

Abstract
Introduction:
Integral membrane proteins and lipids constitute the bilayer membranes that surround cells and sub-cellular compartments, and modulate movements of molecules and information between them. Since membrane protein drug targets represent a disproportionately large segment of the proteome, technical developments need timely review.
Areas covered and literature search strategy:
Publically available resources such as Pubmed were surveyed. Bottom-up proteomics analyses now allow efficient extraction and digestion such that membrane protein coverage is essentially complete, making up around one third of the proteome. However, this coverage relies upon hydrophilic loop regions while transmembrane domains are generally poorly covered in peptide-based strategies. Top-down mass spectrometry where the intact membrane protein is fragmented in the gas phase gives good coverage in transmembrane regions, and membrane fractions are yielding to high-throughput top-down proteomics. Exciting progress in native mass spectrometry of membrane protein complexes is providing insights into subunit stoichiometry and lipid binding, and cross-linking strategies are contributing critical in-vivo information.
Expert commentary:
It is clear from the literature that integral membrane proteins have yielded to advanced techniques in protein chemistry and mass spectrometry, with applications limited only by the imagination of investigators. Key advances toward translation to the clinic are emphasized.
Keywords: proteome, bilayer, FT-ICR, CyTOF, FASP, micelle, nanodisc, HDX, FPOP, LILBID
1. Introduction
Membrane proteins and lipids make up the fluid-mosaic bilayer membranes that surround cells and sub-cellular organelles. Integral membrane proteins modulate movements of molecules and information across membranes making them functionally very important and thus common drug targets. Protocols developed over the last few years have enabled efficient extraction and digestion such that membrane protein coverage in bottom-up, peptide-based proteomics is essentially complete based upon soluble peptides from loop regions. Transmembrane domains of membrane proteins are under-represented in these traditional bottom-up proteomics experiments because their hydrophobicity and lack of charged residues leads to very poor peptide recovery. Top-down mass spectrometry studies that target intact integral membrane proteins provide access to transmembrane regions though high-throughput top-down proteomics on this sector of the proteome has lagged behind peptide-based strategies. Steady progress in analysis of protein-protein interactions is providing insights into complex stoichiometry by native mass spectrometry and cross-linking strategies are gaining ground. Clever chemical approaches are being applied to study cell-surface membrane proteins and the complex glycosylation signatures that decorate them. This review aims to provide insights into the critical experimental progress that has occurred over the last 2–3 years, and to emphasize advances relevant to translation to the clinic.
2. Bottom-up proteomics advances for membrane proteins
For a number of years the ‘filter-assisted sample preparation’ (FASP) method, first developed in Liebler’s laboratory [1,2], became the favored protocol for whole proteome or membrane fraction analysis relying upon the power of 4% SDS to solubilize membrane and other proteins [3]. Mann’s group published a series of papers that established FASP as state-of-the-art for coverage of integral membrane proteins including over 83% mouse known glutamate and GABA receptors from hippocampal membranes [4]. The group extended the power of the technique by using PTMs such as phosphorylation and glycosylation to map plasma membrane protein topology [5]. However, there are disadvantages to the FASP protocol that seem to originate in the unpredictable protein loss associated with filter usage and resulting in problems with quantification. Loo’s group replaced urea with deoxycholate providing an interesting variation that eliminates problems associated with use of urea, and reported other protocol modifications such as pre-passivation of the filters using tween detergent (eFASP) [6]. So while FASP protocols are still in use a common theme in the last few years involves in-solution digests in detergents that do not need to be removed by macro-scale filtration. Mann’s group introduced an ‘in stage-tip’ protocol involving guanidinium and deoxycholate allowing the entire sample preparation to be performed in a pipette tip [7,8]. Krijgsveld introduced ‘SP3’ whereby peptides are immobilized on the hydrophilic surface of carboxylate-coated paramagnetic beads allowing organic rinsing to remove SDS in a variant of hydrophilic interaction chromatography [9]. Membrane proteins are well covered through in-solution digestion in 0.5% sodium deoxycholate, 12 mM N-lauryl sarcosine at pH 8.5 [10] and this protocol is holding up well in comparison to others [11,12]. This mixture becomes denaturing at 95 ˚C enabling full solubilization of membrane proteins yet trypsin-friendly once returned to 37 ˚C. Detergents are removed after digestion via acidification and ether extraction.
So how can bottom-up procedures be further improved ? Many researchers are looking to develop small bio-reactors that go beyond Stage Tips for very quick preparation of samples for bottom-up analysis, with the overall goal of full automation to eliminate human steps and thus improve reproducibility. Zhang extrapolates from a specialized sample preparation pipeline originally set up for hydrogen-deuterium exchange (HDX) analysis of GPCRs that required very rapid proteolysis and analysis to limit peptide back exchange [13]. However, it is not yet clear that use of pepsin will provide the sequence specificity required to address complex mixtures. A bioreactor based upon polydopamine grafted porous graphene showed promising results for membrane proteins [14]. Automated systems that incorporate these themes enabling fast and reproducible sample preparation will become increasingly prevalent.
