells that stably overexpressed WT and mutant TGF-β1 variants. RFP, red fluorescent protein; R45C, TGF-β1 Arg45Cys; R110C, TGF-β1 Arg110Cys; R218C, TGF-β1 Arg218Cys; C387R, TGF-β1 Cys387Arg. e, ELISA determining the TGF-β1 levels in conditioned medium from HEK293T cells that stably overexpressed WT and mutant TGF-β1 variants± HCl treatment for the release of mature growth factor from latent complexes. Samples of ten biologically independent cell culture experiments were analyzed. Box-and-whisker plots: center line, median; box limits, upper and lower quartiles; whiskers, quartile range. P values (indicated in the graphs) were calculated using a two-tailed unpaired t test with Welch’s correction. f, SMAD-dependent luciferase reporter assays in HEK293T cells that were stimulated with conditioned medium from cells stably overexpressing WT or mutant TGF-β1. Samples of nine biologically independent cell culture experiments were analyzed. Box-and-whisker plots: center line, median; box limits, upper and lower quartiles; whiskers, quartile range; individual data points as overlays. P values were calculated using a two-tailed unpaired t test with Welch’s correction. g, Analysis of the re-association capacity of mutant TGF-β1 variants and LAP over time. Data shown represent the means± s.e.m. of 11 independent cell culture experiments. P values were calculated using two-way repeated-measures ANOVA with Dunnett’s correction for multiple comparisons. h, CyTOF analysis of SMAD2/3 phosphorylation (p-SMAD2/3) in lamina propria mononuclear cells derived from patients without IBD (uninflamed, inflamed), a patient with Crohn’s disease (CD) and P1 (A.II-1). Histogram plots of baseline p-SMAD2/3 levels (left) and median expression values (MEV) of p-SMAD2/3 (right) are shown for the indicated hematopoietic cell (CD45+, CD19+ or CD3+) populations. CyTOF analysis was performed once owing to limited availability of patient material.
We also identified a homozygous missense mutation in TGFB1 (c.133C>T, p.Arg45Cys) in a second pedigree with two affected individuals (patients 2 and 3) born to consanguineous parents from Pakistan (Supplementary Fig. 3a,b). The p.Arg45Cys substitution is located in the LAP domain of the pre-pro-TGF-β1 precursor (Fig. 2a). Patient 2 (also referred to as P2 or B.II-1) had a small head circumference (2.5th centile) and bloody diarrhea at 3 months of age. Neurological development reached a plateau at 9–10 months of age and subsequently regressed. At 19 months of age, P2 developed refractory complex partial and myoclonic seizures. EEG analysis showed a pathological pattern similar to that for hypsarrhythmia. Cerebral MRI showed volume loss, cortical atrophy and thinning of the corpus callosum. Between 19 and 20 months of age, P2 started to lose her abilities to communicate and became increasingly spastic. Despite optimization of her diet via a nasogastric tube, P2 failed to thrive and died at the age of 25 months while hospitalized for suspected septicemia.
Patient 3 (also referred to as P3 or B.II-4) had microcephaly at birth (head circumference <2.5th centile). At 3 months of age, he presented with failure to thrive and bloody diarrhea. Colonoscopy and histology showed chronic active inflammation with abscesses and crypt branching (Supplementary Fig. 3c). Psychomotor regression, muscular hypotonia and hyperreflexia were diagnosed at 12 months of age. At 25 months of age, P3 had complex partial seizures and a hypsarrhythmia-like EEG pattern. Cerebral MRI examination showed gross cortical atrophy, delayed myelination and marked thinning of the corpus callosum (Supplementary Fig. 3d). He developed spasticity and contractures and lost visual and social contact, as well as the ability to perform voluntary movements. Moreover, P3 had fungal dermatitis, scabies skin infection and an episode of severe varicella infection. An influenza A viral infection triggered renal and subsequent multi-organ failure, causing his death at the age of 39 months. Immunophenotyping of PBMCs showed normal numbers and distributions for T cells, B cells and NK cells but a reduced ability of CD4+ and CD8+ T cells to proliferate after stimulation with anti-CD3 (data not shown).
