

Abstract
Mouse models are indispensible tools for understanding the molecular basis of cancer. However, despite the invaluable data provided regarding tumor biology, due to inbreeding, current mouse models fail to accurately model human populations. Polymorphism is the essential characteristic that makes each of us unique human beings, with different disease susceptibility, presentation and progression. Therefore as we move closer toward designing clinical treatment based on an individual’s unique biological makeup it is imperative that we understand how inherited variability influences cancer phenotypes, how it can confound experiments and how it can be exploited to reveal new truths about cancer biology.
Introduction
Inherited variability, now called polymorphisms, has been recognized as an important contributor to mouse models of cancer for a hundred years. Early studies using outbred populations of mice established that cancer susceptibility was an inherited trait1, long before DNA was established as the genetic material. However it was recognized early on that variability due to uncontrolled genetic factors segregating in these populations was confounding the ability of researchers to interpret their results. As a result, starting with the pioneering work of C.C. Little, investigators began to develop inbred mouse strains to eliminate uncontrolled inherited variability, which resulted in the dilute brown albino (DBA) strain2, the first inbred strain. Shortly thereafter, additional strains were developed by L.C. Strong and others3.
Subsequently investigators began to use the nascent inbred strains in chemical carcinogenesis and genetic mapping experiments to establish the multigenic nature of cancer susceptibility4. Since much of this work was performed before the advent of molecular biology, the co-segregation of phenotypes of interest with visible markers such as the albino coat color mutation were used to map the first cancer susceptibility genes. Based on results using these strategies investigators not only recognized that different genetic backgrounds displayed significantly different susceptibilities to cancers, but were also able to begin to estimate the number of susceptibility genes and assign them to linkage groups5.
These strategies focusing on inherited factors of cancer susceptibility encompassed much of mouse cancer modeling in the first 60–70 years of the 20th century. With the development of recombinant DNA technology, there was a realization that cancers accumulate somatic mutations of endogenous genes, and the ability to manipulate and engineer the mouse genome radically changed the way that mouse cancer modeling was approached. Rather than looking at how population dynamics results in changes in cancer incidence, the focus is now on molecular mechanisms and modeling individual mutations. While genetically engineered mouse (GEM) models (e.g. transgenics, knockouts) have been extremely valuable for the tremendous advances in our understanding of the molecular etiology of cancer, it has come at a price. Ironically, considering the reason they were originally made was to model human cancer development, inbred GEM models represent, at best, single individuals in the human population. Thus it is difficult to successfully translate information garnered from inbred mouse models back into human populations.
The purpose of this article is therefore to re-examine the need to incorporate polymorphism-based population diversity into our analysis of GEM cancer models. Inherited polymorphism can have profound effects on experimental outcome in animal model studies. It is therefore important to recognize these potential confounds and to design experiments appropriately to account for any unexpected effects. Inherited polymorphism is not only an experimental problem, but also can provide valuable insights into biological mechanisms. Incorporating this aspect of biology into the research portfolio of the average cancer research laboratory would therefore be an important step toward improving the prognostic value of mouse models.
Problems with polymorphisms
An underappreciated problem with GEM models is the unrecognized re-introduction of polymorphisms into the experimental mix. The standard procedure to generate GEM models is to engineer the desired genetic alterations in embryonic stem (ES) cells, and then to introduce the engineered alterations into the germline via chimera with transmission monitored by outcrossing to other strains that have different coat colors (figure 1)6. This strategy can potentially result in animals that have an undefined, mixed genetic background, which will invariably increase genetic variability or even alter the phenotype of interest. The introduced variability does not necessarily result in subtle phenotypic changes. An example is the rescue of the embryonic lethal epidermal growth factor receptor (Egfr)-null mutation. On a CF-1 outbred background, Egfr homozygous mutant embryos die around implantation. However, on a 129/Sv inbred strain background embryos survive until mid-gestation, and when on an ALR/LtJ background, Egfr homozygous mutants survive until birth and can live as long as three weeks after birth7. Unless undefined segregating polymorphisms are bred out of GEM models, variability will continue to be a confounding problem. Another potential example of this problem was identified by a transgene-based study of the p53 arginine-to-serine point mutation seen in aflatoxin-associated hepatocellular carcinoma (HCC). Analysis of aflatoxin-induced HCC susceptibility in mice expressing the mutant p53 protein in a mixed C57BL/6 and DBA/2J background led to the conclusion that this point mutation enhanced the carcinogenic affect of aflatoxin8. What was not considered is that the DBA/2J genome itself is more sensitive than the C57BL/6 genome to aflatoxin-induced carcinogenesis, potentially due to polymorphic differences in xenobiotic metabolizing enzymes9.
