

Abstract
Acute lung injury (ALI) is a common complication of sepsis that often leads to fatal lung disease without effective therapies. It is known that bone marrow derived macrophages are important in resolving the inflammation and maintaining tissue homeostasis. Here, we hypothesize that treatment in combination of DNA methyl transferase inhibitor (DNMTi) 5-Aza 2-deoxycytidine (Aza) and histone deacetylase inhibitor (HDACi) Trichostatin A (TSA) mitigates the inflammation induced pyroptosis and apoptosis during endotoxemia induced ALI. To test this hypothesis, the mice challenged with a sublethal dose of LPS followed by one-hour post-treatment with a single dose of Aza and TSA intraperitoneally showed a substantial attenuation of apoptosis and inflammation. Importantly, we observed significant changes in the mitochondrial membrane structure, and lower levels of DNA fragmentation, reduced expression of apoptotic and pyroptotic genes both transcriptionally and translationally in LPS induced BMDMs treated by a combination of Aza and TSA than in LPS-induced BMDMs treated with either drug alone. The protection was mediated by an inhibition of JNK-ERK and STAT3-JMJD3 activated pathways. Thus, targeting these important signaling pathways with the combination of Aza and TSA would be a good treatment modality for ALI.
Keywords: epigenetic modifiers, acute lung injury, inflammation, STAT3, JMJD3, pyroptosis, apoptosis
Graphical abstract
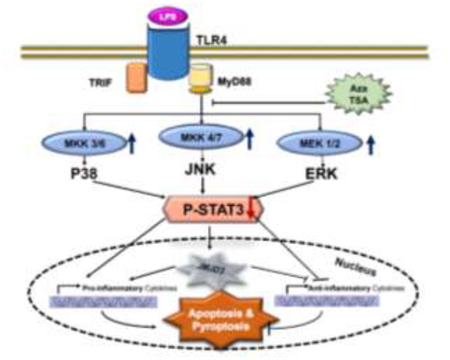
Proposed molecular mechanisms of the combination Aza and TSA treatment mediated protection on endotoxemia induced macrophage apoptosis and pyroptosis. In bone maroow derived macrophages (BMDMs), LPS activates JMJD3- STAT3 signaling pathway by MEK/MAP kinases. Moreover, the increase of JMJD3 by LPS induces pro-inflammatory cytokines, and result in apoptosis of the cells. However, epigenetic modifiers Aza and TSA, which inhibits the activity of MAPK, JMJD3-STAT3 signaling and restores mitochondrial membrane integrity, and thereby reverses the apoptosis and pyroptosis of BMDMs induced by LPS.
1. Introduction
During acute lung injury (ALI), pathogens interact with cells of the immune system and activate an inflammatory response involving sustained changes in gene expression. Even though the inflammatory response is vital for immune defense, unrestricted production of inflammatory proteins can cause substantial damage to the local tissue environment and be deleterious. Successful treatment strategies include mechanical ventilation, antibiotics and fluid-restrictive resuscitation strategies, are the current supportive treatment options (Brower and Fessler, 2000; Dellinger et al., 2008; Wiedemann et al., 2006). However, to date, no specific pharmacological treatment is available that targets the underlying pulmonary inflammation during the development or the clinical course of ALI. Macrophages, neutrophils, and endothelial cells are crucial in mediating the inflammatory response and the loss of lung vascular barrier function in lipopolysaccharide (LPS)-induced ALI (Di et al., 2012; Wang et al., 2011). There are studies showed the significance of circulating monocytes which have derived from bone marrow in the resolution of inflammation (O’Dea et al., 2009). We have therefore selected bone marrow derived macrophages (BMDMs) in our study because of their important role in regulating inflammation (Kochanek et al., 2012; Song et al., 2001). When macrophages are exposed to LPS, a conserved component of the Gram-negative bacteria, macrophages release a large number of regulatory molecules that recruit and activate other immune cells to help in fighting with the bacterial infection. Toll-like receptor 4 (TLR4) is critical for the recognition of LPS and is expressed in various cells, including macrophages. Binding of LPS with the TLR4 activates intracellular signal transduction pathways, including MAPK to produce an inflammatory response. The accompanying inflammatory response is mainly characterized by a massive release of active inflammatory molecules include tumor necrosis factor-α (TNF α), interleukin 1 (IL-1), IL-6, and arachidonic acid metabolites and many others.
The involvement of epigenetic modifiers during lung inflammation, cell remodeling and protection of microvascular permeability is increasingly being appreciated. Severe inflammation has been shown to be associated with histone deacetylase (HDAC)/histone acetyltransferase (HAT) imbalance, which in turn influences the acetylation status of histones in different organs, including lungs and heart (Butt et al., 2009). Combination of Aza and Trichostatin A treatment in hematopoietic stem cells affects methylation patterns of CpG sites and induces changes in the acetylation status of histone at the H4 region (Araki et al., 2007; Milhem et al., 2004). Therefore, we reasoned and examined the combination of Aza and TSA on inflammatory/anti-inflammatory balance during LPS-induced BMDMs.
Studies have shown that inflammation causes macrophages and endothelial cells to undergo apoptosis and pyroptosis during ALI, which is the key cellular event that triggers different physiological processes, including clonal selection of lymphocytes (Albert, 2004; Cheng et al., 2017; Rock and Kono, 2008) and removal of inflammatory cells (Suzuki et al., 2004; Xaus et al., 2000). Apart from macrophages, LPS also causes apoptosis and/or pyroptosis in B memory cells (Yokochi et al., 1996), CD4+8+ thymocytes, lymphoid organs (Kato et al., 1997), and endothelial cells (Cheng et al., 2017; Choi et al., 1998; Munshi et al., 2002). Thus, discovery of pharmacological agents that mitigate the LPS-induced apoptosis and pyroptosis are desperately needed. It is now well established that IL-10 activates the STAT3 signaling pathway responsible for the anti-inflammatory effect both in vivo and in vitro (El Kasmi et al., 2006; Kobayashi et al.,2003; Lang et al., 2002). STAT3 translocate into the nucleus where it binds to the promoter region of target genes and regulates the expression of specific anti-inflammatory genes, including those involved in the differentiation of M2 macrophages. IL-10 mediated STAT3 signaling blocks the inflammatory pathway associated with NFKB and MAPK. It has been reported that in response to inflammatory stimuli, histone lysine demethylases 6B (KDM6B) or JMJD3 is immediately induced by NF-κB in primary mouse macrophages (De Santa et al., 2007). In an early immune response, there is an increased expression of JMJD3 demethylase mRNA after inhibition of STAT3 (Sherry-Lynes et al., 2017). This increased expression of JMJD3 is responsible for the activation of inflammatory genes followed by apoptosis genes and therefore epigenetically regulates the inflammatory response (Das et al., 2012; De Santa et al., 2009). We have already shown that combined treatment with Aza (DNMTi) and TSA (HDACi) attenuates inflammation and promotes anti-inflammatory M2 macrophage phenotype in mice with ALI (Thangavel, J. et al., 2015). However, the molecular mechanisms behind the therapeutic effect of these epigenetic modifiers against inflammation induced cell apoptosis and pyroptosis during ALI are not known. In our present study, we revealed that the inflammatory pathway associated with MAPK is inhibited by treatment with Aza and TSA mediated activation of STAT3 that leads to the suppression of JMJD3. This may explain the anti-inflammatory followed by anti-apoptotic and anti-pyroptotic effects of Aza and TSA in macrophages during inflammation.