We studied the structural consequences of the amino acid substitutions in the mutant TGF-β1 proteins by analyzing the crystal structure of latent TGF-β1 (Protein Data Bank (PDB) accession 3RJR)13. The substitutions we identified may perturb the interaction of TGF-β1 with the pro-domain or with the TGF-β1 cysteine knot (Fig. 2b). Arg110 maps to a region denoted as the ‘fastener, which locks the interaction between the pro-domain and the growth factor domain (Fig. 2c, left). The β-sheet harboring Arg110 forms a ‘super β-sheet’ with the growth factor domain. Thus, both a proper stable interaction, as well as an integrin-binding-mediated release of the growth factor domain, could be affected. Arg45 maps to the interface between the TGF-β1 dimer and the pro-domain, suggesting that the substitution p.Arg45Cys alters the interaction between these two functional elements (Fig. 2c, middle). Correct folding requires the presence of the pro-domain and may therefore be affected by the p.Arg45Cys substitution. We predict that the p.Cys387Arg alteration perturbs the correct formation of the cysteine knot in the growth factor domain, as the substitution will result in an unpaired cysteine and will prevent formation of the disulfide bond to Cys322 (Fig. 2c, right). Furthermore, the unpaired Cys322 might also affect correct formation of the other disulfide bonds by disulfide scrambling. Thus, p.Cys387Arg presumably affects folding or stability of the TGF-β1 growth factor domain dimer.
To validate the predicted consequences of the mutations on the biosynthesis and function of the TGF-β1-LAP complex, we used heterologous HEK293T cells that were transduced with lentiviral particles encoding wild-type (WT) TGF-β1 or mutant TGF-β1 variants. The CED-causing TGF-β1 variant Arg218Cys was used as a control11. Immunoblotting of cell lysates confirmed stable expression of TGF-β1-LAP homodimers in cells that had been transduced with lentivirus expressing either WT or mutant TGF-β1 (Fig. 2d). Latent and mature TGF-β1 could be detected in conditioned medium from HEK293T cells overexpressing the TGF-β1 Arg45Cys, Arg110Cys or Arg218Cys variant. The TGF-β1 variants Arg45Cys and Arg110Cys showed reduced levels of secreted TGF-β1. In contrast, the Cys387Arg mutant could not be detected in supernatants, suggesting that abrogation of the disulfide bond prevented proper assembly and secretion. Correspondingly, ELISAs showed that (i) only the Arg218Cys variant was detected in the mature form under cell culture conditions (without acidification with HCl), indicating a gain of function for this mutant11, (ii) mature TGF-β1 was released from the SLC after HCl treatment in the cases of the Arg45Cys and Arg110Cys variants, although at lower levels than for WT TGF-β1, and (iii) secretion of the Cys387Arg variant was completely abrogated (Fig. 2e). To analyze downstream effects on TGF-β1-mediated signaling, we examined conditioned medium in HEK293T cells expressing a SMAD-sensitive luciferase reporter. Cells expressing the Arg45Cys and Arg110Cys variants exhibited reduced luciferase activity as compared to the activity with WT TGF-β1, whereas no activity could be detected for the Cys387Arg mutant (Fig. 2f). To assess the stability of WT and mutant SLCs, we monitored re-association of LAP and TGF-β1 over time. The SLCs in supernatants from HEK293T cells were destabilized by HCl treatment and subsequently allowed to reassemble after neutralization with NaOH. In contrast to WT TGF-β1, all of the mutants showed compromised re-association capacity, suggesting reduced stability of the SLC (Fig. 2g). To assess TGF-β1 signaling in mucosal tissue, we performed CyTOF analysis on colonic biopsies from P1. As compared to patients without IBD (uninflamed and inflamed controls), the mean expression values of phosphorylated SMAD2 and SMAD3 (p-SMAD2/3) were reduced in lamina propria mononuclear CD45+, CD19+ and CD3+ cells from P1 (Fig. 2h), whereas TGF-β1-independent STAT6 phosphorylation was normal (Supplementary Fig. 4). Reduced levels of phosphorylated SMAD2/3 were also seen in CD45+ and CD19+ cells from an unrelated patient with Crohn’s disease, confirming impaired SMAD3 activity in the mucosal tissue of patients with IBD14. Taken together, all of the newly identified mutated TGFB1 alleles seem to have deleterious consequences with respect to TGF-β1 complex formation, secretion and/or bioavailability for signal transduction, as well as direct effects on downstream SMAD2/3 signaling in vivo.