Figure 1:
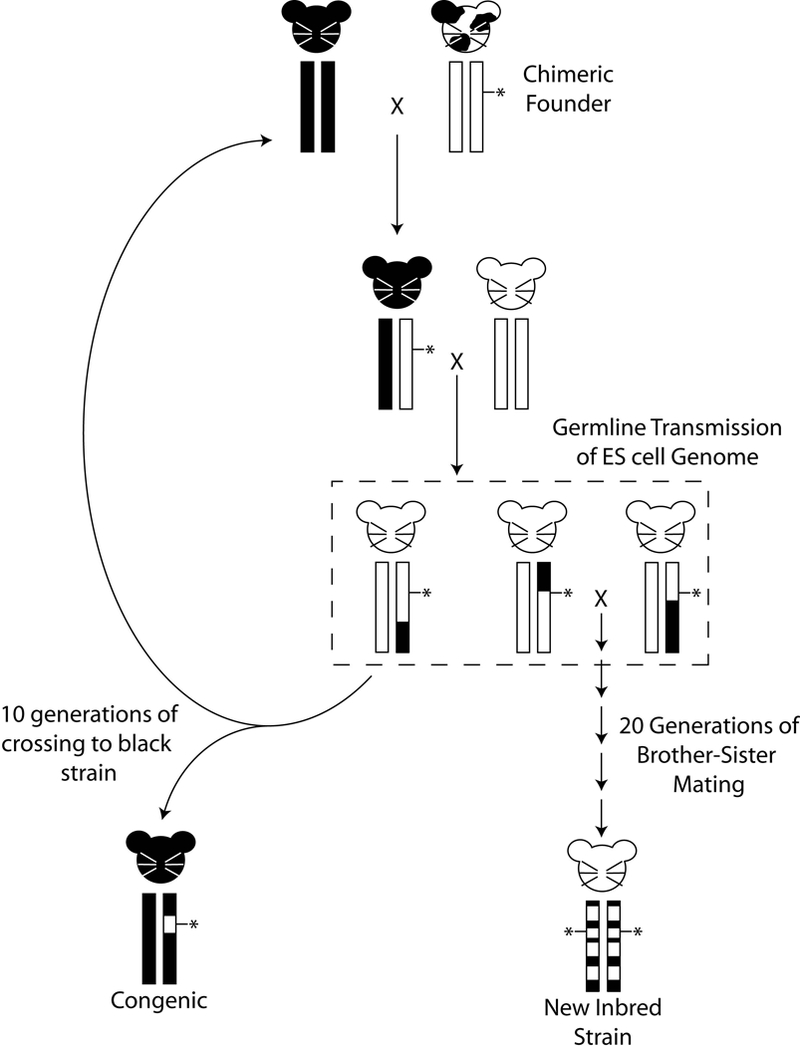
Potential genomic structures of genetically engineered mice, based on an albino embryonic stem (ES) cell line. The chimeric founder, after injection of albino ES cells into a C57BL/6 recipient blastocyst, is shown in the upper right. Germline transmission of the engineered chromosome (vertical rectangles) is performed by breeding the chimeric founder to a mouse with a different coat color. Coat color is indicated by the mouse cartoon. Percentage of the genome from the ES donor strain and recipient strains are depicted by the circles below the chromosome boxes. Since albinism is recessive, the F1 progeny is crossed back to an albino mouse. Generation of white mice in the second generation indicates successful germline transmission of the ES cell genome (dashed box). Note that the coat color mutation is usually not linked to the engineered locus and therefore they will segregate independently in the progeny. These mice will carry not only the engineered locus (indicated by the asterisk) but also 25% of the black genome. Repeated crossing of the engineered locus back to the black strain results in a congenic animal that is homozygous black for the entire genome except the region surrounding the locus of interest. Repeated brother-sister mating to carry the construct of interest homozygously without first generating congenics can result in novel inbred (recombinant congenic) strains that are composites of the ES cell donor and recipient genomes.