3. Organelle localization
The approaches described in section 2 allow all quantification strategies to be used in the context of membrane proteins though important information can be lost if cellular location is ignored. Localization of organelle proteins by isotope tagging or protein correlation profiling are powerful techniques to group proteins based upon their co-migration through gradients as a result of being in the same membrane fraction [15,16]. The Lilley group recently presented a more evolved version of the methodology named hyperplexed LOPIT (hyperLOPIT) [17], to study the subcellular proteome of the mouse pluripotent stem cells. HyperLOPIT introduces novel approaches for sample preparation, mass spectrometry data acquisition and multivariate analysis, benefiting from a number of recent technological improvements, such as the development of neutron-encoded isotopologue variants of tandem mass tags (TMT), which increase the multiplexing capacity of isobaric tagging experiments, enabling larger subcellular fractions to be sampled [18], and the quantitative accuracy of TMT-based applications that significantly improved by mass spectrometry data acquisition using synchronous precursor selection MS3 (SPS-MS3) [19]. Therefore Christoforou et al, incorporated SPS-MS3 acquisition on the Orbitrap Fusion Tribrid mass spectrometer into the hyperLOPIT pipeline, and demonstrated that it significantly improved spatial resolution and the reliability which protein localization may be determined, and then they extended the data analysis platform to facilitate rapid interrogation of the data by providing an easy-to-use graphical user interface.
This method may have implications for characterizing the impact of post-transcriptional and post-translational modification on protein location and studies involving proteome-level locational changes on cellular systems. Furthermore it may provide information on the localization of protein isoforms, although the impact of post-transcriptional modification on protein location has been previously studied [20], however the examination of the differences in subcellular location of closely related isoforms has not been previously possible with LOPIT, as the accuracy of quantification was inadequate to reliably characterize the few peptides, or single peptide, which distinguish protein isoforms [17]. Finally, a related approach was used to define major organellar membrane fractions from a macrophage derived cell line and in parallel empower better localization of membrane lipids [21,22]. Since many gradient fractions must be analyzed with multiple sub-fractions the hyperLOPIT approach may be currently regarded as heavy on instrument usage. However, as high-order multiplexing becomes available and instruments become faster such considerations will diminish in significance.
4. Top-down proteomics
While critical modifications to bottom-up protocols improved access to transmembrane domains at the peptide level [23,24] these methods have not seen widespread adoption. Top-down mass spectrometry whereby the intact membrane protein is analyzed by tandem mass spectrometry provides a rigorous solution to inclusion of transmembrane domains and was first demonstrated for single pass membrane proteins in 2002 [25]. Now that general chromatographic systems have been established for purification of integral membrane proteins analysis by top-down mass spectrometry with both collisionally activated and electron capture/transfer dissociation (CAD, ECD, ETD) is routine [26]. Efforts now have moved to high-throughput analysis of membrane protein fractions with online LC-MS. Coverage of membrane proteins in the first high-throughput top-down analysis was respectable [27] and experiments specifically focused on a mitochondrial-enriched membrane fraction were successful in identifying 83 integral membrane proteins with 46 from the oxidative phosphorylation pathway [28]. The workflow relies upon solubilization of membranes in SDS and a size-based separation on a Gel-Free device (Off-Gel) generating SDS containing fractions that need organic precipitation followed by resolubilization prior to nano-reverse-phase separations of fractions. Perhaps the greatest disadvantage of this approach is that the top-down mass spectrometry experiment is limited by the time available across a chromatographic peak such that lower abundance or poorly ionizing proteins have sparse data coverage. This problem leads to a bias toward smaller proteins and only a handful greater than 30 kDa were identified, and most of these lacked an intact mass measurement [28]. This issue is being addressed as more advanced instrument control systems use collected data to direct on-going tandem mass spectrometry experiments [29]. Disadvantages of online top-down can be addressed using offline separations as originally advocated [25]. Skinner reported that CAD was more efficient than ECD in fragmentation of transmembrane domains in a study of 152 integral membrane proteins from a human cell line [30], though this was probably due to lack of ion activation during, or after, the ECD experiment in order to melt the alpha-helices so that successful electron-induced dissociation events could be observed [31]. It will be interesting to see whether ultra-violet photodissociation (UVPD) [32] improves access to transmembrane domians.
An outstanding question is whether there is a single separation that can accommodate all of the membrane proteins in a specific membrane system. Walker and colleagues have published detailed intact protein analyses of the subunits of the complexes of the inner mitochondrial membrane and it is clear that the integral subunits coded within the mitochondrial genome may represent the greatest challenge in this respect [33]. Mitochondrial ATP synthase and Complex-1 preparations were precipitated with ethanol and redissolved in formic acid, trifluoroethanol, hexafluoro-propan-2-ol, water (60:15:1:24; v/v) with reductant TCEP where desired. The authors used a polymeric column at elevated temperature (PLRP/S, 50˚C)[34] and running buffers containing trifluoroethanol, hexafluoro-propan-2-ol with 50 mM ammonium formate and a propan-2-ol gradient. There is a huge space in stationary and mobile phases for further exploration.
The major limitation of a single separation is that space is limited such that while isolated protein complexes can be resolved to their individual subunits, more diverse mixtures from whole membrane systems need a second dimension of separation. This is why Kelleher and coworkers used an SDS-PAGE based separation followed by reverse-phase chromatography for their high-throughput work [28]. Others are experimenting with capillary electrophoresis coupled to reverse-phase chromatography [35].
Another approach is to combine top-down with bottom-up technology. Pasa-Tolic and coworkers used pepsin in an online reactor taking advantage of reverse-phase separations at low pH [36]. Hunt and coworkers used such an approach to fully characterize antibodies including disulfide assignments [37]. Such a strategy might be especially effective for membrane proteins where middle-down approaches are desirable, though probably more effective where mixtures have been previously simplified.
