

Abstract
Duclauxins are dimeric and heptacyclic fungal polyketides significant bioactivities. We characterized the cascade of redox transformations in the biosynthesis of duclauxin pathway from Talaromyces stipitatus. The redox reaction sequence is initiated by a cupin family dioxygenase DuxM that performs an oxidative cleavage of the peri-fused tricyclic phenalenone and affords a transient hemiketal-oxaphenalenone intermediate. Additional redox enzymes then morph the oxaphenoalenone into either an anhydride or a dihydrocoumarin-containing monomeric building block that are found in dimeric duxlauxins. Oxidative coupling between the monomers to form the initial C-C bond was shown to be catalyzed a P450 monooxygenase, although the enzyme responsible for the second C-C bond formation was not found in the pathway. Collectively, the number and variety of redox enzymes used in the duclauxin pathway showcases Nature’s strategy to generate structural complexity.
INTRODUCTION
Fungal aromatic polyketides comprise a large group of natural products with diverse biological activities, ranging from the beneficial antifungal griseofulvin to the carcinogenic aflatoxin.1 The core of aromatic polyketides are biosynthesized by nonreducing polyketide synthases (NR-PKSs).2 Significant structural diversity and complexity in these polyketides are then introduced by an assortment of enzymes that catalyze redox reactions on the aromatic precursors,3,4 some of which have been used as useful biocatalysts.5 A spectacular example of complexity generation from a relatively simple precursor can be found in the duclauxins family of natural products, which are dimeric oxaphenalenones containing the 6/6/6/5/6/6/6 ring system (Figures 1 and S1).6,7 Duclauxins have diverse biological activities.7–9 For example, duclauxin (1) and cryptoclauxin (2) exhibit tumor suppression by inhibiting RNA synthesis or mitochondrial ATP synthesis,10, 11 and talaromycesone B (3) is an inhibitor of acetylcholinesterase.12 Structural variations among duclauxins are attributed to different oxaphenalenone monomers and connectivities between them (Figures 1 and S1). All duclauxins contain at least one unit of the dihydrocoumarin benzo[de]isochromen-1(3H)-one. Homodimerization of two benzo[de]isochromen-1(3H)-one molecules can afford 1 or bacillisporin (4), while heterodimerization between different oxaphenalenones can lead to 2, 3 and bacillisporin F (5). Dimerization regiospecificities can also be different, as 1-3 are dimerized through C8-C7’ and C8’-C9a bonds to form a bicyclo[3.2.1] octane ring, while dechlorogilmaniellin (6) are dimerized between C8-C9’ and C9a-C9a’ bonds (Figure 1).
Figure 1.
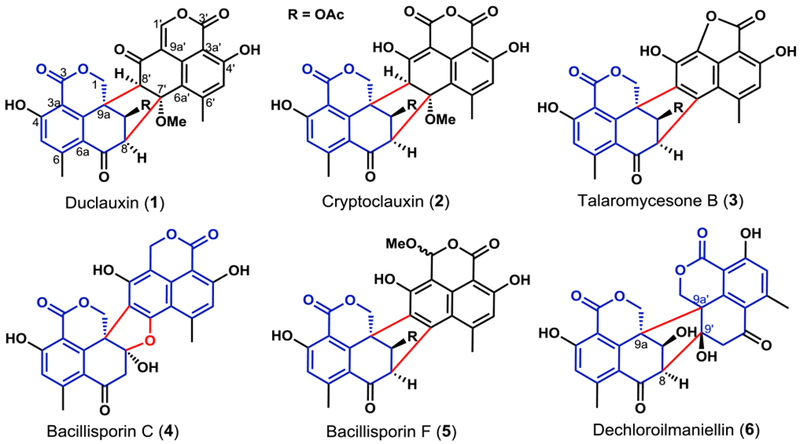
Duclauxins are dimeric oxaphenalenones. The common monomer benzo[de]isochromen-1(3H)-one core is in blue and the C-C bonds between the monomers are in red.
To date, no total synthesis of any of the duclauxins has been reported, while the biosynthetic pathways of these molecules have also remained unknown. The oxaphenalenone monomers in duclauxins, including benzo[de]isochromen-1(3H)-one, were proposed to be derived from extensive redox morphing of the aromatic polyketide phenalenone 7 (Figures S2).6,7 Our recent work showed that 7 is synthesized by an NR-PKS PhnA and a flavin-dependent monooxygenase (FMO) PhnB from Penicillium herquei (Figure 2A).13 From 7, the proposed oxidative modification steps include extrusion of the oxygen-substituted carbon C2 to yield the di-ketone 9, followed by oxygen insertion to generate lamellicolic anhydride (10) (Figure 2A),6,7 which is a natural product isolated from Verticillium lamellicola.14 10 is then proposed to be asymmetrically reduced to the dihydrocoumarin-containing SF226 (11)15, a compound reported from Penicillium purpurogenum, followed by dimerization, methylation and acylation to yield 12. An additional dehydrogenation across C1’-C9a’ of 12 affords 1 (Figure 2A)6. However, the exact sequence of redox modifications and enzymatic basis of these proposed reactions have not been resolved. Here we identified the dux biosynthetic pathway of 1 from Talaromyces stipitatus ATCC 10500. We reconstituted the enzymes that catalyze the cascade of redox reactions transforming 7 into 11, and demonstrated 10 is in fact a branched pathway product. A P450 enzyme catalyzing the first dimerization C-C bond formation is also identified.