Not controlling background leads to common mishandling of GEM models. Investigators frequently propagate models generated after coat color transmission testing without repeated backcrossing to an existing inbred strain, which results in the generation of partially inbred strains. For models that can be carried as homozygotes, it is possible to develop a new strain that is a random mix of the original progenitor strains and without an experimental control (figure 1). In this situation phenotyping all of the littermates provides some level of experimental control. However, since the genotypes of both the experimental and control littermates are not stable, genotypes and phenotypes may shift over time, preventing unambiguous interpretation between sequential experiments. Novel gene-gene interactions within the derived GEM strain may lead to different baseline physiology compared to either progenitor strain. This problem could be exacerbated when combining two GEM models with ill-defined backgrounds. Thus, results obtained by comparing GEM models on uncontrolled backgrounds to the progenitors would be ambiguous since it may not be clear whether the phenotypic variation is due to the genetic background, the engineered gene(s), polymorphisms in the donor DNA flanking the engineered gene(s), or a combination of all of the above.
To reduce this problem, suppliers like The Jackson Laboratory backcross GEM models onto a single genetic background. This results in a congenic strain in which a single subchromosomal fragment of a donor strain is substituted for the same interval in a different inbred recipient strain (see figure 1). These congenic strains usually contain a segment of the ES cell donor genome surrounding the engineered alteration on a C57BL/6J background. While this greatly reduces the potential confounding affect of background polymorphism, it does not completely eliminate it. Appropriate care should therefore be exercised in the use and interpretation of GEM models, particularly when combining different engineered genes by breeding. To properly interpret results, all genotype classes, and in particular background matched controls, should be saved and phenotyped to most accurately assess any phenotype variation that may be due to the engineered genes compared to unknown and unanticipated variation due to genetic background.
The potential promise of polymorphisms
Intriguingly, polymorphisms in GEM models are not just experimental confounds. Genetic variation also represents an opportunity to better understand the complex physiology associated with cellular equilibrium and neoplastic transformation. As evidenced by the incomplete penetrance of breast cancer in BRCA1 and BRCA2 mutation carriers10, polymorphic variants in other genes can have major impacts on tumor incidence and biology even in patients with constitutional mutations in important tumor suppressor genes. Deep sequencing technologies are currently identifying many somatically altered cancer driver genes. Identification and characterization of inherited cancer modifier genes may therefore be a valuable complement, providing important information regarding how cells and organisms have evolved to try to prevent cancers. Even if polymorphic genes themselves are not identified, exploring construction of biological networks based on inherited rather than somatic diversity can provide critical information regarding molecular and cellular processes associated with disease initiation and progression11. Furthermore, since cancer modifying genetic networks may be affecting mechanisms not directly associated with driver mutations, these networks may be more amenable to pharmacological manipulation than the permanently mutated oncogenic genes.
The shift in mouse modeling from inherited to somatic genetics is easy to understand, being driven primarily by changes in technology. The rapid evolution of strategies to identify somatically altered genes has far outpaced our ability to identify naturally occurring variants that modify phenotypes of interest. For example the ability of massive parallel high throughput sequencing to identify potential cancer driver mutations is limited primarily by acquisition of sufficient high quality samples for analysis. Similarly, improvements in genetic mapping capabilities and technologies have resulted in detection of hundreds of modifier or susceptibility loci in both mice (Mouse Genome Informatics, www.informatics.jax.org) (Box 1) and humans12–14 The difficulty has not been identifying the presence of these modifier loci, but rather identifying and validating exactly which genes are responsible for modulating the phenotype of interest. Unlike tumor suppressors and oncogenes that have basically a digital phenotype, i.e. tumor or no tumor, polymorphic genes have an analog output. Thus there are significantly more barriers for the identification and validation these of genetic factors than for somatically altered genes.