5. Native mass spectrometry of membrane proteins
Significant breakthroughs have been made in analysis of integral membrane protein complexes under native conditions that preserve non-covalent associations between subunits as well as bound lipids [38,39]. An important insight emerging from this work was the demonstration that while a range of detergents could be used to solubilize and ionize a number of different micellar complexes, only a subset of those preserved the most compact form of the native state measured as collisional cross section by ion-mobility mass spectrometry [40]. Furthermore, for some complexes specific lipid binding events conferred stability on the complex. This was so for phosphatidylglycerol binding to the ammonia transporter AmtB and a new X-ray crystal structure of the PG-AmtB complex revealed minor structural changes that altered interaction with free bilayer lipids. An extension of this work showed that detergents that promoted lower charge states were most effective at preserving the compact form of the native complex [41]. Such measurements allowed careful review of observed collisional cross sections for a range of membrane protein complexes leading to the conclusion that choice of calibrant native protein complexes is critical [42]. A detailed analysis of the role of lipids in providing interfaces between subunits in a membrane protein complex provided examples where a lipid such as cardiolipin could take on such a role essentially as a structural element supporting parallel molecular dynamics simulations [43,44]. For complexes evolved to bind cardiolipin at the interface its presence was essential for dimerization.
The most commonly used mass spectrometer for native MS work has traditionally been the quadrupole time-of-flight especially those equipped for ion-mobility MS. While the latest Q-TOF instruments are achieving ever improving resolution, there have been notable efforts to translate native MS to high-resolution mass spectrometers including Fourier-transform ion cyclotron resonance (FT-ICR) [45,46] and orbitrap platforms [47–49]. Judging by the inclusion of membrane proteins in nearly all these reports one concludes that the principles of native MS are holding firm across a variety of source and front-end geometries. The extra resolution allows true native top-down MS [47,50] whereby a native complex is sprayed, excited to release individual polypeptide chains that are then subjected to CAD/ECD for full characterization of quaternary and primary structure in one experiment. Alternative activation techniques have also been demonstrated with use of infra-red multi-photon laser for heating gas-phase micelles to release bacteriorhodopsin holoprotein with out disruption of its Schiff-linked retinal chromophore [46] and use of surface-induced dissociation to release monomers of AmtB from the native trimer [51]. With increasing focus on the study of native membrane protein complexes and new investigators joining the field there will undoubtedly be exciting new discoveries emerging. While the native mass spectrometry community has predominantly used detergent micelles as the most common means to solubilize membrane–protein complexes for ionization, arguments remain that at least in some cases the protein needs to be surrounded by a lipid bilayer to achieve faithful recapitulation of function. For this reason, considerable excitement has surrounded development of nanodiscs and that this environment will empower native membrane protein mass spectrometry [52,53].
Nanodiscs are small lipid bilayers bounded and solubilized by an apolipoprotein scaffold that rings the disc providing a small bilayer-like environment for integral membrane-protein complexes, with typically one complex per disc. While spraying nanodiscs does not present significant challenges, activation to selectively remove scaffold protein and lipids while leaving the membrane-protein complex intact has so far presented a barrier to progress. Recently, Morgner’s group reported use of laser-induced-liquid-bead-ion-desorption (LILBID) to study a number of nanodisc-solubilized membrane-protein complexes providing proof of principal for the approach [54]. Other tools in structural proteomics such as fast photochemical oxidation of proteins (FPOP) have been successfully applied to membrane proteins in nanodiscs identifying aqueous accessible surfaces [55]. Hydrogen-deuterium exchange was recently applied to nanodisc preparations of LeuT, a thermostable eubacterial homolog of mammalian neurotransmitter/sodium symporters, enabling local measurements of kinetics in the presence of lipids [56]. Intense study in this area will yield many new insights in the near future.
6. Structural proteomics
While chemical crosslinking mass spectrometry (XL-MS) approaches to study membrane protein structure are not novel there has been a strong resurgence in this strategy over the last few years. This has been fueled by a new generation of carefully designed crosslinking reagents, widespread availability of high-resolution mass spectrometers and improvements in software for crosslink assignment [57,58]. Some approaches have been especially useful in the context of membrane protein structural proteomics. One such strategy is in vivo crosslinking whereby crosslinking chemistry is performed within live cells [59,60]. In vivo distance constraints can be compared to available crystal structures providing valuable insight into structure and function in the cellular context. Furthermore, distance constraint information can be used by other researchers to steer their own work. Marcoux and coworkers used a specific constraint from Bruce’s E. coli dataset [60] to help interpret gas-phase collisional cross-section (CCS) data from ion-mobility native MS of the E. coli outer membrane OmpA [61]. CCS of the OmpA dimer supports a model of paired periplasmic C-terminal domains projecting away from the transmembrane porins where they might interact with peptidoglycans [62]. This study is a fine example of how an existing crystal structure and a published distance constraint from XL-MS, combined with molecular modeling and native MS could yield important insights into a long standing controversy over the function of OmpA in vivo. Goulding and coworkers combined X-ray crystallography of sub-domains with XL-MS to better understand the transmembrane topology of the MmpL proteins of the Mycobacterium inner membrane [63]. Understanding topology of critical substrate binding and transport residues of the MmpL family are pivotal for current anti-tuberculosis drug development efforts. An expansion in cross-linking studies will be reflected in the literature in the coming years.
7. Glycosylation analysis of integral membrane proteomics
Patterns of glycosylation on cell-surface and other membrane proteins are clinically important and a range of approaches has been developed to characterize them using mass spectrometry. Most efforts have been directed at N-linked glycans while O-linked complex glycans are gaining ground. Progress in direct analysis of glycosylated peptides [64–66] and appropriate informatics strategies [67] as well as chemical glycoproteomics strategies [68] have been reviewed recently. Analysis of tryptic peptides that carry their natural glycosylation is particularly attractive since the context of the glycosylation is retained. Analysis is aided greatly by high-resolution MS1 and MS2 scans because product-ions are more accurately assigned. Use of electron-transfer dissociation (ETD) provides a complementary fragmentation mechanism to CAD that often improves characterization of the peptide backbone. Workflows can lead to description of multiple N- and O- glycosylation sites in one experiment [69]. For higher throughput Mann and coworkers used lectin enrichment of N-linked peptides yielding 2383 glycosites on 1321 protein groups, finding them highly enriched in cell membrane proteins [70]. The experiment allowed segregation of ABC from GCB varieties of diffuse large B-cell lymphoma, while prior gene expression analysis had classified this as a single disease entity.
