

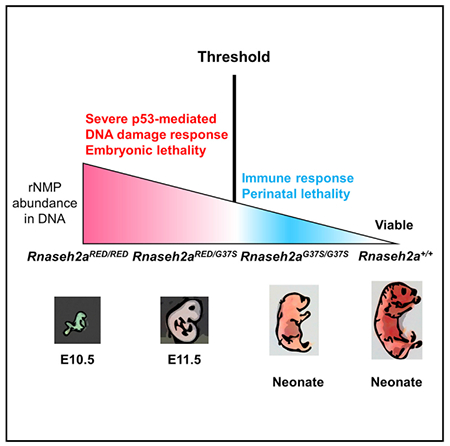
SUMMARY
RNase H2 has two distinct functions: initiation of the ribonucleotide excision repair (RER) pathway by cleaving ribonucleotides (rNMPs) incorporated during DNA replication and processing the RNA portion of an R-loop formed during transcription. An RNase H2 mutant lacking RER activity but supporting R-loop removal revealed that rNMPs in DNA initiate p53-dependent DNA damage response and early embryonic arrest in mouse. However, an RNase H2 AGS-related mutant with residual RER activity develops to birth. Estimations of the number of rNMPs in DNA in these two mutants define a ribonucleotide threshold above which p53 induces apoptosis. Below the threshold, rNMPs in DNA trigger an innate immune response. Compound heterozygous cells, containing both defective enzymes, retain rNMPs above the threshold, indicative of competition for RER substrates between active and inactive enzymes, suggesting that patients with compound heterozygous mutations in RNASEH2 genes may not reflect the properties of recombinantly expressed proteins.
Graphical Abstract
In Brief
Uehara et al. use RNase H2 mice with differing activity levels for removal of rNMPs embedded in DNA. Moderate levels of rNMPs lead to perinatal lethality activating the cGAS-Sting DNA sensing innate immune response. Exceeding a threshold, high abundance of rNMPs activates p53-dependent DNA damage, causing early embryonic lethality.
INTRODUCTION
Errors during DNA replication and repair processes can challenge genome integrity. Cells are well equipped to restore normal duplex DNA, but in some circumstances defects may persist, leading to severe DNA damage. Discrimination against rNTPs by DNA polymerases at the sugar moiety is far from perfect, and the three replicative DNA polymerases α, δ, and ε include ribonucleotide monophosphates (rNMPs) at different frequencies, resulting in about 1 rNMP for every 7,000 deoxyribo-nucleotide monophosphates (dNMPs) in DNA in mouse (Hiller et al., 2012; Reijns et al., 2012) and yeast (Lujan et al., 2012; Williams et al., 2016) cells. The abundance of rNMPs incorporated in DNA is significantly higher than any of the well-studied modified nucleotides produced by, for example, UV irradiation. The highly efficient ribonucleotide excision repair (RER) pathway is initiated by incision at the rNMP by RNase H2 followed by repair synthesis to replace rNMPs by dNMPs (Sparks et al., 2012). From a biochemical standpoint, rNMP incorporation could be described as a replication error, but because these rNMPs are included so frequently and efficiently removed, we and others have suggested that biologically, the transient presence of rNMPs in DNA serves a positive role in DNA replication (Cerritelli and Crouch, 2016; Clausen et al., 2015), perhaps as a mark for mismatch repair as suggested (Ghodgaonkar et al., 2013; Lujan et al., 2012) or to release torsional constrains. RNase H2 is uniquely suited to recognize rNMPs in DNA, but it also cleaves the RNA strand of RNA/DNA hybrids mostly found in cells as part of R-loops (with the non-hybridized DNA in single-stranded form) (Cerritelli and Crouch, 2009). R-loops, which form during transcription, are also processed by RNase H1 (Drolet et al., 1995). Complete absence of RNase H2 eliminates both activities, making it difficult to assign any phenotype to defects in RER or RNA/DNA hydrolysis. We modified the Rnh201 catalytic subunit in yeast RNase H2 to retain RNA/DNA activity, eliminating almost completely its rNMP cleavage activity, resulting in an RNase H2RED (ribonucleotide excision defective) (Chon et al., 2013). We verified that typical 2–5 bp deletions created in the absence of RNase H2 were as prevalent in the RNase H2RED mutant. The RNA/DNA hybrid activity of RNase H2RED rescued the growth defect of top1 Δrnh1 Δrnh201 Δ strain and the synthetic slow growth of sgs1 Δrnh201 Δ strain, both indicative of defects in RNA/DNA processing.
RNase H2 is essential in mice and medically relevant in humans. More than half of patients with the neuroinflammatory Aicardi-Goutières syndrome (AGS) have biallelic mutations in any of the three RNase H2 subunits (Crow et al., 2015). Some AGS-related RNASEH2 mutations have been associated with systemic lupus erythematosus (Günther et al., 2015; Ramantani et al., 2010). Both RER and RNA/DNA hydrolysis activities are absent in mouse in which RNase H2-B or H2-C subunit is ablated, resulting in embryonic lethality at around E10.5 (Hiller et al., 2012; Reijns et al., 2012). It seems reasonable that RNase H2’s unique ability to cleave at single rNMPs in DNA is directly related to developmental arrest in the absence of RNase H2. Reijns et al. (2012) drew such a conclusion, and others (Günther et al., 2015) suggested that the DNA damage caused by unrepaired rNMPs is the main contributor to AGS. However, Chedin’s laboratory examined cell lines from several AGS patients and attributed AGS phenotypes to unresolved R-loops (RNA/DNA hybrids) (Lim et al., 2015).
To examine the contribution of each of RNase H2 activities in mice to viability and genome integrity, we used three Rnaseh2a mutant mice: knockout; Rnaseh2aG37S, a hypomorphic mutation (Pokatayev et al., 2016) producing an enzyme with partial loss of both RNA/DNA hydrolysis and RER; and Rnaseh2aRED, which is ribonucleotide excision defective but active for RNA/DNA hybrid processing. By crossing these three mice with different RNase H2 activities, we observed that accumulation of rNMPs triggered activation of the p53-mediated DNA damage response pathway in Rnaseh2aRED/RED, Rnaseh2aRED/− and Rnaseh2a−/− embryos, resulting in early embryonic developmental arrest. This strongly indicates that the abundance of unrepaired rNMPs is the cause of embryonic lethality. We also found that although Rnaseh2aG37S/− mice survived to birth, they became embryonic lethal at E12.5 in Rnaseh2aG37S/RED mice, suggesting that the RNase H2RED enzyme acts in a dominant manner for limiting the RER pathway. Such differences in activity among RNase H2 mutants may be relevant for AGS patients with biallelic mutations (Crow et al., 2015), which could present complex defects due to competitive binding on genomic DNA substrates.
RESULTS
We expressed different variants of RNase H2 in mice to learn how its two activities contribute to viability and fitness. A mutant strain in which RNase H2RED is the sole source of the enzyme is the centerpiece of the results we report here.
In Vitro, Mouse RNase H2RED Is Unable to Remove Single Ribonucleotides in dsDNA but Retains High Levels of RNA/DNA Degradation Activity
We wanted to confirm that mouse RNase H2A protein with the corresponding substitutions of the RED mutant (P40D and Y211A) exhibits the same enzymatic characteristics as the S. cerevisiae RNase H2RED enzyme. We purified the mutated enzyme expressed in E. coli to near homogeneity, detecting only the three expected protein bands of the RNase H2 subunits by SDS-PAGE (Figure S1A). We examined enzymatic activities of mouse RNase H2RED using uniformly 32P-labeled poly-rA/poly-dT (hereafter noted as Hybrid) (Figure 1A) and 5′ end-labeled DNA 12 bases in length containing a single rNMP 1R (DNA5-RNA1DNA6/DNA12) (Figure 1B) and 12R (RNA12/DNA12) substrates (Figure 1C). RNase H2RED exhibited 130% ± 6.1% of the activity of the RNase H2WT on hybrid substrates (Figure 1D). The degradation of Hybrid by RNase H2RED was slightly faster than that of RNase H2WT. RNase H2WT produced shorter RNA fragments and degraded 12R substrate faster than RNase H2RED (Figure 1C), suggesting that the RNase H2RED enzyme prefers long RNA/DNA hybrids to short substrates, as previously found for the yeast enzyme (Chon et al., 2013). In contrast, RNase H2RED retained only 0.31% ± 0.036% of the RNase H2WT RER activity toward the 1R substrate, demonstrating that mouse RNase H2RED is, like its yeast homolog, an RNA/DNA-specific enzyme. Rnaseh2aG37S is an AGS-related mutation that impairs the catalytic function of RNase H2 in human and yeast (Chon et al., 2009, 2013; Coffin et al., 2011; Crow et al., 2006; Perrino et al., 2009). We found that mouse RNase H2 with the corresponding mutation (RNase H2G37S) retains activities toward Hybrid, 1R, and 12R substrates but greatly reduced to 1.6% ± 0.28%, 17% ± 4.7%, and 3.5% ± 0.14% of wild-type, respectively (Figure 1D).
Figure 1. In Vitro Activities of RNase H2WT, RNase H2RED, and RNase H2G37S.