The role of TGF-β signaling in human disease has been controversial. Although increased TGF-β activity has been linked to cancer, fibrosis and progressive diaphyseal dysplasia, decreased TGF-β activity has been associated with early tumorigenesis, vascular dysplasia, developmental defects and atherosclerosis1. Our studies highlight a nonredundant role of TGF-β1 in controlling intestinal immune homeostasis and CNS function, whereas other organ systems apparently were not affected. These findings are reminiscent of those in patients with IL-10 or IL-10 receptor deficiency who present predominantly with infantile IBD15,16, even though IL-10 is known to mediate pleiotropic stimulatory and suppressive functions in the immune system.
The role of TGF-β1 in immunity has previously been documented in experimental models. Mice that have a constitutive disruption of Tgfb117 or a T cell-specific deletion of Tgfb118 or that express dominant-negative TGFBR219 develop a lethal wasting syndrome, including severe colitis. In patients with Crohn’s disease, intestinal tissue or mucosal T cells are characterized by increased activation of SMAD7, an inhibitor of TGF-β1 signaling14. Treatment with SMAD7-specific antisense oligonucleotides holds promise to alleviate colitis in mice20 and in patients21 by restoring TGF-β1 signaling.
The role of TGF-β1 in the brain is less well understood. Brionne et al. have reported that lack of TGF-β1 expression in mice results in neuronal cell death and microgliosis22. Tissue-specific deletion of Tgfb1 in the mouse CNS prevents lethal hyperinflammation but leads to progressive defects in synaptic plasticity and loss of microglia23. Decreased plasma levels of TGF-β124 and reduced neuronal expression of TGFBR225 have been documented in patients with Alzheimer’s disease. Genetic polymorphisms altering TGF-β1 expression have been associated with increased risk for conversion of mild cognitive impairment in patients with Alzheimer’s disease26. Our studies suggest that TGF-β1 may have a neuroprotective role, but the mechanisms remain unknown.
Human TGF-β1 deficiency is a life-threatening disease, yet clinical management remains challenging. In view of the documented role of TGF-β1 in T cells, allogeneic hematopoietic stem cell transplantation might be considered to alleviate intestinal inflammation. We opted not to pursue this approach given the severe neurological comorbidities. Substitution with recombinant TGF-β1 may represent an alternative experimental approach, but currently no such product is available for therapeutic use, and controlling tissue- and context-specific bioavailability of TGF-β1 is challenging, in particular in the CNS.
In conclusion, our study demonstrates a nonredundant role of TGF-β1-mediated signaling for intestinal immune homeostasis and neurological development in humans.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available at https://doi.org/10.1038/s41588-018-0063-6.
Methods
Patients.
Patients were originally identified by the Departments of Pediatrics at the Faculty of Medicine, University Malaya, Kuala Lumpur, Malaysia (family A) or Oslo University Hospital, Norway (family B), and they were referred for further studies to the Dr. von Hauner Children’s Hospital at the Ludwig-Maximilians-Universität (LMU) München, Germany. Peripheral blood samples and biopsies from patients and their unaffected first-degree relatives and from healthy volunteers for genetic and functional experiments, as well as photographs of patients for publication, were obtained upon written consent. The investigation was performed in accordance with current ethical and legal frameworks, and the study protocols were approved by the Institutional Review Boards at the LMU (#66-14) and by the Health South-East Regional Ethics Committee, Norway.
Whole-exome sequencing.
Genomic DNA from patients and parents was isolated using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s instructions. After enrichment for all coding exons using the SureSelect Human All Exon Kit (Agilent Technologies), sequencing was performed on an Illumina Genome Analyzer II (family A) or Illumina HiSeq 2000 (family B). Short paired sequence reads were mapped to the human reference genome GRCh37 with BWA28. Genome Analysis Tool Kit (GATK)29 was used to analyze the WES data, and functional annotation was performed with snpEff30 and Variant Effect Predictor (VEP) using Ensembl31 release 85 (family A) or 71 (family B). WES data were filtered and analyzed using an in-house SQL database (family A) or FILTUS v.0.99–934 (family B)32. Rare variants were distinguished by incorporating frequency information from the 1000 Genomes Project33, NHLBI GO Exome Sequencing Project (ESP; http://evs.gs.washington.edu/EVS/) and/or ExAC34. Effects of filtered variants were predicted with a multitude of software, including snpEff30, VEP31, SIFT35 and PolyPhen-236. The remaining variants were compiled and filtered for rare homozygous and compound heterozygous mutations following a pattern of autosomal recessive inheritance.