2. Materials and Methods:
2.1. Antibodies and reagents
We used antibodies for p-JNK, total JNK, p-ERK, total-ERK, AceH3K9, H3K917Me3, Bcl-XL, Caspase3, cleaved Caspase 3, Caspase 11, ILβ and cytochrome C (Cell Signaling Technology); β-actin and BAX (Santa Cruz Biotechnology, Inc.). Secondary antibodies were IRDye680 conjugated anti-mouse IgG and IRDye680 conjugated anti-goat IgG, IRDye800 conjugated anti-rabbit IgG and IRDye800 conjugated anti-goat IgG (Rockland, PA) (LICOR); TRITC- and FITC-conjugated donkey anti–mouse, anti–goat, and anti–rabbit (Jackson ImmunoResearch Laboratories, Inc.), and DAPI (Life-Tech); Antibodies CD68 (e-Bioscience), JC-1 staining kit (Thermo Fisher Scientific), and TUNEL assay kit (TMR Tunel staining kit, Roche) JNK-inhibitor SP600125 (Calbiochem) and ERK–inhibitor PD98059 (Calbiochem).
2.2. Mice
Mice (C57BL/J6, 8–10 wks old, male) were obtained from Jackson Laboratory. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD) and were approved by the institutional animal care and use committees of the University of Kansas Medical Center Kansas City, Kansas. All experiments were performed on 8- to 10-week-old mice.
2.3. Primary mouse bone marrow-derived macrophages (BMDMs) culture
BMDM culture was performed in C57BL6 mice as described by us earlier (Thangavel, J. et al., 2015). Briefly, BMDMs were isolated from the tibias and femurs of mice and cultured in DMEM supplemented with 10% FBS along with 10% L929 cell conditioned medium on 10-cm diameter cell culture dishes. After 7 days of culture, the cells were stained with CD11b and analyzed by flow cytometry.
2.4. DNA fragmentation assay: Analysis of DNA fragmentation.
BMDMs (107 cells) were treated with LPS (1 μg/ml) in the presence or absence of the combination of the epigenetic modifiers Aza and TSA for 24 h. After each treatment, cellular DNA was extracted as reported previously (Watabe et al., 1996). Briefly, cells were collected by centrifugation and washed with PBS and then lysed in a solution of 10 mM Tris-HCl (pH 8.0), 10 mM EDTA, 0.5% (wt/vol) sodium dodecyl sulfate (SDS), and 0.1% (wt/vol) RNase A, with incubation for 60 min at 50°C. The lysates were incubated for an additional 60 min at 50°C with proteinase K (1 mg/ml) and then subjected to electrophoresis in a 1% (wt/vol) agarose gel in 40 mM Tris-acetate buffer (pH 7.4) at 50 V. After electrophoresis, DNA was visualized by staining with ethidium bromide.
2.5. Immunofluorescence staining of lung tissue
Paraffin sections (5 μm thick) were deparaffinized, the sections were blocked with goat serum for 30 mins at 37 °C. Sections were immunostained with rabbit polyclonal anti-BAX antibody (1:100; Cell signaling, Beverly, MA USA) and goat polyclonal anti-CD68 antibody (1:100, eBioscience, USA) at 4 °C overnight. The sections were then incubated with fluorescence conjugated secondary antibody for 1 hour at 37 °C. The slides were washed with wash buffer for three times and mounted with DAPI. All immunofluorescence staining was photographed using either confocal and immunofluorescence microscope.
2.6. Determination of mitochondrial membrane potential:
Cells were harvested (at least 2×105) from experimental samples and brought total volume up to 1 ml of complete medium and then 2.5 mg/ml JC-1 added to the cell suspension. Cell suspension vortexed vigorously to mix the dye until is well dissolved, giving a uniform red-violet color. The samples were kept in a dark place at room temperature for 15–20 minutes. Then the samples were washed twice by centrifuging at 500 g for 5 min with a double volume of PBS. Finally, resuspend in 0.3 mL of PBS, and analyze immediately by the flow cytometer, (FACSCalibur, BD Biosciences) using Cell Quest software and the data were analyzed by FlowJo software (Tree Star, Ashland, OR).
2.7. Western Blot analysis:
Western blot analysis of p-JNK, p-ERK, JNK, ERK, Bcl-xl, caspase3 and Bax proteins were performed as described by us earlier (Thangavel et al., 2014; Thangavel, J. et al., 2015). Protein bands were detected by a LiCor Odyssey scanner system. Band intensities were quantified using ImageJ (National Institutes of Health). The ratio of band intensity was calculated by using the value of band intensity of experimental protein divided by its respective loading control. Data are expressed as means ± SEM of three replicated experiments.
2.8. Quantitative RT-PCR
The LPS-induced BMDMs untreated and treated with Aza and TSA from a six-well plate were washed once in PBS and harvested for qRT-PCR analysis as described by us earlier (Thangavel et al., 2014; Thangavel, J. et al., 2015). The relative mRNA expression of target genes was normalized to endogenous GAPDH control gene (Applied Biosystems). The primers are summarized in online Table 1. Results were expressed as fold change in expression, and values were calculated as ratio of induced expression-to-control expression.
Table1:
RT-PCR primer sequence
| Gene | Forward | Reverse |
|---|---|---|
| TLR4 | ATGGCATGGCTTACACCACC | GAGGCCAATTTTGTCTCCACA |
| MyD88 | AGGACAAACGCCGGAACTTTT | GCCGATAGTCTGTCTGTTCTAGT |
| BAX | AAGCTGAGCGAGTGTCTCCGGCG | GCCACAAAGATGGTCACTGTCTGCC |
| BAD | CCAGTGATCTTCTGCTCCACATCCC | CAACTTAGCACAGGCACCCGAGGG |
| Cytochrome C | GGAGGCAAGCATAAGACTGG | TCCATCAGGGTATCCTCTCC |
| Bcl2 | CTCGTCGCTACCGTCGTGACTTCG | CAGATGCCGGTTCAGGTACTCAGTC |
2.9. Preparation of cytosolic extract and immunoblot analysis of cytochrome c release
To carry out cytochrome c translocation studies, cellular sub fractionation was performed as previously reported [39] with minor modifications. At the end of LPS treatment, cells were washed twice with ice-cold PBS. The cell pellet was resuspended in 300 μl of extraction buffer containing 200 mM mannitol, 70 mM sucrose, 20 mM HEPES–KOH, pH 7.4, 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 0.1 mM PMSF and protease inhibitors (Complete Cocktail; Marck bioscience). After 20 min incubation on ice, cells were homogenized by 30–40 strokes with a glass Dounce homogenizer on ice, and resulting homogenates were left on ice for an additional 20 min. Homogenates were centrifuged at 600 g for 15 min at 4°C, and resulting supernatant was further centrifuged at 12000rpm for 30 min at 4°C, to yield cytosolic extract. The pellet was dissolved in lysis buffer to get the mitochondrial fraction.
2.10. TUNEL Assay
To assess the Aza and TSA treatment mediated survival of macrophages that are present in lung tissues, we performed an apoptosis assay on mice lung tissue collected on day 6. Cell death was assessed with the terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) kit according to the manufacturer’s instructions (Roche Applied Science, Indianapolis, IN). After TUNEL staining, sections were examined by confocal microscopy.
2.11. Immunoprecipitation and Western analysis
Immunoprecipitation and Western analyses of STAT3 and JMJD3 proteins were performed as described earlier (Rajasingh and Bright, 2006). Briefly, The proteins were resolved on 10% SDS-PAGE and electrophoretically transferred to a PVDF membrane (EMD Millipore, Billerica, MA). The membranes were incubated with IRDye -conjugated IgG antibody. Protein bands were detected by a LiCor Odyssey scanner system. The blots were stripped and reprobed with antibodies specific to JMJD3 or acH3K9 or H3K27me3 to examine the coexistence with STAT3.