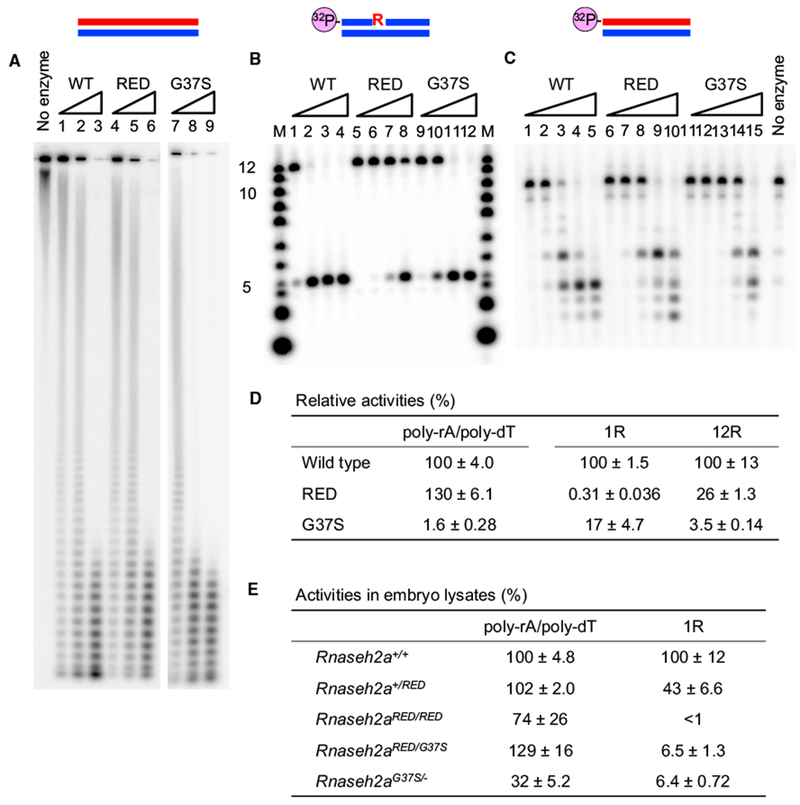
(A) The uniformly 32P-labeled poly-rA/poly-dT substrate (1 μM) was hydrolyzed with increasing amounts of RNase H2WT and its derivatives at 37°C for 30 min. Lanes 1 and 4 contained 10 pM proteins, lanes 2 and 5 contained 20 pM proteins, lanes 3 and 6 contained 50 pM proteins, lane 7 contained 2 nM protein, lane 8 contained 5 nM protein, and lane 9 contained 10 nM protein. Reaction products were analyzed by 12% TBE-urea PAGE.
(B) The 5′-32P-labeled 12-mer 1R (DNA5-RNA1-DNA6/DNA12) substrate (1 μM) was hydrolyzed with increasing amounts of RNase H2 and its derivatives at 37°C for 30 min. Lanes 1,5, and 9 contained 100 pM proteins; lanes 2,6, and 10 contained 1 nM proteins; lanes 3, 7, and 11 contained 10 nM proteins; and lanes 4,8, and 12 contained 100 nM proteins. Reaction products were analyzed by 20% TBE-urea PAGE. The molecular size marker is indicated as M.
(C) The 5′-32P-labeled 12-mer 12R (RNA12/DNA12) substrate (1 μM) was hydrolyzed with increasing amounts of RNase H2 and its derivatives at 37°C for 30 min. Lanes 1,6, and 11 contained 10 pM proteins; lanes 2, 7, and 12 contained 100 pM proteins; lanes 3, 8, and 13 contained 1 nM proteins; lanes 4, 9, and 14 contained 10 nM proteins; and lanes 5, 10, and 15 contained 100 nM proteins. Reaction products were analyzed by 20% TBE-urea PAGE.
(D) Specific activities of mutant enzymes relative to wild-type activity (%).
(E) Relative RNase H2 activities (%) in E11.5 embryos lysates. The mean values of three independent embryos are shown with SD. Enzymatic activities were normalized to the mean value of uterine sibling wild-type embryos.
Rnaseh2a KO Mice Are Early Embryonic Lethal
We wished to examine mice homozygous and heterozygous for RNase H2-defective mutations in the Rnaseh2a gene. We obtained Rnaseh2atm1(KOMP)Wtsi knockout (KO) embryonic stem cells (ESCs) from the Mouse Knockout Project and by standard techniques generated viable heterozygous mice. Rnaseh2a−/− embryos arrested in development at about E10.5 (Figure 2A), consistent with results observed for inactivation of the Rnaseh2b or Rnaseh2c genes (Hiller et al., 2012; Reijns et al., 2012).
Figure 2. Developmental Defect of Embryos.
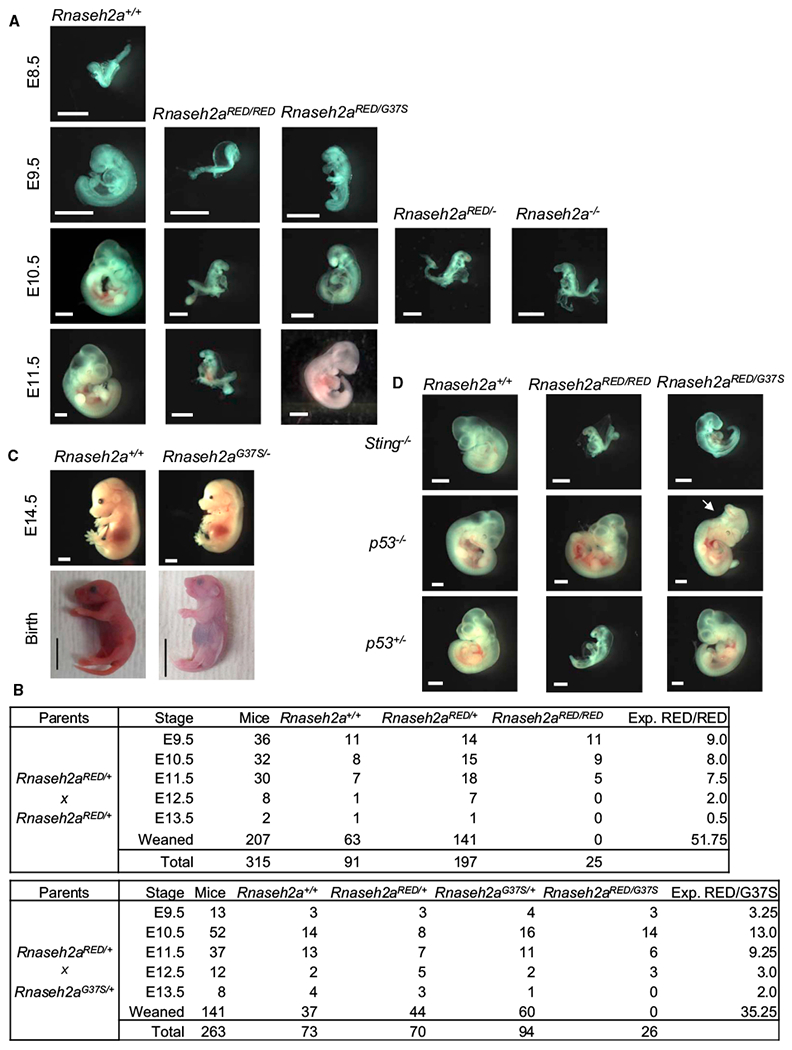
(A) Rnaseh2a+/+, Rnaseh2aRED/RED, Rnaseh2aRED/G37S, Rnaseh2aRED/−, and Rnaseh2a−/− embryos at embryonic days 8.5–11.5. Scale bar, 1 mm.
(B) Survival of mouse embryos from crosses of Rnaseh2aRED/+ × Rnaseh2aRED/+ (top) and Rnaseh2aRED/+ × Rnaseh2aG37S/+ (bottom). Expected number of Rnaseh2aRED/RED and Rnaseh2aRED/G37S at Mendelian ratio is shown in right column.
(C) Rnaseh2a+/+ and Rnaseh2aG37S/− embryos and neonates. Scale bars, 2 mm (E14.5) and 1 cm (birth).
(D) Partial rescue of embryonic lethality by ablation of p53 gene. Images of E10.5 embryos are shown. A morphological rescue of Rnaseh2aRED/RED and Rnaseh2aRED/G37S was observed on p53 background. Rnaseh2aRED/G37S was also rescued on p53+/− background (n = 5), whereas Rnaseh2aRED/RED was not (n = 3). No rescue of Rnaseh2aRED/RED (n = 8) and Rnaseh2aRED/G37S (n = 5) phenotypes was observed on Sting−/− background.