8. Cytometry mass spectrometry and cellular imaging.
A defining feature of higher mammalian species is the wide diversity of cell types present often defined by unique sets of cell surface markers, typically integral membrane proteins of the plasma membrane. Traditionally flow cytometry has been used to sort and classify different cell populations based upon a small number of antibodies to cell surface markers, with modest multiplexing achieved through tagging the antibodies with different fluorophores. A significant advance is cytometry by time-of-flight or CyTOF [71]. This technology replaces fluorophores with rare-earth metals that can be read out with high sensitivity and accuracy using a mass spectrometer. The antibodies used are decorated with polymer chains of chelating groups that bind metal ions predominantly lanthanides, from 141-Pr to 176-Yb. Since crossover between different metals is practically zero a much higher level of multiplexing can be achieved – 30 – 40 different antibodies are possible. For effective single cell detection hundreds of antibodies per target must bind to each cell and each antibody must carry multiple metals, and of course target specificity is only as good as the antibody used. The inductively coupled plasma time-of-flight-mass spectrometry (ICP-MS) readout destroys the sample so no other information can be extracted from the data. Specialized bioinformatics tools have been developed to recognize single cell events as cell suspensions pass through the MS interface [71]. The potential relevance of CyTOF technology as a clinical tool has been highlighted in studies that cover comprehensive profiling of cellular phenotype, epitope screening, signaling state, cytokine/chemokine expression, and viability [72,73]. It is beyond any doubt that CyTOF will help us to optimize strategies for stem cell expansion and tracking. Of perhaps even greater significance is the application of CyTof technology to cellular imaging whereby laser ablation is used to eject metal ions from antibody decorated tissue sections [74,75]. Thirty-two separate proteins and phosphorylation sites within a single paraffin-embedded formalin-fixed breast cancer tumor specimen were analyzed demonstrating heterogeneity among cellular subtypes, and cell–cell crosstalk at the tumor–stromal interface in the form of two phosphorylation sites on S6 protein. These studies clearly indicate that CyTOF is well suited to the task of defining subpopulations and delineating the contextual dependency of their signaling states. A related technology called multiplexed ion beam imaging (MIBI) from the same group has also been used for imaging with promising results [76]. Use of an O- ion beam with sub-micron spot size promises un-paralleled spatial resolution.
9. Expert commentary:
While apprehension of working with membrane proteins has been overcome, the challenges remain real. Bottom-up proteomics of membrane proteins has matured with essentially complete coverage achieved if one accepts that integral membrane proteins are represented by peptides derived from their aqueous accessible loop regions, adequate for global quantification studies and post-translation modifications such as phosphorylation. Unfortunately, transmembrane domains remain largely in the dark in bottom-up proteomics work, leading many to assume there’s nothing interesting going on there. Knowing biology, when scientists do start looking carefully it will turn out that this is not the case. Currently, to look at transmembrane regions it is most effective to perform top-down analysis using the sparing solubility imparted to the intact protein from its soluble domains to get the whole molecule into the mass spectrometer where the power of gas-phase dissociation chemistries can be brought to bare. It is likely that stable features like the transmembrane alpha-helices remain folded in the gas phase raising the potential to obtain structural information from dissociation experiments and requiring thermal excitation for effective electron capture dissociation. Native mass spectrometry of intact integral membrane protein complexes is developing rapidly with an expanding number of groups exploring the role lipid molecules play in their structure and function. Single lipid molecules can control monomer/dimer equilibria and thus function. It is becoming clear that lipid modulation of membrane protein function is a highly dynamic property with tightly binding ‘structural’ lipid cofactors competing with less specific bilayer lipids from the milieu in an ongoing basis. Molecular dynamics simulations are starting to play what will become a central role in guiding experimentation in this respect but practical analyses will continue to be necessary to tease apart bilayer effects on membrane-protein structure and function and the way small molecules can be used to modulate these properties. Specially designed crosslinking molecules that cross cell membranes have opened up the potential of in-vivo crosslinking providing a new avenue to confirm relevance of in vitro experiments and to discover novel interactions. Membrane protein glycosylation, of critical importance at the cell surface and in several internal membrane systems is now accurately addressed by mass spectrometry with the potential to impact clinical diagnoses. New strategies for imaging using ICP-MS coupled with antibodies decorated with heavy metals have enabled much higher levels of multiplexing in both flow cytometry and cellular imaging. The approach can be directed at the cell surface or at internal targets after permeabilization for probe access.
10. Five-year view:
As more young researchers are trained to handle membrane proteins in pioneer laboratories the field will continue to expand. Faster and more sensitive mass spectrometry equipment will drive advances in throughput especially for bottom-up and top-down approaches where chromatography controls sample delivery to the instrument. Study of native complexes will expand beyond the traditional Q-tof platform, though the high resolution afforded by Fourier-transform ICR and orbitrap instruments will only become maximally empowered when extended mass range is combined with ion mobility measurements of collisional cross section. From a practical perspective it is important to remember that nearly all studies of integral membrane proteins are currently being performed using detergent micelles to provide solubility thereby separating the protein from any bilayer influences. It is thus critically important to qualify each new membrane protein target to ensure that any functional features being analyzed in a micelle are faithful to behavior in the bilayer. For this reason current attempts to use small bilayer nanodiscs will undoubtedly be expanded to address technical hurdles that have limited progress in the past. Each jump in instrument technology is accompanied by a wealth of novel opportunities and membrane protein research will benefit in ways as yet unforeseen.
