

Abstract
To enable inexpensive molecular detection at the point-of-care and at home with minimal or no instrumentation, it is necessary to streamline unit operations and store reagents refrigeration-free. To address this need, a multifunctional enzymatic amplification reactor that combines solid-phase nucleic acid extraction, concentration, and purification; refrigeration-free storage of reagents with just-in-time release; and enzymatic amplification is designed, prototyped, and tested. A nucleic acid isolation membrane is placed at the reactor’s inlet, and paraffin-encapsulated reagents are prestored within the reactor. When a sample mixed with chaotropic agents is filtered through the nucleic acid isolation membrane, the membrane binds nucleic acids from the sample. Importantly, the sample volume is decoupled from the reaction volume, enabling the use of relatively large sample volumes for high sensitivity. When the amplification reactor’s temperature increases to its operating level, the paraffin encapsulating the reagents melts and moves out of the way. The reagents are hydrated, just-in-time, and the polymerase reaction proceeds. The amplification process can be monitored, in real-time. We demonstrate our reactors’ ability to amplify both DNA and RNA targets using polymerase with both reverse-transcriptase and strand displacement activities to obtain sensitivities on-par with benchtop equipment and a shelf life exceeding 6 months.
GRAPHICAL ABSTRACT:
Molecular assays utilizing enzymatic amplification of nucleic acids (NA) enable highly specific and sensitive detection, among other things, of infectious diseases, drug-susceptibility, and resistance; genetic disorders; and cancer-related mutant alleles.1–3 NA tests (NATs) are the gold standard in clinical laboratories. Since conventional NATs require multiple unit operations, their use has been mostly confined to well-equipped centralized facilities. Point of care (POC) tests mostly comprise immunoassays carried out with lateral flow strips. However, immunoassays often lack sufficient sensitivity and selectivity and, in some cases, are inadequate.2 For example, an antibody test for HIV of infants born to HIV-infected mothers may result in false positives due to the presence of maternal antibodies.4 Similarly, antibody tests for sexually transmitted diseases or mosquito-borne pathogens5 may result in false positives since a significant fraction of the population carries antibodies to the targeted pathogens while being free of active infection. Moreover, antibody tests cannot discriminate between drug-susceptible and drug-resistant pathogens.6
The ability to detect NAs at the POC and at home can compensate for the lack of centralized facilities, trained-personnel, and logistic networks in resource-poor settings; empower providers to administer evidence-based healthcare; enable personalized therapies; and enhance productivity by saving patients the need to travel to centralized facilities.1 Self-diagnosis at home, such as in the case of chronic diseases, may enable patients to participate in their own treatment, modify behavior, and reduce health-care cost. Given the desirability of NAT for POC use, many researchers have been investing in efforts to develop such tests.
One approach foregoes enzymatic amplification and utilizes biosensors to detect NAs directly, typically by hybridizing target NAs to immobilized probes and detecting the presence of captured NAs with various transduction methods.7 Because target NAs are typically at low concentrations, among an abundance of host’s NAs, these methods require significant sample preparation to reduce background, long incubation times to improve sensitivity, and signal amplification. Although the literature is rich with ingenious ideas for transduction mechanisms, there are few, if any, POC commercial products for enzymatic amplification-free detection of NAs.
In contrast to biosensors, enzymatic amplification takes advantage of nature’s ability to amplify selected NAs, rapidly and with high fidelity. Two approaches are used for enzymatic amplification outside centralize laboratories. One approach uses fully or partially automated instruments that duplicate laboratory procedures in a single “box,” eliminating the need for highly trained personnel, while providing detection sensitivities on par with laboratory equipment. These instruments tend, however, to be complex, expensive, maintenance-heavy, and low throughput—mostly suitable for medical centers;8–10 even the simplest published implementation of NAT requires a centrifuge.11
A second approach, dubbed rapid NAT, sacrifices sensitivity for simplicity, forgoing sample preparation. A small volume of a minimally processed sample is added directly into the reaction mix for enzymatic amplification. To avoid inhibition, the sample volume, typically ranging from 1 to 5 μL, must be much smaller than the reaction volume (typically, 10–50 μL). A small sample volume results in low sensitivity (typically, > 105 target copies/mL). While this sensitivity may be orders of magnitude better than what is achievable with biosensors, it is still insufficient in many cases such as HIV viral-load monitoring that requires detection of a fewer than 103 virions/mL.