Embryonic Lethality of Rnaseh2aRED/RED
Inactivation of any of the three genes encoding RNase H2 subunits leads to complete loss of both RER and Hybrid activities. We wanted to address directly the role of RER activity in embryonic viability in mice by using a knock-in mouse strain carrying an Rnaseh2a allele that exclusively abolishes RER activity, as we demonstrated in vitro for the recombinant enzyme (see above and Figure 1). We generated a Rnaseh2aP40D-Y211A (Rnaseh2aRED) mouse. We obtained viable heterozygous Rnaseh2aRED/+ mice that exhibited no detectable abnormalities. Intercrossing of Rnaseh2aRED/+ mice resulted in live Rnaseh2aRED/+ and wild-type (Rnaseh2a+/+) mice at a ratio of approximately 2:1, but no viable Rnaseh2aRED/RED mouse was found in more than 200 weaned mice (Figure 2B). We subsequently investigated embryos from crosses of Rnaseh2aRED/+ parents and observed Rnaseh2aRED/RED embryos at Mendelian ratios at early stages of development (Figure 2B). At embryonic day 9.5 (E9.5), Rnaseh2aRED/RED embryos were developmentally retarded by approximately 1 day compared with their littermates (Figure 2A). E10.5 Rnaseh2aRED/RED embryos were not resorbed yet but remained morphologically abnormal, with a slight increase in size compared with E9.5 embryos. E11.5 embryos appeared unchanged from day E10.5. No E12.5 Rnaseh2aRED/RED embryo was found. Therefore, Rnaseh2aRED/RED embryos exhibited delayed development and completely arrested at E10.5 and were resorbed after E11.5. We subsequently crossed Rnaseh2a−/+ and Rnaseh2aRED/+ mice and compared Rnaseh2a−/−, Rnaseh2aRED/−, and Rnaseh2aRED/RED embryos at E10.5. They were indistinguishable in size and tissue morphology (Figure 2A). We conclude that Rnaseh2aRED/RED and Rnaseh2aRED/− exhibit the same early embryonic lethality as seen when RNase H2 is completely absent (Hiller et al., 2012; Reijns et al., 2012). In addition, we conclude that a single wild-type Rnaseh2a allele is sufficient to produce normal mice and that the presence in the same cells of RNase H2RED mutant enzyme does not influence normal viability, as previously reported for Rnaseh2a+/G37S mice.
Rnaseh2aG37S Contribution to Mouse Development
We previously reported that the hypomorphic RNase H2G37S enzyme allowed Rnaseh2aG37S/G37S mice to survive only to birth, with a smaller size than the Rnaseh2aG37S/+ and wild-type (WT) mice in the same birthing. Rnaseh2aG37S/G37S embryos remains proportionally smaller through development (Figure S2A). Rnaseh2aG37S/+ mice are not defective in any obvious way. RNase H2G37S retains RER and Hybrid activities but both are significantly decreased (Figure 1D). When we crossed Rnaseh2a−/+ and Rnaseh2aG37S/+ mice, we discovered that Rnaseh2aG37S/− also developed to birth and were similarly retarded in growth only slightly smaller than Rnaseh2aG37S/G37S dead pups (Figures 2C and S2A). At delivery, Rnaseh2aG37S/37S mice weighed about 0.9 g compared with 1.2 g for Rnaseh2aG37S/G37S (each an average of four pups). Rnaseh2aG37S/G37S and even Rnaseh2aG37S/− embryonic development goes beyond that of Rnaseh2aRED/−, Rnaseh2aRED/RED, and Rnaseh2a−/− embryos because of the presence of sufficient RNases H2 activities to reduce DNA damage.
Rnaseh2aG37S/RED Mice Are Early Embryonic Lethal
We anticipated that a mouse having both RNase H2G37S and RNase H2RED might be viable because of increased Hybrid activity of RNase H2RED and RER activity of RNase H2G37S. Surprisingly, we found no viable or perinatal-lethal Rnaseh2aG37S/RED pups when we crossed Rnaseh2aG37S/+ with Rnaseh2aRED/+ mice; rather, they were early embryonic lethal. However, the Rnaseh2aRED/G37S embryos survived a bit longer than embryos from Rnaseh2a−/− or Rnaseh2aRED/RED mice (Figures 2A and 2B). At E9.5 the Rnaseh2aRED/G37S embryos were larger and more developed than Rnaseh2aRED/RED embryos. We saw progression of three Rnaseh2aRED/G37S embryos to E12.5 with various sizes at E11.5 and E12.5 stages. However, they arrested development at E11.5 some 7 or 8 days before Rnaseh2aG37S/G37S and Rnaseh2aG37S/− become perinatal lethal. We propose that either RNase H2RED is expressed at a higher rate and competes with RNase H2G37S for assembly with the RNase H2-B and H2-C subunits, or that RNase H2RED competes with RNase H2G37S in the RER pathway. We address these possibilities in the following sections.
RNase H Activities in RNase H2RED and RNase H2G37S Embryo Extracts
Instability of RNase H2REDin vivo could account for the defects we saw during embryonic development of Rnaseh2aRED/RED mice. We measured RNase H2 Hybrid and RER activities in crude lysates of E11.5 embryos using Hybrid and 1R substrates. We found that Hybrid activity in Rnaseh2aRED/RED embryo extracts was high, and RER activity was undetectable (Figure 1E). The relative ex vivo activities were quite similar to those we found with recombinantly expressed purified enzymes (Figure 1D), indicating stability and efficient assembly of the mutant enzyme in vivo. Additionally, immunoblotting of RNase H2-A and H2-C from mouse embryonic fibroblast (MEF) cell homogenates implied that the cellular amount of RNase H2A and H2C, which forms a 1:1:1 heterotrimer with H2B, is almost equivalent in Rnaseh2a+/+, Rnaseh2aRED/+, and Rnaseh2aRED/RED (Figure S2B). These results led us to conclude that loss of RER activity accounts for early arrest of development of Rnaseh2aRED/RED embryos.
Rnaseh2aG37S/− mice survive to birth with about 6% of RER activity and 30% of Hybrid activity (Figure 1E). The significantly higher Hybrid activity in Rnaseh2aG37S/− embryo extracts with respect to recombinantly expressed proteins could be due to stimulation of the RNase H2G37S activity or to RNase H1 degradation of the substrate. The former seems more likely because RNase H1 activity is at best only 15% of that of RNase H2 (Büsen and Hausen, 1975). However, the patterns of products are those produced by both RNases H1 and H2 (Figure S2C). Therefore, we cannot rule out that RNase H1 is contributing to activity in Rnaseh2aG37S/− embryo extracts.
RNase H2RED Competes with RNase H2G37S in the RER Pathway
Similar RER activity was found in Rnaseh2aRED/G37S embryo extracts as in extracts of Rnaseh2aG37S/− embryos (Figure 1E), although the former arrest development much earlier. Of course, these ex vivo assays were performed with excess added substrates. We suggest that in vivo RNase H2RED retains Hybrid activity and capability to bind to RER substrates, resulting in blocking the removal of genomic rNMPs. Because Rnaseh2aRED/G37S embryos survived only modestly further than Rnaseh2aRED/RED embryos, we predict that RNase H2RED might bind better than RNa-seH2G37S to substrates with a single rNMP embedded in DNA. We first tested for differences in association of the two enzymes to an RER substrate immobilized on the sensor chips using surface plasmon resonance. We found that the WT enzyme bound very well to the substrate, but the RNase H2RED and RNase H2G37S both bound very poorly; so poorly that we were unable to fit the binding curves to those of a 1:1 binding model (Figure S3A). Therefore, we performed competition for incision of a single rNMP in double-stranded DNA (dsDNA) in vitro using RNase H2G37S and 10- or 100-fold excess of RNase H2RED or RNase H2D34A, the latter being totally inactive because of the substitution of the catalytic aspartate residue by alanine. When added in excess, we found that RNase H2RED or RNase H2D34A neither inhibited nor stimulated RNase H2G37S incision activity in these assays (Figure S3B). Even when we added double mutant RNase H2D34A-RED or RNase H2D34A-G37S, we did not observe any inhibition of RNase H2G37S activity. Therefore, we conclude that the enzyme-substrate complexes formed by mutant proteins are unstable and cannot compete for binding and catalysis by RNase H2G37S. On the basis of these results, we propose that competition occurs later in the RER pathway involving RNase H2’s interaction with PCNA via its PCNA-interacting peptide located on the RNase H2B subunit (see Discussion and Figure S3C).
Partial Rescue of Embryonic Lethality of Rnaseh2aRED/RED and Rnaseh2aRED/G37S Embryos by p53−/− but Not by Sting−/−
We previously reported that Rnaseh2aG37S/G37S p53−/− mice failed to alter the perinatal lethality of Rnaseh2aG37S/G37S mice, but we did find that Rnaseh2aG37S/G37S in a Sting−/− background eliminates the innate immune response (Pokatayev et al., 2016). We considered the possibility that RNase H2G37S and perhaps RNase H2RED might lead to induction of the innate immune response in Rnaseh2aRED/G37S embryos. We obtained embryos of Rnaseh2aRED/RED and Rnaseh2aRED/G37S in a Sting−/− background and found that these embryos had identical characteristics as embryos in a Sting+/+ strain (Figure 2D). They all arrest at the same stage and embryos exhibited poor morphology.