2.12. Induction of ALI
ALI in C57BL/6J mice was induced by intraperitoneal administration of LPS (10 mg/g BW). One hour after the LPS induction, the mice received PBS or Aza (1 mg/g BW) or TSA (1 mg/g BW) or in combination of Aza and TSA. The lung tissues were harvested for histological and gene expression analyses.
2.13. Statistics
All data was expressed with the mean±SD. Student’s t-test was conducted for the statistical analysis to identify significant differences. Data was considered as statistically significance at p<0.05. GraphPad Prism version 5.0 (San Diego, CA, USA) and JPEG software were applied for our statistical analyses.
3. Results:
3.1. Induction of apoptosis by LPS is rescued by the combinatorial treatment of Aza and TSA
To understand the effect of combination of treatment of epigenetic modifiers Aza and TSA on LPS-induced cell death, BMDMs were challenged with LPS (1μg/ml) and treated with Aza and TSA for 24 h in culture and assessed for DNA fragmentation. It was found that reduced level of DNA fragmentation was observed in the BMDM cells after treatment with the epigenetic modifiers, Aza and TSA (Fig. 1A). Apoptotic cell death caused by LPS was also assessed and confirmed by DAPI staining where the apoptotic body formations were counted and represented graphically (Fig. 1B). Further, to analyze the typical morphological signs of apoptosis, the ultra-thin sections of the LPS-induced BMDMs and LPS induced BMDM treating with Aza and TSA for 24 hours were observed under the electron microscope. The chromatin of apoptotic macrophages appeared condensation, approaching to nuclear membrane, gathering at the edge and forming typical apoptotic bodies. Consistently, discontinuities of the nuclear envelope are visible by electron microscopy in the LPS induced BMDMs when compared to the untreated control. The nuclear membrane structure was restored when the LPS-induced BMDMs were treated with Aza and TSA (Fig. 1C). These data indicate that the treatment with both Aza and TSA significantly reduced the effect of LPS-induced DNA fragmentation followed by cell death in BMDMs.
Figure 1. Effects of LPS-induce BMDMs treated with Aza and TSA on DNA fragmentation, mitochondrial membrane structure and nuclear fragmentation.

In all experiments, BMDMs were treated with Aza (50nM)and TSA (25nM) in the presence and absence of LPS (1μg/ml). (A) For DNA gel analysis, cells were washed with PBS, lysed, run electrophoresis in a 1% (wt/vol) agarose gel and stained in ethidium bromide. DNA fragmentation assay showed that treatment with Aza and TSA has less fragmented DNA in the LPS challenged cells when compared to the LPS challenged BMDMs untreated or treated with Aza alone or TSA alone after 48 hours. (B) To count the fragmented nuclei, BMDMs were cultured on chambered slides and following treatment, cells were fixed in 4% paraformaldehyde and stained with DAPI. Graphic representation of the number of cells having nuclear fragmentation after LPS induced BMDMs untreated or treated with Aza alone or TSA alone. (C) For electron microscopic (EM) study, cells were washed with PBS followed by fixing the cells with 4% glutaraldehyde and then used for EM preparation. The EM image showed that the LPS-induced BMDMs treated with Aza and TSA showed a distinct lipid bilayer structure of nuclear membrane whereas the LPS challenged untreated BMDMs showed a ruptured nuclear membrane and disturbed lipid bilayers. *p < 0.05 CTL vs. LPS and LPS vs. LPS+Aza+TSA. Each bar represents mean ± SEM of three triplicate experiments. A, Aza; T, TSA, L, LPS.
3.2. Reduction of elevated mitogen activated protein kinase (MAPK) in LPS-induced apoptosis by treating with the combination of Aza and TSA
To understand the mechanisms underlying LPS-mediated cell death, we further investigated the involvement of MAPK in death signaling pathway. For this, we cultured BMDMs in the presence and absence of LPS and treated with the combination of Aza and TSA or Aza alone or TSA alone for 30 min. Then the cells were harvested and subjected to protein analysis for activated pERK and activated pJNK. Our data showed an increased activation of pERK as well as pJNK in LPS-induced BMDMs, which was significantly inhibited by treatment with Aza and TSA when compared to untreated or treated with either drug alone (Fig. 2A p<0.001, p<0.01). In our previous study, we have already established that the combination of Aza and TSA inhibits LPS-induced p-38 mediated inflammatory response in macrophages. It is also important to understand the possible role of other two MAPK isoforms, p-ERK and p-JNK in LPS mediated inflammatory response. To understand their involvement, we treated the cells with selective inhibitor for p-JNK and p-ERK. Western blot analysis was performed on LPS-challenged BMDMs cultured for 30 minutes with the combination of Aza and TSA in the presence and absence of a selective inhibitor of JNK (SP600125), or selective inhibitor for ERK (PD98059). Treatment with SP600125 reversed the suppressive effect of Aza and TSA in LPS-induced p-JNK activation in BMDMs (Fig. 2B). Moreover, when PD98059 was used as a selective inhibitor for p-ERK, the reversal effect was more prominent than that of JNK (Fig. 2C). All these data suggest that LPS causes activation of p-JNK and p-ERK (Fig. 2) and thereby induced inflammatory pathway associated cell death in BMDMs. Furthermore, treatment with Aza and TSA inhibits p-JNK and p-ERK signaling, which reduces the inflammation associated death in BMDMs. These results, together with our previous study strongly support that the effect of Aza and TSA could be multi facet and overall blocks the activation of MAPK pathway mediated inflammatory response.
Figure 2. Aza and TSA together abrogates LPS induced activation of p-JNK and p-ERK in BMDMs.

(A) Western analysis and the graphical representation data showed that the LPS-challenged BMDMs treated with Aza (50nM) and TSA (25nM) have lesser p-JNK and p-ERK expression than the cells treated with Aza alone and TSA alone. (B, C) Western analysis data showed that, in presence of the inhibitors of p-JNK (SP600125) and p-ERK (PD98059), there is an increase in the expression of p-JNK and p-ERK respectively in the LPS induced BMDMs treated with Aza and TSA. Total JNK and ERK were used as a loading control. All the protein expressions were analysed from a single blot reprobed with different antibodies. Bands intensities were quantified using ImageJ. The ratio of band intensity was calculated by using the value of band intensity of experimental protein divided by its respective loading control. Data were expressed as means ± SEM of three replicated experiments. The numbers in the graph represent the corresponding lanes in the Western blots. *p < 0.05 LPS vs. LPS+TSA; **p< 0.01 LPS vs. LPS+Aza+TSA. A, Aza; T, TSA; L, LPS.
