

Abstract
Objective:
Apolipoprotein (apo)CIII inhibits lipoprotein lipase (LpL)-mediated lipolysis of very low density lipoprotein (VLDL) TG and decreases hepatic uptake of VLDL remnants. The discovery that 5% of Lancaster Old Order Amish are heterozygous for the APOC3 R19X null mutation provided the opportunity to determine the effects of a naturally occurring reduction in apoC-III levels on the metabolism of atherogenic containing lipoproteins.
Approach and Results:
We conducted stable isotope studies of VLDL-TG and apoB100 in five individuals heterozygous for the null mutation APOC3 R19X (CT) and their unaffected (CC) siblings. Fractional clearance rates (FCRs) and production rates (PRs) of VLDL-TG and apoB100 in VLDL, intermediate density lipoprotein (IDL), low density lipoprotein (LDL), apoCIII and apoCII were determined. Affected (CT) individuals had 49% reduction in plasma apoCIII levels compared to CCs (p<0.01) and reduced plasma levels of TG (35%, p<0.02), VLDL TG (45%, p<0.02), and VLDL-apoB100 (36%,p<0.05). These changes were due to higher FCRs of VLDL-TG and VLDL-apoB100 with no differences in PRs. CTs had higher rates of the conversion of VLDL remnants to LDL compared to CCs. In contrast, rates of direct removal of VLDL remnants did not differ between the groups. As a result, the flux of apoB100 from VLDL to LDL did not change, and the plasma levels of LDL-chol and LDL-apoB100 were not lower in the CT group. ApoCIII PR was lower in CTs compared to CCs, whereas apoCII PR was not different between the two groups. The FCRs of both apoCIII and apoCII were higher in CTs than CCs.
Conclusions:
These studies demonstrate that 50% reductions in plasma apoCIII, in otherwise healthy subjects, results in a significantly higher rate of conversion of VLDL to LDL, with little effect on direct hepatic uptake of VLDL. When put in the context of studies demonstrating significant protection from cardiovascular events in individuals with loss of function variants in the APOC3 gene, our results provide strong evidence that therapies which increase the efficiency of conversion of VLDL to LDL, thereby reducing remnant concentrations, should reduce the risk of cardiovascular disease.
Keywords: ApoCIII, triglycerides, human stable isotope atudies, lipoproteins, cardiovascular disease.
Introduction
Apolipoprotein (apo)CIII was first isolated and characterized nearly 50 years ago by Brown et al. 1. Soon after, it was shown that apoCIII was an inhibitor of lipoprotein lipase (LpL) 2, an action opposing the activity of another apoprotein; apo CII, the necessary activator of LpL 3. The role of apoCIII in lipoprotein metabolism was later expanded by studies in perfused rat livers demonstrating that apoCIII inhibits uptake of triglyceride (TG) rich lipoproteins and remnants 4, 5. The identification of two sisters with complete absence of apoCIII due to a homozygous chromosomal deletion that also included apoA-I, apoA-IV, and apoA-V 6, allowed us to demonstrate, in vivo, that absence of apoCIII resulted in a dramatic increase in lipolysis of very low density lipoprotein (VLDL)-TG 7. Studies in mice overexpressing apoCIII or with targeted deletions of the APOC3 gene confirmed the human findings and also supported the role of apoCIII as an inhibitor of hepatic uptake of apoB100-containing lipoproteins 8–11. In addition, mouse- and hepatoma-based studies suggested that apoCIII may increase the incorporation of TG into nascent VLDL particles 10, 12.
Although interest in apoCIII’s role in lipid metabolism has continued, enthusiasm for apoCIII as a therapeutic target was limited by uncertainty regarding the relationship between hypertriglyceridemia and risk for cardiovascular disease (CVD) 13, 14. Recent genetic studies using both genome wide association (GWAS) and Mendelian randomization approaches have, however, established the relationship between loss of function of apoCIII, which results in lower plasma TG concentration, and reduced CVD risk. 15, 16 We found that 5% of the Lancaster Old Order Amish are heterozygous carriers of a null mutation, R19X (HGVS NM 000040.2 c.55C>T p.Arg19Ter rs76353204) in the APOC3 gene, which converts an arginine to a termination codon, resulting in a 50% reduction of plasma apoC-III levels 17. In addition to having lower fasting and postprandial TG, higher levels of high density lipoprotein (HDL)-cholesterol, and lower levels of low density lipoprotein (LDL)-cholesterol, heterozygous deficient individuals have less subclinical atherosclerosis, as determined by coronary artery calcification 17. Our report was followed by large cohort studies of APOC3 loss of function mutations, including R19X, which demonstrated a 40% reduction in myocardial infarction in carriers 18, 19.
Based on the non-human mechanistic studies described above, heterozygous loss of function of APOC3 might reduce risk for CVD by any or all of the following: (a) reducing secretion of VLDL from the liver, (b) increasing hepatic uptake of VLDL and chylomicron remnants by the liver, and (c) increasing lipolytic conversion of VLDL to LDL. We used stable isotope tracers to determine which of these three possible results of heterozygous loss of function of APOC3 would be most important in determining the differences in plasma lipids between R19X affected Amish and their unaffected siblings. Our findings support the development of therapies that lower plasma apoCIII levels as a means of treating moderate to severe hypertriglyceridemia 20, 21 which may reduce risk for CVD.
Methods
The data that support the findings of this study are available within the article and its online supplementary files.
Study Subjects:
We recruited five participants heterozygous for the APOC3 R19X mutation, hereafter denoted by CT, and five gender-matched, unaffected siblings, hereafter denoted by CC, ages 35 to 71 years, from the Lancaster Old Order Amish population 17. Participants were not receiving lipid altering medications. All study participants provided written informed consent and the studies were approved by the Institutional Review Boards of the University of Maryland School of Medicine and Columbia University Medical Center.
Stable Isotope Kinetic Studies:
The two sibs, in each pair, were studied on the same day at the Amish Research Clinic in Lancaster, PA. The protocol for these studies was one that we have used previously 22, with minor modifications to allow subjects to complete some visits in their homes. On day 1, participants fasted for 12 hours, after which a nurse visited their homes and drew baseline bloods for safety and fasting lipid and lipoprotein measurements. After 6pm on Day 1, they were NPO until 11pm when they started a liquid, isocaloric, 18% fat diet that was provided every two hours for the next 30 hours. At 5:30am (Day 2), they arrived at the Amish Research Clinic, where two IV’s were placed in antecubital veins of each arm and baseline bloods were drawn (time 0hr). Immediately after, boluses of ²H₃-L-leucine (10 μmol/kg BW), Ring-¹³C₆-L-phenylalanine (29.4 μmol/kg BW), and ²H₅-glycerol (100 μmol/kg BW) were administered over a 10-minute period, followed by a constant infusion of ²H₃-L-leucine (10 μmol/kg BW per hour) over 15 hours. Additional blood samples were collected at 20 and 40 minutes, and at 1, 2, 4, 6, 8, 10, 12, 14, 15, 15.2, 15.4, 16 hours after the administration of tracers and processed to isolate plasma and serum. Following the16hr blood sample, the subjects returned to their homes where they continued to consume the liquid meal protocol. Eight hours later a nurse drew the final 24hr blood sample in their homes. VLDL, intermediate density lipoprotein (IDL), LDL, and HDL were obtained from the 16 plasma samples via sequential density ultracentrifugation.
Determination of stable isotope enrichment of apoB100 and TG:
The isolated lipoprotein fractions were used to determine stable isotopic enrichments of ²H₃-L-leucine and Ring-¹³C₆-L-phenylalanine in apoB100 in VLDL, IDL, and LDL. ApoB100 was isolated from VLDL, IDL, and LDL by SDS-polyacrylamide gel electrophoresis. The isolated apoB100 bands were excised from the gels, hydrolyzed, and the amino acids derivatized. Plasma free amino acids were recovered from 0.25 mL plasma after precipitation of proteins with acetone and extraction of the aqueous phase with hexane. The aqueous phase was dried under vacuum, amino acids were derivatized. Enrichments of [5,5,5-2H3]-leucine and [13C6]-phenylalanine tracers in apoB100-lipoproteins and plasma free leucine and phenylalanine were measured by gas chromatography - mass spectrometer using an Agilent 6890 gas chromatography and a 5973 mass spectrometer with negative chemical ionization. Additionally, kinetic analysis of TG in VLDL was performed with ²H₅-glycerol. TG was separated from phospholipid by zeolite binding. The TG was resolubilize with chloroform. We performed transesterficiation with methanolic HCL. Glycerol was isolated by liquid/liquid extraction (hexane and water added) and derivatized to triacetin (glycerol tri-acetate) through incubation with acetic anhydride. We performed gas chromatography - mass spectrometer positive chemical ionization with selective ion monitoring of m/z 159 and 164.