In contrast to lack of effect of Sting−/−, Rnaseh2aRED/RED, and Rnaseh2aRED/G37S E10.5 embryos in a p53−/− background were normal in size, with some morphological abnormalities (Figure 2C, arrow). Rnaseh2aRED/RED p53+/− E10.5 embryos were small and defective (bigger than or same as p53+/+) but Rnaseh2aRED/G37S p53+/− E10.5 embryos were normal size. For Rnaseh2aRED/RED we saw the same effect of p53−/− as reported for Rnaseh2b- and Rnaseh2c-null embryos (Hiller et al., 2012; Reijns et al., 2012). We conclude that the p53−/− background effect is consistent with p53-induced DNA damage response, causing very early lethality.
p53-Mediated DNA Damage Response Caused by Loss of RER Activity
Our results, which showed improved growth in embryos from Rnaseh2aRED/RED and Rnaseh2aRED/G37S in a p53−/− background, demonstrate that p53 response is activated in E10.5 embryos. We determined the expression levels of p53 target genes (p21 and Cyclin G1) by qRT-PCR analyses, and as seen in Figure 3A, p21 was increased more than 20-fold in Rnaseh2a−/−, Rnaseh2aRED/RED, and Sting−/− Rnaseh2aRED/RED embryos but only 6-to 9-fold in Rnaseh2aRED/G37S, Sting−/− Rnaseh2aRED/G37S, and Rnaseh2aG37S/G37S embryos. The E10.5 embryos, in which we saw most upregulation, were arresting growth, while the embryos with less p21 mRNA activation were still in a growth phase. Expression of cyclin G1 transcripts were also elevated 3- to 10-fold in all embryos, but there was no clear pattern of increases as seen for p21 activation. Furthermore, we found no indication of a cGAS-Sting-dependent immune response for interferon-stimulated genes (ISGs), Ifit1, Ifit3, and Cxcl10. We observed some variations in the expression levels of ISGs, but they were not consistent with the genotypes (e.g., Ifit1 and Cxcl10 expression levels in Rnaseh2aRED/RED Sting−/− were considerably elevated, whereas Rnaseh2aRED/RED exhibited moderate upregulation of all three ISGs). We examined the stimulation of these same three ISGs in MEFs from embryos from Rnaseh2aG37S/− at E14.5 and we saw no activation of p21 and CyclinG1 but consistent small increases of the ISG mRNA (Figure 3B). Taking these results together, we concluded that Rnaseh2aRED/RED and Rnaseh2aRED/G37S embryos, whether in a Sting−/− or Sting+/+ background, are embryonic lethal because of p53-dependent DNA damage response as reported here also for Rnaseh2a−/− and previously for Rnaseh2b-null and Rnaseh2c−/− embryos (Hiller et al., 2012; Reijns et al., 2012). However, p21 expression in Rnaseh2aRED/G37S embryos was significantly lower than that of Rnaseh2aRED/RED embryos, indicative of less DNA damage in the former.
Figure 3. qRT-PCR Analysis of p53-Target and Interferon-Stimulated Genes.
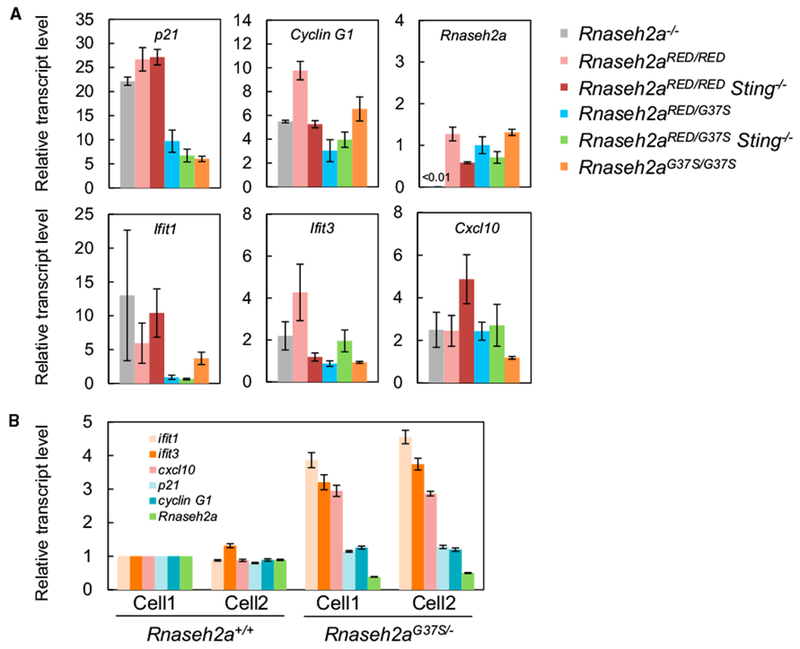
(A) Expression levels of two p53 target genes (Cdkn/p21 and Ccng1/Cyclin G1) and three interferon-stimulated genes (Ifit1, Ifit3, and Cxcl10) were measured by RT-qPCR of RNA from E10.5 whole embryos. The mean values of four Rnaseh2aRED/RED, three Rnaseh2a−/−, three Rnaseh2aG37S/G37S, and three Rnaseh2aRED/G37S embryos were normalized to that of Rnaseh2a+/+. The mean values of Rnaseh2aRED/RED Sting−/− (n = 4) and Rnaseh2aRED/G37S Sting−/− (n = 5) embryos were normalized to that of Rnaseh2a+/+ Sting−/−. Error bars represent SEM.
(B) Expression levels ofp21, cyclin G1, ISGs, and Rnaseh2a in Rnaseh2aG37S/− E14.5 MEFs. MEF cell lines were established from two embryos. Transcript levels were normalized to that of Rnaseh2a+/+.
Increased rNMPs in Rnaseh2aRED/RED, Rnaseh2aRED/G37S, and Rnaseh2aG37S/− MEFs
To study ribonucleotide presence in DNA of various mutants, we prepared MEFs from embryos at different stages. We readily obtained Rnaseh2aG37S/− MEFs from E14.5 embryos. In contrast, we were unable to establish MEFs from Rnaseh2aRED/RED embryos, unless crossed with p53−/− mice. We established Rnaseh2aRED/G37S p53−/− cell lines as well. Rnaseh2aRED/G37S cells in p53+/− and p53+/+ background, like Rnaseh2aRED/RED, grew poorly in culture medium. To examine the extent to which incorporated ribonucleotides remain in Rnaseh2aRED/RED p53−/− MEFs, we isolated genomic DNA, treated with 0.3 M NaOH, and analyzed by agarose gel electrophoresis at alkaline pH. DNA mobility of Rnaseh2aRED/RED was increased after alkaline treatment, giving a substantial peak shift of DNA intensity tracing curve from those of WT and Rnaseh2aRED/+ (Figures 4A and S2D). The mobility shift of DNA fragments in alkaline agarose gels for Rnaseh2b-null and Rnaseh2aRED/RED DNA (both p53−/− background) with respect to Rnaseh2a+/+ indicates that a considerable number of rNMPs accumulate in Rnaseh2b-null and Rnaseh2aRED/RED genomic DNA. The number of rNMPs per cell in Rnaseh2b-null DNAs was estimated to be about 970,000/cell (Figure 4C), slightly less than reported by Reijns et al. (2012). Rnaseh2aRED/RED DNA contained about 630,000 ± 40,000 rNMPs/cell, fewer than Rnaseh2b−/− but still well over the background strand breaks seen in Rnaseh2a+/+ MEFs (Figure 4). We suggest that the reason for fewer DNA breaks in Rnaseh2aRED/RED compared with Rnaseh2b−/− cells is the presence of persistent RNA/DNA hybrids in the latter cells, which contribute to replicative stress and DNA damage. However, an alternative explanation could be that the Rnaseh2aRED/RED cells are early-passage MEFs, whereas the Rnaseh2b−/− cells are greater than 20 cell passages and could have accumulated greater numbers of rNMPs. We found large variation of rNMPs frequency in Rnaseh2aRED/G37S DNA from two independent cell lines, both of which contained significantly fewer rNMPs than Rnaseh2aRED/RED. In contrast, accumulation of rNMPs in Rnaseh2aRED/+ was undetectable as reported for Rnaseh2b+/− (Reijns et al., 2012), suggesting that 50% RER activity of WT is sufficient to remove rNMPs from genomic DNA (Figure S2D). MEFs from Rnaseh2aG37S/− embryos (on p53+/+ background) showed a distribution of DNA fragments similar to WT (Figure 4B) and contained about a third of the amount of rNMPs present in Rnaseh2b-null DNAs. Because Rnaseh2aG37S/− embryos develop to birth, it suggests that 260,000 ± 40,000 rNMPs per cell, which is slightly but significantly less than the frequency found in Rnaseh2aRED/G37S, can be tolerated at least during embryogenesis.
Figure 4. Accumulation of Unrepaired rNMPs Causes Serious DNA Damage.

(A and B) Differences in abundance of rNMPs in embryonic fibroblast DNA were analyzed using alkaline gel electrophoresis. The genotypes of cell lines are shown in the inset of figures.
(A) Increased migration of Rnaseh2aRED/RED and Rnaseh2aRED/G37S DNA in alkaline agarose gel. Gel image after electrophoresis and densitometry curves ofeach DNA are shown. Total MEF nucleic acids were isolated, treated with 0.3 M NaOH and analyzed by 0.7% agarose gel electrophoresis in alkaline running buffer.
(B) Low but significant number of rNMPs is retained in Rnaseh2aG37S/−.
(C) The estimated numbers of rNMPs in RER-defective cells.
(D) Alkaline comet assay of MEFs. Representative images of comets after single-cell electrophoresis are shown on the right. DNA was unwound under alkaline condition followed by electrophoresis. Damaged DNA fragments migrated out of the nucleus, forming a comet tail. DNA damage levels of cells were quantified by olive tail moment and shown in graph. More than 100 comets were randomly chosen and analyzed. The values are expressed in mean ± SEM from three independent experiments. *p < 0.02, **p < 0.005, and ***p < 0.0001 (t test).