Figure 1.
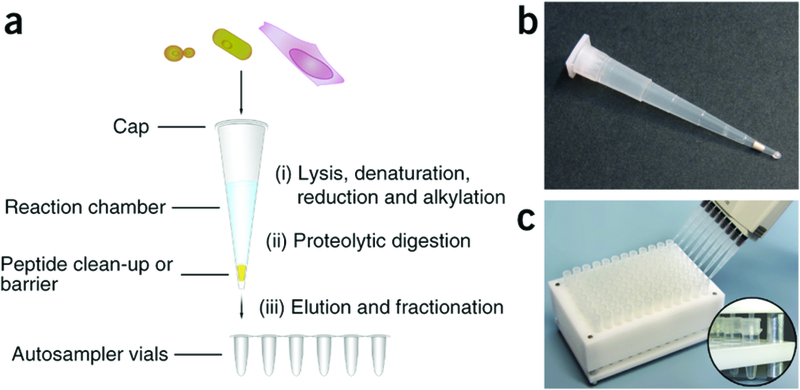
Sample-preparation bioreactors. Stop and go extraction tips (STAGE tips) have become the favored sample preparation platform for bottom-up proteomics. Use of deoxycholate/lauryl sarcosine may be superior to dodecyl sulfate for membrane proteome coverage. Small bioreactors that can be automated for robotic sample preparation will gain prevalence. Reproduced from ‘Minimal, encapsulated proteomic-sample processing applied to copy-number Nils A Kulak, Garwin Pichler, Igor Paron, Nagarjuna Nagaraj, Matthias Mann. Nature Methods 11, 319–324 (2014)’. Reprinted by permission from Macmillan Publishers Ltd (http://www.nature.com/nmeth/index.html).
Figure 2.
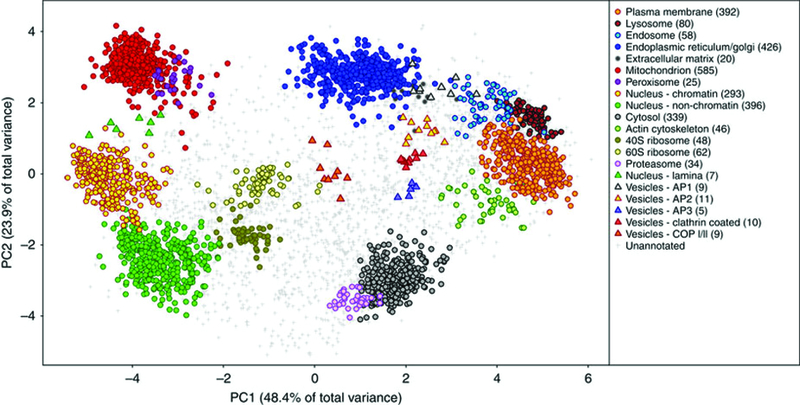
The spatial proteome. Clustering of proteins to individual organelles and machines was achieved using correlation profiling (HyperLopit). Cellular maps such as this one can be used to study perturbations in health, disease and treatment. Reproduced from ‘A draft map of the mouse pluripotent stem cell spatial proteome. Andy Christoforou, Claire M. Mulvey, Lisa M. Breckels, Aikaterini Geladaki, Tracey Hurrell, Penelope C. Hayward, Thomas Naake, Laurent Gatto, Rosa Viner, Alfonso Martinez Arias, Kathryn S. Lilley. Nature Communications 7, Article number: 9992 (2016)’. Reprinted by permission Nature Publishing Group (doi: 10.1038/ncomms9992).
Figure 3.
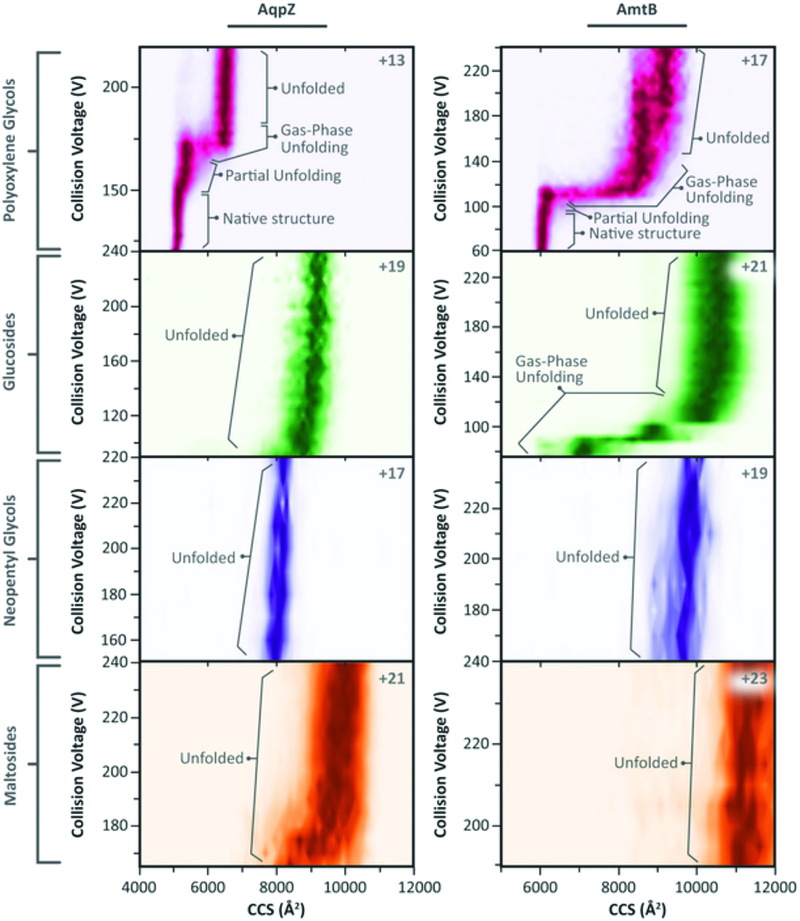
Ion mobility of native membrane-protein complexes. While these complexes all retain native oligomeric state, certain detergents favor lower charge states (top right of each panel) and minimal collisional cross sections consistent with X-ray crystal structures. Ion mobility mass spectrometry appears essential for true native MS. Reproduced from ‘Membrane proteins bind lipids selectively to modulate their structure and function. Arthur Laganowsky, Eamonn Reading, Timothy M. Allison, Martin B. Ulmschneider,Matteo T. Degiacomi, Andrew J. Baldwin, Carol V. Robinson. Nature 510, 172–175 (05 June 2014)’. Reprinted by permission from Macmillan Publishers Ltd: Nature (510, 172–175), copyright (2014) (https://www.nature.com/ncomms/).