In prior work,12 we demonstrated paraffin encapsulation of PCR reagents in a chip. Here, we go much further. We develop a multifunctional reaction chamber that contains a NA isolation membrane at its inlet to bind NAs and to decouple sample volume from the reaction volume, enabling target concentration. Instead of PCR, we use isothermal amplification to reduce processor complexity and a robust polymerase enzyme with both reverse-transcriptase and strand displacement activities to eliminate the need for a separate reverse transcription step. All reagents for the enzymatic amplification are lyophilized, encapsulated with paraffin, and prestored in the reaction chamber, refrigeration-free for automated, heat-activated, just-in-time release. Shelf life exceeding at least 6 months is demonstrated without a significant deterioration in enzyme activity. Using this approach, we demonstrate that a rapid NA test is possible without sacrificing sensitivity for both DNA and RNA targets.
EXPERIMENTAL SECTION
Our reaction chamber is fabricated with a plastic substrate. Here, we use poly(methyl methacrylate) (PMMA). Panels (i) and (ii) of Figure 1A show, respectively, a three-dimensional drawing and a photograph of a plastic chip housing two 55 μL reaction chambers (including 15 μL reagent storage wells). For additional details, see Section S1 of Supporting Information
Figure 1.

(A) A plastic chip with two multifunctional amplification reactors. (i) A three-dimensional rendering of the multifunctional reactor’s cross-section, showing two amplification chambers with a cut through one of the chambers. Each chamber houses a NA binding membrane at its inlet and stores paraffin-encapsulated dry reagents (OmniAmp Pol polymerase, primers and dye). (ii) A photograph of the plastic chip. (B) The operating principle of reagent release illustrated with food coloring. (i) Prior to heating. (ii) As the chip is heated to its operating temperature, molten paraffin floats up and away, allowing reagents to hydrate. (iii) Food dye dissolves in water. (iv) The dye diffuses to the NA binding membrane. Video S2 features paraffin melting and food coloring release.
A silica NA isolation membrane is located at each reaction chamber’s inlet. We13–15 and others16 have demonstrated that the silica membrane captures ~80% of 100–50 000 bp long NAs in the sample. The silica membrane can be replaced with a membrane from a different material, such as Q-Sepharose resin, when capture of shorter (50–150bp) DNA fragments is needed.16
All reagents needed for the polymerase reaction and for detection, including dye and enzymes, were lyophilized (BIOLYPH, MN), placed in the prefabricated recess in the reaction chamber, and coated with paraffin that filled the reagent storage well, leaving a reaction chamber with 40 μL volume. Video S1 shows cartoons depicting reagent storage and chip operation.
In our experiments to detect DNA, we used saliva (50 μL) spiked with HPV-16 DNA. The sample was mixed with 50 μL chaotropic salt (6 M guanidinium chloride) lysis buffer (QIAamp MinElute Virus Spin Kit, Qiagen, Germantown MD) and 62.5 μL ethanol; and then filtered through the NA isolation membrane. The NAs bound to the membrane while the filtrate discharged to waste. Following the manufacturer’s protocol, the membrane was washed with 150 μL of an ethanol-based solution (AW1, Qiagen, Germantown, MD), with 150 μL of a second solution (AW2, Qiagen, Germantown, MD), and dried in air for 30 s to remove any remaining ethanol, which is an inhibitor. After the reactors were dried, they were filled with DNA-grade water through the NA capture membranes, and the inlet and exit ports were sealed with a PCR tape.
In our experiments to detect RNA, we spiked plasma (140 μL) with HIV virus and mixed the plasma with 560 μL of lysis buffer (QIAamp Viral RNA Mini Kit, Qiagen) and 560 μL of ethanol, and we pipetted into the multifunctional reactors. Then the membrane was washed and dried using the sequence described in the previous paragraph.
The plastic chip was placed vertically in our custom-made, battery-operated portable processor14,17 (Supporting Information, Section S2) with the isolation membrane located at the lower part of the chip. Upon heating the chip to its operating temperature, the paraffin melted and moved out of the way by buoyancy and surface tension forces. The reaction mix hydrated and reconstituted, and the polymerase reaction proceeded. The vertical orientation of the reaction chamber helped remove the paraffin encapsulation and prevented bubbles from accumulating in the imaged part of the reaction chamber and interfering with signal acquisition. The paraffin motion also induces advection in the reaction chamber assisting in stirring and homogenizing the reaction mix.