Figure 2.
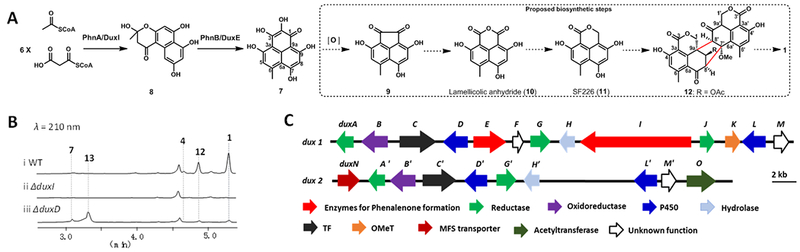
Gene clusters and proposed biosynthetic pathways of duclauxins. A) Proposed biosynthetic pathways of oxaphenalenone 11 and dimerization to 1; B) LC-MS analysis of organic extracts obtained from the wild type strain and mutants (ΔduxI and ΔduxD) of T. stipitatus; C) Schematic representation of the two separate gene clusters (dux1 and dux2) from T. stipitatus.
RESULTS
Identification of Two Separate Gene Clusters dux1 and dux2.
We grew T. stipitatus in MEPA medium and observed the production of three known phenalenone derived compounds 1 16, 7 13, and 4 17, as well as a new duclauxin derivative 12 (Figure 2B, trace i). Subsequent isolation and NMR characterization confirmed the structure of 12 to match the proposed dimerization product as shown in Figure 2A (Table S4 and Figures S25–S28). Based on the hypothesis that the monomeric units, such as 11, are synthesized from 7 (Figure 2A), we searched the genome sequence of T. stipitatus for biosynthetic gene clusters containing homologs to PhnA (NR-PKS) and PhnB (FMO).13 BLAST search led to a candidate cluster (dux1), which consisted of 13 open reading frames containing a NR-PKS encoding gene (duxI, Figure 2C). DuxI (237 kDa) has an overall amino acid sequence identity of 49% compared to PhnA in P. herquei. A gene encoding an FMO (duxE) with 68% amino acid sequence identity to PhnB (Figure 2C) was also found in the cluster. Deletion of duxI completely abolished the production of 1, 4, 7, and 12, thereby establishing the link between the dux1 cluster and the biosynthesis of 1 (Figure 2B, trace ii, Figure S3). We also expressed duxI in Saccharomyces cerevisiae, which produced 8 after 72 h of culture (Figure S4), further confirming DuxI shares the same function with PhnA from the herqueinone pathway. We then supplemented 4, 7 and 12 to the ΔduxI strain to determine if these are on pathway products (Figures S5). Whereas feeding either 7 or 12 could restore the biosynthesis of 1, feeding 4 did not. Therefore, 4 represents a shunt product, while 7 and 12 are on pathway as proposed in Figure 2A.
To map the steps between 7 and 12, we turned our attention to the assortment of redox enzymes present in the gene cluster. These include predicted P450 monooxygenases (DuxD and DuxL), oxidoreductase (DuxB), NAD(P)H-dependent reductases (DuxA, DuxG and DuxJ) and a cupin-family oxygenase (DuxM). Other genes in the cluster include transcription factor (DuxC), hydrolase (DuxH) and O-methyltransferase (DuxK), etc (Figure 2C and Table S2). We generated individual knockout mutants of dux1 genes and characterized the products (Figures S6–S7). Deletions of duxH (hydrolase), duxJ (reductase) and duxM (cupin) resulted in the complete abolishment of 1, without accumulation of any new products (Figure S7). In contrast, deletions of duxA (reductase) and duxL (P450) had no effects on the production of 1 (Figure S7). New peaks were only identified in the deletion of duxD (P450) and duxG (reductase), while small amount of 1 remained (Figures 2B and S7). This raised the possibility of enzymes encoding redundant genes in the genome. Further BLAST search for the homologs of proteins in the dux1 cluster resulted in the discovery of a separate cluster encoding DuxA’, DuxB’, DuxC’, DuxD’, DuxG’, DuxH’, DuxL’ and DuxM’ all of which show over 70% sequence identity to the corresponding proteins in the first cluster. Accordingly, we named this cluster dux2 (Figure 2B and Table S2), which might account for the residue biosynthesis of 1 in ΔduxA, ΔduxD, ΔduxG, and ΔduxL. A more detailed bioinformatic analysis of dux2 revealed the presence of two more adjacent genes that might be involved in duclauxin 1 biosynthesis: duxN encoding a transporter and duxO encoding an acetyltransferase. RT-PCR analysis showed transcriptions of the dux1 and dux2 genes are synchronized with the production of 1 in T. stipitatus cultured in MEPA medium (Figure S8).