Figure 4.
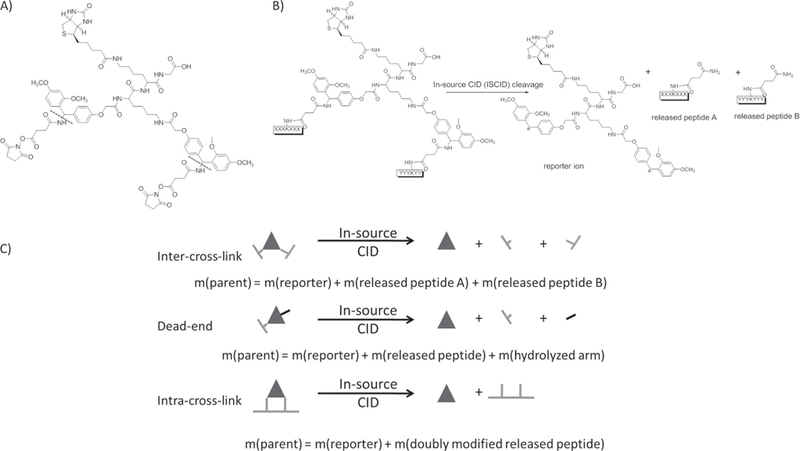
In-vivo crosslinking of membrane protein complexes. The PIR strategy uses specially designed reagents that cross biological membranes to access membrane proteins in their native environment. Lability of the linker during ionization is exploited to reveal low-abundance productive crosslinked pairs of peptides. Reproduced from ‘Cross-linking Measurements of In Vivo Protein Complex Topologies. Chunxiang Zheng, Li Yang, Michael R. Hoopmann, Jimmy K. Eng, Xiaoting Tang, Chad R. Weisbrod, James E. Bruce. Mol. Cell. Proteomics. 10(10):M110.006841. 2011’. Reprinted by permission American Society for Biochemistry and Molecular Biology 2011.
11. Key issues:
The membrane proteome is now well covered in bottom-up proteomics experiments with a shift from filter-assisted digestion protocols to small bio-reactors such as ‘stage tips’ that minimize sample loss.
Quantitative analysis of density gradients using the HyperLopit mass spectrometry protocol is providing a powerful tool with which to group proteins including the membrane fraction into different organelles.
Transmembrane domains are poorly represented in bottom-up experiments but can be accessed using top-down mass spectrometry. High-throughput top-down analysis of membrane fractions has been demonstrated and for smaller proteins will become routine over the next few years.
Membrane-protein complexes are being more widely analyzed by native mass spectrometry allowing measurements of collisional cross-section using ion mobility mass spectrometers. Details of specific lipid binding are providing new insights into their role in structure and function of these complexes.
Native ionization of membrane protein complexes is being combined with top-down high-resolution mass spectrometry for more complete analysis of structure.
Great strides have been made in chemical crosslinking combined with high-resolution mass spectrometry as well as dedicated software platforms in order to provide structural constraints. In vivo crosslinking is providing insights that are impossible to obtain after solubilization of membranes in vitro.
New approaches to define cell surface integral membrane proteins are improving better precision in this arena and glycosylation patterns of these proteins are becoming tractable.
Heavy metal coding of antibodies with readout by flow cytometry and inductively coupled plasma mass spectrometry is establishing itself in the clinic for considerably improved multiplexing during flow analyses. The technology has been extended to imaging providing a powerful new tool for pathologists.
Acknowledgments
Funding
This manuscript was supported by grants from the United States Department of Health, National Institutes of Health: P30 DK063491 (JPW), R01AI101888 and U19AI067769.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Manza LL, Stamer SL, Ham AJ, et al. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics, 5, 1742–5 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Liebler DC, Ham AJ. Spin filter-based sample preparation for shotgun proteomics. Nat Methods, 6, 785–6 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Wisniewski JR, Zougman A, Nagaraj N, et al. Universal sample preparation method for proteome analysis. Nat Methods, 6, 359–62 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Wisniewski JR, Zougman A, Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J Proteome Res, 8, 5674–8 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Wisniewski JR, Nagaraj N, Zougman A, et al. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res, 9, 3280–9 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Erde J, Loo RR, Loo JA. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J Proteome Res, 13, 1885–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc, 2, 1896–906 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Kulak NA, Pichler G, Paron I, et al. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods, 11, 319–24 (2014).*Stage tips surpass FASP for membrane protein coverage in bottom-up proteomics.