Box 1
Useful online resources for Mouse Genetics and Cancer Modeling
Origins of Inbred Mice (reference 3) is available online at http://www.informatics.jax.org/morsebook/
Mouse Genome Informatics (MGI) (www.informatics.jax.org): This website is maintained by The Jackson Laboratory and integrates access to several databases providing genetic, genomic and biological data for the laboratory mouse to aid its use as a model of human diseases.
Mouse Tumor Biology Database (http://tumor.informatics.jax.org/mtbwi/index.do): This database is part of the MGI database. It integrates data on tumor frequency, incidence, genetics and pathology in mice to support the use of the mouse as a cancer model.
Mouse Phenome Database (http://phenome.jax.org): This database is also maintained by The Jackson Laboratory and contains strain characterization data (phenotype and genotype) for the laboratory mouse, to facilitate translational research.
electronic Models Information, Communication, and Education (eMICE) (http://emice.nci.nih.gov/): This database is maintained by the US National Cancer Institute and provides information about a wide variety of animal models of cancer, including mice.
Collaborative Cross (CC) Status (http://csbio.unc.edu/CCstatus/index.py): This website contains information on the current status of the CC project.
Recombinant inbred (RI) strains (http://www.jax.org/smsr/ristrain.html): This website contains information on available RI panels.
Kent Hunter received a B.Sc. in Biochemistry from the Pennsylvania State University in 1985. He did his graduate work in murine retrovirology with Nancy Hopkins at MIT and, subsequently, a postdoctoral study in murine genetics and genomics with David Housman, also at MIT. In 1996, he joined the faculty at the Fox Chase Cancer Center as an associate member. In 1999, he joined Laboratory of Population Genetics at the National Cancer Institute before moving to the Laboratory of Cancer Biology and Genetics at NCI in 2007. The focus of his laboratory is the use of systems genetics tools in mouse models to investigate the mechanisms of metastasis.
Despite these challenges significant progress has been made toward identifying low-penetrance modifier genes. Polymorphic genes affecting phenotypes (modifiers) have been identified in a number of mouse models of neoplasia. The first modifier identified was in the Apcmin model of dominant familial adenomatous polyposis. The modifier, known as modifier of Min1 (Mom1), was first detected by a reduction of intestinal adenomas when the C57BL/6-based Apcmin animal was breed to either AKR/J or MA/MyJ inbred strains15. Subsequent studies demonstrated that the modifier was a polymorphism in the gene encoding secretory type II phospholipase A2 (PLA2G2A) ADDIN EN.CITE 16. Modifiers have also been identified from chemical carcinogenesis screens. Investigators have taken advantage of the inherent difference in carcinogen-sensitivity of different mouse strains and subspecies to map and subsequently clone a polymorphic variant of the aurora kinase A (AURKA) gene that functions as a cancer susceptibility gene in both mouse and man17. Incorporation of GEM models in polymorphism screens permits the investigation of particular pathways or processing by using mouse strains selected for or engineered to express a phenotype of interest. For example genes and loci have been identified that modify the latency5, 6, growth7 and metastatic progression18 of transgene-induced tumors. Similarly, modifier loci have been identified using genetic engineering technologies to model the effects of inherited polymorphism on tumor suppressor biology19. Epidemiology studies for some of the low-penetrance mouse susceptibility genes suggest similar roles in human disease17, 20, 21.
The GEM models have a number of attractive features for screens for inherited variants that modify phenotypes of interest. First, they represent specific mutational events and tumor subtypes. Second, screens can be performed in controlled environments to reduce the experimental “noise” that cannot be controlled or even fully described in human populations. Third, the screens are relatively rapid, due to the short lifespan of the mouse. Fourth, susceptibility loci can be identified with relatively small sample sizes compared to large multi-institution, genome-wide association studies in humans because the lineage and breeding history of the population is known. In addition, because the breeding structure and environment of the experimental subjects can be controlled mouse genetics screens usually require many fewer individuals than human studies to achieve statistically significant results. Finally, unlike epidemiology studies, it is possible to directly validate any gene in the mouse by generating new GEM models to test the role of specific genes in the phenotype of interest.