DNA sequencing.
Genomic DNA from patients, parents and healthy siblings was isolated using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s instructions. Segregation of identified mutations in TGFB1 was confirmed in available family members in families A and B by DNA Sanger sequencing. Primer sequences are listed in Supplementary Table 1.
Sanger sequencing was done in house on a Hitachi 3130× genetic analyzer or by GATC Biotech, Konstanz, Germany. The sequence reads were analyzed using the DNASTAR Lasergene software.
Electroencephalography and magnetic resonance imaging.
24-channel EEG recordings using Xltek hardware and software equipment (Natus DBA, Excel-Tech Corp.) was performed using standard adjustments (0.5-Hz low-frequency filter, 70-Hz high-frequency filter, resistance 5–10 kΩ). MRI of the brain was obtained using a 3-Tesla high-resolution scanner (1.0- to 1.5-mm slices, T1 (longitudinal relaxation time) with and without gadolinium contrast enhancement, T2 (transverse relaxation time) and fluid attenuated inversion recovery techniques) in axial, sagittal and coronal planes (Philips Ingenia).
Structural analysis of TGF-β1 mutants.
Structural visualization and modeling of the amino acids encoded by the identified TGFB1 mutations was performed with PyMol (Schrödinger, LLC).
Construction of expression vectors, cell culture, transfection and lentiviral transduction.
Human WT TGFB1 was amplified from a Mammalian Gene Collection (MGC) sequence-verified cDNA clone (cat. no. MHS6278-202757887, accession: BC022242, Dharmacon GE Healthcare). Mutations in TGFB1 (encoding Arg45Cys, Arg110Cys, Arg218Cys or Cys387Arg) were introduced by site-directed PCR mutagenesis using corresponding primer pairs. WT and mutated TGFB1 cDNAs were cloned into the IRES-EGFP or IRES-RFP bicistronic lentiviral pRRL vectors.
Biochemical assays were performed on HEK293T cells (ATCC) that routinely tested negative for Mycoplasma contamination. For production of WT or mutant TGF-β1, HEK293T cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin and 200 mM l-glutamine (all from Gibco, Life Technologies) in a humidified incubator at 37°C with 5% CO2 and were transduced with lentiviruses according to previously published protocols37. Briefly, vesicular stomatitis virus G glycoprotein (VSV-g)-pseudotyped lentiviral particles were generated by transfection of HEK293T cells. Using polyethyleneimine (PEI; Polysciences) as a transfection agent, cells were incubated with 5 μg lentiviral vector, 12 μg pcDNA3.GP4xCTE (which expresses HIV-1 gag–pol), 5 μg pRSV-Rev and 1.5 μg pMD.G (which encodes VSV-g) in the presence of 25 μM chloroquine (Sigma) for 12 h. Supernatants containing viral particles were collected every 24 h for 72 h and concentrated by ultracentrifugation. Viral titration was performed on HEK293T cells and viral concentrations were determined by flow cytometry. Next, HEK293T cells (at 60–80% confluency) were transduced with lentiviral particles in the presence of polybrene (8 μg/ml) for 6–12 h. To establish stable cell lines, transduced cells were sorted based on EGFP or RFP expression using a BD FACSAria cell sorter (BD Bioscience).
FACS and immunophenotyping.
For immunophenotypic analysis, blood samples were washed with PBS and stained with monoclonal antibodies, as indicated in Supplementary Table 2. Red blood cells were lysed by 1× BD FACS Lysing Solution (BD Biosciences) according to the manufacturer’s instructions. The samples were acquired using a LSRFortessa Flow Cytometer (BD Bioscience), and data were analyzed using FlowJo Software (TreeStar). Gating strategies are shown in Supplementary Fig. 5.
CyTOF analysis.