Modification of Phenalenone 7 by DuxM and DuxJ.
Because of the partial duplication of the dux genes in the native host, we decided to reconstitute the pathway using Saccharomyces cerevisiae. Starting with a S. cerevisiae strain expressing PhnA and PhnB, which produces 7 at a moderate level, additional dux genes were introduced. When the cupin-family homolog DuxM was coexpressed, the emergence of a new compound 10 with m/z 259 [M-H]− was observed (Figure 3A, trace ii). The product was purified and characterized as the known lamellicolic anhydride (10) by MS and NMR (Table S5 and Figures S18, and S29–S32). DuxM is therefore involved in removing the C2 carbon atom from 7. However, feeding of 10 to the ΔduxI mutant T. stipitatus strain did not restore biosynthesis of 1 (Figure S5), suggesting 10 is a shunt product rather than an on-pathway intermediate of 1.
Figure 3.
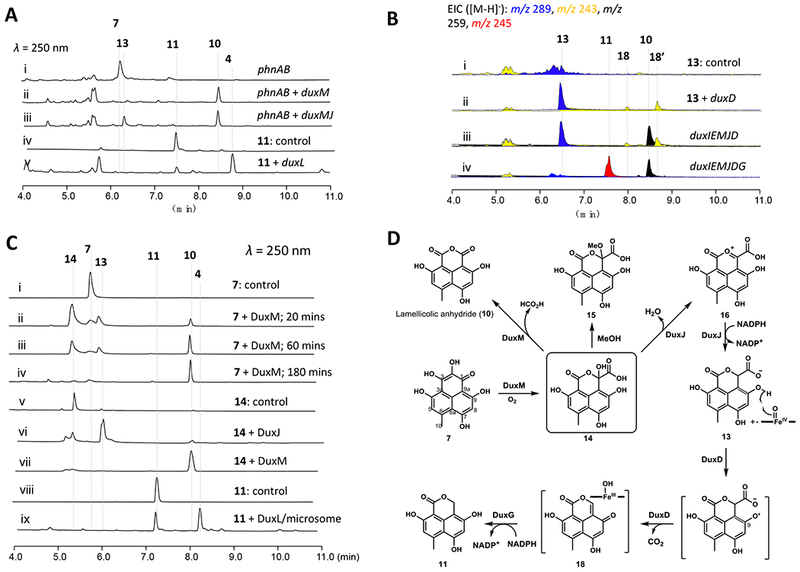
Biosynthesis of oxaphenalenone 11. A) Product profiles of S. cerevisiae transformed with the combinations of phnA, phnB and dux genes and chemical complementation studies; B) Product profiles of A. nidulans transformed with combinations of dux genes and chemical complementation studies; C) In vitro reactions of dux enzymes; D) Biosynthetic pathways of 10 and 11 via a common precursor 7.
Additional genes were then coexpressed with phnA, phnB, and duxM in yeast, followed by metabolite analysis to identify possible functions. With the addition of duxJ, encoding a maleylacetate reductase homolog, we observed the production of a new compound 13 (m/z 289 [M-H]−, Figures 3A, trace iii, and S22) together with 10. 13 was purified and characterized (Table S6 and Figures S30–S33) to be the C1-carboxylated version of the anticipated monomer 11. Unlike 10, 13 can readily restore the biosynthesis of 12 and 1 when supplied to the ΔduxI mutant (Figure S5), establishing its identity as an on-pathway intermediate to duclauxins. Coexpressing PhnA, PhnB and DuxJ (without DuxM) in yeast, however, did not lead to formation of 13, suggesting DuxJ must function downstream of DuxM. Furthermore, feeding of 10 to yeast expressing DuxJ alone also did not lead to transformation to 13, which points to the possibility that an intermediate generated by DuxM prior to 10 may be the substrate of DuxJ (Figure 3D).
To identify the proposed intermediate of DuxM reaction, we expressed and purified DuxM and DuxJ from Escherichia coli BL21 (DE3) and S. cerevisiae BJ5456 (Figure S9), respectively. The substrate 7 was added to the enzymes together with the required cofactors. Incubation of 7 with DuxM alone led to the full conversion to 10 in 3 hrs (Figure 3C, trace iv). At earlier time points a new peak 14 (m/z 305 [M-H]−) that gradually converted to 10 (Figure 3C, traces ii-iv) was detected. 14 was purified from a 300-mL in vitro reaction using a low-temperature isolation procedure (see the methods for details). 14 was characterized to contain a carboxylic acid group at C1, which was confirmed by a long-range J4 HMBC correlations from H8 (δH 6.45) to C1 (δC 100.3) (Table S7 and Figures S37–S40). Upon dissolving 14 in methanol for 30 min, a new product (15) (Figures 3D and S10), with molecular weight of the methoxy derivative of 14 was formed. A key HMBC correlation from the 1-O-methyl group (δH 3.08) to C1 (δC 104.3) in 15 was observed (Figure S24), confirming the hemiketal structure at C1 of 14 (referred to as the hemiketal-oxaphenalenone).