As a complementary means to examine rNMPs in MEFs, we performed a single-cell gel electrophoresis (comet) assay under alkaline conditions, which detects both single- and double-strand breaks as well as alkaline-labile sites, including rNMPs in DNA. We used the same cell lines at the same cell passage, except for Rnaseh2b−/−, as for alkaline gel analysis. Greater than 90% of Rnaseh2aRED/RED MEF and Rnaseh2b-null cells’ DNAs migrated from the nucleus. With MEF cells from Rnaseh2a+/+ and Rnaseh2aRED/+ DNAs, in which little or no rNMPs are present, we found that only a small amount of DNA migrated from the nucleus (Figure 4D). Quantification of DNA damage by olive tail moment indicated that Rnaseh2aRED/RED DNA is as seriously damaged as Rnaseh2b-null compared with Rnaseh2a+/+ and Rnaseh2aRED/+ (Figure 4D), consistent with alkaline gel results. Similarly, we observed elevated but significantly less DNA damage in Rnaseh2aG37S/− and Rnaseh2aG37S/G37S. Importantly, we found that the comet assay of Rnaseh2aRED/G37S MEFs indicates a high level of rNMP retention but less than the Rnaseh2aRED/RED and Rnaseh2b-null cells. The results from alkaline gel and comet assays show clearly an inverse correlation between levels of rNMPs in DNA and embryonic progression.
DISCUSSION
Abundant Unrepaired rNMPs in DNA Produce Sufficient DNA Damage to Cause Early Embryonic Lethality
RNase H2 performs two distinct functions: hydrolysis of RNA/DNA hybrids formed during transcription and incision at rNMPs incorporated during DNA replication and repair. These two substrates are present simultaneously during the S and G2/M phases of the cell cycle and could compete for binding RNase H2. We are interested in establishing how these two activities contribute to cellular and organism fitness and viability. We have examined mice carrying a mutant form of RNase H2 (RNase H2RED) lacking RER of single rNMPs embedded in DNA and found that Rnaseh2aRED/RED embryos are similarly defective at the same stage of development as mice with null mutations in Rnaseh2b or Rnaseh2c. Rnaseh2aRED/RED MEFs retain about 65% of the null mutants’ unrepaired rNMPs in DNA, similarly inducing a p53 DNA damage response, which caused early embryonic arrest. We concluded that, as previously suggested (Hiller et al., 2012; Reijns et al., 2012), accumulation of rNMPs in DNA causes early embryonic lethality in null embryos, as well as in Rnaseh2aRED/RED.
A Threshold of rNMPs in DNA for Survival of Rnaseh2-Defective Embryos
We found the abundances of rNMPs in DNA of Rnaseh2aG37S/−, Rnaseh2aRED/G37S, and Rnaseh2aRED/RED MEFs are approximately 27%, 37%, and 65%, respectively, of Rnaseh2b-null cells (Figure 4C). We believe that development to birth of Rnaseh2aG37S/− pups having fewer, but still abundant, rNMPs indicates a threshold of ribonucleotides in DNA, above which the p53 damage response is activated, leading to early embryonic lethality. This order of relative abundance of rNMPs is confirmed by comet assay results (Figure 4D).
Support for this comes from our finding that we obtained MEFs from Rnaseh2aRED/RED embryos only in a p53−/− background, not in a p53+/+ or p53+/− background, whereas MEFs were readily obtained from Rnaseh2aG37S/− embryos in a p53+/+ strain. We interpret these results to indicate that DNA damage in Rnaseh2a−/−, Rnaseh2aRED/RED, and Rnaseh2aRED/G37S MEF is so severe that p53 activation leads to irreversible genome instability and apoptotic death. Rnaseh2aG37S/− and Rnaseh2aG37S/G37S embryos and MEFs do exhibit DNA damage but at a level that is tolerated, allowing their survival to birth (Figures 2 and 4). However, DNA damage in Rnaseh2aG37S/− and Rnaseh2aG37S/G37S embryos activates an innate immune response (Pokatayev et al., 2016) (Figure 3B).
The fact that the absence of the p53 DNA damage response extends the viability of Rnaseh2aRED/RED embryos is an indication that dsDNA breaks may be the major contributor to early embryonic lethality. In yeast, topoisomerase 1 has been shown to generate breaks at rNMPs when RER is defective, and deletion of Top1 allows viability in the presence of these increased rNMPs (Williams et al., 2013). Similarly, Top1-mediated nicks could induce DNA damage in mouse MEFs in the presence of rNMPs above the threshold. Replication-transcription collisions may also be contributing to dsDNA breaks. Perhaps different organisms have variable thresholds of ribonucleotides in DNA, or even different tissue and cell types could be more sensitive than others to rNMPs. Embryonic development, when DNA is being synthesized rapidly, seems to be a particularly susceptible stage. It would be interesting to determine whether after birth or in postmitotic tissues the RNase H2G37S and or the RNase H2RED mutants could become viable.
RNase H2RED Prohibits Completion of RER in the Presence of RNase H2G37S
The initial description of the RER pathway (Sparks et al., 2012) used purified WT S. cerevisiae enzymes in which the interaction between RNase H2 and the substrate did not require PCNA, thus describing the RER first step as incision of the rNMP by RNase H2. Our SPR data indicate that affinity of the RNase H2RED and RNase H2G37S enzymes for RER substrates is very low, and the competition experiments between the two mutant enzymes showed little or no interference for substrate binding (Figure S3A). We suggest that PCNA binding to either mutated enzyme aids in directing to or stabilizing the enzyme-1R-substrate interactions. In a Rnaseh2aRED/G37S strain, both mutant enzymes could compete for binding PCNA with only the RNase H2G37S:PCNA complex resulting in RER activity. RNase H2RED might sequester PCNA or form stronger complexes, preventing RER. We hypothesize that an RNase H2G37S:PCNA interaction could promote RER, as demonstrated by the survival to birth of Rnaseh2aG37S/− and Rnaseh2aG37S/G37S embryos (Figures 2 and S3C).
First Structural Sign of Embryonic Defects Occurs at the Epiblast-to-Gastrulation Transition
Reijns et al. (2012) reported that differences in sizes of Rnaseh2b-null and WT embryos were first observed at E7.5. Between embryonic stage E6.5 and E7.5, cell numbers double every 5 hr, increasing 23-fold from 660 at E6.5 to 15,000 at E7.5 (Kojima et al., 2014). At this stage, almost 60% of the cell cycle is S phase, with short G1 and shorter G2/M phases. Embryos for Rnaseh2a−/−, Rnaseh2aRED/RED, and Rnaseh2aRED/G37S would be challenged to complete replication and repair of rNMPs in DNA in the context of epiblast expansion and gastrulation. Accumulation of rNMPs creates replication stress responses triggering a p53-dependent cascade of events that induced apoptosis and cell death, as indicated by much larger size of Rnaseh2aRED/RED embryos in a p53−/− background.
Interestingly, we found that except for their smaller size, the mutant embryos of all RNase H2G37S defective mice appear normal. At E10.5, WT embryos weigh about 10 mg, and at birth, pups weigh slightly more than 1 g. We estimate the weight of Rnaseh2aRED/G37S E10.5 embryos as ~1–2 mg (Figure 2A). At birth, the actual weight of perinatal-lethal Rnaseh2aG37S/− pups approximates that of an E17 WT embryo (~900 mg) (Kojima et al., 2014; Macdowell et al., 1927) (Figure 2C). In the case of Rnaseh2aG37S/− or Rnaseh2aG37S/G37S mice, the small size is observed as early as E10.5 and continues thereafter as if development were delayed by 1 day (Figure S2A), neither becoming relatively smaller nor able to catch up to normal size. We suggest that the smaller size results from fewer, yet normal, cells produced in the transition from epiblast to gastrulation.
Are rNMPs in DNA the Cause of Perinatal Lethality in RnasehG37S Mice?
What, if any, is the contribution of R-loops to early embryonic death and to innate immune response induction? Formally, we cannot exclude the need for the hybrid activity of RNase H2 at any stage of development. Because the large numbers of rNMPs in DNA in Rnaseh2a-null, Rnaseh2b-null or Rnaseh2c-null, Rnaseh2aRED/RED, and Rnaseh2aRED/G37S stimulate the DNA damage response early in development, it may mask any defect due to hybrid accumulation. Does the perinatal lethality of Rnaseh2aG37S/G37S mice depend on the decreased abundance of rNMPs, or is there a role for Hybrid activity?
Our studies of functions of RNase H2 in S. cerevisiae showed that RNase H1 and H2 can share certain substrates, while only RNase H2 can recognize rNMPs. The synthetic sickness of rnh201Δ sgs1Δ strain requires the RNase H2 hybrid activity, which cannot be supplied by RNase H1 (Chon et al., 2013). Subsequent studies using the yeast RED mutation have permitted assignment of which of the two activities is relevant to alter a specific phenotype. For example, the Argueso lab (Conover et al., 2015; Cornelio et al., 2017) has shown in S. cerevisiae that both activities contribute to genome stability depending on the relative abundance of each substrate; when rNMPs are abundant, there is a greater requirement for RER activity, and when high frequencies of transcription of a particular sequence occur, there can be a larger need of the Hybrid activity. The Klein lab described accumulation of contiguous embedded rNMPs in the absence of RNase H2 that are RNA/DNA hybrids but not R-loops and are recognized by RNase H2 but not by RNase H1(Epshtein et al., 2016). In our Rnaseh2aG37S and Rnaseh2aRED mice, such differential requirement for the two activities could vary from tissue to tissue.