Colonic tissue was digested overnight on a shaker at 37°C in complete RPMI medium (Gibco, Life Technologies) with 2 μl of collagenase and 2 μl of DNase per 10 ml of medium. Undigested material was filtered out using a 10-μm filter. Single cells were resuspended in CyTOF staining buffer, and 1 × 106 to 2 × 106 cells/sample were prepared for CyTOF analysis according to the Fluidigm protocol with minor modifications. Briefly, cells were stained with Rh103 as a viability dye, washed, blocked with Fc Block and incubated with the cocktail of metal-coupled antibodies specific for surface molecules for 30 min. Next, cells were fixed in 1.6% formaldehyde and treated with isopropanol for detection by the phospho-specific antibodies or were permeabilized with the FOXP3/Transcription Factor Staining Buffer Set (eBioscience) for staining with a cocktail of intracellular antibodies. Cells were then re-fixed in 1.6% formaldehyde and stained with Ir-DNA intercalator solution (Fluidigm). Finally, cells were resuspended in water containing a 1:10 dilution of EQ beads and run on a Helios CyTOF machine, Fluidigm, at the Harvard Medical School (HMS) CyTOF Core. Antibodies used for CyTOF analysis are summarized in Supplementary Table 3. Antibodies not purchased from Fluidigm were conjugated at the HMS CyTOF core. Data were analyzed using the Premium CyTOBANK cloud-based software. Gating strategies are shown in Supplementary Fig. 6.
Protein blot analysis and ELISA.
To study TGF-β1-LAP biosynthesis and secretion, cell lysates and supernatants of HEK293T cells overexpressing WT and mutant TGF-β1 were analyzed by immunoblotting and ELISA following standard protocols. Briefly, 0.5 × 106 HEK293T cells and their derivatives were cultured in 1 ml of FBS-containing DMEM. After incubation for 12 h, cell lysates or supernatants were fractionated under reducing conditions by SDS–PAGE. Proteins were blotted onto polyvinylidene difluoride membranes using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked in 5% skim milk before staining. Antibodies used for detection are indicated in Supplementary Table 4. Membranes were developed using a chemiluminescent substrate (Thermo Fisher Scientific). Images were captured using a ChemiDoc XRS+ System (Bio-Rad). Uncropped immunoblots are shown in Supplementary Fig. 7.
TGF-β1 levels in serum samples and cellular supernatants were measured by using the Human TGF-β1 ELISA DuoSet (DY240, R&D Systems) according to the manufacturer’s instructions. To release the mature TGF-β1 from latent complexes, conditioned medium was treated with 1 N HCl for 10 min, followed by neutralization with a solution containing 1.2 N NaOH and 0.5 M HEPES. Supernatants were analyzed in duplicate by using a Synergy H1 microplate reader (BioTek Instruments).
TGF-β1-sensitive firefly luciferase reporter assays.
A lentiviral TGF-β1-sensitive firefly luciferase reporter plasmid was designed by insertion of the SMAD response elements (CAGA)38 into the pGreenFire1-mCMV vector (#TR010PA-1-SBI, Biocat) between the EcoRI and SpeI restriction sites. HEK293T cells were transduced with the reporter system as described above.
HEK293T cells overexpressing WT or mutant TGF-β1 were plated at a density of 1 × 106 cells per well of a 12-well plate in 1 ml of serum-supplemented DMEM. After 4–6 h of incubation, cells were rinsed with PBS, and medium was replaced with 1 ml of serum-free minimal essential medium (MEM; Gibco, Thermo Fisher) to avoid the potential influence of TGF-β1 contained in FBS. Conditioned medium from cultured cells was harvested after 12 h. To measure TGF-β1-mediated SMAD signaling activity of the identified mutants, 0.5 × 106 HEK293T cells encoding the firefly luciferase reporter were plated in 0.5 ml of serum-supplemented DMEM in each well of a 48-well plate. After 4–6 h of incubation, cells were rinsed with PBS, and medium was replaced with 1 ml of serum-free MEM. Conditioned medium was added to the reporter cell line in both the native and HCl-activated forms. Stimulated reporter cells were incubated for 12 h at 37 °C and subsequently lysed and assayed for firefly luciferase activity using the Firefly and Renilla Dual Luciferase Assay Kit (#30005, Biotium, USA) according to the manufacturer’s instructions. Briefly, 45 μl of lysate from samples was transferred in duplicate to a 96-well luminometry plate (NUNC) and mixed with 80 μl of firefly working solution. Luminescence signals were measured for a period of 10 s.