Compartmental modeling of apoB100 and TG metabolism:
Fractional clearance rates (FCR) and production rates (PR) of TG and apoB100 in VLDL, and of apoB100 in IDL and LDL were determined using a compartmental model to fit stable isotope enrichment data23–25. In our general model, apoB100 and TG are required to have the same pool structure and the same rate constants for each VLDL pool, but with different mass distributions. With more than one pool in the VLDL fraction, the different mass distributions lead to different VLDL FCRs for TG and apoB, since VLDL FCR is obtained as a weighted average of the individual FCRs (the weights given by the mass distribution). However, if there is only one pool in the VLDL fraction, TG and apoB necessarily have the same FCR. For each study, the minimum number of pools needed to simultaneously fit the nine sets of data (two tracers, leucine and phenylalanine, in VLDL-, IDL-, and LDL-apoB100 and plasma amino acids, and one tracer, glycerol, in VLDL-TG) is chosen for the final model. In the present study, the final model had one pool each for VLDL, IDL, and LDL. The data were fitted by least squares, giving equal weight to all data points (that is, assuming a constant error variance for all measurements) using a computer program, Pool fit 23, which solves the differential equations in closed form and computes the fits and parameter sensitivities as sums of exponentials. The fits yielded fractional catabolic rates (FCRs) of apoB100 in VLDL, IDL, and LDL, and TG in VLDL. The model also estimated rates of conversion of apoB100 between VLDL, IDL, and LDL. Production rates (PRs in mg/kg/day) were calculated by multiplying FCRs (in pools/day) by the appropriate lipoprotein pool sizes of apoB100, which were calculated as each lipoprotein’s concentration of apoB100 in mg/dl multiplied by an estimate of each individual’s plasma volume (45 ml/kg). Schematics of the models used to analyze apoB100, TG-glycerol and apoCIII in the present study are included in the supplement (Supplemental Fig I). Based on the best fit of the present data, we have only one VLDL pool for apoB and TG, direct conversion of VLDL apoB to LDL as well as conversion from VLDL to LDL via IDL, no direct out pathway from IDL, and a small component of direct secretion of LDL from the liver.
Determination of stable isotope enrichment of apoCII and apoCIII:
ApoCIII enrichment with ²H₃-L-leucine was measured using Ultraperformance liquid chromatography-mass spectrometry . VLDL and HDL fractions were digested with trypsin as previously described 26. A multiple reaction monitoring method was used to determine the following precursor-product ion transitions for a peptide specific to apoCIII (GWVTDGFSSLK; M0: 599.0>854.5; M3: 600.5>857.6) and apoCII (TYLPAVDEK; M0: 518.4>658.4; M3: 519.9>658.4).
Compartmental modeling of apoCII and apoCIII metabolism:
The 2H3-leucine enrichment data for apoCII and apoCIII in HDL or VLDL was fitted by a single pool with the VLDL apoB100 enrichment plateau used as the best available estimate of the liver leucine pool enrichment. Separate FCRs were estimated for HDL and VLDL. While the enrichment of apoCIII was different between HDL and VLDL, there was a constant ratio across time points in any single study. This means that the FCR of apoCIII is estimated to be the same in VLDL and HDL; any difference is solely due to random measurement error. We ascribe this to apoCIII moving freely among lipoproteins 27. The situation was similar with apoCII. Therefore, we a calculated single plasma FCR for apoCIII and apoCII. ApoCIII and apoCII PRs were calculated using the FCR for each apolipoprotein and its plasma pool size, as described above for apoB.
Quantitation of ApoCIII in VLDL and HDL Fractions:
ApoCIII was quantitated by Ultraperformance liquid chromatography-mass spectrometry, using a Waters Xevo TQS triple quad mass spectrometer coupled with an Acquity UPLC (Waters, Milford, MA). VLDL and HDL fractions were digested with trypsin as previously described 28. In brief, 200μl of ultracentrifuged VLDL or HDL from each time point was desalted, reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin overnight. A multiple reaction monitoring method was used to determine the following precursor product ion transitions for a peptide specific to apoCIII (GWVTDGFSSLK; M0: 599.0>854.5) and a deuterated internal standard (M8: 603.0>862.5). Lower limit of quantitation, defined as the level at which the residual of the calibration line is <20% of the expected concentration was determined to be 1nm. The intra-assay precision for the assay was 4.00%. ApoCII was similarly quantitated using the following precursor product ion transitions for a peptide specific to apoCII (TYLPAVDEK; M0: 518.4>658.4; M3: 519.9>658.4). The proportions of plasma apoCIII and apoCII residing in VLDL and HDL were then calculated.
Biochemical and immunologic assays:
Day 1 blood was collected after a 12-hr overnight fasting period. Additional timed blood samples were collected while the subjects were consuming the liquid diet, both before (0hr) and at various time points after the stable isotope infusion was started (20min, 40min, 1, 2, 4, 6, 8, 10, 12, 14, 15, 15.2, 15.4, 16, 24 hrs.). Plasma cholesterol, TG and HDL cholesterol were measured by Integra400plus (Roche). Plasma LDL cholesterol levels were estimated using the Friedewald formula. Cholesterol and TG were also measured enzymatically in VLDL, IDL, LDL and HDL isolated by ultracentrifugation. Plasma apoCII, apoCIII and apoE were measured by human enzyme-linked immunosorbent assay kits (ab168549 [apoCII]; ab154131 [apoCIII]; ab108813 [apoE], Abcam, Cambridge, MA). ApoB100 in plasma and in VLDL, IDL, and LDL was measured using an apoB100 ELISA kit (A70102 AlerCheck, Inc.) .
Statistical Analysis
The data are presented as means and standard deviations (SD). The mutation effects were assessed by analyzing within-pair differences for statistical significance using paired t-tests. The primary endpoint was the percent difference in FCR of VLDL-TG and VLDL-apoB100 between affected and unaffected sib-pairs, and p=0.05 was considered significant. A key secondary endpoint was the partition of VLDL between conversion to LDL and hepatic uptake, and p=0.01 was considered significant. All other comparisons were exploratory.
Results
Study Population:
We enrolled 10 participants (5 CT and 5 CC), including 3 affected males, 2 affected females, and sex- and age- (within ten years) matched unaffected siblings. Mean ages (CT 50±11.1, CC 52.4±12.4) and BMIs (CT 28.6±4.3, CC 26.9±5.9,) were similar between the groups (Table 1).
Table1:
Baseline Characteristics and Plasma Lipid and Apolipoprotein Levels ofAPOC3 R19X Carriers and Unaffected Sib Pairs.
| ID# | Sex | Age yrs. |
BMI | Chol. mg/dl | TG mg/dl | LDL-C mg/dl |
HDL-C mg/dl | apoB100 mg/dl | ApoC-III ug/ml |
ApoC-II ug/ml |
ApoE ug/ml |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CC1 | F | 55 | 34 | 181 | 62 | 92 | 78 | 105 | 140 | 45 | 52 |
| CC2 | F | 71 | 18 | 167 | 120 | 100 | 44 | 70 | 138 | 94 | 223 |
| CC3 | M | 53 | 26 | 303 | 117 | 219 | 60 | 142 | 85 | 101 | 172 |
| CC4 | M | 37 | 30 | 179 | 88 | 98 | 63 | 66.2 | 160 | 63 | 236 |
| CC5 | M | 48 | 27 | 137 | 73 | 84 | 39 | 66 | 92 | 53 | 94 |
| Mean±SD | 52±12 | 27±6 | 193±64 | 92±26 | 119±56 | 57±16 | 90±34 | 123±33 | 71±25 | 155±81 | |
| CT1 | F | 46 | 27 | 145 | 25 | 76 | 63 | 53 | 30 | 22 | 165 |
| CT2 | F | 63 | 32 | 215 | 103 | 127 | 68 | 87 | 75 | 73 | 160 |
| CT3 | M | 50 | 34 | 220 | 67 | 138 | 69 | 120 | 69 | 70 | 154 |
| CT4 | M | 35 | 28 | 160 | 61 | 92 | 57 | 64 | 63 | 45 | 164 |
| CT5 | M | 57 | 23 | 173 | 48 | 93 | 70 | 105 | 66 | 28 | 92 |
| Mean±SD | 50±11 | 29±4 | 183±33 | 61±29 | 105±5 | 65±26 | 86±28 | 61±18 | 48±23 | 147±31 | |
| %Diff. (SD) | -2.3 22.1 | 13.7 40.9 | -1.6 20.8 | -35.2 20.1* | -4.9 ±22.9 | 22.3 34 | -2.6 17.2 | -49.1 18.5† | -35.1 13.8† | 17.0 55.5 | |
Participants are defined as CC (unaffected siblings) and CT (affected siblings), in order of sib-pairs. BMI: body mass index. Chol: plasma cholesterol, TG: plasma triglycerides, LDL-C: plasma low density lipoprotein cholesterol, HDL-C: plasma high density lipoprotein cholesterol, apo: plasma apolipoprotein. All lipid and lipoprotein levels were obtained from five time-points during the 24-hr period of the stable isotope studies. Data are presented as means and standard deviations (SD) of absolute concentrations as well as means and SD of the percent differences between the two groups. Statistical significance of the percent differences was assessed using paired t-tests.
significance at p<0.02,
significant at p<0.01.