The amplification process was monitored, in real time, with an inexpensive USB fluorescent microscope (AM4113T-GFBW Dino-Lite Premier, AnMo Electronics, Taipei, Taiwan) interfacing with our custom-made processor.14,17 The images were analyzed with custom-written software that computed spatially averaged fluorescent emission intensity as a function of time and the threshold time (T1/2) required for the intensity to reach half its saturation value.
RESULTS AND DISCUSSION
Reagent Release.
To better understand reagent release and hydration, we dried food coloring in the reagent storage well, filled the well with paraffin, filled the reaction chamber with plain water, and monitored the reaction chamber during heating. Figure 1B and Video S2 show the dye release and hydration. When the chamber is heated, the paraffin melts. Since the paraffin is lighter than water, it floats to the top of the vertically positioned chip (Figure 1B (ii)), occupying the chamber’s upper part. The dye hydrates (Figure 1B (iii)) and spreads rapidly, driven by advection. The paraffin serves here a second function; its motion helps stir and homogenize the reaction mix. The chamber is darker at the position of the recess and the position of the NA capture membrane due to the greater liquid thickness at these locations. Here, we oriented the chamber vertically; it suffices, however, to tilt the chamber with respect to the horizontal.
Next, we monitored the effects of sample and wash solutions flows on the paraffin-encapsulated lyophilized reagents placed in the chamber’s recess (Video S3, bright field) and verified that these liquid flows do not disturb the paraffin-encapsulated reagents.
DNA Detection with our Multifunctional Amplification Reactor.
To demonstrate our multifunctional reaction chamber’s operation, we used 50 μL saliva spiked with HPV-16 gDNA. Detection of HPV-16 in oral fluids is of interest for early oral cancer diagnostics - the frequency of which is increasing.18,19 We carried out Loop Mediated Isothermal Amplification (LAMP)20 with the polymerase enzyme OmniAmp (Lucigen) that has both reverse transcriptase and strand displacement activities.21 We used the six primers (Lucigen) listed in Table S1, newly and specifically designed for the temperature range 66–74 °C. Additional details and the workflow associated with this test are described in Supporting Information Section S4.
After DNA binding to the NA isolation membrane and membrane washes, the reaction chamber was filled with DNA-grade water, sealed, placed vertically in our portable custom-made processor,14,17 and monitored. Prior to heating, the paraffin encapsulation reflects green light (Figure 2A, time = 0, and Video S4). See Supporting Information section S3 for discussion. When the reaction chambers were heated to above ~45 °C, the paraffin melted and floated away, the reagents were hydrated and stirred, and the green reflection from the paraffin disappeared (Figure 2A, 3 min). Our reagent-storage method provides “hot-start” amplification. No reaction can take place until the reagents have been constituted. In the presence of targets, amplification proceeds and the test chamber lights up, typically within 10–50 min (depending on target concentration). In the absence of targets (negative-control), the chamber remains dark (Figure 2A, 35 min).
Figure 2.

(A) Fluorescence images illustrating reagent hydration and release (Video S5). Before the beginning of the heating process (t < 0), the lyophilized reagents are encapsulated with paraffin and isolated from the liquid in the reaction chamber. In 3 min after the start of heating, the paraffin encapsulation melts and floats away, the reagents are hydrated, and the polymerase reaction proceeds. At 35 min, clear fluorescence emission is evident in the test chamber (20 HPV-16 DNA copies/μL in 50 μL saliva) and absent in the negative (no target) control chamber. (B) Real time amplification curves with 200, 20, and 2 HPV-16 DNA/μL spiked in 50 μL saliva, obtained with our multifunctional reactor. (C) Threshold time as a function of target concentration. (D) Rapid test (see Section S4). Amplification curves of LAMP with 24000, 2400, 240, and 24 HPV-16 DNA /μL in saliva with minimal sample preparation and processed with a benchtop PCR machine. (E) The threshold time of the rapid test as a function of target concentration.
Figure 2B depicts the normalized fluorescence emission intensity as a function of time when the sample comprises 0 (no-target control), 2, 20, and 200 HPV-16 gDNA per μL. In the above, I is the emission intensity (au). With the exception of the negative control, all tests were positive, producing a signal. Our experiment indicates assay sensitivity better than 2 DNA copies per μL of sample. When desired, sensitivity can be improved by increasing sample volume.