The production of 1 and 12 were restored when 14 was fed to the ΔduxI mutant (Figure S5). Incubation of 14 directly with DuxM lead to complete conversion to 10 as expected (Figure 3C, trace vii). Adding 14 to DuxJ in the presence of NADPH led to the complete conversion to 13 (Figure 3C, trace vi), thereby confirming 14 is the proposed intermediate between DuxM and DuxJ. DuxJ therefore catalyzes both the dehydration of 14 and the reduction of the resulting oxonium 16 to yield 13 (Figure 3D). Similar cascaded dehydration and reduction was reported in the penigequinolone pathway and was shown to be catalyzed by a short-chain dehydrogenase/reductase (SDR)-like reductase, PenD.18 Based on these results, the functions of DuxM and DuxJ are shown in Figure 3D: i) DuxM can oxidatively rearrange the northern ring in 7 to yield the hemiketal lactone 14; ii) in the absence of the reductase DuxJ, 14 can be oxidatively decarboxylated, losing C2 as CO2 to yield 10. 14 therefore represents a bifurcating point for the redox reactions in this pathway. While 10 is not an on-pathway intermediate to 1 in T. stipitatus, it is a building block for other duclauxins such as 2; and iii) DuxJ is a bifunctional reductase that catalyzes the reduction of 14 to 16, followed by reduction of 16 to 13.
Proposed Mechanism of DuxM:
DuxM is annotated as a cupin family protein (Figures S11 and S12), which is characterized by a conserved six stranded β-barrel fold (or jelly roll motif).19,20 At the center of the conserved β-barrel domain are conserved residues (His, Glu) that can coordinate to different metal ions.19 ICP-MS measurement of purified, FLAG-tagged DuxM indicates coordination of iron (70%) and zinc (30%, Table S9). Structural analysis by Phyre2 showed that DuxM resembles quercetinase from Aspergillus japonicus (Accession no. Q7SIC2, Figure S12A), which catalyzes the oxidative cleavage of quercetin.21,22 Sequence alignment of DuxM with fungal quercetinases indicates conserved residues at ligand-binding sites, including three histidines and one glutamic acid residue (Figure S12B). Quercetinase was also characterized as a cambialistic enzyme, capable of using Fe2+, Ni2+, Cu2+, or Co2+ as metal cofactors. It was concluded that identity of the metal ion is not essential for the catalytic activity of quercetinase; and catalysis may involve direct electron transfer from the metal-bond flavonolate anion to O2 without a valence change of the metal to activate O2.19,23–25
Based on these observations, we proposed a possible mechanism for DuxM-catalyzed reactions (Figure 4). The metal ion can serve as the coordinating counterion to stabilize developed negative charges during the reaction. The active site MII-binding glutamate, where M represents the metal ion, is proposed to act as a general base to generate the substrate anion (I) at C2 that coordinates to metal. Then, activation of O2 occurs when dioxygen coordinates to MII (M represents the metal ion) and substrate dissociation promotes an electron transfer to yield the superoxide anion radical (II). The superoxide anion can then perform a nucleophilic attack at the C3 position, leading to the formation of an end-on η1 alkylperoxo intermediate (III), followed by the subsequent nucleophilic attack to form the C2-C3 peroxide complex (IV). A final step involving the simultaneous cleavage of O-O and C2-C3 bonds results in the formation of the dicarboxylate 17. This compound can then undergo general acid catalyzed lactone formation to arrive at the hemiketal 14. 14 can diffuse out and reenter the DuxM active site as shown in the in vitro assays. In the absence of DuxJ catalyzed reduction, 14 can be decarboxylated to 10, via either a concerted or step-wise mechanism, aided by coordination of the carboxylate group to the metal ligand, to generate formate during the process (Figure S13). Density functional theory (DFT) calculations based on a model system showed the plausibility of these processes (Figure S14).
Figure 4.