3.3. Change in mitochondrial membrane potential and release of apoptotic signature genes in LPS-induced BMDMs is reversed by the combination of Aza and TSA
It has been already reported that LPS induces cell death in macrophages (Ma et al., 2015). To investigate the cell death mechanism, we further induced BMDMs with LPS for 24 hours in the presence and absence of Aza and TSA. We have observed that after 24 hours of treatment, LPS stimulated BMDMs and LPS induced BMDMs treated with Aza alone showed a significant damage in the mitochondria as evident from the electron microscopic (EM) study. Whereas, the LPS-induced BMDMs treated with Aza and TSA or TSA alone showed more restructured mitochondrion in respect to its shape and the membrane integrity (Fig. 3A). To further characterize the involvement of mitochondria in LPS mediated cell death, we performed mitochondrial membrane potential (ΔΨm) assay by JC-1 staining. It was observed that, LPS induced BMDMs has significant reduction of membrane potential by shifting of the spectra from red (PE) to green (FITC) (Supplementary Fig. S1A). Whereas, the LPS-induced BMDMs treated with the combination of Aza and TSA or TSA alone, shows a significant restoration of the membrane potential as indicated by the increased PE/FITC ratio (Fig. 3B). This was further supported by the Western blot analysis of mitochondrial cytochrome-c availability in BMDMs. Our data showed that lesser level of mitochondrial cytochrome-C in mitochondrial fraction from LPS-induced BMDMs untreated or treated with Aza alone and TSA alone, when compared to LPS-induced BMDMs treated with both Aza and TSA (Fig. 3C). Furthermore, the higher level of cytochrome-c in cytosolic fraction from LPS-induced BMDMs untreated or treated with Aza alone when compared to TSA alone or combination of Aza and TSA (Supplementary Fig. S1B). The lesser level of cytochrome C in mitochondrial fraction and higher level of cytochrome-c in cytosolic fraction from LPS induced BMDMs is due to mitochondrial membrane disruption and signature of cell death. This LPS-induced mitochondrial membrane disruption and cell death was abrogated by treating with the combination of Aza and TSA. In addition, to investigate the role of mitochondria in the death pathway induced by LPS, we performed reactive oxygen species (ROS) production assay by flowcytometric analysis using 2’,7’ – dichlorofluorescin diacetate (DCFDA, also known as H2DCFDA). Flow cytometry data revealed that LPS induced BMDMs produce a significantly higher amount of ROS which was brought down a significant level when BMDMs were treated with the combination of Aza and TSA (Fig. 3D). We also measured the mean intensity of DCFDA by the cells and were plotted on a graph. Our data show that the mean intensity was higher in LPS-induced BMDMs and it was significantly reduced in LPS-induced BMDMs treated with Aza and TSA (Fig. 3E). These data strongly suggest that Aza and TSA suppress LPS-induced mitochondrial changes associated with increased apoptosis.
Figure 3. Epigenetic modifiers Aza and TSA restore mitochondrial membrane integrity after LPS stimulation.

(A) The electron microscopy (EM) image showed that the mitochondrial membrane integrity was compromised after LPS induction (marked with arrows) as compare to the LPS-induced BMDMs treated in combination of Aza (50nM) and TSA (25nM). (B) Mitochondrial depolarization was calculated by the red/green fluorescence intensity ratio from the flow cytometry analysis of JC1 staining. High red/green ratio indicates better mitochondrial membrane potential. The data showed the loss of mitochondrial membrane potential (low ratio) after LPS challenges for 24 hours in the BMDMs which were rescued after treatment with Aza and TSA. FCCP (uncoupler, Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) was used as a positive control. (C) Western blot data and the graphical representation of mitochondrial membrane fraction showed that cytochrome C is more in the Aza and TSA treated BMDMs after LPS challenges when compared to the LPS induced BMDMs untreated or treated with either Aza alone or TSA alone. (D) Reactive oxygen species (ROS) was estimated by flow cytometry after macrophages were challenged with LPS (24h) and further treated with Aza+TSA. Flow cytometry results show that LPS has induced ROS in 99.8% of cells which was reduced to 9.6% when the cells were treated with Aza and TSA together. (E) Graphical data represent the mean intensity was higher in LPS-induced BMDMs and it was significantly reduced in LPS-induced BMDMs treated with Aza and TSA. The numbers in the graph represent the corresponding lanes in the Western blots. Data are expressed as means ± SEM, n =3 **P<0.01, ***P<0.001. A, Aza; T, TSA; L, LPS.
3.4. Treatment with Aza and TSA significantly reduced LPS-induced pyroptosis in macrophages
Recent studies have shown that Caspase 11 plays an important role in LPS-induced macrophage pyroptosis, a programmed cell death caused by rapid cell lysis (Cheng et al., 2017; Jorgensen and Miao, 2015). To see the protective effect of Aza and TSA in LPS induced pyroptosis in macrophages, we cultured the LPS challenged macrophages in the presence and absence of Aza and TSA for 6h and 24h. In the BMDMs, after 6 h of LPS stimulation, the protein expression of IL1β was significantly less treated with Aza and TSA (Fig. 4A). Caspase11 was found to be more after 24 hours which has also a similar reduced expression after treating the LPS challenged BMDMs with the Aza and TSA (Fig. 4B). Similar result is reflected in the Western analysis done in lung tissues collected from mice 24 hours after LPS injection and drugs treatment (Fig. 4C). Our data also show that Aza and TSA treatment significantly suppressed the expression of both caspase 11 and ILβ at their transcriptional level (Fig. 4D, E). These data together suggest that the treatment with Aza and TSA not only suppressed the apoptotic effect but also suppressed the pyroptotic effect caused by LPS.
Figure 4. Aza and TSA together inhibits pytoptosis in LPS-induced BMDMs.

(A, B) Western blot data and the graphical representation in the BMDMs showed that the LPS-challenged BMDMs treated with Aza (50nM) and TSA (25nM) have lesser IL1β and caspase 11 after 6h and 24 h of treatment respectively. (C) Western blot data and the graphical representation also showed that the lung tissues collected from LPS challenged mice treated with Aza alone or TSA alone or a combination of Aza and TSA has lesser IL1β and caspase 11 when compared to the untreated mice. All the protein expression was analyzed from a single blot reprobed with different antibodies. (D, E) qRT-PCR data show that Aza and TSA treatment significantly reduced the mRNA expression of IL1β and caspase11 genes 6 h after LPS stimulation when compared with no treatment or treatment with Aza alone or TSA alone in BMDMs. *p < 0.05 LPS vs. LPS+TSA; **p< 0.01 LPS vs. LPS+Aza+TSA. A, Aza; T, TSA; L, LPS
3.5. In vivo LPS-induced release of Cytochrome C and BAX expression was inhibited by the combination of Aza and TSA through the activation of anti-apoptotic genes
We performed immunofluorescence study of BAX protein expression on lung tissue collected from mice treated with a sub lethal dose of LPS and untreated or treated with Aza and TSA for 24 hours. Our immunofluorescence images show that the BAX expression in macrophages was assessed by the colocalization (yellow) of Bax (red) and CD68 (green) (Fig. 5A). A total of 100 cells (DAPI positive) were counted for each case and were represented graphically as a percentage of BAX positive macrophages (Fig. 5B). The number of BAX positive macrophages were significantly lower in mice that received both Aza and TSA group than in untreated or treated either with Aza alone or TSA alone (Fig. 5A, B). The immunostaining image analysis of BAX protein expressions in lung tissues were supported by Western analysis (Fig. 5C). Moreover, treatment with Aza and TSA increased the expression of Bcl-2 and Bcl-xL (Fig. S2A), and thereby inhibits the release of cleaved caspase 3, caspase 3 and caspase 9 (Fig. S2B) thus, reduced apoptosis.
Figure 5. Aza and TSA treatment reduced apoptosis in lung tissues 24 hours after the mice exposed to LPS.