Exploiting polymorphism in mouse models
To fully exploit the potential of inherited polymorphism it is necessary to rapidly and efficiently identify the variant genes of interest. As alluded to above, a number of strategies have been developed to achieve this goal (ex.22–24). Due to space limitations the intent of this article is not to comprehensively review the relative strengths and weaknesses of these approaches. Instead the focus will be on a relatively new resource that can be adapted for use by the non-geneticist for not only performing population studies on their favorite mouse models, but also to easily integrate their results with independent studies from other laboratories.
One way to rapidly and efficiently identify variant genes of interest would be for individual investigators to do the population analysis and genetic mapping using a common genetic mapping resource, based on a recombinant inbred (RI) panel. RI panels are developed from intercrosses of established strains (figure 2A)25. F1 progeny from progenitor strains are then bred by strict brother-sister mating for 20 or more generations to yield new sublines that are genetic mixes of the original parents25. If sufficient numbers of sublines are generated, inherited traits that differ between the progenitor strains can be readily mapped by screening the trait across the sublines and comparing the phenotype to the segregation of the parental genomes. Importantly, since the sublines are inbred, genotyping only needs to be performed once. Subsequently, all additional traits can be mapped by simply using the pre-existing genetic mapping information. Furthermore, multiple animals with the identical genotype can be screened. For phenotypes with significant variation due to random or uncontrollable factors the ability to phenotype multiple animals of the same genotype enables a more precise measurement of the influence of genotype versus random fluctuation on a complex trait, which improves genetic mapping and resolution. Moreover, since the RI panels are inbred they represent a stable source of identical segregating genotypes. In standard genetic mapping panels based on intercross or backcross strategies each animal is genetically unique and can therefore only be used to study a single phenotype. In contrast, RI panels, based on panels of inbred strains, are an infinite source of identical animals that segregate different segments of the original donor genomes. This feature permits in at least some instances the opportunity for multiple investigators with similar experimental designs to assay and integrate the genetic screens on a single genetic mapping panel. Furthermore, the inbred nature of RI lines provides an immortalized, virtually unlimited tissue resource, which facilitates the incorporation of new analyses with historical data as novel technologies are developed.
Figure 2:
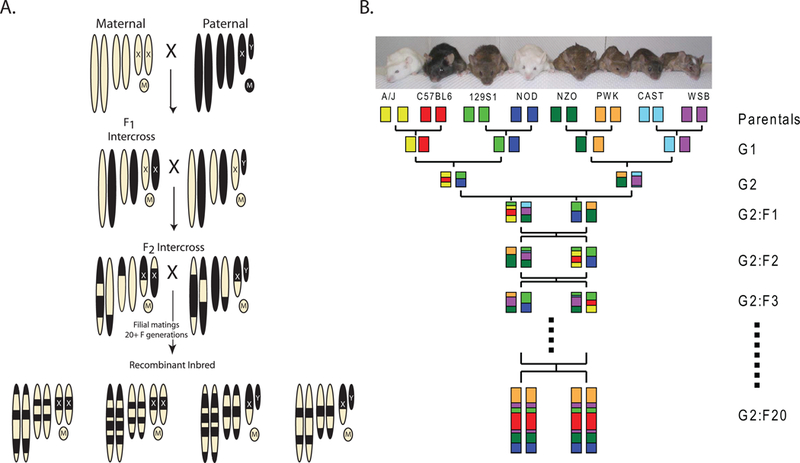
Strategies to generate Recombinant Inbred (RI) panels and the Collaborative Cross. A) Mating strategy to generate a standard RI panel. The chromosomes of the maternal strain are depicted by the tan ovals. The paternal strain chromosomes are black. Mitrochondrial genomes are depicted by circles with M’s. The resulting RI panel substrains are inbred chimeras of the original two parental strains, as indicated at the bottom of the figure. RI panels usually consist of 13–75 substrains (RI strains – The Jackson Laboratory, http://www.jax.org/smsr/ristrain.html) (Box 1). B) The eight-way funnel breeding design of the Collaborative Cross. One example of the funnel design is shown here. Additional funnels are generated by changing the position of the parental strains at the top of the funnel. The genomes of each of the eight progenitor strains are indicated by colored boxes. The funnel design incorporates all eight genomes randomly until inbreeding begins after the G2:F1 generation. The ultimate goal of the CC is to generate more than a hundred sublines. Current status of the CC can be found at the Collaborative Cross Status website (http://csbio.unc.edu/CCstatus/index.py).