Recent reports demonstrate that defects in RNase H2 result in micronuclei that provide cytoplasmic DNA to stimulate the innate immune response via the cGAS-Sting pathway (Bartsch et al., 2017; Mackenzie et al., 2017). DNA damage leading to cytoplasmic DNA could occur in RNase H2-defective mice and humans either via dsDNA breaks, as seen in E2-stimulated estrogen transcription induced R-loops (Stork et al., 2016), or resulting from rNMPs in DNA (Cerritelli and Crouch, 2016; Epshtein et al., 2016; Williams et al., 2016). Radiotherapy and chemotherapeutics can cause DNA strand breaks and formation of micronuclei, which also activate the cGAS-Sting pathway (Harding et al., 2017).
Both RNA/DNA hybrids and rNMPs in DNA are known to cause dsDNA breaks, and each could activate the cGAS-Sting DNA immune-activating pathway or otherwise induce p53-dependent DNA damage response. The RNase H2RED enzyme both in vitro and in vivo has no significant activity cleaving at rNMPs in DNA. Recent studies in human tissue culture cells have exploited the differential activities of RNase H2RED to assign which of its two activities is required for (1) retrotransposition of Line-1 (Benitez-Guijarro et al., 2018) and (2) providing sites in DNA for interaction with PARP1 (Zimmermann et al., 2018). The former needs RNA/DNA-resolving activity and the latter the retention of rNMPs in DNA. These cultured cells can survive without RNase H2, unlike our Rnaseh2aRED/RED mice, which arrest during early embryonic development. It will be interesting to see the properties of specific tissues when RNase H2RED is the only RNase H2 present. A mutant defective in Hybrid hydrolysis but fully capable of RER activity may reveal which nucleic acid is responsible for the autoimmune response found in Rnaseh2aG37S/G37S mice and ultimately in AGS patients with the G37S mutation as well as aid in defining roles for RNA/DNA hybrids that are exclusively recognized by RNase H2.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-beta actin | Abcam | ab8227; RRID:AB_2305186 |
| Rabbit polyclonal anti-RNase H2 subunit A | Proteintech | Cat#16132-1-AP; RRID:AB_2269729 |
| Rabbit polyclonal anti-RNase H2 subunit C | Abcam | ab89726; RRID:AB_2042815 |
| Goat polyclonal anti-rabbit IgG | Abcam | ab205718 |
| Bacterial and Virus Strains | ||
| One Shot BL21(DE3) competent cells | Invitrogen | C600003 |
| One Shot MAX Efficiency DH5α-T1R competent cells | Invitrogen | Cat#12297016 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse RNase H2 | This study | N/A |
| E. coli RNase HII | New England Biolabs | M0288S |
| T4 Polynucleotide Kinase | New England Biolabs | M0201S |
| Phosphodiesterase I | Worthington Biochemical | LS003926 |
| Streptavidin | Sigma-Aldrich | S4762 |
| Pierce Bovine Serum Albumin Standard’ | Thermo Scientific | Cat#23209 |
| HBS-EP Buffer | GE Healthcare | BR-1001-88 |
| Newborn Calf Serum | GIBCO | Cat#16010-159 |
| DMEM | GIBCO | Cat#12430-054 |
| L-Glutamate | GIBCO | Cat#25030-081 |
| Penicillin Streptomycin | GIBCO | Cat#15140-148 |
| DPBS | GIBCO | Cat#14190144 |
| 10×Tris/Glycine/SDS running buffer | BIORAD | Cat#161-0732 |
| Blotting-Grade Blocker | BIORAD | Cat#1706404 |
| PureLink DNase | Invitrogen | Cat#12185-010 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#04707516001 |
| CometSlides | Trevigen | Cat#4250-200-03 |
| Pierce ECL Western Blotting Substrate | Thermo Scientific | Cat#32106 |
| Critical Commercial Assays | ||
| Quikchange II site-directed mutagenesis kit | Agilent technologies | Cat#200524 |
| Histrap crude FF columns | GE Healthcare | Cat#11-0004-58 |
| Hitrap Heparin HP columns | GE Healthcare | Cat#17-0406-01 |
| HiLoad 16/600 Superdex 200 pg columns | GE Healthcare | Cat#28989335 |
| Quick-DNA Universal kit | Zymo Research | D4068 |
| QIAprep Spin Miniprep kit | QIAGEN | Cat#27106 |
| DNeasy Blood and Tissue kit | QIAGEN | Cat#69506 |
| Trizol | Life technologies | Cat#15596018 |
| PureLink RNA Mini kit | Invitrogen | Cat#12183018A |
| Omniscript RT Kit | QIAGEN | Cat#205113 |
| Comet assay kit | Trevigen | Cat#4250-050-K |
| Experimental Models: Cell Lines | ||
| Mouse: embryonic primary fibroblasts | This study | N/A |
| Mouse: ES Cells Rnaseh2a−/− | KOMP | Rnaseh2a tm1(KOMP)Wtsi |
| Mouse: Rnaseh2b-null cell line | Rejins and Jackson | N/A |
| Mouse: Rnaseh2a−/− | This study | N/A |
| Mouse: Rnaseh2aG37S/− | This study and Pokatayev et al., 2016 | N/A |
| Mouse: Sting−/− | KOMP | Tmem173 tm1.1(KOMP)Mbp |
| Mouse: p53 | Jackson Lab Mice | B6.129S2-Trp53 tm1Tyj/J |
| Oligonucleotides | ||
| Primers for genotyping, see Table S1 | This study | N/A |
| Primers for RT-qPCR, see Table S1 | This study | N/A |
| RNase H2 substrate oligos, see Table S1 | This study | N/A |
| Short hairpin DNA for Biacore, see Table S1 and Figure S6 | This study | N/A |
| Oligo(dT)18 Primer | Thermo Scientific | SO131 |
| Recombinant DNA | ||
| Plasmid: pET15b | Novagen | S4762 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Opencomet plugin | Gyori et al., 2014 | http://www.cometbio.org/index.html |
| Math Works SLM tool | NIH | http://www.mathworks.com/matlabcentral/fileexchange/24443 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert J. Crouch (crouchr@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice and cell lines used in this study are listed in Key Resources Table. Rnaseh2aKO, STING−/−, RnasehG37S and Rnaseh2aRED were all originally produced in C57BL6/N ES cells. B6.129S2-Trp53tm1Tyj/J mice 6J. Mice for removal of drug-resistant markers employed were also C57BL/6 but if 6N or 6J not known. Thus, the mice studied in this investigation most likely contain mixtures of 6N and 6J genomes but with a strong bias for 6N genes.
All animal care and experimental procedures were performed in accordance with the ethical guidelines approved by NIH’s Office of Animal Care and Use 15-029 (OACU). The day when we confirmed vaginal plugs was defined as E0.5, and the day of birth was as P0. Primary MEF cell lines were prepared from E10.5 or E14.5 embryos of either sex and cultured in DMEM at 37°C in humidified incubator under 5% CO2.
Rnaseh2aRED (P40D Y211A) ES cells were first made for the P40D mutation by modification by Quikchange of the plasmid used to produce the RnasehaG37S mouse (Pokatayev et al.,2016) by neomycin selection. These ES cells were then used to introduce the Y211A mutation by hygromycin selection. Each cassette for drug selection contained an I-Scel site. To ensure the mutations resided on the same allele, DNA from neomycin-hygromycin selected cells was digested by I-Scel and a ~10kbp DNA fragment was detected by Southern analysis resistance genes were used for positive selection of double mutation and subsequently deleted from knock-in mice by Cre- and Flip-dependent recombination. Rnaseh2aG37S knock-in mice were as described previously (Pokatayev et al., 2016) except the region between the loxP site was removed. Rnaseh2atm1(KOMP)Wtsi ES cells and Tmem173tm1.1(KOMP)Mbp (Sting) KO mice were obtained from the Knockout Mouse Project (KOMP) repository. Rnaseh2atm1(KOMP)Wtsi ES cells were used to generate heterozygous Rnaseh2a+/−. B6.129S2-Trp53tm1Tyi/J (p53) mice were obtained from The Jackson Laboratory. Rnaseh2aKO, STING−/−, RnasehG37S and Rnaseh2aRED were all originally produced in C57BL6/N ES cells. B6.129S2-Trp53tm1Tyj/J mice are 6J. Mice for removal of drug-resistant markers employed were also C57BL/6 but if 6N or 6J not known. Thus, the mice studied in this investigation most likely contain mixtures of 6N and 6J genomes but with a strong bias for 6N genes. Sex of embryos were not determined. All mouse studies were approved by the NICHD Animal Use Committee protocol 15-029.
METHODS DETAILS
Genotyping
For mouse genotyping, DNA was isolated from mouse tail-clips or embryo yolk sacs with DNeasy Blood & Tissue kit (QIAGEN) according to manufacturer’s instructions. Genotypes were verified by PCR with Taq polymerase (Roche).