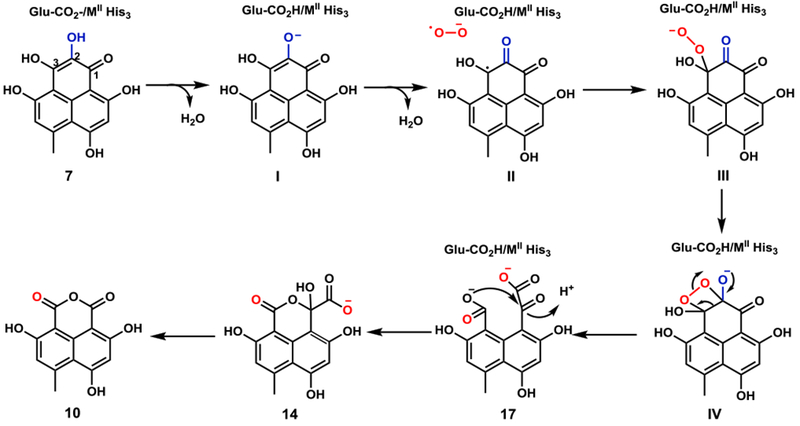
Proposed catalytic mechanism for the cupin family protein DuxM. Substrate and O2 binding modes to the metal ion (MII) are omitted due to the lack of available structural data.
Formation of the duclauxin monomeric building block 11.
Having identified 13 is an on pathway intermediate in the biosynthesis of 1, we reexamined the genetic knock out strains to see if any strains accumulated 13, which can provide clues to the next enzyme in the pathway en route to the building block 11. Inactivation of the P450 oxygenase DuxD indeed accumulated 13 (Figure 2B, trace iii). Expression of DuxD in yeast failed to show any activity, which led us to reconstitute the enzyme in an engineered A. nidulans A1145 ΔSTΔEM heterologous host.26 When 13 was fed to A. nidulans expressing duxD, two new peaks (18 and 18’) with MW = 244 were observed (Figure 3B, traces i and ii). We also transformed all five genes duxD, duxE, duxI, duxJ, and duxM into the A. nidulans host, which led to the de novo production of 10, 13, 18, and 18’ (Figure 3B, trace iii). Attempts to purify this pair of interchangeable compounds failed. Based on the molecular weight difference between 18/18’ and 13, we propose they are keto-enol tautomers of the C1 decarboxylated product (Figure 3D). The mechanism of this oxidative decarboxylation by P450 resembles the reactions catalyzed by OleT in terminal alkene synthesis 27 and HemQ in heme b maturation.28 In both cases, decarboxylation generates an olefin between the α, and β carbons. We propose the mechanism as shown in Figures 3D and S15, during which the first electron in 13 is transferred from the C9-phenol to Compound I and generate a radical that can be resonance delocalized at C9a. As demonstrated in the OleT mechanistic study 27, two routes may be proposed to result in loss of CO2: i) decarboxylation is coupled to the transfer of the C9a radical to Compound II; or ii) Compound II abstracts an electron from the carboxylic acid to generate an unstable substrate diradical followed by olefin formation (Figure S15).
Since 18 is an α–β enoylreduction away from the monomer 11, we further extended the pathway through coexpression of reductase candidates in the dux pathway. A likely candidate enzyme is DuxG, which is a 42 kDa protein that shows 47% amino acid sequence identity to isoflavone reductase that reduces 4-keto-pyran ring to 4-keto-dihyropyran ring in polyaromatic plant metabolite 2’-hydroxyformononetin.29 A. nidulans expressing DuxI, DuxE, DuxM, DuxJ, and DuxD, indeed led to the emergence of a new compound in the extract (Figure 3B, trace iv) (m/z 245 [M-H]−). The new product 11 was purified and characterized to be identical to SF226 (Figure 2A) based on NMR analysis (Table S8 and Figures S41–S44). The key HMBC correlations from 1-CH2 to the C3, C9 and C9a established that 11 was saturated at C3. With 11 now in hand, we fed 11 to the ΔduxI blocked mutant and restored biosynthesis of 1 and 12 (Figure S5), confirming the original proposal that it is a monomeric building block of duclauxins.
Collectively, we showed that four redox enzymes belonging to different superfamilies (DuxM: cupin; DuxJ: maleylacetate reductase; DuxD: P450; and DuxG: isoflavone reductase) transform the known precursor 7 into one of the duclauxin building block 11, consuming two molecules of molecular oxygen and two molecules of NAPDH (three if counting regeneration of the DuxD Fe(II)-heme). The net result is the conversion of a perinaphthenone structure into a benzo[de]isochromen, extruding one of the carbon as CO2 along the way.
DuxL Mediated Homodimeric Coupling and Late Stage Modifications.
