

Abstract
Hepatic ischemia reperfusion (I/R) is a clinically relevant model of acute sterile inflammation leading to a reverberating, self-sustaining inflammatory response with resultant necrosis. We hypothesized that computerized dynamic network analysis (DyNA) of 20 inflammatory mediators could help dissect the sequence of post-I/R mediator interactions that induce injury. Although the majority of measured inflammatory mediators become elevated in the first 24 hours, we predicted that only a few would be secreted early in the process and serve as organizational centers of downstream intermediator complexity. In support of this hypothesis, DyNA inferred a central organizing role for IL-17A during the first 3 hours of reperfusion. After that, DyNA revealed connections among almost all the inflammatory mediators, representing an ongoing cytokine storm. Blocking IL-17A immediately after reperfusion disassembled the inflammatory networks and protected the liver from injury. Disassembly of the networks was not achieved if IL-17A blockage was delayed 2 or more hours post-reperfusion. Network disassembly was accompanied by decrease in neutrophil infiltration and Neutrophil Extracellular Trap (NET) formation. By contrast, administration of recombinant IL-17A increased neutrophil infiltration, NET formation, and liver necrosis. The administration of DNAse, a NET inhibitor, significantly reduced hepatic damage despite prior administration of IL-17A, and also DNAse disassembled the inflammatory networks. In vitro, IL-17A was a potent promoter of NET formation. Therefore, computational analysis identified IL-17A’s early, central organizing role in the rapid evolution of a network of inflammatory mediators that induce neutrophil infiltration and NET formation responsible for hepatic damage after liver I/R.
Keywords: Ischemia reperfusion injury, Interleukin-17, computational analysis, mathematical modeling, neutrophil extracellular traps
INTRODUCTION
Sterile inflammation is a pathophysiologic response orchestrated by innate immune cells and their products in response to a wide variety of perceived non-infectious threats. This response is fundamental to repair and regeneration, but when excessive, it can contribute to severe organ damage and dysfunction (1). This is of particular significance for the liver where sterile inflammation plays a critical role in alcoholic and nonalcoholic steatohepatitis, drug-induced hepatotoxicity, and liver ischemia reperfusion (I/R) injury (2, 3). During surgery, the liver is subjected routinely to ischemic injury due to both mechanical manipulation and interruption of the hepatic blood supply necessary to control blood loss. Moreover, the initial damage is amplified following restoration of blood flow by a chain of reactive and dysregulated inflammatory processes that contribute to further injury.
The complex of mediator and cellular responses to hepatic I/R has been difficult to unravel. We and others have previously shown that neutrophil extracellular traps (NETs) play a central role in driving inflammatory damage to liver tissue after I/R (4). Neutrophil infiltration is an early response to the products of damaged parenchymal and non-parenchymal cells. Neutrophil influx favors the production of NETs, a process in which nuclear chromatin with embedded damage associated molecular patterns (DAMPs) and cytoplasmic toxic enzymes is extruded into the extracellular microenvironment(4–6). What triggers NETosis in vivo is not clear but likely resides in the complex molecular microenvironment of damaged liver cells.
Following liver I/R, the damaged hepatocytes and non-parenchymal cells release a complex array of interacting molecular mediators that contribute to NETosis (4). Such cytokine storms, however, should be susceptible to computerized dynamic network analysis in order to reveal interactions among inflammatory mediators over time. Such network analysis has been found useful in trauma, hemorrhage, wound healing, allograft rejection, and other self-perpetuating inflammatory consequences of acute insults (7–11). The present study was designed to define the dynamic evolution of the inflammatory mediator networks which emerge rapidly within a few hours of liver I/R and contribute to the formation of NETs, the elimination of which have been shown to ameliorate tissue damage in this and analogous models (4, 12). We hypothesized that a few inflammatory mediators would be secreted early in the process and serve as organizational centers or nodes of downstream complexity. The identification of such organizational centers would allow rational therapeutic modulation, as shown in the current advances in treatment of a variety of autoimmune disorders(13, 14). In the present study, IL-17A, was shown to serve as one such major organizational center of downstream cytokine storm after liver I/R. Indeed, the early administration of anti-IL-17A neutralizing antibodies led to a partial disassembly of the pathological cytokine networks, the inhibition of further NET formation, and prevention of some of the damaging effects of the cytokine storm.