Figure 2C depicts the threshold time (T1/2), defined as the time it takes for emission intensity to reach half its saturation value (Imax), as a function of target concentration. The threshold time decreases linearly with the log of target concentration. When the sample volume is 50 μL, the data correlates with the expression:
| (1) |
where c is the number of DNA molecules in a μL of sample. The variance of the threshold time decreases as the concentration increases. This is partially due to the difficulty of preparing reproducible samples at very low target concentrations. The results obtained with our chip are comparable to the ones obtained with benchtop experiments (Supporting Information Section S6).
HPV-16 gDNA Detection: Comparison with a Rapid-Test.
Next, we compare the performance of our multifunctional amplification reactor with that of a rapid-test, wherein 40 μL diluted, filtered, and preheated saliva sample was directly added to lyophilized reagents.22 The rapid-test workflow is described in Supporting Information Section S7.
Figure 2D depicts the rapid-test normalized fluorescence emission intensity as a function of time when the sample consists of 0 (no-target, negative control), 24, 240, 2400, and 24 000 HPV-16 gDNA per μL of saliva. Samples with 24 HPV-16 gDNA or less per μL tested negative, suggesting sensitivity between 24 and 240 target molecules per μL sample–at least 1 order of magnitude worse than our multifunctional reactor. This is hardly surprising given that the rapid test operates with a diluted sample while our multifunctional amplification reactor does the opposite; it concentrates the sample.
Figure 2E depicts the rapid-test threshold time as a function of target concentration. The threshold time increases linearly as the log of the target concentration decreases. Rapid-test’s threshold times are significantly greater than that of our multifunctional amplification reactor. For example, in the presence of ~200 target molecules per μL of sample, the rapid-test’s and the multifunctional amplification reactor threshold times are, respectively, 35 and 15 min (Figure 2C,E). The better performance of the multifunctional reactor is attributable to (i) the concentration function of the NA isolation membrane that allows larger sample volumes than possible with the rapid-test and (ii) the multifunctional amplification reactor allows us to wash the membrane to remove inhibitors—something that cannot be done with the rapid-test.
To assess the rapid-test’s reduced amplification efficiency as compared to the multifunctional reactor, we consider a sample with 240 molecules per μL. The rapid-test’s threshold time is 33 min (Figure 2E). If the same sample was processed with our multifunctional reactor, the threshold time would be 14.8 min (eq 1). The extra 18.2 min in the rapid-test’s threshold time is attributable to the lower rapid-test’s amplification efficiency, plausibly due to the presence of inhibitors. In summary, our multifunctional reactor has several advantages over rapid-tests. It can operate with much greater sample volumes, providing greater number of target molecules for the amplification process, and it provides higher amplification efficiency by enabling target purification.
Detection of RNA Targets with our Multifunctional Amplification Reactor.
In this section, we demonstrate that the multifunctional reactor can be used to detect RNA targets. Since the OmniAmp enzyme (Lucigen) has both reverse-transcriptase and strand-displacement activities, we can process RNA targets in a single step without a separate reverse-transcription prior to amplification.21,23,24 We experimented with two RNA targets: HIV-1 clade C (common in Africa) and MS2 bacteriophage RNA that is often used as a positive RNA control in molecular tests.
For HIV RNA detection, we used our custom-designed universal (strain-independent) primers25 (ACeIN-26, Table S1) developed to detect HIV subtypes A, B, C, D, and G. We spiked 105, 104, and 103 HIV-1 subtype C virions (Zambia, ZAM18, GenBank: L03705.1, SeraCare Life Science) in 140 μL human plasma.
Figures 3A and 3B depict, respectively, the amplification curves and the threshold time as functions of target concentration. The threshold time increases linearly as the log of the number of targets decreases. The assay can readily detect less than 1000 virions in the sample (140 μL). This sensitivity is not a limitation of our multifunctional reactor but the result of our use of a “universal” assay25 that was purposely designed to amplify a plethora of different HIV strains. Additionally, our assay’s efficiency suffers because we used primers (available in our lab) optimized for the lower LAMP temperature of the OptiGene enzyme (63 °C) instead of primers optimized for our actual reaction temperature of 68 °C (OmniAmp enzyme, Lucigen). Because in some cases the actual HIV strain may not be known a priori, there is a benefit to an assay that can detect multiple strains even at the expense of sensitivity. Previously, we demonstrated a higher sensitivity of about 10 virions per reaction with optimized and specific primers to a particular HIV strain.26
Figure 3.