Plasma Lipid and Apolipoprotein Levels:
Plasma TG levels were lower in the CT subjects (61±29 mg/dl) versus their CC siblings (92±26 mg/dl) (Table 1). The mean (±SD) percent difference for TG between the pairs was 35±20 (p<0.02). Total plasma cholesterol levels did not differ between sib-pairs. Although we have previously shown that APOC3 R19X carriers have higher HDL and lower LDL cholesterol levels 17, in this much smaller sample, the levels of cholesterol in these lipoproteins were not significantly different between the pairs. Plasma levels of apoB100 and apoE were not different between the two groups, but, as expected, apoCIII levels were significantly reduced in the CT group (CT 61±18 μg/ml vs.CC 123±33 μg/ml) with a mean percent difference between pairs of 49±19 (p<0.01). Of interest, apoCII levels were also lower in the affected (48±23 μg/ml) compared to the unaffected siblings (71±25 μg/ml), with a mean percent difference of 35±14 (p<0.01). The percent of plasma apoCIII in VLDL was similar in the CT (23±9%) and CC (28±9%) groups. The percent of plasma apoCII in VLDL was also similar in the CT (30±14%) and CC (35±10%) groups.
The cholesterol and TG levels in the isolated VLDL fractions (Table 2) were lower in the CT (4±2.0 mg/dl and 18±12 mg/dl, respectively) than in their CC siblings(7±3 mg/dl and 32±11 mg/dl, respectively) (p<0.03 for cholesterol and p<0.02 for TG). The cholesterol and TG levels in the IDL and LDL fractions were not different between the sib-pairs. Despite similar plasma apoB100 levels, VLDL-apoB100 levels were lower in the CT (2±0.5 mg/dl) compared to the CC (4±2 mg/dl), with a mean percent difference between pairs of 36±48 (p<0.05). There were no differences in IDL- or LDL-apoB100 levels between the sib-pairs.
Table 2:
Effects of APOC3 R19X on Mean Levels of Lipoprotein Lipids and ApoB100 Concentrations.
| CC (mean±SD) (mg/dl) |
CT (mean±SD) (mg/dl) |
Percent Differences (mean±SD) |
|
|---|---|---|---|
| VLDL-Chol | 6.9±3.4 | 3.5±1.9 | -47.9±13.3* |
| IDL-Chol | 3.07±2.9 | 1.47±1.0 | -37.3±35.8 |
| LDL-Chol | 61.7±24.0 | 56.6±13.6 | -4.1±22.7 |
| VLDL-TG | 31.6±11.0 | 17.7±11.7 | -45.3±26.6* |
| IDL-TG | 3.12±2.0 | 1.89±1.1 | -36.1±24.5 |
| LDL-TG | 9.6±3.8 | 8.6±2.2 | -6.1±26.0 |
| VLDL-apoB100 | 4.29±2.1 | 2.0±0.47 | -36.4±47.6* |
| IDL-apoB100 | 1.96±1.9 | 1.07±0.66 | -24.6±41.7 |
| LDL-apoB100 | 51.2±15.3 | 58.0±22.6 | -13.6±32.9 |
Participants are defined as CC (unaffected siblings) and CT (affected siblings). Chol: cholesterol, TG: triglycerides, VLDL: very low density lipoprotein, IDL: intermediate density lipoprotein, LDL: low density lipoprotein, apoB100: apolipoprotein B100. Data were obtained from five time-points during the 24-hr period of the stable isotope studies. Data are presented as means and standard deviations (SD) of absolute concentrations as well as the means and SDs of the percent differences between CC and CT groups. Statistical significance of the percent differences was assessed using paired t-tests.
significant at p<0.05.
ApoB100 and TG Metabolism:
As noted in Methods, we modeled VLDL- TG and VLDL-apoB100 metabolism jointly, and the best fits of the stable isotope enrichment data required only a single VLDL pool. Thus, the VLDL-apoB100 FCR and VLDL-TG FCR in each subject were the same. The results demonstrate that the lower levels of VLDL-TG and VLDL-apoB100 in the CT group were due to a significantly greater FCR of VLDL-TG and VLDL-apoB100 (% increase 116±31, p<0.001) (Table 3). The VLDL-apoB100 PRs were similar in the CT and CC siblings (27±7 and 26±12 mg/kg/day, respectively), as were the VLDL-TG PRs (790±291 and 555±156 mg/kg/day). The ratio of VLDL-TG PR to VLDL-apoB100 PR, an indicator of the size of newly secreted VLDL particles, did not differ between the groups (CT 32±13 vs. CC 27±19).
Table 3:
Effects of APOC3 R19X Mutation on Kinetic Parameters for TG and apoB100 Metabolism.
| CC ±SD | CT ±SD | %Difference±SD |
|
|---|---|---|---|
| VLDL-TG and VLDL-apoB100 FCR (pools/day) | 15.0±6.0 | 31.0±8.9 | 116±31† |
| IDL-apoB100 FCR (pools/day) | 5.9±3.1 | 12.2±2.6 | 203±223* |
| LDL-apoB100 FCR (pools/day) | 0.63±0.31 | 0.77±0.18 | 37±43 |
| VLDL-TG PR (mg/kg/day) | 555±156 | 790±291 | 45.9±56.9 |
| VLDL-apoB100 PR (mg/kg/day) | 26.0±12.4 | 26.8±6.7 | 34±93 |
| IDL-apoB100- PR (mg/kg/day) | 4.0±3.1 | 5.6±3.5 | 122±222 |
| LDL-apoB100 PR (mg/kg/day) | 14.8±8.7 | 19.1±5.6 | 58±72 |
| LDL-apoB PR from VLDL via IDL (mg/kg/day) | 4.0±3.1 | 5.6±3.5 | |
| LDL-apoB PR directly from VLDL (mg/kg/day) | 8.4±9.8 | 11.9±4.8 | |
| LDL-apoB PR from the liver (mg/kg/day) | 2.4±5.3 | 1.5±2.3 | |
| VLDLTG PR/VLDLapoB100 PR | 27.3±19.1 | 31.6±13.2 | 37.7±71.7 |
Participants are defined as CC (unaffected siblings) and CT (affected siblings). VLDL: very low density lipoprotein, IDL: intermediate density lipoprotein, LDL: low density lipoprotein, TG: triglycerides, apoB: apolipoprotein B100. FCR: Fractional Clearance rate, PR: production rate. Data derived from compartmental modeling of stable isotope enrichments of samples obtained over a 24 hr. period. Data are presented as means and standard deviation (SD) of the absolute FCRs and PRs as well as the means and SD of the percent difference in each parameter between the CT and CC groups. Statistical significance of the percent differences was assessed using paired t-tests.
significant at p<0.02,
p<0.001.
We also found that the FCR of IDL-apoB100 was greater in the CT individuals compared to their CC siblings (% increase 203±223; p<0.02). We did not observe differences between sib-pairs in the FCR of LDL-apoB100 or in the PRs of IDL- or LDL-apoB100. There were three sources of LDL-apoB: conversion of VLDL to LDL via IDL, direct conversion of VLDL to LDL, and direct secretion of LDL from the liver. These made up, respectively on the average, 31%, 62% and 7% of LDL PR in the CT siblings and 33%, 55%, and 12% in the CC siblings; none of the sources differed significantly between the groups (Table 3).