To examine the dimerization steps starting with 11, the remaining uncharacterized dux genes were individually cloned for expression in yeast, accompanied by feeding of 11. In the yeast strain expressing the remaining P450 monooxygenase DuxL, 90% of 11 was converted to a new product with a MW of 490 (m/z 489 [M-H]−; Figure 3A, trace v). The product was purified and confirmed to be bacillisporin C (4), a known product isolated from T. stipitatus (Figure 2B, trace i).17 Microsomal fractions containing the overexpressed DuxL were purified from a 3-day culture of the yeast expression strain. In the in vitro assay where 0.5 mM 11 and 2 mM NADPH were incubated with microsomes (20 mg/mL total protein concentration), a clear product that corresponds to 4 emerged, with 60% conversion (Figure 3C, trace ix). The structure of 4 contains the C9a-C8’ bond present in most duclauxins (Figures 1 and S1), implying that DuxL catalyzes this homodimerization step. However, the second C8-C7’ found in 1 is not formed in 4, thereby suggesting that DuxL is not involved. A proposed pathway involving radical-radical coupling between two monomers is shown in Figure 5. We propose that DuxL catalyzes the hydrogen abstraction of 11 at the C9-phenol to generate the radical 19. Delocalization of the radical via resonance can yield the two carbon radicals 20 and 21, which can undergo coupling in the active site of DuxL to form the C9a-C8’ carbon bond in 22, generating two vicinal stereocenters. In the absence of the second C-C bond forming enzyme, tautomerization and aromatization of the northern fragment of 22 can take place readily to give 22’. This can then result in nonenzymatic conversion to 4 through nucleophilic addition of the C7’-phenol to the C9-carbonyl. As confirmed previously in this work, 4 cannot be converted to 1 and represents a shunt product (Figure S5).
Figure 5.
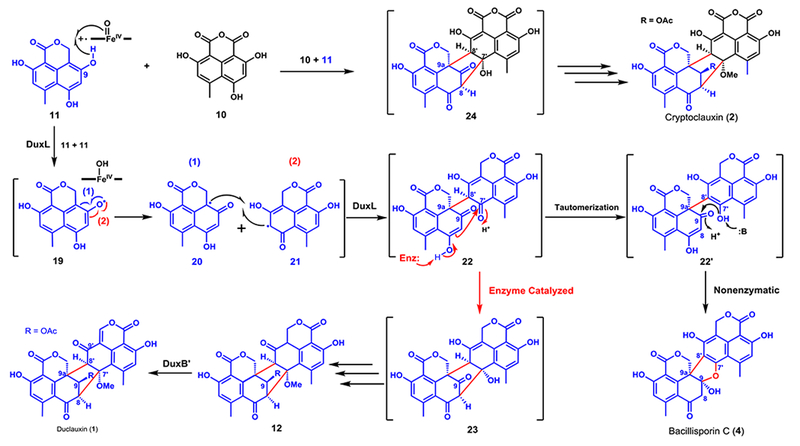
Proposed divergent biosynthetic pathways of duclauxin (1), cryptoclauxin (2), and bacillisporin C (4).
To go from 22 to 1, we propose that the intermediate 23 can be synthesized from 22 via an aldol-like addition with C8 serving as the nucleophile to attack C7’ prior to aromatization (Figure 5). Unfortunately, further efforts to identify an enzyme that can catalyze this reaction have not been successful. We have performed bioconversion of 11 in yeast strains that co-expressed DuxL with the rest of the uncharacterized enzymes in the cluster (DuxA, B, G, H, and K) individually. However, no new product could be generated other than 4 (Figure S20). Once 23 is formed, we anticipate sequential tailoring reactions, including 7’-O-methylation, 9-keto reduction and 9-O-acetylation would generate 12 (Figure 5). The final step in the biosynthesis of 1 is dehydrogenation of C1’-C9a’ in 12. Our yeast expression and bioconversion experiment showed that DuxB’ from the dux2 cluster could convert 12 to 1, albeit with low conversion (2%, Figure S16). In contrast, no modification of 12 by DuxB was observed despite ~85% sequence identity between the two enzymes.
DISCUSSION
In this study, we characterized the redox steps required for the biosynthesis of oxaphenalenone 11, which includes oxidative ring cleavage, deoxygenation, oxidative decarboxylation, and enoylreduction. DuxM is the key enzyme during the conversion of the single substrate 7 into different oxaphenalenone building blocks, which can then oxidatively dimerized to duclauxins. Among these transformations, the DuxM catalyzed formation of lamellicolic anhydride (10) is particularly interesting. It was thought to be synthesized from a di-keto precursor 9, via an unprecedented “Baeyer-Villiger”-like oxygen insertion, which can disrupt two adjacent keto groups by addition of an oxygen atom (Figures 2A and S2)6,7,30. This study, using a combination of genetic knockout, heterologous reconstitution and biochemical assays, demonstrated that the formation of a hemiketal-oxaphenalenone 14, rather than a di-keto product, is the key step en route from 7 to 10. Therefore, DuxM not only provides a starting intermediate 14 for structural diversification of duclauxins, but also plays a second role and converts 14 to 10, a building block for 2 and other duclauxin. 7
Oxidative morphing of aromatic scaffolds is common in natural products3,4 with one notable example being post-PKS tailoring modification in chartreusin biosynthesis. Chartreusin is a DNA-binding antitumor agent produced by Streptomyces chartreusis 31 and a cupin-like dioxygenase ChaP is proposed to mediate the last step, an oxygenative removal of one of the carbonyl carbons in the cyclohexadienone by successive reactions including hydroxylation, ring opening and lactonization as CO2 is released to yield an isocoumarin moiety. However, the real function of ChaP was not characterized and according to the DuxM logic illustrated in this study, ChaP might only catalyze the formation of a hemiketal intermediate, while additional enzymes are required to finish the remaining biosynthetic steps, including reduction and decarboxylation.