TGF-β1-LAP re-association assays.
To examine the stability of latent complexes for the proteins encoded by the identified TGFB1 mutations, we assessed reassociation of TGF-β1 and LAP after complex disruption in a time-dependent manner, as described previously39. To release the mature TGF-β1 from latent complexes, conditioned medium from transduced HEK293T cells was acidified with 1 M HCl for 10 min at room temperature and neutralized with 1.2 M NaOH and 0.5 M HEPES. After neutralization, samples were incubated at 37 °C for 5, 30, 60, 120 and 240 min, and levels of free TGF-β1 were analyzed by ELISA. These were plotted by applying a one-phase exponential decay data transformation using GraphPad Prism software (GraphPad Software).
Statistical analysis.
Statistical evaluation of experimental data was performed using Prism version 6 (GraphPad Software). No method of randomization or blinding was used, and no samples were excluded from analysis. Data in Fig. 2e,f are reported as box-and-whisker plots, with the median (center line), upper and lower quartiles (box limits) and quartile range (whiskers) indicated. Data in Fig. 2g are means ± s.e.m. To analyze quantitative datasets, either a two-tailed unpaired t test with Welch’s correction to account for unequal variances (Fig. 2e,f) or two-way repeated-measures ANOVA with Dunnett’s correction for multiple comparisons (Fig. 2g) was performed. All tests were two-tailed, and P values <0.05 were considered to be statistically significant. Sample numbers are referred to as n unless indicated otherwise. Gaussian distribution of the data was confirmed by DAgostino and Pearson’s omnibus normality test. No statistical method was used to predetermine sample size for analyses.
Life Sciences Reporting Summary.
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability.
The identified TGFB1 mutations have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) with accessions SCV000678250 [c.328C>T], SCV000678251 [c.1159T>C] and SCV000622112 [c.133C>T]. Information on the raw whole-exome sequencing data supporting the findings of this study are available from the corresponding author upon request. These data will not be publicly available as they contain information that could compromise research participant privacy.
Supplementary Material
Acknowledgements
We are very grateful to our patients and their parents for allowing us to study their diseases. We thank the medical staff at the Dr. von Hauner Children’s Hospital, Oslo University Hospital and University Malaya Medical Center. In particular, we would like to acknowledge pathologist D. Klotz (Oslo University Hospital) for the histology of colonic biopsies. Whole-exome sequencing of family A was conducted at the Next-Generation Sequencing facility at the Dr. von Hauner Children’s Hospital under the supervision of M. Rohlfs. The sequencing service of family B was provided by the Norwegian Sequencing Centre, a national technology platform supported by the Functional Genomics and Infrastructure Programs of the Research Council of Norway and the Southeastern Regional Health Authorities, and the sequencing data of family B were analyzed by A. Holmgren. We acknowledge the assistance of the Flow Cytometry Core Facility at the Dr. von Hauner Children’s Hospital and of the Harvard Medical School CyTOF Core. Samples from the patient with CED were provided with support of the Oxford Gastrointestinal Illness Biobank and Biomedical Research Center Oxford. We gratefully acknowledge our bioinformatician, J. Puchalka, who died in a tragic accident during the course of the investigations. This work has been supported by The Leona M. and Harry B. Helmsley Charitable Trust, the Collaborative Research Consortium SFB1054 (DFG), PID-NET (BMBF), BioSysNet, the European Research Council, the Gottfried-Wilhelm-Leibniz Program (DFG), the DAAD network on ‘Rare Diseases and Personalized Therapies’, the German Center for Infection Research (DZIF) and the Care-for-Rare Foundation. W.S.L. was partly funded by University Malaya High Impact Research (UM.C/625/HIR/MOHE/CHAN/13/1). D.K. has been a scholar funded by the Else Kroner-Fresenius-Stiftung, the Daimler und Benz Stiftung and the Reinhard Frank-Stiftung.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41588-018-0063-6.