A key secondary goal of the study was to determine the effects of reduced apoCIII levels on the partitioning of VLDL-apoB100 flux between conversion of VLDL to LDL and direct removal of VLDL by the liver. Figure 1 depicts a model of apoB100-lipoprotein transport from the liver to circulating VLDL, and then to either LDL or back to the liver. Inherent to this model is the concept that the initial LpL-mediated lipolysis of VLDL-TG generates a smaller particle with less TG than present at the time of secretion into the circulation, and at some point in this process, the particle has lost enough TG that it has a density greater than 1.006 and, therefore, leaves the VLDL pool. This particle can be be found in the IDL and LDL pools or be irreversibly taken up by the liver. It is important to understand that although we show a single VLDL pool in Figure 1, it comprises a range of VLDL of differing size and number of TG molecules: the largest and most TG-rich VLDL are those that have just entered the circulating VLDL pool and the smallest and most TG-poor are those that have undergone lipolysis and are about to either leave the circulation directly or become IDL or LDL. We show a single pool, despite the heterogeneous nature of the population of VLDL within this pool, because when we used a model with more than one VLDL pool, it did not improve the fit of our kinetic data.As noted above, the rates of secretion of newly synthesized VLDL from the liver (equal to VLDL PR) were similar for CT and CC siblings and, therefore, the blue arrows from the liver to VLDL in each group are the same. However, because lipolysis of VLDL-TG was much faster in the CTs (depicted by the much thicker black arrow coming out from VLDL in that group), the size of the VLDL pool in CTs was about 36% smaller than the VLDL pool of CCs (Table 2). The green and red arrows represent the number of VLDL plasma pools each day that, after lipolysis of TG, move either to LDL or to the liver, respectively. Our compartmental analysis indicated that there was a significant 12.3±6.3 pools/day difference in the FCRs between CT and CC sib-pairs for the conversion of VLDL-apoB100 to IDL and LDL (p =0.01), but only a 3.6±7.3 pools/day difference in FCRs between the two groups for direct hepatic removal of those particles (p= 0.33). Thus, only the conversion rate of VLDL to LDL, eg, the lipolytic pathway, was significantly greater in the group with partial loss of apoCIII. This is depicted by the thicker green arrow in CT compared to CC, whereas the red arrows are similar in the two groups (Figure 1). The individual and mean data for these parameters are presented in Table 4. This greater rate of conversion of VLDL to IDL and LDL versus uptake by the liver was reflected by a non-significant increase in LDL-apoB100 PR in the CT compared to the CC group (Table 3).
Figure 1.
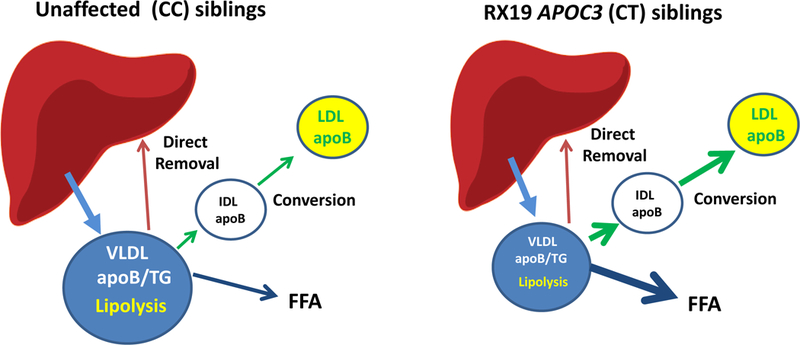
The figures depict the effects of partial loss of apoCIII synthesis of the metabolism of VLDL. Although the rates of secretion of VLDL-apoB100 and VLDL-TG (blue arrows) are similar in CC (unaffected siblings) and CT (affected siblings), the size of the VLDL plasma pool (blue circles) is reduced by 36% in CT because an increase in LpL mediated lipolysis of VLDL-TG (black arrows) leads to a doubling of the fraction clearance rate of apoB100 and TG from the VLDL pool. The lipolysis of VLDL-TG generates particles that either undergo conversion to IDL and LDL (green arrows) after additional lipolysis of TG by LpL or HL or direct removal by the liver (red arrows). The rate of conversion of VLDL to LDL was significantly greater in CT compared to CC whereas the rates of direct removal were similar in the two groups (Table 4). These data indicate that, in individuals heterozygous for loss of function of APOC3, who have normal levels of lipoprotein lipase, reduced levels of apoCIII in plasma significantly affect the lipolytic but not the hepatic uptake pathways for metabolism of VLDL. VLDL: very low density lipoprotein, apoB: apolipoprotein-B100, TG: triglyceride, FFA: free fatty acids, LDL: low density lipoprotein.
Table 4.
Effects of APOC3 R19X Mutation on Individual and Mean Fractional Rates of VLDL-apoB100 Clearance, Conversion of VLDL apoB100 to LDL, and Direct Removal of VLDL apoB100
| Participant | VLDL-apoB100 FCR (pools/day) |
Conversion of VLDL apoB100 to LDL FCR (pools/day) |
Conversion of VLDL apoB100 to LDL directly FCR (pools/day) |
Conversion of VLDL apoB100 to LDL via IDL FCR (pools/day) |
Direct Removal of VLDL apoB100 FCR (pools/day) |
|---|---|---|---|---|---|
| CC1 | 18.7 | 18.7 | 7.24 | 11.42 | 0.00 |
| CC2 | 7.82 | 2.15 | 1.64 | 0.51 | 5.67 |
| CC3 | 10.1 | 3.73 | 0.9 | 2.83 | 6.32 |
| CC4 | 22.4 | 15.2 | 14.66 | 0.57 | 7.16 |
| CC5 | 16.1 | 4.98 | 3.04 | 1.94 | 11.1 |
| Mean±SD | 15.0±6.0 | 9.0±7.5 | 5.5±5.7 | 3.5±4.6 | 6.05±3.98 |
| CT1 | 37.8 | 37.8 | 28.43 | 9.32 | 0.00 |
| CT2 | 20.5 | 11.9 | 5.22 | 6.7 | 8.56 |
| CT3 | 22.8 | 22.8 | 11.41 | 11.41 | 0.00 |
| CT4 | 40.3 | 22.0 | 19.14 | 2.83 | 18.3 |
| CT5 | 33.5 | 11.9 | 9.90 | 2.05 | 21.5 |
| Mean±SD | 31.0±8.87 | 21.3±10.6 | 14.8±9.1 | 6.5±4.0 | 9.67±10.0 |
| Difference±SD | 16.0±3.02† | 12.3±6.28* | 9.3±7.2* | 3.0±4.4 | 3.6±7.3 |
Participants are defined as CC (unaffected siblings), CT (affected siblings), in order of sib pairs. VLDL: very low density lipoprotein, Data derived from compartmental modeling of stable isotope enrichments obtained over a 24 hr. period. Data are presented as means and standard deviation (SD) of the absolute FCRs of VLDL-apoB100, the conversion of VLDL apoB100, and the direct removal of VLDL apoB 100, as well as the means and SD of the absolute differences in each parameter between the CT and CC groups. Conversion of VLDL-apoB100 to LDL includes both conversion of VLDL directly to IDL as well as VLDL to IDL and IDL to LDL. There was no direct conversion of VLDL to LDL required to fit the kinetic data; all the conversion occurred through IDL. Statistical significance assessed using paired t-tests.
significant p<0.05
significant p<.001.
As noted above, the generation of LDL from VLDL occurred via direct conversion of VLDL to LDL or conversion of VLDL to IDL followed by conversion of the latter to LDL, with direct conversion accounting for the largest proportion of LDL generated. We were able, using our model, to determine the FCRs of each of these pathways in the CT and CC groups, and these data are presented in Table 4. The FCR for direct conversion of VLDL to LDL was significantly greater (14.8±9.1 pools/day) in the CT siblings than in the CC group (5.5±5.7 pools/day) (p=0.04). There was no difference in the FCRs for conversion of VLDL to LDL via IDL between the two groups (CT: 6.5±4.0; CC: 3.5±4.6 pools/day; p=0.2). These results, together with those for the FCRs of overall conversion to LDL versus direct uptake of VLDL, support much greater lipolytic activity in the affected vs the non-affected sib-pairs.