RT-LAMP of HIV clade C with our multifunctional reactor. (A) Amplification curves of 140 μL human plasma laden with 105, 104, 103, and 0 (negative control) HIV (subtype C) virions. (B) The threshold time as a function of target concentration. The error bars indicate the scatter of the data (N = 3).
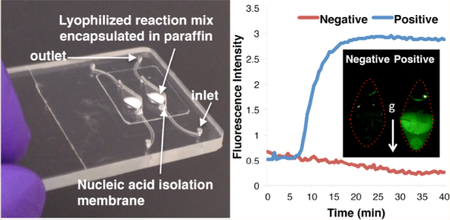
As a second target, we experimented with MS2 bacteriophage RNA. We prestored MS2 LAMP primer set (Table S1) with our reaction mix. Figure 4 depicts an example of an amplification curve when the reactor operated with ~270 target copies added directly into the reaction chamber. The corresponding threshold time is about 11 min. In the next section, we use the MS2 RNA target to study the shelf life of the reaction mix.
Figure 4.

Evaluation of on-chip dry reagents’ shelf life. (A) RT-LAMP of MS2 in a multifunctional chip after 180 days of storage. Inset: a photograph of a test (positive control) and a negative (no target) control after 6 months of storage. (B) The threshold time T1/2 for MS2 RNA (0.5 fg, ~ 270 copies) as a function of storage duration (weeks) in a multifunctional chip stored at ambient conditions (blue rhombus) and benchtop (red squares) operating with wet reagents stored at −20 °C.
Shelf Life.
Commonly used reverse-transcriptase enzymes are labile and may lose activity when dry-stored.21 We examined the amplification efficiency of our paraffin-encapsulated, lyophilized reagents as a function of storage duration, using the threshold-time as a figure of merit. Lyophilized reagents, including the enzyme, MS2 LAMP primer (Table S1), and dye were encapsulated with paraffin in our microfluidic chips and put aside for storage under ambient conditions (without refrigeration or humidity control and with our chips inlet and exit open). The chips were retrieved after various storage durations and tested with purified MS2 RNA.
Figure 4A depicts the amplification curves of 0.5 fg (~270 copies) of MS2 RNA and negative (no-target) control obtained with our multifunctional reactors and 180 days on-chip dry-storage. The presence of MS2 RNA is readily detectable in ~11 min. Figure 4B depicts the threshold-time T1/2 (blue-rhombus) of the amplification curves of 0.5 fg (~270 copies) MS2 RNA as a function of storage duration (weeks). For comparison, we also carried out a few experiments with refrigerated reagents, a benchtop PCR machine, and the same target concentration (red-squares). The threshold-time of both chip and benchtop are similar, indicating no obvious diminution of reverse-transcriptase and strand displacement activities, resulting from lyophilization, paraffin-encapsulation, storage, and rehydration. Our data indicates that our dry reagents are stable during a relatively long storage. In the absence of the paraffin-cladding, the dry reagents hydrate rapidly when in air (Supporting Information Section S8).
CONCLUSIONS
Centralized laboratories routinely use enzymatic amplification of NAs as the method of choice to detect causative agents, drug-resistance, and genetic disorders. It is desirable to facilitate molecular tests at the POC to alleviate shortages of centralized facilities in resource-poor settings; enable health-personnel to provide evidence-based disease management; and enable patients to participate in their own care.
In centralized laboratories, molecular tests require numerous unit operations with benchtop instruments, dedicated space, and highly skilled personnel. To adapt molecular testing for POC, it is necessary to reduce the number of unit operations, remove cold chains, and enable operation by minimally trained personnel. Various strategies have been proposed to meet these objectives: enzymatic amplification-free biosensors, automated-equipment, and rapid NAT that forego sample preparation at the expense of reduced sensitivity.
As an attractive alternative, we propose a relatively inexpensive NAT for use at the POC. We integrate multiple functions into the amplification reaction chamber to streamline unit operations and simplify flow control. Our multifunctional chamber includes NA isolation-membrane at its inlet to capture NA from the sample and decouple sample-volume from reaction-volume. One can operate with sample volumes far exceeding the reaction volume to achieve desired sensitivity while retaining a small reaction volume and small reagent consumption.