(A) Immunofluorescence staining of macrophage markers CD68 (pan marker-green) and BAX (apoptosis marker-red) in lung tissues exposed to LPS for 24 hours followed by respective treatment as shown in the images. Our data showed that treatment with Aza (50nM) and TSA (25nM) consistently decreased LPS-induced co-localization of BAX positive cells (yellow). (B) Quantification of CD68/BAX double positive cells in the lung tissue. (C) Western Blotting analysis and graphical representation data show an increased expression of BAX in lung tissue exposed to LPS and was significantly reduced in lung tissues received from LPS induced mice treated with the combination of Aza and TSA. The numbers in the graph represent the corresponding lanes in the Western blots. Data are expressed as means ± SEM, n =3. *P<0.05. A, Aza; T, TSA; L, LPS.
3.6. Treatment with Aza and TSA together decreased the LPS-induced apoptotic cell death in the lung tissues
It has been shown previously that LPS induces cell death by apoptosis (Nolan et al., 2003). To assess the survival of macrophages in lung tissues and the effects of Aza and TSA, we performed TUNEL assay on lung tissue collected from sublethal dose of LPS induced mice and untreated or treated with Aza and TSA for 24h. We examined the tissue sections by immunofluorescence microscopy. Cell death was assessed by counting the number of double positive (pink) of TUNEL (red) and DAPI (blue) specifically, macrophage death is identified by the green staining of CD68 (Fig. 6A). A total of 100 cells were counted for each case and were represented as a percentage of apoptotic cells. The number of apoptotic cells were significantly lower in mice that received both Aza and TSA group than in untreated or treated either Aza alone or TSA alone (Fig. 6B). In addition, we performed qRT-PCR analysis on lung tissues for the gene expression of the apoptotic genes cytochrome C, BAD and BAX, and anti-apoptotic gene Bcl2. We observed increased expression of proapototic genes in lung tissue from LPS-induced mice, which was significantly reduced in LPS-induced mice treated with Aza and TSA. Furthermore, the antiapoptotic gene Bcl2 was significantly increased in lung tissues from LPS-induced mice treated with Aza and TSA (Fig. 6C). Collectively, these data indicate that combined treatment with Aza and TSA protects macrophages from LPS-induced death.
Figure 6. The effect of Aza and TSA treatment decreases the number of TUNEL positive cells in lung tissues from LPS challenged mice.

(A) Cell death was measured by TUNEL staining, and macrophages were identified by the macrophage surface marker, CD68. The TUNEL positive BMDMs were assessed by counting the number of triple-positive cells as a result of co-localization of TUNEL (red), CD68 (green) and DAPI (blue). (B) Quantification of triple-positive apoptotic cells. LPS group shows an increased number of TUNEL positive BMDMs. These numbers are significantly reduced in the control group, and treatment with Aza and TSA groups. (C) qRT-PCR data show that Aza and TSA treatment significantly reduced LPS-induced apoptotic gene expression (cytochrome C, BAD and BAX) when compared with no treatment or treatment with Aza alone or TSA alone in BMDMs challenged with LPS after 24 hours. There is an increased expression of anti-apoptotic genes (Bcl2) in BMDMs treated with Aza and TSA after LPS challenges. Data are expressed as means ± SEM, n =3 (C). *P<0.05, **P< 0.01, ***P < 0.001. A, Aza; T, TSA; L, LPS.
3.7. Mechanism of JMJD3-STAT3 mediated macrophage cell death and the prevention by Aza and TSA
JMJD3, a JmjC family histone demethylase is known to be induced by TLR activation caused by microbial stimuli. Recent discoveries stated that JMJD3 would be induced via NFκB-dependent (De Santa et al., 2009) as well as STAT-dependent pathways (Przanowski et al., 2014) while serving itself as a transcription factor for the induction of proinflammatory cytokines and chemokines (Hanisch, 2014). We wanted to test the hypothesis that Aza and TSA together suppress the activation of JMJD3 that plays a major role in LPS-induced inflammation associated cell death in BMDMs. Furthermore, we wanted to examine the interaction of STAT3 along with JMJD3 in mediating the inflammatory process during ALI. For this, LPS-challenged BMDMs were treated with Aza alone or TSA alone or in combination of Aza and TSA for 2h. Then the cells were harvested for immunoprecipitation assay against STAT3. Interestingly, we found that JMJD3 is expressed more in the LPS induced BMDMs whereas JMJD3 expression is reduced when treated with Aza and TSA. Thus, JMJD3 physically interacts with STAT3 and is responsible for the inflammatory signaling in LPS treated cells. Moreover, we examined the status of AcH3K9 the activator for JMJD3 and found that the expression of AcH3K9 was increased in the LPS induced BMDMs, whereas it decreases significantly when the LPS-induced BMDMs were treated with Aza and TSA (Fig. 7A, B). Furthermore, to confirm the role of Aza and TSA upon STAT3 activation, we have inhibited STAT3 by Cucurbitacin-I (500nM) a known inhibitor of STAT3 (Rajasingh et al., 2007) for 6 h and perform Western analysis. Our data showed that inhibition of STAT3 phosphorylation impedes the activity of the drugs Aza and TSA (Fig. 7C). These data clearly suggest that the action of Aza and TSA mediated through STAT3 in reducing the inflammation which was not happened in presence of STAT3 inhibitor.
Figure 7. Aza and TSA treatment mediated JMJD3-STAT3 signaling on LPS-induced BMDMs.

(A, B) To examine the interaction between STAT3 and JMJD3 proteins, we performed immunoprecipitation analysis. This was done by immunoprecipitating the cell lysate against STAT3 followed by immunoblotting against JMJD3. Expression of JMJD3 was high in LPS challenged cells compared with the cells treated by the combination of Aza (50nM) and TSA (25nM). The expression of the activator AcH3K9 and the repressor-H3K27Me3 for JMJD3 was analysed. The expression of AcH3K9 was high in the LPS challenged cells and decreases significantly when cells treated with Aza and TSA. Conversely, there was an overall less expression of H3K27Me3 in the LPS treated cells compared to the cells treated with Aza and TSA. All the protein expressions were analysed from a single blot reprobed with different antibodies. (C) Western blot data and its graphical representation showed treatment with Aza and TSA increased the expression of p-STAT3, but its expression has decreased after the treatment with cucurbitacin-I. The numbers in the graph represent the corresponding lanes in the Western blots. Data are expressed as means ± SEM, n =3. *P<0.05, **P< 0.01, ***P < 0.001. A, Aza; T, TSA; L, LPS.
4. Discussion
Sepsis is often a life-threatening complication caused by bacterial infection, leads to a whole-body inflammatory response and may be often fatal. The lungs are the most susceptible tissue to sepsis-induced organ failure, and more than 50% of patients with sepsis develop acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) (Ahmad, 2011; Methe, 2012). Overall due to high incidence of mortality, ALI/ARDS is considered as an unmet medical need. Therefore, more improvement in a treatment regimen is needed to decrease mortality and morbidity from ALI and ARDS (Johnson and Matthay, 2010). In this manuscript, we tried to explore a novel epigenetic therapy that modulates LPS-induced lung injury and inflammation. The dose of LPS (1ug/ml) was selected based upon our previous studies as well as other studies have shown that LPS at this dose caused a significant release of ROS responsible for inflammation (Hsu and Wen, 2002). Here, we also wanted to see whether our drugs have the protecting effect even at the higher dose of LPS which is responsible for generating greater amount of inflammation and cell death than the lower dose. For this, we have used a combination of two epigenetic drugs Aza and TSA to abrogate several aspects of cellular injury during ALI. This is the first study to show that Aza and TSA treatment lowering the apoptotic and pyroptotic effects of macrophage’s death associated with inflammation both in vitro and in vivo.