ApoCIII and apoCII Metabolism:
Previous studies by several groups have demonstrated that the kinetics of rapidly exchangeable apolipoproteins such as apoCIII and apoCII 29 can only be characterized by plasma FCRs and PRs 30–32. However, because of a lack of unanimity on this issue 33, we determined enrichments of each apolipoprotein in both VLDL and HDL, which together transport nearly all of these two proteins in plasma. We found essentially identical FCRs for both apoCIII and apoCII in VLDL and HDL (Supplemental Table I) and, therefore, have presented only a single FCR and PR for each apolipoprotein in Table 5. In the CC group, the FCRs of apoCIII (1.3±0.7 pools/day) and apoCII (1.4±0.8 pools/day) were similar, consistent with a common clearance pathway of each of these apolipoproteins 34 .On the other hand, the PR of apoCIII (7.4±6 mg/kg/d) was almost double that of apoCII (3.9±2 mg/kg/day), indicative of unique regulation of the synthesis of these two apolipoproteins. Similar differences in the PRs of apoCIII and apoCII have been reported previously 34. In the CT group, the FCRs of apoCIII (1.8±0.5 pools/day) and apoCII (2.3±0.9 pool/day) were also similar but, in contrast to the CC group, the PRs of apoCIII (4.9±1.7 mg/kg/day) and apoCII (4.2±1.2 mg/kg/day) in the CT group were also similar, reflecting the reduced rate of synthesis of apoCIII in the CT versus the CC subjects, resulting from the APOC3 R19X null mutation. For both apoCIII and apoCII, neither the FCR nor the PR was significantly different between the CT and CC subjects.
Table 5:
Effects of APOC3 R19X Mutation on Kinetic Parametersfor apoCIII and apoCII Metabolism.
| CC (±SD) | CT (±SD) | Difference | ||
|---|---|---|---|---|
| apoCIII | FCR | 1.3±0.7 | 1.8±0.5 | 0.59±0.38 |
| PR | 7.4±6 | 4.9±2 | -2.54±4.83 | |
| apoCII | FCR | 1.4±0.8 | 2.3±1 | 0.90±0.74 |
| PR | 3.9±2 | 4.2±1 | 0.33±1.34 | |
Participants are defined as CC (unaffected siblings) and CT (affected siblings). FCR: Fractional Clearance rate, PR: production rate. Data are presented as means and standard deviation (SD). There were no significant differences for any of the variables between CC and CT groups or apoCIII and apoCII.
Discussion
We used stable isotopes to investigate, in vivo, the effects of heterozygosity for the APOC3 R19X null mutation and the associated 50% lower levels of plasma apoCIII on apoB100 and TG metabolism. Our purpose was to determine the relative importance of haplodeficiency for three proposed physiologic roles of apoCIII in VLDL metabolism: 1) inhibition of LpL-mediated lipolysis of VLDL-TG, 2) inhibition of hepatic uptake of VLDL or TG-rich remnant particles, and 3) stimulation of the incorporation of TG into VLDL in the liver. Each of these roles, which have been demonstrated by in vitro assays or by studies in cells and mouse models, could have a significant impact on the efficacy of therapeutic agents that partially inhibit apoCIII synthesis for treatment of hypertriglyceridemia and for prevention of CVD. The most significant differences we observed between affected CT and unaffected CC sib-pairs were the higher in FCRs for VLDL-TG and VLDL-apoB100 in the CT. The 50% lower circulating apoC-III levels in the R19X carriers resulted from the doubling of the rate of clearance of TG-rich apoB100-lipoproteins from the bloodstream. The clearance of VLDL occurs as a two-stage process. First, LpL hydrolyzes the TG in nascent VLDL as these particles circulate through the vascular beds of adipose tissue and muscle 35, generating what is typically designated as a remnant that still contains less TG, but enough to keep its density <1.006 (within the VLDL range). These particles circulate back to the liver, which is believed to be the site of the second stage. The latter actually has two components; one comprises additional lipolysis of remnant TG by LpL and probably more importantly HL, resulting in conversion of VLDL to IDL and LDL 36. The second component involves interaction of apoB100 with one or more receptors and proteoglycans on the cell surface of the liver, resulting in internalization of the remnant particle 37–39 . Importantly, it is not clear whether these are completely independent parallel pathways or if some additional lipolysis is necessary before remnant uptake by the liver occurs. Our kinetic data do not allow us to differentiate between these possibilities and, therefore, we have made them independent, parallel pathways.
Our compartmental modeling of stable isotope enrichments of TG and apoB100 in VLDL, and of apoB100 in IDL and LDL, enabled us to determine the effect of partial loss of apoCIII on each of the second stage pathways. Indeed, our results indicate that the rates of conversion of VLDL particles to LDL were significantly greater in the carriers of R19X. The findings that the FCR of IDL-apoB100 (which was not removed directly) was also significantly greater in the CT group supports higher rates of conversion of VLDL to LDL, as does a non-significant, numerical increase in the PR of LDL-apoB100. All of the present results are in accord with those from our previous study in two sisters with complete loss of apoCIII due to a large homozygous chromosomal deletion, where we demonstrated marked increases in FCRs of VLDL-apoB100 and VLDL-TG as well as normal or increased rates of conversion of VLDL-apoB100 to LDL 7. In the present study, we did not find a significant effect of partial loss of apoCIII synthesis on the uptake of VLDL remnants by the liver. This result might seem to conflict with results of recent studies of the efficacy of an apoCIII antisense in patients with no LpL, where the significant reductions in plasma TG levels observed would have required increased direct removal of TG-rich lipoproteins, presumably by the liver 2133. However, whereas hepatic removal of TG-rich lipoproteins was the only pathway that would be susceptible for inhibition by apoCIII in patients lacking LpL, our participants would have both of the apoCIII-susceptible pathways available for participation in the metabolism of VLDL. A simple explanation for our results might be that more efficient LpL-mediated lipolysis of nascent VLDL resulted in smaller VLDL that were more depleted of TG than post-lipolysis VLDL in non-affected individuals and, therefore, more likely to be converted to IDL and LDL by additional lipolytic activities during the second stage of VLDL metabolism than to be internalized by LDL family receptor-mediated pathways present in the liver 38, 39. Our results also contrast with recent studies of the effects of apoCIII antisense in mice with targeted deletion of LpL, LDL receptors, the LDL receptor related protein-1, and heparin sulfate proteoglycan receptors 41. Their results led the authors to conclude that apoCIII inhibited hepatic uptake of remnant lipoproteins, but had little or no effect on LpL-mediated lipolysis of TG. Although those studies were convincing, they were in mice, where apoB48-apoE-enriched lipoproteins are predominant, and where hepatic removal of remnants is much greater than in humans. Direct comparisons between the two studies are, therefore, very limited.