All reagents needed for polymerase amplification and detection are lyophilized, encapsulated in paraffin, and prestored, refrigeration-free in the reaction chamber for prolonged shelf life. When the microfluidic chip is heated to its operating temperature, the paraffin melts, moves out of the way, and the reagents are hydrated. Because the paraffin is lighter than water, it induces circulation in the reaction chamber, assisting in the homogenization of the reaction mix without a need for external stirrers. Although microfluidic chips with dry-stored reagents have been reported before,27–30 they typically dry store reagents on membranes and require flow streams to controllably reconstitute the reagents.28 Our method enables temperature-triggered release of reagents, just-in-time, streamlining flow control.
Here, we carried out the reagent-encapsulation in situ. The reagents can also be encapsulated outside the cassette to form paraffin-coated beads. This allows the flexibility of maintaining a bead library targeting diverse pathogens and an inventory of generic cassettes to be customized prior to use for any desired test.
Our multifunctional chamber can operate with various sample matrices. When the sample is saliva, it can be mixed with a lysis buffer and introduced directly into the cassette. When the sample is blood, it is desirable to filter only plasma through our reaction chamber. This can be accomplished with an appropriate frontend such as our high-capacity, electricity-free, POC plasma-separator.31,32
For our experiments with HPV, we designed new HPV LAMP primers for the OmniAmp enzyme and demonstrated that we can detect 100 HPV DNA molecules per reaction. This compares favorably with FDA-approved commercial tests such as Qiagen (reported sensitivity of 1000 DNA virions/reaction).33 Our HIV tests with our universal, assay, capable of detecting multiple HIV strains, demonstrated a sensitivity of 1000 copies per reaction. This is a high sensitivity for HIV screening, enabling infection detection during the seroconversion period and in infants born to HIV-infected mothers.4 Our sensitivity suffers, however, because of the use of universal primers designed to detect multistrains of HIV at a lower LAMP temperature (OptiGene enzyme, 63 °C) than we used here (68 °C). For viral load monitoring when the HIV strain is known, we can achieve much greater sensitivity (e.g., 10 virions/reaction) by operating with primers specific to the known HIV strain.26 In summary, our POC system can provide performance nearly on par with benchtop equipment.
The reproducibility of our point of care device is somewhat lower than that of our benchtop equipment. In the presence of 100 templates in the sample, the relative scatters in the threshold times of the benchtop (Figure S4) and chip experiments (Figure 2C) are, respectively, ±10% and ±33%. These scatters in the threshold times decline as the template concentration in the sample increases. This scatter in threshold time can be attributed, in part, to statistical variations in target concentration in the sample, in particular, at low concentrations. In the future, we will investigate whether improvements in heating uniformity and in reagent stirring can improve reproducibility. It is worth noting, however, that 100 templates/(50 μL sample) is a very low viral load. To put things in perspective, the FDA-approved laboratory-based test for carcinogenic HPV is reported33 to have analytical sensitivity ranging from 1000 to 5000 copies/reaction. In most cases of clinical significance, the template concentration is likely to be orders of magnitude greater and the scatter in the data much smaller than at low target concentrations. And, of course, we can improve sensitivity by increasing sample volume.
In its current implementation, our microfluidic chip requires a few pipetting operations (Figure S3). In the future, we will prestore wash solutions and water in blisters mounted on the cassette. Because the wash solutions and water do not require refrigeration, long shelf life is possible. The blisters can be activated either manually or with a mechanical or electrical actuator to discharge liquids through the reaction chamber.
Once sample introduction and wash steps have been concluded, the cassette needs to be incubated. Here, we used an inexpensive battery-powered, homemade processor. The processor can be further simplified or eliminated altogether. To reduce cost, the ubiquitous smartphone can be used for fluorescent dye excitation (with the phone’s flashlight); for signal monitoring (with the phone’s camera), analysis, and reporting;34 and to power our heater. If necessary, the smartphone can also provide vibrations to improve reconstituted reagents’ mixing.35
For additional cost savings, the need for excitation can be eliminated by replacing the fluorescent reporter with excitation-free reporters such as visible light-emitting luciferous molecules fueled by polymerase byproducts.36 Furthermore, visual-monitoring of amplification products is also possible, eliminating the need for a detection device. One can use leuco crystal violet (LCV) dye that changes from colorless to violet in the presence of dsDNA.15 Alternatively, since the polymerase process releases protons, amplicon production can be correlated with a change in reactor’s pH and detected as a change in color of litmus paper or litmus dye, either of which can be included with the reaction mix.37 Both change of color of the LCV and the litmus paper/dye are detectable with the unaided eye. When we detect amplicons by eye, a processor is needed only for temperature control.