These advantages were the basis for the recent development of a novel RI panel known as the Collaborative Cross (CC; Collaborative Cross Status website, http://csbio.unc.edu/CCstatus/index.py; see figure 2B and Box 1)26. The CC reference panel was built upon this foundation as an easy to use population genetics tool. To better model the diversity observed in human beings, the CC is being generated from eight progenitor mouse strains, including three wild-derived strains. A randomized breeding scheme and goal to generate a panel of hundreds of sublines combined with computational strategies to identify segregating haplotypes will theoretically permit mapping down to the megabase level. This level of resolution, combined with whole genome sequencing of the eight progenitor strains and other system biology tools will enable rapid identification of candidate genes for validation. Thus, in many ways, CC RI panel was designed to reduce much of the tedious and expensive steps to achieve high resolution mapping by conventional backcross or intercross analysis, in a format that can easily be utilized by investigators without extensive experience in meiotic genetics.
Utilization of this resource would be relatively simple. For cancer models with a dominant phenotype, like transgenics and some knockouts, investigators would examine population diversity by simply generating F1 progeny between their models and some or all of the CC lines (figure 3A). Since half of the chromosomes from F1 cross will be from the GEM model, any loci that would modify the normal GEM model phenotype would have to be attributed to DNA from the RI strain. Thus identification of modifier loci can be performed by comparing the phenotypes of all of the GEM x RI sublines outcrosses with the previously known RI genotypes. In our hands, using one of the original RI panels27, this strategy has significantly contributed to the identification of metastasis-related susceptibility genes18, 28 as well as being the basis for further systems genetic analyses20, 29, 30.
Figure 3:
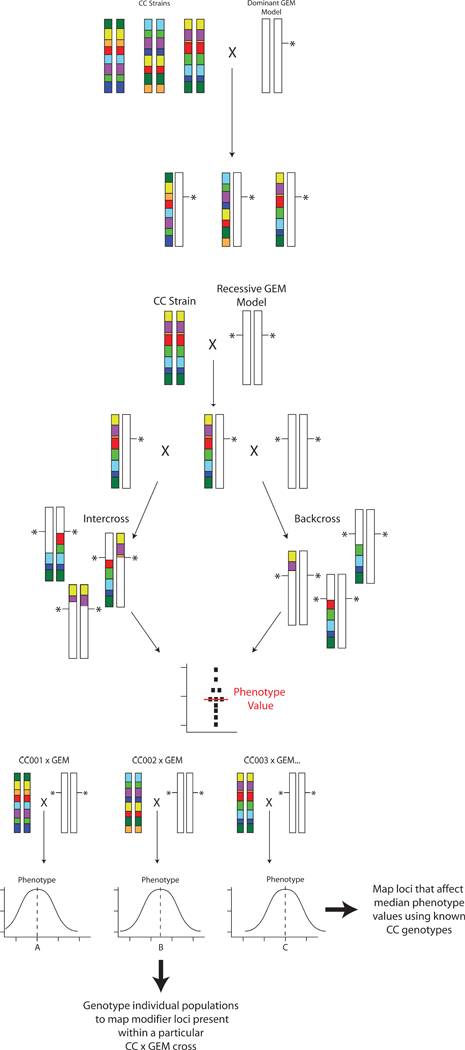
Mating strategies for mapping cancer modifiers of GEM models using the Collaborative Cross. A) Dominant transgene or haploinsufficient GEM strategy. GEM models are bred to the individual CC lines and phenotyped. Modifier genes are identified by comparing the phenotypes of each F1 to the known haplotypes of each CC line. B) Mating strategy for recessive GEM models. The GEM model is bred to CC lines to generate F1 animals, which are then intercrossed (left) or backcrossed (right) to the GEM model to produce homozygous knockouts. Due to the segregation of the CC genome in these animals, a small population is phenotyped to generate an median phenotype value for that population, based on both the segregating background and the engineered locus for each CC line. C) Genetic mapping for recessive GEM models. Each of the CC x GEM crosses produced as shown in figure 3B would produce a population with a distribution of the phenotype in question. The median phenotypic value of each CC x GEM cross, however, would likely be different depending on the complement of modifiers introduced by each CC line. These median values for each cross can then be used as a “meta-phenotype” to map modifiers that influenced the median phenotype value by comparison to the CC parental genotypes. No additional genotyping is required for this. Additional linkage information can be obtained by performing genotyping any of the CC x GEM crosses to further map and/or refine modifiers present in any particular CC subline of interest.