In contrast to the significant effect of reduced apoCIII on clearance of VLDL-TG and VLDL-apoB100, we did not find any evidence supporting a role for apoCIII in the incorporation of TG into nascent VLDL within the liver 10, 12. Our result agrees with data showing that treatment of apoCIII transgenic mice with an apoCIII-antisense had no effect on rates of VLDL secretion 42, and with our previous finding of normal VLDL-apoB100 and VLDL-TG PRs in the two patients with complete absence of apoCIII 7. Our results, indicating that partial loss of apoCIII has a significant impact on the lipolytic pathway but not on direct removal of VLDL remnants, raise obvious questions about the potential of therapies that lower apoCIII levels. On the one hand, it is clear that the marked increase in VLDL TG FCR is consistent with the dramatic reductions in TG levels observed in a study where patients with marked hypertriglyceridemia without mutations in LpL were treated with an antisense to apoCIII43. Those results indicate the utility of apoCIII-lowering therapy to prevent pancreatitis in such patients. In contrast, if therapies that reduce apoCIII synthesis result in higher rates of conversion of VLDL remnants to LDL with no change or modest increases in LDL-cholesterol or apoB100 levels 43, 44, how could that translate to risk for CVD? Specifically, how do our findings relate to the decreased levels of coronary calcification in R19X carriers17 and very significant reductions in CVD risk observed in cohorts with loss of function variants in the APOC3 gene 15,16 A simple answer is that reducing the number of VLDL remnants, which carry more cholesterol per particle than LDL, is beneficial regardless of the mechanism involved. Individuals with dyslipidemia and the apoE2/2 genotype accumulate both apoB100 and apoB48 remnants and have significantly increased risk for CVD 45, 46. Amish individuals heterozygous for the APOC3 R19X mutation and individuals with the combined deletion of APOA1/APOC3/APOA4/APOA5 had decreased postprandial TG levels 7, 47, which have been shown to be atherogenic 48, 49. More dramatic reductions in postprandial lipid levels were reported recently in a small number of individuals homozygous for the R19X mutation in APOC3 50. Of note, individuals in that study who were either heterozygotes or homozygoutes for R19X had LDL-cholesterol levels that were similar to non-carriers 49. A recently published study comparing the roles of remnant cholesterol vs LDL cholesterol in reduced CVD risk in 137,895 individuals with loss-of-function of APOC3 found that lower concentrations of remnant cholesterol accounted for nearly all of the benefit of lower apoCIII levels 51,51Importantly, genetic studies have demonstrated that gain of function variants in the LPL gene are associated with reduced risk for CVD, whereas loss of function variants in this gene are associated with increased risk 52, 53. Our stable isotope kinetic studies of apoCIII and apoCII showed the expected lower apoCIII PR in the CT compared with the CC group, but no difference in apoCII PR between affected and unaffected siblings. These results are in accord with the isolated R19X mutation in the CT group and indicative of independent regulation of the expression of the APOC3 and APOC2 genes which are on different chromosomes. In contrast, the FCRs of both apoCIII and apoCII were greater in the affected compared to the unaffected siblings, suggestive of a common pathway for the clearance of these two apolipoproteins from the plasma, possibly along with VLDL remnants. The FCRs of apoCIII and apoCII are greater than those of both LDL-apoB100 and HDL apoA-I, and slower than the FCRs of VLDL- and IDL-apoB100. However, as both apoCIII and apoCII are associated with all the major lipoproteins, a model that has reservoirs of apoCIII and apoCII on HDL that would feed a catabolic pathway through VLDL remnants seems reasonable to explain our results. A recent paper describing the effects of a rare mutation in APOC3 that affected the binding of the apolipoprotein to lipoproteins resulted in clearance of apoCIII via the kidney 54. We previously demonstrated a role of the kidney in the catabolism of apoA-I in hypertriglyceridemic individuals 55. While we cannot rule out involvement of the kidney in the increased catabolism of apoCIII and apoCII in the CT group, evidence for a significant pool of free apoCIII or free apoCII in plasma is lacking 56, 57, and the mutation in APOC3 in our CT subjects results in decreased secretion with no evidence of altered lipoprotein distribution of apoCIII synthesized by the normal allele. Of note, the lower levels of apoCII in the CT group did not appear to restrict or limit the effect of low apoCIII on VLDL-TG clearance. This is consistent with the lack of any alteration in plasma lipid levels in heterozygotes for apoCII deficiency 58. Despite uncertainties about the effects on CVD risk, which will require large randomized, placebo controlled CVD outcome trials to resolve, the very large reductions of plasma TG levels observed with apoCIII antisense indicate that therapies to significantly lower apoCIII concentrations in the circulation could add a new, potent approach for the prevention of pancreatitis in patients with severe hypertriglyceridemia 59. We also realize that other pro-atherogenic or pro-inflammatory effects of apoCIII have been reported, and these may also be reversed by therapies that reduce apoCIII levels 60, 61.
Study Limitations
This study included a small number of participants, a shortcoming that we attempted to alleviate by using sib-pairs. We also did not address the heterogeneous nature of apoB100-lipoproteins, particularly related to the presence of apoCIII on only a portion of VLDL, IDL and LDL, demonstrated first by Alaupovic et al 62 and then in a series of stable isotope kinetic studies by Sacks and colleagues 63–65. The finding that there are apoB100-lipoproteins with and without apoCIII could have an important implication for the present study. We would note, however, that Sacks and colleagues have reported that 40–80% of VLDL from individuals with normal TG levels contain 60–100 apoCIII molecules per particle 65. Thus, it seems quite likely that a 50% reduction of apoCIII in R19X carriers would not significantly alter the proportion of VLDL containing significant numbers of apoCIII molecules, and that the affected and unaffected siblings would have relatively similar proportions of VLDL with and without apoCIII. Our finding that the proportions of both apoCIII and apoCII in VLDL were similar in affected and unaffected siblings suggests similar proportions of apoCIII containing VLDL in both groups. Importantly, we acknowledge that the metabolic differences we are reporting between affected and unaffected siblings may not completely mimic the effects of a therapeutic agent that reduces levels of plasma apoCIII.
Conclusion
The present studies demonstrate the physiologic effects of 50% reductions in plasma apoCIII resulting from heterozygosity for a naturally occurring loss of function mutation in the APOC3 gene. Lower apoCIII levels in the circulation resulted in higher rates of lipolysis of VLDL-TG and higher rates of conversion of VLDL to LDL in the affected siblings. We did not observe changes in the rate of direct remnant removal of VLDL remnants by the liver or in rates of secretion of VLDL. Our results, together with those from cohorts with loss of function variants in the APOCIII gene, provide evidence for the increased atherogenicity of VLDL (and chylomicron) remnants as well as support for therapies that would reduce remnant concentrations regardless of the mechanism involved. The impact of loss of function variants in APOCIII might be of particular importance in the postprandial period.
Supplementary Material
{kind=link}
Highlights.
APOC3 R19X null mutation causes 50% lower levels of plasma apoCIII.
The changes in ApoCIII levels were due to a doubling of the rate of clearance of TG-rich apoB100-lipoproteins from the bloodstream.
These results provide strong evidence that therapies which increase the efficiency of conversion of VLDL to LDL, thereby reducing remnant concentrations, should reduce the risk of cardiovascular disease.
Acknowledgements:
We are grateful for the efforts and support of the Amish Research Clinic nurses, technicians, and staff in Lancaster, PA. This study would not have been possible without the outstanding support of the Amish research participants. We also thank Drs. Maryam Khavandi and Marie Maraninchi for their assistance in the laboratory processing of the samples and the Irving Institute for Clinical and Translational Research Bionutrition Unit for developing and providing all study meals.
Sources of Funding:
This study was funded by the National Institute of Health: R01-HL104193 (Pollin), R35 HL135833 (Ginsberg), and KL2TR001874 (Reyes-Soffer). Additional support was provided by National Institutes of Health/National Center for Advancing Translational Science: 1UL1TR001873, the Mid-Atlantic Nutrition Obesity Research Center (P30DK072488), and the Geriatric Research, Education and Clinical Center, Baltimore Veterans Affairs Health Care Center.