Present study is more focused on the mechanism of action of our drugs that has any role in the other two major MAPK pathways, JNK and ERK mediated pro-inflammatory signaling and apoptosis. Our data clearly shows that when the LPS-induced BMDMs treated with the combination of Aza and TSA the phosphorylation of JNK and ERK was significantly reduced. When we chemically inhibited the LPS-induced phosphorylation of JNK and ERK individually, we have noticed that there is an increased phosphorylation of JNK and ERK and thereby generate more cellular inflammation and apoptosis. These data clearly suggest that Aza and TSA together act through the MAPK pathway to mitigate the LPS-induced inflammation and apoptosis. Overall, these data indicate that the combination of Aza and TSA treatment inhibits all the major three MAPK signaling pathways (p38, JNK amd ERK) and therefore, reduce inflammation associated cell death in macrophage cells. The three major MAPK cascade component’s p38, ERK and JNK play an important role in TLR signalling and are differentially involved in cell maturation and production of pro-inflammatory mediators (Chang and Karin, 2001; Peroval et al., 2013). Our previous findings showed that the combination of Aza and TSA promoting anti-inflammatory gene activation and switching of macrophages to the alternative anti-inflammatory phenotype by the increased expression of M2 surface markers CD23, CD124, and CD206 and significant reduction in M1 markers CD40, CD14, and NOS2 (Thangavel, J. et al., 2015). It has been shown that the induction of anti-inflammatory activity and expression of M2 macrophages causes an overall reduction of cell death (Genin et al., 2015). Thus, combination of Aza and TSA might be considered as an effective anti-inflammatory drug, which modulates LPS-stimulated inflammatory responses and activates anti-inflammatory signaling pathways and thereby protects the tissues from cell death.
The cell fate is largely dependent on the expression of the Bcl-2 family of anti- and pro-apoptotic members. The expression of two anti-apoptotic members of this family, Bcl-2 and Bcl-xL, have been found to inhibit cell death by inhibiting the expression of cytochrome C (Zucchini et al., 2005). The pro-apoptotic Bcl-2 family protein, BAD reside in the cytosol and translocate to mitochondria following death signaling, forms a proapoptotic complex with Bcl-xL, and promotes the release of cytochrome C in LPS challenged BMDMs. We showed that the combination of Aza and TSA inhibits apoptosis and restores mitochondrial membrane structure by inhibiting pro-apoptotic genes and activating the anti-apoptotic genes. These findings will have a significant impact on the field, in part because Aza is already approved for cancer treatment and could potentially be repurposed against ALI and ARDS.
We used BMDM as our model for studying the molecular mechanism of apoptosis. We know that during ALI, the lung residential macrophages may not be sufficient to overcome inflammation after huge bacterial load. Therefore, there would bea migration of monocytes/macrophages from bone marrow in presence of colony stimulating factor (CSF) to the injured site to compensate for the function of residential alveolar macrophages (O’Dea et al., 2009). Thus, it is quite relevant to use BMDMs in studying the mechanism of inflammation in a mouse model of ALI. We wanted to study whether this anti-inflammatory state of macrophages makes the environment healthier with the reduction in cell death. Moreover, mitochondrial dysfunction has been shown to take part in the induction of apoptosis and has been known to be central to the apoptotic pathway. Our EM image analysis clearly shown that in LPS, the mitochondrial membrane integrity was completely lost and was recovered with the administration of combination of Aza and TSA. This is also evident from JC1 staining which shows formation of J-monomers more in LPS treated cells suggested the less mitochondrial membrane intigrity and potential. Changes in the membrane potential are supposed to be due to the opening MPTP, resulting release of cytochrome C into the cytosol. Our Western blot data showed an increased release of cytochrome C into the cytosol in LPS treated cells whereas the combination of Aza and TSA has markedly reduced release of cytochrome C in the cytosol which also justified the fact of restoration of mitochondrial membrane integrity by the electron microscopic study. In agreement with earlier findings, release of cytochrome C further results in activation of procaspase-9 which consecutively activate caspase 3 LPS-induced lung tissues (Cai et al., 1998). This active caspase-3 subsequently cleaves a variety of substrates resulting in characteristic morphologic changes within the nucleus, including DNA fragmentation. Our data clearly show that the treatment in combination of Aza and TSA, results an increased expression of anti-apoptotic gene Bcl-xL with decrease activity of caspase 3, 9 and 11 as well as a reduction in the quantity of fragmented DNA.
There are also several reports explaining mitochondrial alterations during apoptosis, including production of reactive oxygen species, the depletion of ATP, and the opening of the mitochondrial permeability transition pore (MPTP) (Bonora and Pinton, 2014). In fact, opening of the MPTP has been proved to induce depolarization of the transmembrane potential (Δψm), release of apoptogenic factors and loss of oxidative phosphorylation. In some cases of apoptotis, loss of Δψm may be an early event whereas in some cases may be a consequence of the apoptotic-signaling pathway (Ly et al., 2003). Therefore, mitochondria play a critical role in cell death (Wang and Youle, 2009). Opening of the MPTP causes a dissipation of the inner mitochondrial membrane potential (ΔΨm), which results in an increase in the matrix volume, and a consequential disruption of the outer mitochondrial membrane, leading to the release of intermembrane factors as well as proapoptotic genes (Norenberg and Rama Rao, 2007).
This study also unravels the Aza and TSA treatment mediated inhibitory mechanism of LPS-induced apotosis and pyroptosis in BMDMs. In our study, we have observed that combination of Aza and TSA protects pyroptosis that has started after 6 h of LPS treatment as well as apoptosis that has started after 24h of LPS treatment. Thus, our findings indicate that Aza and TSA together inhibit the activity of IL1β, caspase 3, caspase 9 and caspase 11 and thereby block the inflammatory signaling mediated apaptosis and pyroptosis. It has been studied that both caspase-1 and caspase-11 are responsible for the host defense against inflammation, whereas caspase-11 mostly involved during bacterial infection (Ming Man et al., 2017). Recent studies have shown that pyroptosis is an inflammaotory form of cell death process invloving Capase 1 and/or11 mediated cleavge of IL-1 beta (Py et al., 2014). Different studies have been done so far to understand the excat mechanism of pyroptosis. Our studies clearly show that pyroptosis is an early event taking place for the induction of inflammation whereas apoptosis is a late event that takes place after the onset of inflammation. Therefore, in our present study, we have shown that LPS induces inflammation mediated apoptosis and pyroptosis in macrophages which were significantly reduced after the use of Aza and TSA in combination. Although we have observed that treatment with TSA alone can be controlling some of the apoptotic proteins expression better than the combination of Aza and TSA. However, our earlier mice survivability data clearly shows that the treatment with the combination of Aza and TSA works better than that of individual drugs. Overall, most of our transcriptional and translational analyses of different apoptotic and inflammatory genes from LPS-induced macrophages or lung tissues, the combination of Aza and TSA works better than the either drugs alone.