Can we operate instrument-free? The isothermal amplification process can be incubated by placing the cassette on a hotplate. A single hot-plate can incubate many cassettes. Alternatively, energy sources other than a battery or grid power can be used. Incubation heat can be provided, for example, with an exothermic chemical reaction such as the interaction of magnesium alloy with water.34 This heating method has been long-used by hikers to warm ready-to-eat-meals. Temperature-control can be provided with a material that changes phase (e.g., from solid to liquid) at the desired incubation temperature. Nature mandates that as long as the phase-change material coexists in two-phases, the temperature remains fixed—independent of the ambient temperature. Since the magnesium alloy and phase change material are inexpensive and can be housed in a disposable Styrofoam cup or in the cassette itself,38 the entire molecular detection system (cassette and processor) can be rendered disposable, like the ubiquitous lateral flow strips, but with much greater capabilities.
Another highly desired attribute for POC systems is multiplexing. Most current POC molecular detection devices focus on single target detection. In many endemic regions, there are multiple causative agents that induce similar initial symptoms but require diverse disease management strategies. Moreover, coinfections may mask symptoms and may require a therapy different than the one mandated by a single infection. Evidence-based health care requires testing for multiple causative agents. Our multifunctional reactor provides the core elements for a two stage isothermal amplification assay capable of codetecting many different targets in a single sample.39
Supplementary Material
ACKNOWLEDGMENTS
This work was supported, in part, by NIH grant 1R43DE025466–01 to Lucigen (subcontract to the University of Pennsylvania), NIH NIDCR 1R21DE026700–01, and NIH NCI R01CA214072 to the University of Pennsylvania.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Niemz A; Ferguson TM; Boyle DS Trends Biotechnol 2011, 29, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hans R; Marwaha N Asian J. Transfus. Sci 2014, 8, 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tatsumi K; Mitani Y; Watanabe J; Takakura H; Hoshi K; Kawai Y; Kikuchi T; Kogo Y; Oguchi-Katayama A; Tomaru Y; Kanamori H; Baba M; Ishidao T; Usui K; Itoh M; Cizdziel PE; Lezhava A; Ueda M; Ichikawa Y; Endo I; Togo S; Shimada H; Hayashizaki YJ Mol. Diagn 2008, 10, 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).World Health Organization (WHO). WHO Recommendations on the Diagnosis of HIV Infection in Infants and Children; WHO: Geneva, Switzerland, 2010. [PubMed] [Google Scholar]
- (5).Steinhagen K; Probst C; Radzimski C; Schmidt-Chanasit J; Emmerich P; van Esbroeck M; Schinkel J; Grobusch MP; Goorhuis A; Warnecke JM; Lattwein E; Komorowski L; Deerberg A; Saschenbrecker S; Stocker W; Schlumberger W Euro. Surveill 2016, 21, DOI: 10.2807/1560-7917.ES.2016.21.50.30426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Centers for Disease Control and Prevention. MMWR Recomm. Rep 2014, 63, 1–19. [Google Scholar]
- (7).Smith SJ; Nemr CR; Kelley SO J. Am. Chem. Soc 2017, 139, 1020–1028. [DOI] [PubMed] [Google Scholar]
- (8).Poritz MA; Blaschke AJ; Byington CL; Meyers L; Nilsson K; Jones DE; Thatcher SA; Robbins T; Lingenfelter B; Amiott E; Herbener A; Daly J; Dobrowolski SF; Teng DH; Ririe KM PLoS One 2011, 6, DOI: 10.1371/annotation/468cfdcd184c-42f7-a1d0-3b72a2f6a558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wahrenbrock MG; Matushek S; Boonlayangoor S; Tesic V; Beavis KG; Charnot-Katsikas AJ Clin. Microbiol 2016, 54, 1902–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).LaBarre P; Boyle D; Hawkins K; Weigl B Proc. SPIE 2011, 8029, 802902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bissonnette L; Bergeron M Micromachines 2016, 7, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kim J; Byun D; Mauk MG; Bau HH Lab Chip 2009, 9, 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mauk M; Song J; Bau HH; Gross R; Bushman FD; Collman RG; Liu C Lab Chip 2017, 17, 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Song J; Liu C; Bais S; Mauk MG; Bau HH; Greenberg RM PLoS Neglected Trop. Dis 2015, 9, e0004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Song J; Mauk MG; Hackett BA; Cherry S; Bau HH; Liu C Anal. Chem 2016, 88, 7289–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shekhtman EM; Anne K; Melkonyan HS; Robbins DJ; Warsof SL; Umansky SR Clin. Chem 2009, 55, 723–729. [DOI] [PubMed] [Google Scholar]