NONSTANDARD ABBREVIATIONS AND ACRONYMS:
- Apo
Apolipoprotein
- LpL
Lipoprotein Lipase
- TG
Triglyceride
- VLDL
Very low density lipoprotein
- IDL
Intemediate Density Lipoprotein
- LDL
Low density Lipoprotein
- HDL
High Density Lipoprotein
- CVD
Cardiovascular disease
- GWAS
Genome Wide Association Studies
- FCR
Fractional Clearance Rates
- PR
Production Rates
- SD
Standard Deviation
Footnotes
Disclosures: The authors do not have relationships that are in confict with the current manuscript
References
- 1.Brown WV, Levy RI and Fredrickson DS. Studies of the proteins in human plasma very low density lipoproteins. J Biol Chem. 1969;244:5687–5694. [PubMed] [Google Scholar]
- 2.Brown WV and Baginsky ML. Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem Biophys Res Commun. 1972;46:376–382. [DOI] [PubMed] [Google Scholar]
- 3.LaRosa JC, Levy RI, Herbert P, Lux SE and Fredrickson DS. A specific apoprotein activator for lipoprotein lipase. Biochem Biophy Res Commun. 1970;41:57–62. [DOI] [PubMed] [Google Scholar]
- 4.Windler E, Chao Y and Havel RJ. Regulation of the hepatic uptake of triglyceride-rich lipoproteins in the rat: opposing effects of homologeous apolipoprotein E and individual C apoproteins. J Biol Chem. 1980;255:8303–8307. [PubMed] [Google Scholar]
- 5.Quarfordt S, Michalopoulos G and Schrimer B. The effect of human C apolipoproteins on the in vitro hepatic metabolism of triglyceride emulsions in rats. J Biol Chem. 1982;257:14642–14647. [PubMed] [Google Scholar]
- 6.Norum RA, Lakier JB, Goldstein S, Angel A, Goldberg RB, Block WD, Noffze DK, Dolphin PJ, Edelglass J, Bogorad DD and Alaupovic P. Familial deficiency of apolipoproteins A-I and C-III and precocious coronary-artery disease. N Engl J Med. 1982;306:1513–9. [DOI] [PubMed] [Google Scholar]
- 7.Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, Norum R and Brown WV. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J Clin Invest. 1986;78:1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito Y, Azrolan N, O’Connell A, Walsh A and Breslow JL. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. 1990;249:790–793. [DOI] [PubMed] [Google Scholar]
- 9.de Silva HV, Lauer SJ, Wang J, Simonet WS, Weisgraber KH, Mahley RW and Taylor JM. Overexpression of human apolipoprotein C-III in transgenic mice results in an accumulation of apolipoprotein B48 remnants that is corrected by excess apolipoprotein E. J Biol Chem. 1994;269:2324–2335. [PubMed] [Google Scholar]
- 10.Aalto-Setala K, Fisher EA, Chen X, Chajek-Shaul T, Hayek T, Zechner R, Walsh A, Ramakrishnan R, Ginsberg H and Breslow JL. Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice: Diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apoE on the particles. J Clin Invest. 1992;90:1889–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeda N, Li H, Lee D, Oliver P, Quarfordt SH and Osada J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J Biol Chem. 1994;269:23610–23616. [PubMed] [Google Scholar]
- 12.Sundaram M, Zhong S, Bou KM, Links PH, Zhao Y, Iqbal J, Hussain MM, Parks RJ, Wang Y and Yao Z. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res. 2010;51:150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hulley SB, Rosenman RH, Bawol RD and Brand RJ. Epidemiology as a guide to clinical decisions. The association between triglyceride and coronary heart disease. N Engl J Med. 1980;302:1383–9. [DOI] [PubMed] [Google Scholar]
- 14.Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A and Watts GF. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musunuru K KS. Surprises From Genetic Analyses of Lipid Risk Factors for Atherosclerosis. Circ Res 2016;118:579–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pare G and Anand SS. Mendelian randomisation, triglycerides, and CHD. Lancet. 2010;375:1584–6. [DOI] [PubMed] [Google Scholar]
- 17.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, Post W, McLenithan JC, Bielak LF, Peyser PA, Mitchell BD, Miller M, O’Connell JR and Shuldiner AR. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG and Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. [DOI] [PubMed] [Google Scholar]
- 19.Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, Masca N, Stirrups K, Kanoni S, Do R, Jun G, Hu Y, Kang HM, Xue C, Goel A, Farrall M, Duga S, Merlini PA, Asselta R, Girelli D, Olivieri O, Martinelli N, Yin W, Reilly D, Speliotes E, Fox CS, Hveem K, Holmen OL, Nikpay M, Farlow DN, Assimes TL, Franceschini N, Robinson J, North KE, Martin LW, DePristo M, Gupta N, Escher SA, Jansson JH, Van Zuydam N, Palmer CN, Wareham N, Koch W, Meitinger T, Peters A, Lieb W, Erbel R, Konig IR, Kruppa J, Degenhardt F, Gottesman O, Bottinger EP, O’Donnell CJ, Psaty BM, Ballantyne CM, Abecasis G, Ordovas JM, Melander O, Watkins H, Orho-Melander M, Ardissino D, Loos RJ, McPherson R, Willer CJ, Erdmann J, Hall AS, Samani NJ, Deloukas P, Schunkert H, Wilson JG, Kooperberg C, Rich SS, Tracy RP, Lin DY, Altshuler D, Gabriel S, Nickerson DA, Jarvik GP, Cupples LA, Reiner AP, Boerwinkle E and Kathiresan S. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaudet D, Drouin-Chartier JP and Couture P. Lipid Metabolism and Emerging Targets for Lipid-Lowering Therapy. Can J Cardiol. 2017;33:872–882. [DOI] [PubMed] [Google Scholar]
- 21.Bauer RC, Khetarpal SA, Hand NJ and Rader DJ. Therapeutic Targets of Triglyceride Metabolism as Informed by Human Genetics. Trends Mol Med. 2016;22:328–40. [DOI] [PubMed] [Google Scholar]
- 22.Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, Karmally W, Nandakumar R, Fontanez N, Obunike JC, Marcovina SM, Lichtenstein AH, Matthan NR, Matta J, Maroccia M, Becue F, Poitiers F, Swanson B, Cowan L, Sasiela WJ, Surks HK and Ginsberg HN. Effects of PCSK9 Inhibition with Alirocumab on Lipoprotein Metabolism in Healthy Humans. Circulation. 2016;135:352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Studying RR apolipoprotein turnover with stable isotope tracers - correct analysis is by modeling enrichments. Jour Lipid Research. 2006;47:2738–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramakrishnan R and Ramakrishnan JD. Using mass measurements in tracer studies--a systematic approach to efficient modeling. Metabolism. 2008;57:1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagashima K, Lopez C,Donovan D,Ngai C,Fontanez N,Bensadoun A,Fruchart-Najib J,Holleran S,Cohn JS,Ramakrishnan R,Ginsberg HN Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest. 2005;115:1323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan Y, Zhou H, Mahsut A, Rohm RJ, Berejnaia O, Price O, Chen Y, Castro-Perez J, Lassman ME, McLaren D, Conway J, Jensen KK, Thomas T, Reyes-Soffer G, Ginsberg HN, Gutstein DE, Cleary M, Previs SF and Roddy TP. Static and turnover kinetic measurement of protein biomarkers involved in triglyceride metabolism including apoB48 and apoA5 by LC/MS/MS. J Lipid Res. 2014;55:1179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ginsberg HN and Ramakrishnan R. Kinetic studies of the metabolism of rapidly exchangeable apolipoproteins may leave investigators and readers with exchangeable results. Arterio Thromb Vasc Biol. 2008;28:1685–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lassman ME MT, Lee A,Chappell DA,Wong O,Zhou H,Reyes-Soffer G,Ginsberg HN,Millar JS,Rader DJ,Gutstein DE,Laterza O Practical immunoaffinity-enrichment LC-MS for measuring protein kinetics of low-abundance proteins. Clin Chem. 2014;60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Havel RJ, Kane JP and Kashyap ML. Interchange of apoliproteins between chylomicrons and high density lipoproteins during alimentary lipemia in man. J Clin Invest. 1973;52:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huff MW, Fidge NH, Nestel PJ, Billington T and Watson B. Metabolism of C-apolipoproteins: kinetics of C-II, C-III1 and C-III2, and VLDL-apolipoprotein B in normal and hyperlipoproteinemic subjects. J Lipid Res. 1981;22:1235–1246. [PubMed] [Google Scholar]
- 31.Chan DC, Watts GF, Ooi EM, Ji J, Johnson AG and Barrett PH. Atorvastatin and fenofibrate have comparable effects on VLDL-apolipoprotein C-III kinetics in men with the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ooi EM CD, Watts GF, Chan DC, Ng TW, Dogra GK, Irish AB, Barrett PH. Plasma apolipoprotein C-III metabolism in patients with chronic kidney disease. J Lipid Res. 2011;52:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sacks FM ZC, Cohn JS. Complexities of plasma apolipoprotein C-III metabolism. Journal of Lipid Research. 2011;52:1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huff MW and Nestel PJ. Metabolism of apolipoproteins CII, CIII2, CIII2 and VLDL-B in human subjects consuming high carbohydrate diets. Metabolism. 1982;31:493–499. [DOI] [PubMed] [Google Scholar]