Generation of MEFs
For generation of Rnaseh2aRED/RED p53−/−, Rnaseh2aRED/G37S p53−/−, Rnaseh2aRED/+ p53−/− and Rnaseh2a+/+ p53−/− MEFs, whole embryos at E10.5 were minced and cultured in DMEM with 10% FBS, 2 mM L-Glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (GIBCO), 0.1 mM β-Mercaptoethanol, initially in 12-well plate at 37°C, 5% CO2. Rnaseh2aG37S/− p53+/+ and Rnaseh2a+/+ p53+/+ MEFs were also established using E14.5 embryos as described above after removing head, limbs, tail and red organs from the embryos. After incubation for 24 h, culture medium was exchanged with fresh medium to remove non-adherent cells. Adherent cells were subsequently maintained and passaged as appropriate under the same condition.
Western blotting
Harvested MEFs were washed twice in ice-cold PBS, resuspended in buffer containing 50 mM Tris-HCl (pH 8.0), 60 mM KCl, 0.1% Triton X-100 and 100-fold diluted protease inhibitor cocktail (Calbiochem) and disrupted by sonication. Insoluble proteins were removed by centrifugation. The concentration of soluble proteins was determined with Pierce BCA Protein Assay kit (Thermo Scientific) using bovine serum albumin as a standard. Equal amount of protein homogenates was loaded onto 10%–20% SDS-PAGE gel and separated. Upon electrophoresis, gel was soaked with 20% ethanol for 5 min and proteins were transferred to PVDF membrane with iBlot 2 Dry Blotting System (Invitrogen). Membrane was blocked with 5% milk (BIORAD) in TBST (50 mM Tris-HCl pH7.5,150 mM NaCl, 0.2% Triton X-100) at RT for 1 h and subsequently incubated with diluted antibodies (1:1000 for anti-RNase H2A antibody (Proteintech) or 1:2000 for anti-RNase H2C and anti-β-actin antibodies (Abcam)) in TBST containing 0.5% milk at RT for 1 h with agitation. After three washes with TBST, the membrane was incubated with 20000-fold diluted anti-rabbit IgG secondary antibody (Abcam) in TBST containing 0.5% milk at RT for 1 h and further washed three times with TBST plus 0.5% milk. HRP activity from antibody was detected by soaking membrane with Pierce ECL western blotting substrate (Thermo Scientific) at RT for 1 min and subsequently scanning chemiluminescent signal on membrane with Azure C600 (Azure Biosystem).
Preparation of recombinant mouse RNase H2
Three genes encoding mouse RNase H2 subunits (Rnaseh2a, Rnaseh2b and Rnaseh2c) were cloned from cDNA and inserted into pET15b expression vector as a polycistronic construct as was done for expression of human RNase H2 (Chon et al., 2009). All proteins bear the N-terminal His tag removable with thrombin. The pET15b derivatives for overproduction of RNase H2RED and RNase H2G37S were constructed with Quikchange mutagenesis kit (Stratagene) by introducing point mutations into Rnaseh2a.
Mouse RNase H2 was overproduced in E. coli BL21(DE3) and purified as described for human RNase H2 (Chon et al., 2009). When further purification is required, the protein was loaded to Hiload 16/60 Superdex 75pg column (GE Healthcare) equilibrated with 20 mM Tris-HCl (pH 7.5) containing 1 mM EDTA and 150 mM NaCl and the fractions containing 1:1:1 complex of three RNase H2 subunits were collected. The purity of proteins was confirmed by 10%–20% gradient SDS-PAGE (Bio-Rad), followed by Coomassie Brilliant Blue staining. Protein concentrations were determined by measuring the absorption at 280 nm (A280) and the A280 values for 0.1% (1.0 mg/ml) protein solution are 0.85 for RNase H2 and RNase H2G37S and 0.83 for RNase H2RED. These values were calculated by using absorption coefficient of 1576 M−1 cm−1 for Tyr and 5225 M−1 cm−1 for Trp at 280 nm (Goodwin and Morton, 1946).
RNase H activity assays
We employed 32P-labeled substrates because we found the fluorescent substrates previously used (Crow et al., 2006; Reijns et al., 2012) were less well hydrolyzed by RNase H2RED. When we used the same 18 bp hybrid substrates labeled with 32P-5′-RNA or with the fluorescent label, we found RNase H2RED to be 20% as active as RNase H2WT by autoradiography, compared to ~10% with autofluorography (Figure S1B). When we measured by fluorescence emission, the activity of RNase H2RED was even lower (~4%) (Figure S1B). Interestingly, RNase H2RED failed to cleave at several sites, one of them near the 3′ end with the FAM tag. This modest change in site selection was independent of the FAM moiety and explains the difference in product sizes seen by autofluorography (Figure S1B). Two recent reports have made similar observations for human RNase H2RED, including reduced affinity toward the same FAM-modified 18R substrate (Benitez-Guijarro et al., 2018; Zimmermann et al., 2018). Taken together, these data indicate that RNase H2RED activity is compromised toward short RNA/DNA duplexes, but it is proficient in removing long RNA/DNA hybrids, which are presumably present in R-loops.
Enzyme activity assays were performed using 5′-32P-labeled 12 base DNA5-RNA1-DNA6 (5′-GACACcTGATTC-3′) hybridized with 2 molar equivalents of the complementary DNA. Other 5′-32P-labeled RNAs were similarly treated. Uniformly labeled poly-32P-rA/poly-dT to determine Hybrid activity was synthesized as described (Crouch et al., 2001). The reaction buffer was 50 mM Tris-HCl (pH 8.0) containing 50 mM NaCl, 10 mM MgCl2,1 mM dithiothreitol, 100 μg/ml BSA and 5% (w/v) glycerol. Products of 1 μM substrates were applied to a 20% or 12% TBE-urea polyacrylamide gel and quantified by Typhoon imager upon electrophoresis.
For preparation of embryo homogenates, whole embryos at E11.5 were washed twice in ice-cold PBS and sonicated on ice in the buffer containing 50 mM Tris-HCl (pH 8.0), 60 mM KCl, 0.1% Triton X-100 and 100-fold diluted protease inhibitor cocktail. The supernatant was collected by centrifuge and used for activity assays. The concentration of total proteins was determined with Pierce BCA Protein Assay kit as described for western blot (see above).
Alkaline gel analysis of rNMPs in DNA
Genomic DNA was isolated from MEFs using Quick DNA Universal kit (Zymo Research). For alkaline hydrolysis, 1 μg of the genomic DNA was incubated with 0.3 M NaOH at 55°C for 2 h. After alkaline treatment, 6 × loading buffer (300 mM NaOH, 6 mM EDTA, 18% (w/v) Ficoll PM400 and Orange G) was added to treated DNA samples. 0.7% agarose gel was equilibrated by incubating in running buffer (50 mM NaOH and 1 mM EDTA) for 1 h. Electrophoresis was performed at 1 V cm−1 for 18 h. Upon electrophoresis, the gel was neutralized by soaking with 0.7 M Tris-HCl (pH 8.0) containing 1.5 M NaCl and stained with SYBR Gold. Gel images were obtained using UVP BioSpectrum AC Imaging System. Densitometric quantification of DNA in gel was performed with ImageQuant TL software (GE Healthcare).
Comet assay
The comet assay was performed with Comet Assay Electrophoresis System (Trevigen) following manufacturer’s instructions with some modifications. MEFs were synchronized by serum starvation for 24 h in serum-free DMEM medium and subsequently harvested, washed and suspended in ice-cold PBS at 2.0 × 105 cells/ml. LMAgarose (Trevigen) incubated at 37°C was added to suspended cells at a ratio of 10:1 (v/v) and the mixture was dispersed on comet slides (Trevigen). After gelling at 4°C, slides were soaked with 10 mM Tris-HCl (pH 9.5) containing 2.5 M NaCl, 0.1 M EDTA and 1% (v/v) Triton X-100 at 4°C for 16–18 h. Slides were then incubated in running buffer (0.2 M NaOH, 1 mM EDTA) for 20 min at room temperature. Electrophoresis was performed at 21 V for 15 min. Upon electrophoresis, slides were washed twice in dH2O and 70% ethanol and dried at 37°C. The DNA was stained with SYBR-Gold for 30 min at room temperature and briefly rinsed with dH2O. Comet images were captured with epifluorescence microscopy. Olive tail moments were determined by analyzing at least 100 comets/slide with OpenComet software (Gyori et al., 2014).
RT-qPCR
For RNA isolation, mouse embryos were washed in ice-cold PBS and homogenized in Trizol reagent (Invitrogen). Upon addition of chloroform, total RNA was isolated from water phase and purified with PureLink RNA Mini Kit columns (Ambion) and PreLink DNase (Invitrogen). cDNA was reverse transcribed from isolated RNA with Omniscript RT kit (QIAGEN) using oligo-dT primers (Thermo Scientific). Quantitative PCR was performed with SYBR Green I Master reagent (Roche) using Lightcycler 480 system (Roche). The expression of target genes was calculated by E-method (Tellmann, 2006) and normalized to the housekeeping gene (β-actin). All primer sequences are listed in Table S1.