- (17).Liu C; Sadik MM; Mauk MG; Edelstein PH; Bushman FD; Gross R; Bau HH Sci. Rep 2015, 4, DOI: 10.1038/srep07335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhao M; Rosenbaum E; Carvalho AL; Koch W; Jiang WW; Sidransky D; Califano J Int. J. Cancer 2005, 117, 605–610. [DOI] [PubMed] [Google Scholar]
- (19).Chai RC; Lim Y; Frazer IH; Wan YX; Perry C; Jones L; Lambie D; Punyadeera C BMC Cancer 2016, 16, DOI: 10.1186/s12885-016-2217-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Notomi T; Okayama H; Masubuchi H; Yonekawa T; Watanabe K; Amino N; Hase T Nucleic Acids Res 2000, 28, 63e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chander Y; Koelbl JA; Puckett J; Moser MJ; Klingele AJ; Liles MR; Carrias A; Mead DA; Schoenfeld TW Front. Microbiol 2014, 5, DOI: 10.3389/fmicb.2014.00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Benzine JW; Brown KM; Agans KN; Godiska R; Mire CE; Gowda K; Converse B; Geisbert TW; Mead DA; Chander YJ Infect. Dis 2016, 214, S234–S242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Schoenfeld T; Patterson M; Richardson PM; Wommack KE; Young M; Mead D Appl. Environ. Microbiol 2008, 74, 4164–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Moser MJ; DiFrancesco RA; Gowda K; Klingele AJ; Sugar DR; Stocki S; Mead DA; Schoenfeld TW PLoS One 2012, 7, e38371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ocwieja KE; Sherrill-Mix S; Liu C; Song J; Bau H; Bushman FD PLoS One 2015, 10, e0117852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu C; Geva E; Mauk M; Qiu X; Abrams WR; Malamud D; Curtis K; Owen SM; Bau HH Analyst 2011, 136, 2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ahlford A; Kjeldsen B; Reimers J; Lundmark A; Romani M; Wolff A; Syvanen AC; Brivio M Analyst 2010, 135, 2377–2385. [DOI] [PubMed] [Google Scholar]
- (28).Liu RH; Yang J; Lenigk R; Bonanno J; Grodzinski P Anal. Chem 2004, 76, 1824–1831. [DOI] [PubMed] [Google Scholar]
- (29).Laouenan F; Monsalve LG; Goiriena A; Agirregabiria M; Ruano-Lopez JM Procedia Eng 2012, 47, 1484–1490. [Google Scholar]
- (30).Stumpf F; Schwemmer F; Hutzenlaub T; Baumann D; Strohmeier O; Dingemanns G; Simons G; Sager C; Plobner L; von Stetten F; Zengerle R; Mark D Lab Chip 2016, 16, 199–207. [DOI] [PubMed] [Google Scholar]
- (31).Liu C; Mauk M; Gross R; Bushman FD; Edelstein PH; Collman RG; Bau HH Anal. Chem 2013, 85, 10463–10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu C; Liao SC; Song J; Mauk MG; Li X; Wu G; Ge D; Greenberg RM; Yang S; Bau HH Lab Chip 2016, 16, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Burd EM Clin. Microbiol. Rev 2016, 29, 291–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liao SC; Peng J; Mauk MG; Awasthi S; Song J; Friedman H; Bau HH; Liu C Sens. Actuators, B 2016, 229, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nemiroski A; Christodouleas DC; Hennek JW; Kumar AA; Maxwell EJ; Fernandez-Abedul MT; Whitesides GM Proc. Natl. Acad. Sci. U. S. A 2014, 111, 11984–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gandelman OA; Church VL; Moore CA; Kiddle G; Carne CA; Parmar S; Jalal H; Tisi LC; Murray JA PLoS One 2010, 5, e14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tanner NA; Zhang YH; Evans TC BioTechniques 2015, 58, 59–68. [DOI] [PubMed] [Google Scholar]
- (38).Liu CC; Mauk MG; Hart R; Qiu XB; Bau HH Lab Chip 2011, 11, 2686–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Song JZ; Liu CC; Mauk MG; Rankin SC; Lok JB; Greenberg RM; Bau HH Clin. Chem 2017, 63, 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.