- 35.Wang H and Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–88. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi J, Miyashita K, Nakajima K and Mabuchi H. Hepatic Lipase: a Comprehensive View of its Role on Plasma Lipid and Lipoprotein Metabolism. J Atheroscler Thromb. 2015;22:1001–11. [DOI] [PubMed] [Google Scholar]
- 37.Foley EM and Esko JD. Hepatic heparan sulfate proteoglycans and endocytic clearance of triglyceride-rich lipoproteins. Progress in molecular biology and translational science. 2010;93:213–33. [DOI] [PubMed] [Google Scholar]
- 38.Ishibashi S, Perrey S, Chen Z, Osuga J, Shimada M, Ohashi K, Harada K, Yazaki Y and Yamada N. Role of the low density lipoprotein (LDL) receptor pathway in the metabolism of chylomicron remnants. A quantitative study in knockout mice lacking the LDL receptor, apolipoprotein E, or both. J Biol Chem. 1996;271:22422–7. [DOI] [PubMed] [Google Scholar]
- 39.Rohlmann A,MG, Hammer RE and Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM and Witztum JL. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6. [DOI] [PubMed] [Google Scholar]
- 41.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL and Esko JD. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. 2016;126:2855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham MJ, Lee RG, Bell TA III, Fu W, Mullick AE, Alexander VJ, Singleton W, Viney N, Geary R, Su J, Baker BF, Burkey J, Crooke ST and Crooke RM Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–1490. [DOI] [PubMed] [Google Scholar]
- 43.Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, Geary RS, Hughes SG, Viney NJ, Graham MJ, Crooke RM, Witztum JL, Brunzell JD and Kastelein JJ. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N Engl J Med. 2015;373:438–47. [DOI] [PubMed] [Google Scholar]
- 44.Pechlaner R, Tsimikas S, Yin X, Willeit P, Baig F, Santer P, Oberhollenzer F, Egger G, Witztum JL, Alexander VJ, Willeit J, Kiechl S and Mayr M. Very-Low-Density Lipoprotein-Associated Apolipoproteins Predict Cardiovascular Events and Are Lowered by Inhibition of APOC-III. J Am Coll Cardiol. 2017;69:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gregg RE, Zech LA, Schaefer EJ and Brewer HBJ. Tpe III hyperlipoproteinemia: Defective metabolism of an abnormal apolipoprotein E. Science. 1981;211:584–586. [DOI] [PubMed] [Google Scholar]
- 46.CB B Type III Hyperlipoproteinemia: Still Worth Considering? Prog Cardiovasc Dis. 2016;59:119–124. [DOI] [PubMed] [Google Scholar]
- 47.Norum RA, Forte TM, Alaupovic P and Ginsberg HN. Clinical syndrome and lipid metabolism in hereditary deficiency of apolipoproteins A-I and C-III, variant I. In: Angel A and Frohlich J, eds. Lipoprotein deficiency syndromes: Plenum Publishin0067; 1986. [DOI] [PubMed] [Google Scholar]
- 48.Langsted A, Freiberg JJ, Tybjaerg-Hansen A, Schnohr P, Jensen GB and Nordestgaard BG. Nonfasting cholesterol and triglycerides and association with risk of myocardial infarction and total mortality: the Copenhagen City Heart Study with 31 years of follow-up. J Intern Med. 2011;270:65–75. [DOI] [PubMed] [Google Scholar]
- 49.Mora S, Rifai N, Buring JE and Ridker PM. Fasting compared with nonfasting lipids and apolipoproteins for predicting incident cardiovascular events. Circulation. 2008;118:993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won HH, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, Weisburd B, Gupta N, Zaidi M, Samuel M, Imran A, Abbas S, Majeed F, Ishaq M, Akhtar S, Trindade K, Mucksavage M, Qamar N, Zaman KS, Yaqoob Z, Saghir T, Rizvi SNH, Memon A, Hayyat Mallick N, Ishaq M, Rasheed SZ, Memon FU, Mahmood K, Ahmed N, Do R, Krauss RM, MacArthur DG, Gabriel S, Lander ES, Daly MJ, Frossard P, Danesh J, Rader DJ and Kathiresan S. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wulff AB, Nordestgaard BG and Tybjaerg-Hansen A. APOC3 Loss-of-Function Mutations, Remnant Cholesterol, Low-Density Lipoprotein Cholesterol, and Cardiovascular Risk: Mediation- and Meta-Analyses of 137 895 Individuals. Arterioscler Thromb Vasc Biol. 2018;38:660–668. [DOI] [PubMed] [Google Scholar]
- 52.Rip J, Nierman MC, Ross CJ, Jukema JW, Hayden MR, Kastelein JJ, Stroes ES and Kuivenhoven JA. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol. 2006;26:1236–45. [DOI] [PubMed] [Google Scholar]
- 53.Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators SN, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, König IR, Weeke PE, Webb TR, Auer PL, Schick UM, Lu Y, Zhang H, Dube MP, Goel A, Farrall M, Peloso GM, Won HH, Do R, van Iperen E, Kanoni S, Kruppa J, Mahajan A, Scott RA, Willenberg C, Braund PS, van Capelleveen JC, Doney AS, Donnelly LA, Asselta R, Merlini PA, Duga S, Marziliano N, Denny JC, Shaffer CM, El-Mokhtari NE, Franke A, Gottesman O, Heilmann S, Hengstenberg C, Hoffman P, Holmen OL, Hveem K, Jansson JH, Jöckel KH, Kessler T, Kriebel J, Laugwitz KL, Marouli E, Martinelli N, McCarthy MI, Van Zuydam NR, Meisinger C, Esko T, Mihailov E, Escher SA, Alver M, Moebus S, Morris AD, Müller-Nurasyid M, Nikpay M, Olivieri O, Lemieux Perreault LP, AlQarawi A, Robertson NR, Akinsanya KO, Reilly DF, Vogt TF, Yin W, Asselbergs FW, Kooperberg C, Jackson RD, Stahl E, Strauch K, Varga TV, Waldenberger M, Zeng L, Kraja AT, Liu C, Ehret GB, Newton-Cheh C, Chasman DI, Chowdhury R, Ferrario M, Ford I, Jukema JW, Kee F, Kuulasmaa K, Nordestgaard BG, Perola M, Saleheen D, Sattar N, Surendran P, Tregouet D, Young R, Howson JM, Butterworth AS, Danesh J, Ardissino D, Bottinger EP, Erbel R, Franks PW, Girelli D, Hall AS, Hovingh GK, Kastrati A, Lieb W, Meitinger T, Kraus WE, Shah SH, McPherson R, Orho-Melander M, Melander O, Metspalu A, Palmer CN, Peters A, Rader D, Reilly MP, Loos RJ, Reiner AP, Roden DM, Tardif JC, Thompson JR, Wareham NJ, Watkins H, Willer CJ, Kathiresan S, Deloukas P, Samani NJ, Schunkert H. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med. 2016;374:1134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khetarpal SA ZX, Millar JS, Vitali C, Somasundara AVH, Zanoni P, Landro JA, Barucci N, Zavadoski WJ, Sun Z, de Haard H, Toth IV, Peloso GM, Natarajan P, Cuchel M, Lund-Katz S, Phillips MC, Tall AR, Kathiresan S, DaSilva-Jardine P, Yates NA, Rader DJ. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. 2017;23:1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horowitz BS, Goldberg IJ, Merab J, Vanni T, Ramakrishnan R and Ginsberg HN. Increased plasma and renal clearance of an exchangeable pool of apolipoprotein A-I in subjects with low levels of high density lipoprotein cholesterol. J Clin Invest. 1993;91:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ginsberg HN, Le N- A, Goldberg IJ, Wang-Iverson P, Gibson JC, Rubinstein A, Norum RA and Brown WV. Apolipoprotein B metabolism in subjects with deficiency of apolipoprotein C-III and A-I: Evidence that apolipoprotein C-III inhibits lipoprotein lipase in vivo. J Clin Invest. 1986;78:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gibson JC, Rubinstein A, Brown WV, Ginsberg HN, Greten H, Norum R and Kayden H. Apolipoprotein E containing lipoproteins in low density or high density lipoprotein-deficient states. Arteriosclerosis. 1985;5:371–380. [DOI] [PubMed] [Google Scholar]
- 58.Gabelli C BC, Santamarina-Fojo S, Martini S, Brewer HB Jr, Crepaldi G, Baggio G. Heterozygous apolipoprotein C-II deficiency: lipoprotein and apoprotein phenotype and RsaI restriction enzyme polymorphism in the Apo C-IIPadova kindred. Eur J Clin Invest. 1993;23:522–8. [DOI] [PubMed] [Google Scholar]
- 59.Brunzell JD and Schrott HG. The interaction of familial and secondary causes of hypertriglyceridemia: role in pancreatitis. J Clin Lipidol. 2012;6:409–12. [DOI] [PubMed] [Google Scholar]
- 60.Jensen MK, Rimm EB, Furtado JD and Sacks FM. Apolipoprotein C-III as a Potential Modulator of the Association Between HDL-Cholesterol and Incident Coronary Heart Disease. J Am Heart Assoc. 2012;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendivil CO, Rimm EB,Furtado J,Chiuve SE,Sacks FM Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation. 2011;124:2065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alaupovic P, McConathy WJ, Fesmire J, Tavella M and Bard JM. Profiles of apolipoproteins and apolipoproteins B-containing lipoportein particles in dyslipoproteinemias. Clin Chem. 1988;34:B13–B27. [PubMed] [Google Scholar]
- 63.Zheng C, Khoo C, Ikewaki K and Sacks FM. Rapid turnover of apolipoprotein C-III-containing triglyceride-rich lipoproteins contributing to the formation of LDL subfractions. J Lipid Res. 2007;48:1190–203. [DOI] [PubMed] [Google Scholar]
- 64.Mendivil CO, Zheng C, Furtado J, Lel J and Sacks FM. Metabolism of very-low-density lipoprotein and low-density lipoprotein containing apolipoprotein C-III and not other small apolipoproteins. Arterioscler Thromb Vasc Biol. 2010;30:239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.FM. S The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr Opin Lipidol. 2015;26:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.