- 9.Hughes CS, Foehr S, Garfield DA, et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol, 10, 757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masuda T, Tomita M, Ishihama Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J Proteome Res, 7, 731–40 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Leon IR, Schwammle V, Jensen ON, et al. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol Cell Proteomics, 12, 2992–3005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capri J, Whitelegge JP. Full membrane protein coverage digestion and quantitative bottom-up mass spectrometry proteomics. Methods Mol Biol, 1550, 61–7 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Zhang X Less is More: Membrane protein digestion beyond urea-trypsin solution for next-level proteomics. Mol Cell Proteomics, 14, 2441–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang X, Zhao J, Zhang K, et al. Polydopamine Grafted porous graphene as biocompatible nanoreactor for efficient identification of membrane proteins. ACS Appl Mater Interfaces, 8, 6363–70 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Forner F, Foster LJ, Campanaro S, et al. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics, 5, 608–19 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Dunkley TP, Watson R, Griffin JL, et al. Localization of organelle proteins by isotope tagging (LOPIT). Mol Cell Proteomics, 3, 1128–34 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Christoforou A, Mulvey CM, Breckels LM et al. A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun, 7, 8992 (2016).**The most detailed correlation profiling study to date clusters families of proteins within their organelle or machine.
- 18.McAlister GC, Huttlin EL, Haas W et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem, 84, 7469–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAlister GC, Nusinow DP, Jedrychowski MP et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem, 86, 7150–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmad Y, Boisvert FM, Lundberg E, et al. Systematic analysis of protein pools, isoforms, and modifications affecting turnover and subcellular localization. Mol Cell Proteomics, 11, M111 013680 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreyev AY, Shen Z, Guan Z et al. Application of proteomic marker ensembles to subcellular organelle identification. Mol Cell Proteomics, 9, 388–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreyev AY, Fahy E, Guan Z et al. Subcellular organelle lipidomics in TLR-4-activated macrophages. J Lipid Res, 51, 2785–97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Speers AE, Blackler AR, Wu CC. Shotgun analysis of integral membrane proteins facilitated by elevated temperature. Anal Chem, 79, 4613–20 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Fischer F, Wolters D, Rogner M, et al. Toward the complete membrane proteome: high coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics, 5, 444–53 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Whitelegge JP, Zhang H, Aguilera R, et al. Full subunit coverage liquid chromatography electrospray ionization mass spectrometry (LCMS+) of an oligomeric membrane protein: cytochrome b(6)f complex from spinach and the cyanobacterium Mastigocladus laminosus. Mol Cell Proteomics, 1, 816–27 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Whitelegge JP. Integral membrane proteins and bilayer proteomics. Anal Chem, 85, 2558–68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran JC, Zamdborg L, Ahlf DR et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature, 480, 254–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catherman AD, Durbin KR, Ahlf DR et al. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol Cell Proteomics, 12, 3465–73 (2013).*The first high-throughput top-down proteomics analysis of a membrane enriched fraction.
- 29.Durbin KR, Fellers RT, Ntai I, et al. Autopilot: an online data acquisition control system for the enhanced high-throughput characterization of intact proteins. Anal Chem, 86, 1485–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skinner OS, Catherman AD, Early BP, et al. Fragmentation of integral membrane proteins in the gas phase. Anal Chem, 86, 4627–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zabrouskov V, Whitelegge JP. Increased coverage in the transmembrane domain with activated-ion electron capture dissociation for top-down Fourier-transform mass spectrometry of integral membrane proteins. J Proteome Res, 6, 2205–10 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Shaw JB, Li W, Holden DD et al. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc, 135, 12646–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carroll J, Fearnley IM, Wang Q, et al. Measurement of the molecular masses of hydrophilic and hydrophobic subunits of ATP synthase and complex I in a single experiment. Anal Biochem, 395, 249–55 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Whitelegge JP, Jewess P, Pickering MG, et al. Sequence analysis of photoaffinity-labelled peptides derived by proteolysis of photosystem-2 reaction centres from thylakoid membranes treated with [14C]-azidoatrazine. Eur J Biochem, 207, 1077–84 (1992). [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Roth MJ, Chang AN et al. Top-down mass spectrometry on tissue extracts and biofluids with isoelectric focusing and superficially porous silica liquid chromatography. Anal Chem, 85, 10377–84 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Ferrer D, Petritis K, Robinson EW et al. Pressurized pepsin digestion in proteomics: an automatable alternative to trypsin for integrated top-down bottom-up proteomics. Mol Cell Proteomics, 10, M110 001479 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, English AM, Bai DL et al. Analysis of Monoclonal Antibody Sequence and Post-translational Modifications by Time-controlled Proteolysis and Tandem Mass Spectrometry. Mol Cell Proteomics, 15, 1479–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laganowsky A, Reading E, Hopper JT, et al. Mass spectrometry of intact membrane protein complexes. Nat Protoc, 8, 639–51 (2013).*Highly detailed protocols for performing native mass spectrometry on integral membrane protein complexes.
- 39.Hopper JT, Yu YT, Li D et al. Detergent-free mass spectrometry of membrane protein complexes. Nat Methods, 10, 1206–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laganowsky A, Reading E, Allison TM et al. Membrane proteins bind lipids selectively to modulate their structure and function. Nature, 510, 172–5 (2014).**Binding of specific lipids can stabilize some membrane protein structures and modulate function.