It has been previously studied that H3K27 demethylase, Jmjd3 (also known as KDM6B) is responsible for the induction of most of the inflammatory genes assoiciated with LPS stimulation (De Santa et al., 2009). Recent findings also suggest that JMJD3 may be involved in acute inflammatory response and is transcriptionally regulated by STAT1 and STAT3 which are a novel mechanism critical for initiating inflammatory responses Studies have also shown that inhibition of STATs leads to the increased activity of JMJD3 (Przanowski et al., 2014; Sherry-Lynes et al., 2017). In this study, we have shown that macrophages in response to LPS stimulation, there is an increased expression of JMJD3, which causes the induction of inflammatory cyto- and chemokines as well as the induction of MAPK signaling pathway.. Acetylation of histone 3 at lysine 9 (H3K9) is associated with gene activation, whereas trimethylation of histone 3 at lysine 27 (H3K27) is associated with gene repression (Li et al., 2014; Lopez-Atalaya et al., 2013). Recently, it has been found that JMJD3 catalyses the demethylation of H3K27me2/3 in vitro. We therefore, have checked the status of H3K27Me3 and found that there was an overall lesser expression of H3K27Me3 in the LPS induced BMDMs when compared to the LPS-challenged BMDMs treated with Aza and TSA. Moreover, the data support our hypothesis that the increased expression of JMJD3 is responsible for the demethylation of H3K27Me3, a histone mark associated with transcriptional repression of the anti-inflammatory genes in the LPS treated cells. Also, we have checked the overall status of acH3K9 and found that there was an increased expression in the LPS induced macrophages. This observation supports that LPS induced cells have an increased expression of proinflammatory genes which was reduced after the treatment with combination of Aza and TSA.
Although STAT3 is reported to be mostly activated in inflammatory responses, but there are findings that STAT3 plays a crucial role in IL10 mediated anti-inflammatory responses (El Kasmi et al., 2006). Previous studies have shown that IL10 is responsible for the anti-inflammatory state that exerts its activity through the activation of STAT3 pathways (Sharma et al., 2011). Our study shows that there is reduced expression of STAT3 in LPS challenged BMDMs. Here, we have shown that STAT3 physically interacts with JMJD3 and is responsible for the activation of inflammatory cytokines which causes LPS mediated cell death. In the present study, we found that treatment with Aza and TSA reduced the expression of JMJD3 in the LPS challenged BMDMs and increased the expression of STAT3 thereby reduces the inflammatory response mediated apoptosis. Previously, we have also shown that STAT3 promoter is acetylated with the addition of Aza and TSA followed by LPS treatment, which makes the macrophages more polarized to M2 or anti-inflammatory status (Thangavel, Jayakumar et al., 2015). These results therefore suggest that STAT3 plays a significant role in JMJD3 mediated inflammatory responses and cell death during ALI.
5. Conclusion
Overall, our data suggest that combination of Aza and TSA having better treatment effect when compared to the LPS-induced BMDMs treated with either Aza alone or TSA alone. We believe that our epigenetics-based approach using in vitro and in vivo models may lead to further development of safe and successful therapy for ALI/ARDS. This study also yields valuable mechanistic insights and identifies a novel effective epigenetic-based drug therapy for ALI that will improve, prolong and finally eliminate their diseases form the lives of millions of people.
Supplementary Material
Highlights:
Combinatory treatment of Aza and TSA reduces the LPS induced mitochondrial DNA fragmentation, reduced expression of apoptotic and pyroptotic genes in bone marrow derived macrophages.
LPS induced inflammatory pathway is mainly mediated through the activation of JNK-ERK and STAT3-JMJD3.
Treatment with Aza and TSA together abrogate the inflammatory pathway through their epigenetic modification.
Acknowledgments
The electron microscopy research laboratory of the Kansas University Medical Center was used for electron microscopy analyses.
Funding
This work was supported, in part, by the American Heart Association Grant-in-Aid 16GRNT30950010 and the National Institutes of Health COBRE grant P20GM104936 (to JR), and R01HL128374 (to VS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare no competing or financial interests.
References
- Ahmad S, 2011. Novel Peptide to Treat Sepsis-Induced Acute Lung Injury. Science translational medicine 3(66), 66ec67. [Google Scholar]
- Albert ML, 2004. Death-defying immunity: do apoptotic cells influence antigen processing and presentation? Nat Rev Immunol 4(3), 223–231. [DOI] [PubMed] [Google Scholar]
- Araki H, Yoshinaga K, Boccuni P, Zhao Y, Hoffman R, Mahmud N, 2007. Chromatin-modifying agents permit human hematopoietic stem cells to undergo multiple cell divisions while retaining their repopulating potential. Blood 109(8), 3570–3578. [DOI] [PubMed] [Google Scholar]
- Bonora M, Pinton P, 2014. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Frontiers in Oncology 4, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower RG, Fessler HE, 2000. Mechanical ventilation in acute lung injury and acute respiratory distress syndrome. Clinics in chest medicine 21(3), 491–510, viii. [DOI] [PubMed] [Google Scholar]
- Butt MU, Sailhamer EA, Li Y, Liu B, Shuja F, Velmahos GC, DeMoya M, King DR, Alam HB, 2009. Pharmacologic resuscitation: cell protective mechanisms of histone deacetylase inhibition in lethal hemorrhagic shock. J Surg Res 156(2), 290–296. [DOI] [PubMed] [Google Scholar]
- Cai J, Yang J, Jones D, 1998. Mitochondrial control of apoptosis: the role of cytochrome c. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1366(1–2), 139–149. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M, 2001. Mammalian MAP kinase signalling cascades. Nature 410(6824), 37–40. [DOI] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, Miao EA, Rehman J, Malik AB, 2017. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. The Journal of clinical investigation 127(11), 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KB, Wong F, Harlan JM, Chaudhary PM, Hood L, Karsan A, 1998. Lipopolysaccharide mediates endothelial apoptosis by a FADD-dependent pathway. J Biol Chem 273(32), 20185–20188. [DOI] [PubMed] [Google Scholar]
- Das ND, Jung KH, Choi MR, Yoon HS, Kim SH, Chai YG, 2012. Gene networking and inflammatory pathway analysis in a JMJD3 knockdown human monocytic cell line. Cell Biochem. Funct 30(3), 224–232. [DOI] [PubMed] [Google Scholar]
- De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola S, Testa G, Sung WK, Wei CL, Natoli G, 2009. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 28(21), 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G, 2007. The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell 130(6), 1083–1094. [DOI] [PubMed] [Google Scholar]
- Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson BT, Townsend S, Vender JS, Zimmerman JL, Vincent JL, 2008. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 36(1), 296–327. [DOI] [PubMed] [Google Scholar]
- Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB, 2012. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nature immunology 13(1), 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y, Suda T, Donnelly RP, Myers MG Jr., Alexander W, Vignali DA, Watowich SS, Ernst M, Hilton DJ, Murray PJ, 2006. General nature of the STAT3-activated anti-inflammatory response. J. Immunol 177(11), 7880–7888. [DOI] [PubMed] [Google Scholar]
- Genin M, Clement F, Fattaccioli A, Raes M, Michiels C, 2015. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 15, 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U-K, 2014. Linking STAT and TLR signaling in microglia: a new role for the histone demethylase Jmjd3. Journal of Molecular Medicine 92(3), 197–200. [DOI] [PubMed] [Google Scholar]
- Hsu HY, Wen MH, 2002. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J. Biol. Chem 277(25), 22131–22139. [DOI] [PubMed] [Google Scholar]