References
- 1.Blobe GC, Schiemann WP & Lodish HF Role of transforming growth factor-β in human disease. N. Engl. J. Med 342, 1350–1358 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Wu MY & Hill CS TGF-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 16, 329–343 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Derynck R et al. Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature 316, 701–705 (1985). [DOI] [PubMed] [Google Scholar]
- 4.Principe DR et al. TGF-β: duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst 106, djt369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silberstein GB & Daniel CW Reversible inhibition of mammary gland growth by transforming growth factor-β . Science 237, 291–293 (1987). [DOI] [PubMed] [Google Scholar]
- 6.Li MO, Wan YY, Sanjabi S, Robertson AK & Flavell RA Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol 24, 99–146 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Pohlers D et al. TGF-β and fibrosis in different organs—molecular pathway imprints. Biochim. Biophys. Acta 1792, 746–756 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Miyazono K, Hellman U, Wernstedt C & Heldin CH Latent highmolecular-weight complex of transforming growth factor-β1. Purification from human platelets and structural characterization. J. Biol. Chem 263, 6407–6415 (1988). [PubMed] [Google Scholar]
- 9.Rifkin DB Latent transforming growth factor-β (TGF-β )-binding proteins: orchestrators of TGF-β availability. J. Biol. Chem 280, 7409–7412 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Li MO & Flavell RA TGF-β: a master of all T cell trades. Cell 134, 392–404 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janssens K et al. Camurati–Engelmann disease: review of the clinical, radiological and molecular data of 24 families and implications for diagnosis and treatment. J. Med. Genet 43, 1–11 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loeys BL et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet 37, 275–281 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Shi M et al. Latent TGF-β structure and activation. Nature 474, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteleone G et al. Blocking SMAD7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Invest 108, 601–609 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glocker EO et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med 361, 2033–2045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotlarz D et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 143, 347–355 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Shull MM et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 359, 693–699 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MO, Wan YY & Flavell RA T cell–produced transforming growth factor-β1 controls T cell tolerance and regulates TH1- and TH17 cell differentiation. Immunity 26, 579–591 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Gorelik L & Flavell RA Abrogation of TGF-β signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 12, 171–181 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Boirivant M et al. Inhibition of SMAD7 with a specific antisense oligonucleotide facilitates TGF-β1-mediated suppression of colitis. Gastroenterology 131, 1786–1798 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Monteleone G et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med 372, 1104–1113 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Brionne TC, Tesseur I, Masliah E & Wyss-Coray T Loss of TGF-β1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 40, 1133–1145 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Koeglsperger T et al. Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-β1 in the CNS. Glia 61, 985–1002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Servi B, La Porta CA, Bontempelli M & Comolli R Decrease of TGF-β1 plasma levels and increase of nitric oxide synthase activity in leukocytes as potential biomarkers of Alzheimer’s disease. Exp. Gerontol 37, 813–821 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Tesseur I et al. Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Invest 116, 3060–3069 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arosio B et al. + 10 T/C polymorphisms in the gene of transforming growth factor-β1 are associated with neurodegeneration and its clinical evolution. Mech. Ageing Dev 128, 553–557 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Amir ED et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol 31, 545–552 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H & Durbin R Fast and accurate short-read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cingolani P et al. A program for annotating and predicting the effects of single-nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLaren W et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vigeland MD, Gjotterud KS & Selmer KK FILTUS: a desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics 32, 1592–1594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar P, Henikoff S & Ng PC Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Nat. Protoc 4, 1073–1081 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Adzhubei I, Jordan DM & Sunyaev SR Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet 76, 7–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kotlarz D et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J. Exp. Med 210, 433–443 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dennler S et al. Direct binding of SMAD3 and SMAD4 to critical TGF-β-inducible elements in the promoter of human plasminogen activator inhibitor type 1 gene. EMBO J 17, 3091–3100 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walton KL et al. Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-β1 complex. J. Biol. Chem 285, 17029–17037 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The identified TGFB1 mutations have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) with accessions SCV000678250 [c.328C>T], SCV000678251 [c.1159T>C] and SCV000622112 [c.133C>T]. Information on the raw whole-exome sequencing data supporting the findings of this study are available from the corresponding author upon request. These data will not be publicly available as they contain information that could compromise research participant privacy.