Cytochrome P450 oxygenases that catalyze regio- and stereoselective intermolecular couplings have been reported, exemplified by KtnC,32 DtpC,33 CnsC,34 ClaM,35 Bmp7,36 and CYP158,37 which play key roles in dimerization of napthoquinones, diketopiperazine alkaloids, indole alkaloids, nataloe-emodins, bromophenols, bromopyrrole, and flaviolins, respectively, whereas CYP121 catalyzed the intramolecular C-C bond couplings between the tyrosyl side chains of the cyclodipeptide cyclo(L-Tyr-L-Tyr).38 CnsC was demonstrated to show promiscuous substrate recognition and incorporates ω-N-methyltryptamine and tryptophol in place of tryptamine to synthesize isocommunesin compounds.34 Similarly, we proposed that DuxL also possesses some flexible substrate selectivity and is capable of catalyzing heterodimerization by using both of 10 and 11 to generate the intermediate 24 for the biosynthesis of 2 (Figure 5). However, the LC-MS trace of the in vitro reaction containing 10 and 11 using DuxL microsomal fractions is identical to that containing only 11, which suggests that the formation of C8-C9a’ linkage in the biosynthesis of 2 might be mediated by a different P450 other than DuxL in the producing host.
In summary, this study characterized oxaphenalenone construction and duclauxin biosynthesis. The study was initiated by the identification of two separate gene clusters dux1 and dux2 for the biosynthesis of 1. Seven genes are present in both clusters as two copies, which might provide additional transcripts to complete the biosynthesis of 1. We clarified the enzymes and reactions involved in the oxidative steps during the synthesis and the relationships between oxaphenalenone 10, 11, 13 and 14. The catalytic mechanism for the key enzyme DuxM was studied based on the ICP-MS data and DFT calculations. Further downstream tailoring modifications, including C9a-C8’ homodimerization and C1’-C9a’ desaturation were also characterized. Based on this study, the divergent nature of the biosynthesis of 1, 2 and 4 was proposed (Figures 3D, 4, and 5).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (1R35GM118056 to Y.T.), the NSF (CHE-1361104 to K.N.H.), as well as the NSFC (No. 81522043), CAMS Initiative for Innovative Medicine (2017-I2M-4-004), and the Thousand Young Talents Program of China. T.Z was supported by China Postdoctoral Science Foundation (2016M591119). Y.H. and J.M.B. are supported by the NIH predoctoral training grant T32 GM067555. M.G.-B. thanks the Ramón Areces Foundation for a Postdoctoral Fellowship. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575).
Footnotes
Supporting Information
Experiment details and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Present Address
State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, 100101 Beijing, China.
The authors declare no competing financial interest.
REFERENCES
- (1).Chooi YH; Tang YJ Org. Chem 2012, 77, 9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zhou H; Li Y; Tang Y Nat. Prod. Rep 2010, 27, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cox R Nat. Prod. Rep 2014, 31, 1405. [DOI] [PubMed] [Google Scholar]
- (4).Tang MC; Zou Y; Watanabe K; Walsh CT; Tang Y Chem. Rev 2017, 117, 5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baker Dockrey SA; Lukowski AL; Becker MR; Narayan AR H. Nat. Chem 2018, 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cao P; Yang J; Miao CP; Yan Y; Ma YT; Li XN; Zhao LX; Huang SX Org. Lett 2015, 17, 1146. [DOI] [PubMed] [Google Scholar]
- (7).Elsebai MF; Saleem M; Tejesvi MV; Kajula M; Mattila S; Mehiri M; Turpeinen A; Pirttila AM Nat. Prod. Rep 2014, 31, 628. [DOI] [PubMed] [Google Scholar]
- (8).Dethoup T; Manoch L; Kijjoa A; Nascimento MS; Puaparoj P; Silva AM; Eaton G; Herz W Planta Med 2006, 72, 957. [DOI] [PubMed] [Google Scholar]
- (9).Kawai K; Shiojiri H; Nakamaru T; Nozawa Y; Sugie S; Mori H; Kato T; Ogihara Y Cell Biol. Toxicol 1985, 1, 1. [DOI] [PubMed] [Google Scholar]