For GEM models that require homozygosity to generate a cancer phenotype the situation is somewhat more complex. The investigators would need to generate F1 hybrids with multiple CC lines and then intercross the F1 progeny or backcross them to the GEM parental line. The resulting intercross F2 or backcross N2 progeny would display a new distribution of the phenotype for each GEM x CC subline combination due to the segregation of the CC genome. The median phenotype value from each GEM x CC subline cross could be determined and this value used as a “meta”-phenotype (figure 3B) for mapping using the known CC parental genotypes to identify loci that alter the median phenotype across the CC panel (figure 3C). Additional complementary linkage information could also be obtained by performing high density genotyping of interesting GEM x CC subline combinations to map individual modifiers present within a particular GEM x CC pair (figure 3C). Genetic mapping and identification of potential candidate modifier genes would be performed computationally by analyzing F1 phenotypes with the pre-existing CC genotype data.
The cost to understand how underlying polymorphism may be impacting the interpretation of GEM experiments for the average investigator would therefore be limited to animal breeding and housing costs. Although these are not insubstantial expenses, information gleaned from these experiments may re-direct time, effort and resources toward avenues of research more representative of the human population rather than experimental systems that represent only a fraction of the human cancer population. Furthermore, elucidating the variability in cancer that is encoded by inherited polymorphism may reveal unexpected insights and interconnections that could be exploited clinically to prevent or treat neoplastic disease.
In summary, as the cancer research and oncology communities continue toward treatment based on an individual’s unique characteristics, all the factors that influence tumor biology must be considered. Technology and computational capabilities have advanced to the point where systems wide interactions can be constructed and examined. Our mouse models of cancer need to fully embrace this complexity by reintroduction of population diversity into cancer modeling rather than relying on single variable systems based on inbred strains. Incorporating the effect of inherited polymorphism with that of somatic mutation should not only better inform us of who is susceptible to cancer, but also aid for example in identifying patients susceptible to specific drug toxicities, which therapies may be most effective in a given individual, and which individuals have tumors that are most likely to progress. Just as polymorphism makes each of us unique individuals, being aware of, and exploiting population diversity will improve our ability to more accurately model cancer in animal systems to improve cancer outcomes.
Acknowledgements
I would like to thank Drs. Jude Alsarraj and David Threadgill for their insightful comments on this manuscript. I would also like to apologize to the many investigators whose work was not discussed in this manuscript due to space constraints. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
These reviews cover multiple methods (22) or specific methods (23,24)
Go ahead an leave out the dead mice.
The phenotype could be something like tumor burden, or metastasis counts. I wonder whether it might be better to have a scatterplot with different medians, rather than a distribution curve. I drew the figure to be rather generic on purpose, since I did not want to imply any limitation on the type of phenotype involved. I can alter this if necessary.