Biosensor analysis
The binding properties of mouse RNase H2 to single ribonucleotide in DNA duplex were examined by surface plasmon resonance (SPR) analysis using Biacore 3000 instrument (GE Healthcare Life Science). All experiments were performed at 25°C in triplicate and repeated on three different sensor chips. The running buffer was HBS (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.005% Surfactant P20). A hairpin ligand DNA containing a single ribonucleotide in the stem of the hybrid and a biotinylated T in the loop (Figure S3A) (Gaidamakov et al., 2005) was immobilized on a streptavidin-coated CM4 sensor chip through biotin-streptavidin interaction. CM4 sensor chips were preconditioned by two times alternating injections of 10 mM HCl, 50 mM NaOH and 0.1% (w/v) SDS and then washed with water. Streptavidin was immobilized using amine coupling method by injection of 0.1 mg/ml streptavidin in HBS for 5 min. The surface was activated by injecting 1:1 mixture of 0.4 M 1-ethyl-3-(3-dimethylamino-propyl)-carbodiimide and 0.1 M N-hydroxysuccinimide at 5 μl/min of flow rate for 7 min, followed by injection of 0.01 mg/ml streptavidin in 10 mM sodium acetate (pH 5.0) for 7 min followed by 1 M ethanolamine hydrochloride for 7 min to deactivate excess reactive groups. Non-covalently bound streptavidin was eliminated by four alternate injections of 100 μL of 1 M NaCl and 50 mM NaOH at 50 μl/min. The single-stranded loop ligand DNA was diluted to 2 nM in HBS, heated at 90°C for 1 min and immediately transferred on ice. Then, ligand DNA was injected at 10 μl/min until about 40-50 RU of ligand was captured on the surface. Proteins were serially diluted at final concentrations in HBS. To minimize mass transport effects, proteins were injected at fast flow rate 60 μl/min for 90 s and then dissociation was monitored for 5 min. Surface regeneration was performed by duplicate injections of 20 μL of 10 mM HCl. To avoid protein contamination on surface, the binding of one protein was continuously measured at different concentrations and the surface was washed with 20 μL of 50 mM NaOH before injection of another protein. Data processing and analysis was performed using BIAevaluation 4.1 software (BIACORE).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical details in experiments were described in figures and figure legends. Comet assay data was analyzed by Student’s t test using Microsoft Excel. Also see above for the details of quantification of ribonucleotides in genomic DNA.
Ribonucleotide incorporation abundance were estimated through a slightly modified version of the method described in (Reijns et al., 2012). Starting from the densitometric histograms of electrophoresis lanes corresponding to alkali-treated genomic DNA, the background intensity was subtracted and a smoothing spline with 15 optimally placed internal knots was applied to the densitometric profiles by running the SLM tool (D’Errico, August 10, 2017(http://www.mathworks.com/matlabcentral/fileexchange/24443)) in MATLAB version 9.2. The smoothened intensity curves were resampled at intervals of width Δd = 1mm. For each interval, the characteristic fragment size (sz) was calculated following the equation d = a + b × log(sz), where d represents the electrophoretic distance in the middle point of the interval. Parameters a and b were inferred by fitting the linear model lm(d ~ log (sz)) to the peaks in the size reference lane. Because the conversion from densitometric intensity to fragment count is highly sensitive to small, noisy fluctuations in the far end of the electrophoretic gel, a cutoff at an electrophoretic distance dmax was introduced. The value of dmax was determined under supervision, as the point where fluctuations in the original (non-smoothened) intensities of the loaded lanes became similar in magnitude to those observed in empty lanes, indicating a poor signal-to-noise ratio. The fragment count associated to each interval was estimated as nsz = Isz/sz, being Isz the densitometric intensity in the interval. From these numbers, a preliminary estimate of the mean fragment size was obtained as , where the sum extends over all the intervals. To account for small fragments that had migrated beyond dmax, the mean fragment size was corrected under the assumption that break points are randomly distributed along the genome. Thus, the corrected estimate for the mean fragment size became , where szmin is the minimum size cutoff corresponding to dmax.
Given a genome of size G, the number of breakpoints is readily obtained as . Using the size of the mouse genome as reference, the number of break points was calculated for every sample. The excess of break points associated to a mutation was obtained by subtracting the number of break points in the WT to the number of break points in the mutant. Finally, the increase in the ribonucleotide incorporation rate associated to the mutation was computed as (N_WT – N_mutant)/G.
Supplementary Material
Highlights.
Excess ribonucleotides in DNA cause mouse early embryonic death
Above a threshold of tolerance, ribonucleotides in DNA induce apoptotic cell death
Below the threshold, ribonucleotides in DNA trigger an innate immune response
Competition between RNases H2 mutants affects rNMP removal and cell survival
ACKNOWLEDGMENTS
We acknowledge the National Cancer Institute (NCI) transgenic facility for development of the Rnaseh2aRED mice. We thank Dr. Reijns and Jackson for providing the Rnaseh2b-null cell line. R.U. is a Japan Society for the Promotion of Science Research Fellow in Biomedical and Behavioral Research at the NIH. This work was supported by the Division of Intramural Research (DIR) at the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Library of Medicine (NLM).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.019.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris A, and Rabe B (2017). Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum. Mol. Genet 26, 3960–3972. [DOI] [PubMed] [Google Scholar]
- Benitez-Guijarro M, Lopez-Ruiz C, Tarnauskaitė Ž, Murina O, Mian Mohammad M, Williams TC, Fluteau A, Sanchez L, Vilar-Astasio R, Garcia-Canadas M, et al. (2018). RNase H2, mutated in Aicardi-Goutieres syndrome, promotes LINE-1 retrotransposition. EMBO J 37, e98506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büsen W, and Hausen P (1975). Distinct ribonuclease H activities in calf thymus. Eur. J. Biochem 52, 179–190. [DOI] [PubMed] [Google Scholar]
- Cerritelli SM, and Crouch RJ (2009). Ribonuclease H: the enzymes in eukaryotes. FEBS J 276, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, and Crouch RJ (2016). The balancing act of ribonucleotides in DNA. Trends Biochem. Sci 41, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Vassilev A, DePamphilis ML, Zhao Y, Zhang J, Burgers PM, Crouch RJ, and Cerritelli SM (2009). Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res 37, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, and Cerritelli SM (2013). RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41, 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, and Kunkel TA (2015). Tracking replication enzymology in vivo by genomewide mapping of ribonucleotide incorporation. Nat. Struct. Mol. Biol 22, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin SR, Hollis T, and Perrino FW (2011). Functional consequences of the RNase H2A subunit mutations that cause Aicardi-Goutieres syndrome. J. Biol. Chem 286, 16984–16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover HN, Lujan SA, Chapman MJ, Cornelio DA, Sharif R, Williams JS, Clark AB, Camilo F, Kunkel TA, and Argueso JL (2015). Stimulation of chromosomal rearrangements by ribonucleotides. Genetics 201, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelio DA, Sedam HN, Ferrarezi JA, Sampaio NM, and Argueso JL (2017). Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair (Amst.) 52, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch RJ, Arudchandran A, and Cerritelli SM (2001). RNase H1 of Saccharomyces cerevisiae: methods and nomenclature In Methods in Enzymology, Nicholson AW, ed. (Academic Press; ), pp. 395–413. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. (2006). Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet 38, 910–916. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 167A, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolet M, Phoenix P, Menzel R, Masse E, Liu LF, and Crouch RJ (1995). Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. U S A 92, 3526–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein A, Potenski CJ, and Klein HL (2016). Increased spontaneous recombination in RNase H2-deficient cells arises from multiple contiguous rNMPs and not from single rNMP residues incorporated by DNA polymerase epsilon. Microb. Cell 3, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidamakov SA, Gorshkova II, Schuck P, Steinbach PJ, Yamada H, Crouch RJ, and Cerritelli SM (2005). Eukaryotic RNases H1 act processively by interactions through the duplex RNA-binding domain. Nucleic Acids Res 33, 2166–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Borán M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi-Falconi M, and Jiricny J (2013). Ribonucleotides misincorporated into DNA act as strand discrimination signals in eukaryotic mismatch repair. Mol. Cell 50, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin TW, and Morton RA (1946). The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem. J 40, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, et al. (2015). Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Invest 125, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement M-V (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, and Roers A (2012). Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med 209, 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Tam OH, and Tam PPL (2014). Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol 34, 65–75. [DOI] [PubMed] [Google Scholar]
- Lim YW, Sanz LA, Xu X, Hartono SR, and Chédin F (2015). Genomewide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. eLife 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Pursell ZF, Abdulovic-Cui AA, Clark AB, Nick McElhinny SA, and Kunkel TA (2012). Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet 8, e1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdowell EC, Allen E, and Macdowell CG (1927). The prenatal growth of the mouse. J. Gen. Physiol 11, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino FW, Harvey S, Shaban NM, and Hollis T (2009). RNaseH2 mutants that cause Aicardi-Goutieres syndrome are active nucleases. J. Mol. Med. (Berl.) 87, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, and Crouch RJ (2016). RNase H2 catalytic core Aicardi-Goutières syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J. Exp. Med 213, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, et al. (2010). Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum 62, 1469–1477. [DOI] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. (2012). Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, and Burgers PM (2012). RNase H2-initiated ribonucleotide excision repair. Mol. Cell 47, 980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, and Cimprich KA (2016). Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellmann G (2006). The E-method: a highly accurate technique for gene expression analysis. Nat. Methods 3, i. [Google Scholar]
- Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, and Kunkel TA (2013). Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Mol. Cell 49, 1010–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Lujan SA, and Kunkel TA (2016). Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat. Rev. Mol. Cell Biol 17, 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.