- Johnson ER, Matthay MA, 2010. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. Journal of Aerosol Medicine and Pulmonary Drug Delivery 23(4), 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Miao EA, 2015. Pyroptotic cell death defends against intracellular pathogens. Immunological reviews 265(1), 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Morikawa A, Sugiyama T, Koide N, Jiang GZ, Lwin T, Yoshida T, Yokochi T, 1997. Augmentation of lipopolysaccharide-induced thymocyte apoptosis by interferon-gamma. Cellular immunology 177(2), 103–108. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Kweon MN, Kuwata H, Schreiber RD, Kiyono H, Takeda K, Akira S, 2003. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. The Journal of clinical investigation 111(9), 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek AR, Fukudome EY, Li Y, Smith EJ, Liu B, Velmahos GC, deMoya M, King D, Alam HB, 2012. Histone deacetylase inhibitor treatment attenuates MAP kinase pathway activation and pulmonary inflammation following hemorrhagic shock in a rodent model. J Surg Res 176(1), 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ, 2002. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol 169(5), 2253–2263. [DOI] [PubMed] [Google Scholar]
- Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, Zhao W, Wei G, Cui J, Zhao K, Wang HY, Wang RF, 2014. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat Commun 5, 5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Atalaya JP, Ito S, Valor LM, Benito E, Barco A, 2013. Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 41(17), 8072–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly JD, Grubb DR, Lawen A, 2003. The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis 8(2), 115–128. [DOI] [PubMed] [Google Scholar]
- Ma B, Yu J, Xie C, Sun L, Lin S, Ding J, Luo J, Cai H, 2015. Toll-Like Receptors Promote Mitochondrial Translocation of Nuclear Transcription Factor Nuclear Factor of Activated T-Cells in Prolonged Microglial Activation. The Journal of neuroscience: the official journal of the Society for Neuroscience 35(30), 10799–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methe H, 2012. Unmasking Lung Injury in Sepsis. Science translational medicine 4(145), 145ec135. [Google Scholar]
- Milhem M, Mahmud N, Lavelle D, Araki H, DeSimone J, Saunthararajah Y, Hoffman R, 2004. Modification of hematopoietic stem cell fate by 5aza 2’deoxycytidine and trichostatin A. Blood 103(11), 4102–4110. [DOI] [PubMed] [Google Scholar]
- Ming Man S, Karki R, Briard B, Burton A, Gingras S, Pelletier S, Kanneganti T-D, 2017. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci. Rep 7, 45126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi N, Fernandis AZ, Cherla RP, Park IW, Ganju RK, 2002. Lipopolysaccharide-induced apoptosis of endothelial cells and its inhibition by vascular endothelial growth factor. J Immunol 168(11), 5860–5866. [DOI] [PubMed] [Google Scholar]
- Nolan Y, Vereker E, Lynch AM, Lynch MA, 2003. Evidence that lipopolysaccharide-induced cell death is mediated by accumulation of reactive oxygen species and activation of p38 in rat cortex and hippocampus. Experimental neurology 184(2), 794–804. [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Rama Rao KV, 2007. The Mitochondrial Permeability Transition in Neurologic Disease. Neurochemistry international 50(0), 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dea KP, Wilson MR, Dokpesi JO, Wakabayashi K, Tatton L, van Rooijen N, Takata M, 2009. Mobilization and Margination of Bone Marrow Gr-1(high) Monocytes during Sub-clinical Endotoxemia Predisposes the Lungs towards Acute Injury. Journal of immunology (Baltimore, Md.: 1950) 182(2), 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroval MY, Boyd AC, Young JR, Smith AL, 2013. A Critical Role for MAPK Signalling Pathways in the Transcriptional Regulation of Toll Like Receptors. PloS one 8(2), e51243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przanowski P, Dabrowski M, Ellert-Miklaszewska A, Kloss M, Mieczkowski J, Kaza B, Ronowicz A, Hu F, Piotrowski A, Kettenmann H, Komorowski J, Kaminska B, 2014. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. Journal of molecular medicine (Berlin, Germany) 92(3), 239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, Dietrich A, Lipinski MM, Henry T, Clapham DE, Yuan J, 2014. Caspase-11 Controls Interleukin-1β Release through Degradation of TRPC1. Cell Rep. 6(6), 1122–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasingh J, Bord E, Hamada H, Lambers E, Qin G, Losordo DW, Kishore R, 2007. STAT3-dependent mouse embryonic stem cell differentiation into cardiomyocytes: analysis of molecular signaling and therapeutic efficacy of cardiomyocyte precommitted mES transplantation in a mouse model of myocardial infarction. Circulation research 101(9), 910–918. [DOI] [PubMed] [Google Scholar]
- Rajasingh J, Bright JJ, 2006. 15-Deoxy-delta12,14-prostaglandin J2 regulates leukemia inhibitory factor signaling through JAK-STAT pathway in mouse embryonic stem cells. Exp Cell Res 312(13), 2538–2546. [DOI] [PubMed] [Google Scholar]
- Rock KL, Kono H, 2008. The inflammatory response to cell death. Annual review of pathology 3, 99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI, 2011. IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res. 1373, 189–194. [DOI] [PubMed] [Google Scholar]
- Sherry-Lynes MM, Sengupta S, Kulkarni S, 2017. Regulation of the JMJD3 (KDM6B) histone demethylase in glioblastoma stem cells by STAT3. 12(4), e0174775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Ao L, Raeburn CD, Calkins CM, Abraham E, Harken AH, Meng X, 2001. A low level of TNF-alpha mediates hemorrhage-induced acute lung injury via p55 TNF receptor. American journal of physiology. Lung cellular and molecular physiology 281(3), L677–684. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kobayashi M, Isatsu K, Nishihara T, Aiuchi T, Nakaya K, Hasegawa K, 2004. Mechanisms Involved in Apoptosis of Human Macrophages Induced by Lipopolysaccharide from Actinobacillus actinomycetemcomitans in the Presence of Cycloheximide. Infection and Immunity 72(4), 1856–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel J, Malik AB, Elias HK, Rajasingh S, Simpson AD, Sundivakkam PK, Vogel SM, Xuan YT, Dawn B, Rajasingh J, 2014. Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am J Pathol 184(8), 2237–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel J, Samanta S, Rajasingh S, Barani B, Xuan Y-T, Dawn B, Rajasingh J, 2015. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. Journal of Cell Science 128(16), 3094–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel J, Samanta S, Rajasingh S, Barani B, Xuan YT, Dawn B, Rajasingh J, 2015. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J Cell Sci 128(16), 3094–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Youle RJ, 2009. The Role of Mitochondria in Apoptosis(). Annual review of genetics 43, 95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Malik AB, Sun Y, Hu S, Reynolds AB, Minshall RD, Hu G, 2011. Innate immune function of the adherens junction protein p120-catenin in endothelial response to endotoxin. J Immunol 186(5), 3180–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K, 1996. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J. Biol. Chem 271(24), 14067–14072. [DOI] [PubMed] [Google Scholar]
- Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, Connors AF Jr., Hite RD, Harabin AL, 2006. Comparison of two fluid-management strategies in acute lung injury. The New England journal of medicine 354(24), 2564–2575. [DOI] [PubMed] [Google Scholar]
- Xaus J, Comalada M, Valledor AF, Lloberas J, Lopez-Soriano F, Argiles JM, Bogdan C, Celada A, 2000. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 95(12), 3823–3831. [PubMed] [Google Scholar]
- Yokochi T, Kato Y, Sugiyama T, Koide N, Morikawa A, Jiang GZ, Kawai M, Yoshida T, Fukada M, Takahashi K, 1996. Lipopolysaccharide induces apoptotic cell death of B memory cells and regulates B cell memory in antigen-nonspecific manner. FEMS immunology and medical microbiology 15(1), 1–8. [DOI] [PubMed] [Google Scholar]
- Zucchini N, de Sousa G, Bailly-Maitre B, Gugenheim J, Bars R, Lemaire G, Rahmani R, 2005. Regulation of Bcl-2 and Bcl-xL anti-apoptotic protein expression by nuclear receptor PXR in primary cultures of human and rat hepatocytes. Biochim. Biophys. Acta 1745(1), 48–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.