Reference
- 1.Slye M The Incidence and Inheritability of spontaneous tumors in Mice : (Second report.). J Med Res 30, 281–98 (1914). [PMC free article] [PubMed] [Google Scholar]
- 2.Little C & Tyzzer E Further experimental studies on the inheritance of susceptibiilty to a transplantable tumor, carcinoma (J. w. A.) of the Japanese waltzing mouse. J Med Res 33, 393–453 (1916). [PMC free article] [PubMed] [Google Scholar]
- 3.Strong LC in Origins of Inbred Mice (ed. H CM III) (Academic Press, 1978). [Google Scholar]
- 4.Heston WE Genetic analysis of susceptibilty to induced pulmonary tumors in mice. JNCI 3, 69–78 (1942). [Google Scholar]
- 5.Heston WE Relationship between susceptibility to induced pulmonary tumors and certain known genes in mice. J Natl Cancer Inst 2, 127–132 (1942). [PubMed] [Google Scholar]
- 6.Capecchi MR Altering the genome by homologous recombination. Science 244, 1288–92 (1989). [DOI] [PubMed] [Google Scholar]
- 7.Strunk KE, Amann V & Threadgill DW Phenotypic variation resulting from a deficiency of epidermal growth factor receptor in mice is caused by extensive genetic heterogeneity that can be genetically and molecularly partitioned. Genetics 167, 1821–32 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghebranious N & Sell S The mouse equivalent of the human p53ser249 mutation p53ser246 enhances aflatoxin hepatocarcinogenesis in hepatitis B surface antigen transgenic and p53 heterozygous null mice. Hepatology 27, 967–73 (1998). [DOI] [PubMed] [Google Scholar]
- 9.McGlynn KA et al. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Res 63, 4594–601 (2003). [PubMed] [Google Scholar]
- 10.Struewing JP et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336, 1401–8 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Quigley DA et al. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature 458, 505–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber JL & May PE Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 44, 388–96 (1989). [PMC free article] [PubMed] [Google Scholar]
- 13.Pletcher MT et al. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol 2, e393 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlson CS et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 74, 106–20 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moser AR, Dove WF, Roth KA & Gordon JI The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol 116, 1517–26 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacPhee M et al. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell 81, 957–66 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Ewart-Toland A et al. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat Genet 34, 403–12 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Park YG et al. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat Genet 37, 1055–1062 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reilly KM et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci U S A 101, 13008–13 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crawford NP et al. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet 3, e214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crawford NP et al. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res 8, R16 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darvasi A Experimental strategies for the genetic dissection of complex traits in animal models. Nat. Genet. 18, 19–24. (1998). [DOI] [PubMed] [Google Scholar]
- 23.Nadeau JH, Singer JB, Matin A & Lander ES Analysing complex genetic traits with chromosome substitution strains. Nat Genet 24, 221–5 (2000). [DOI] [PubMed] [Google Scholar]
- 24.de Koning JP, Mao JH & Balmain A Novel approaches to identify low-penetrance cancer susceptibility genes using mouse models. Recent Results Cancer Res 163, 19–27; discussion 264–6 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Bailey D in The Mouse in Biomedical Research (ed. Fox J) 223–239 (Academic, New York, 1981). [Google Scholar]
- 26.Churchill GA et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36, 1133–7 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Mucenski ML, Taylor BA, Jenkins NA & Copeland NG AKXD recombinant inbred strains: models for studying the molecular genetic basis of murine lymphomas. Mol Cell Biol 6, 4236–43 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hunter KW et al. Predisposition to efficient mammary tumor metastatic progression is linked to the breast cancer metastasis suppressor gene Brms1. Cancer Res 61, 8866–72 (2001). [PubMed] [Google Scholar]
- 29.Crawford NP et al. Bromodomain 4 activation predicts breast cancer survival. Proc Natl Acad Sci U S A 105, 6380–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crawford NP et al. The Diasporin Pathway: a tumor progression-related transcriptional network that predicts breast cancer survival. Clin Exp Metastasis 25, 357–369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]