- 41.Mehmood S, Marcoux J, Hopper JT et al. Charge reduction stabilizes intact membrane protein complexes for mass spectrometry. J Am Chem Soc, 136, 17010–2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allison TM, Landreh M, Benesch JL, et al. Low charge and reduced mobility of membrane protein complexes has implications for calibration of collision cross section measurements. Anal Chem, 88, 5879–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta K, Donlan JA, Hopper JT et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature, 541, 421–4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landreh M, Marklund EG, Uzdavinys P et al. Integrating mass spectrometry with MD simulations reveals the role of lipids in Na+/H+ antiporters. Nat Commun, 8, 13993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Cui W, Gross ML. Native electrospray ionization and electron-capture dissociation for comparison of protein structure in solution and the gas phase. Int J Mass Spectrom, 15, 354–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campuzano ID, Li H, Bagal D et al. Native MS Analysis of Bacteriorhodopsin and an Empty Nanodisc by Orthogonal Acceleration Time-of-Flight, Orbitrap and Ion Cyclotron Resonance. Anal Chem, 88, 12427–36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belov ME, Damoc E, Denisov E et al. From protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal Chem, 85, 11163–73 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Dyachenko A, Wang G, Belov M et al. Tandem native mass-spectrometry on antibody-drug conjugates and submillion Da antibody-antigen protein assemblies on an Orbitrap EMR equipped with a high-mass quadrupole mass selector. Anal Chem, 87, 6095–102 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Gault J, Donlan JA, Liko I et al. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat Methods, 13, 333–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Y, Zhang H, Cui W et al. Top-down mass spectrometry analysis of membrane-bound light-harvesting complex 2 from Rhodobacter sphaeroides. Biochemistry, 54, 7261–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harvey SR, Liu Y, Liu W, et al. Surface induced dissociation as a tool to study membrane protein complexes. Chem Commun (Camb), 53, 3106–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borch J, Hamann T. The nanodisc: a novel tool for membrane protein studies. Biol Chem, 390, 805–14 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Marty MT, Zhang H, Cui W, et al. Native mass spectrometry characterization of intact nanodisc lipoprotein complexes. Anal Chem, 84, 8957–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henrich E, Peetz O, Hein C et al. Analyzing native membrane protein assembly in nanodiscs by combined non-covalent mass spectrometry and synthetic biology. Elife, 6, e20954 (2017).**The first native mass spectrometry analysis of integral membrane proteins in nanodiscs.
- 55.Lu Y, Zhang H, Niedzwiedzki DM, et al. Fast photochemical oxidation of proteins maps the topology of intrinsic membrane proteins: light-harvesting complex 2 in a nanodisc. Anal Chem, 88, 8827–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adhikary S, Deredge DJ, Nagarajan A, et al. Conformational dynamics of a neurotransmitter:sodium symporter in a lipid bilayer. Proc Natl Acad Sci U S A, 114, E1786–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leitner A, Faini M, Stengel F, et al. Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem Sci, 41, 20–32 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Sinz A, Arlt C, Chorev D, et al. Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology. Protein Sci, 24, 1193–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H, Tang X, Munske GR, et al. Identification of protein-protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry. Mol Cell Proteomics, 8, 409–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng C, Yang L, Hoopmann MR et al. Cross-linking measurements of in vivo protein complex topologies. Mol Cell Proteomics, 10, M110 006841 (2011).*Novel crosslinking molecules distinguish productively crosslinked peptide pairs by mass spectrometry and target membrane proteins in vivo.
- 61.Marcoux J, Politis A, Rinehart D et al. Mass spectrometry defines the C-terminal dimerization domain and enables modeling of the structure of full-length OmpA. Structure, 22, 781–90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitelegge J Gas-phase structure of the E. coli OmpA dimer. Structure, 22, 666–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chim N, Torres R, Liu Y et al. The structure and interactions of periplasmic domains of crucial MmpL membrane proteins from Mycobacterium tuberculosis. Chem Biol, 22, 1098–107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao L, Qu Y, Zhang Z, et al. Intact glycopeptide characterization using mass spectrometry. Expert Rev Proteomics, 13, 513–22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chandler KB, Costello CE. Glycomics and glycoproteomics of membrane proteins and cell-surface receptors: Present trends and future opportunities. Electrophoresis, 37, 1407–19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mulagapati S, Koppolu V, Raju TS. Decoding of O-Linked glycosylation by mass spectrometry. Biochemistry, 56, 1218–26 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Hu H, Khatri K, Zaia J. Algorithms and design strategies towards automated glycoproteomics analysis. Mass Spectrom Rev, 36, 475–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palaniappan KK, Bertozzi CR. Chemical glycoproteomics. Chem Rev, 116, 14277–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Medzihradszky KF, Kaasik K, Chalkley RJ. Tissue-specific glycosylation at the glycopeptide Level. Mol Cell Proteomics, 14, 2103–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deeb SJ, Cox J, Schmidt-Supprian M, et al. N-linked glycosylation enrichment for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol Cell Proteomics, 13, 240–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bendall SC, Simonds EF, Qiu P et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science, 332, 687–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bjornson ZB, Nolan GP, Fantl WJ. Single-cell mass cytometry for analysis of immune system functional states. Curr Opin Immunol, 25, 484–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bendall SC, Davis KL, Amir el AD et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell, 157, 714–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giesen C, Wang HA, Schapiro D et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods, 11, 417–22 (2014).*Tissue imaging using highly multiplexed antibody-metal complexes and ICP-MS
- 75.Bodenmiller B Multiplexed Epitope-Based Tissue Imaging for Discovery and Healthcare Applications. Cell Syst, 2, 225–38 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Angelo M, Bendall SC, Finck R et al. Multiplexed ion beam imaging of human breast tumors. Nat Med, 20, 436–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]