- (10).Kawai K; Nozawa Y; Ito T; Yamanaka N Res. Commun. Chem. Pathol. Pharmacol 1982, 36, 429. [PubMed] [Google Scholar]
- (11).Fuska J; Kuhr I; Nemec P; Fuskova AJ Antibiot. (Tokyo) 1974, 27, 123. [DOI] [PubMed] [Google Scholar]
- (12).Wu B; Ohlendorf B; Oesker V; Wiese J; Malien S; Schmaljohann R; Imhoff JF Mar. Biotechnol. (NY) 2015, 17, 110. [DOI] [PubMed] [Google Scholar]
- (13).Gao SS; Duan A; Xu W; Yu P; Hang L; Houk KN; Tang YJ Am. Chem. Soc 2016, 138, 4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McCorkindale NJ; Mcritchie A; Hutchinson SAJ Chem. Soc., Chem. Commun 1973, 4, 108. [Google Scholar]
- (15).Li CW; Xia MW; Cui CB; Peng JX; Li DH RSC Adv 2016, 6, 82277. [Google Scholar]
- (16).Kuhr I; Fuska J; Sedmera P; Podojil M; Vokoun J; Vanek ZJ Antibiot. (Tokyo) 1973, 26, 535. [DOI] [PubMed] [Google Scholar]
- (17).Zang Y; Genta-Jouve G; Escargueil AE; Larsen AK; Guedon L; Nay B; Prado SJ Nat. Prod 2016, 79, 2991. [DOI] [PubMed] [Google Scholar]
- (18).Zou Y; Garcia-Borras M; Tang MC; Hirayama Y; Li DH; Li L; Watanabe K; Houk KN; Tang Y Nat. Chem. Biol 2017, 13, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fetzner S Appl. Environ. Microb 2012, 78, 2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Stipanuk MH; Simmons CR; Karplus PA; Dominy JE Jr. Amino Acids 2011, 41, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Oka T; Simpson FJ Biochem. Biophys. Res. Commun 1971, 43, 1. [DOI] [PubMed] [Google Scholar]
- (22).Fusetti F; Schroter KH; Steiner RA; van Noort PI; Pijning T; Rozeboom HJ; Kalk KH; Egmond MR; Dijkstra BW Structure 2002, 10, 259. [DOI] [PubMed] [Google Scholar]
- (23).Steiner RA; Kalk KH; Dijkstra BW Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Saito T; Kawakami T; Yamanaka S; Okumura M J. Phys. Chem. B 2015, 119, 6952. [DOI] [PubMed] [Google Scholar]
- (25).Jeoung JH; Nianios D; Fetzner S; Dobbek H Angew. Chem. Int. Ed 2016, 55, 3281. [DOI] [PubMed] [Google Scholar]
- (26).Li L; Yu P; Tang MC; Zou Y; Gao SS; Hung YS; Zhao M; Watanabe K; Houk KN; Tang YJ Am. Chem. Soc 2016, 138, 15837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Grant JL; Hsieh CH; Makris TM J. Am. Chem. Soc 2015, 137, 4940. [DOI] [PubMed] [Google Scholar]
- (28).Dailey HA; Gerdes S; Dailey TA; Burch JS; Phillips JD Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tiemann K; Inze D; Van Montagu M; Barz W Eur J Biochem 1991, 200, 751. [DOI] [PubMed] [Google Scholar]
- (30).Ayer WA; Pedras MS; Ward DE Can. J. Chem 1987, 65, 760. [Google Scholar]
- (31).Xu Z; Jakobi K; Welzel K; Hertweck C Chem. Biol 2005, 12, 579. [DOI] [PubMed] [Google Scholar]
- (32).Gil Girol C; Fisch KM; Heinekamp T; Gunther S; Huttel W; Piel J; Brakhage AA; Muller M Angew. Chem. Int. Ed 2012, 51, 9788. [DOI] [PubMed] [Google Scholar]
- (33).Saruwatari T; Yagishita F; Mino T; Noguchi H; Hotta K; Watanabe K Chembiochem 2014, 15, 656. [DOI] [PubMed] [Google Scholar]
- (34).Lin HC; McMahon TC; Patel A; Corsello M; Simon A; Xu W; Zhao M; Houk KN; Garg NK; Tang YJ Am. Chem. Soc 2016, 138, 4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Griffiths S; Mesarich CH; Saccomanno B; Vaisberg A; De Wit PJ; Cox R; Collemare J Proc. Natl. Acad. Sci. U. S. A 2016, 113, 6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Agarwal V; El Gamal AA; Yamanaka K; Poth D; Kersten RD; Schorn M; Allen EE; Moore BS Nat. Chem. Biol 2014, 10, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhao B; Guengerich FP; Bellamine A; Lamb DC; Izumikawa M; Lei L; Podust LM; Sundaramoorthy M; Kalaitzis JA; Reddy LM; Kelly SL; Moore BS; Stec D; Voehler M; Falck JR; Shimada T; Waterman MR J. Biol. Chem 2005, 280, 11599. [DOI] [PubMed] [Google Scholar]
- (38).Fonvielle M; Le Du MH; Lequin O; Lecoq A; Jacquet M; Thai R; Dubois S; Grach G; Gondry M; Belin PJ Biol. Chem 2013, 288, 17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.