METHODS
Animals
Male wild-type (C57BL/6) mice (8–10 weeks-old) were purchased from Jackson Laboratories. Animal protocols were approved by the Animal Care and Use Committee of the University of Pittsburgh.
Liver I/R model
An established nonlethal model of segmental (70%) hepatic warm ischemia and reperfusion was used (4). Sham animals underwent anesthesia, laparotomy, and exposure of the portal triad without hepatic ischemia. After 60 minutes of liver I/R the livers were unclamped and reperfused. The mice were sacrificed at different time points after reperfusion (1, 3, 6, and 24 h). Several experimental groups of mice received 100 µg of neutralizing monoclonal anti-IL-17A antibody (BioXcell) diluted in PBS to a total volume of 100 µL via intraperitoneal injection either at the initiation of clamping, 2 hours after reperfusion or 4 hours after reperfusion. A control group received an injection of 100 µL PBS intraperitoneally. The experimental and control mice were sacrificed at 6 hours after reperfusion. Other groups of mice were treated with either DNAse 1 (50 mg/ mouse, Roche) or recombinant IL-17 (500ng/mouse, BioXcell) at the initiation of clamping.
Assessment of Liver Damage and Inflammation Biomarkers in Mouse Liver Samples
Liver damage was assessed 6 hours after initiation of reperfusion in all experiments. Serum alanine aminotransferase (ALT) levels were measured using the DRI-CHEM 4000 Chemistry Analyzer System (HESKA). The extent of parenchymal necrosis in the ischemic lobes was evaluated using H&E stained histological sections, as previously described. For the inflammatory biomarkers, A Luminex™ 100 IS analyzer (Luminex, Austin, TX) was used to measure liver tissue levels of interleukin (IL)-1α, IL-1β, IL-2, IL-4, IL5, IL-6, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17A, IFN-γ inducible protein (IP)-10 (CXCL10), KC (CXCL1), monocyte chemotactic protein (MCP)-1 (CCL2), macrophage inflammatory protein (MIP)-1α (CCL3), monokine induced by gamma interferon (MIG; CXCL9), interferon (IFN)-γ, vascular endothelial growth factor (VEGF), granulocyte-macrophage colony stimulating factor (GM CSF), and tumor necrosis factor alpha (TNF-α).
Western Blot
The reagents and antibodies used for Western Blots were as follows: anti-citrullinated histone H3 (Abcam); β-actin (Sigma).
Confocal microscopy
For immunofluorescence staining, liver sections were fixed, stained, and imaged using confocal microscopy. The specific primary antibodies used are as follows: Ly6G (1:100, BD Bioscience), cit-H3 (Abcam). All slides were scanned under the same conditions for magnification, exposure time, lamp intensity and camera gain. Confocal images were acquired with a PlanApo N (×20 with and without a 2.0 digital zoom)
Quantification of NETs using MPO-DNA
To quantify NETs in mouse sera collected 6 hours after reperfusion, a capture ELISA myeloperoxidase (MPO) associated with DNA was performed as described previously (4).
In vitro NET formation
Mouse and Human neutrophils were isolated from the peripheral blood as described (15). In addition, mouse neutrophils from the bone marrow were isolated from the Tibia and Femurs as described previously (6). Briefly, complete white blood cells (CD45+) were stained and sorted for neutrophils (CD11b+ and Ly6G+). The isolated purity of the neutrophils was 97.4% (Supplementary Figure 3D). Cells (2×106) were then plated in the 6 well plate pre-treated with either Sytoxgreen or DHR staining following treatment. Neutrophils were plated to adhere in coated plates for 1 hour before stimulation for 4 hours with A23187 (5uM, Sigma Aldrich) (16) or rIL-17 (50ng/ml, BioXcell) in 37°C 5% CO2. After 4 hours, the media was collected and used in subsequent experiments as outlined below.
Flow Cytometry
Ischemic liver lobes were harvested from WT mice at 6 h of reperfusion after 1 h of ischemia. Nonparenchymal cells (NPCs) were separated from the hepatocytes by one cycle of differential centrifugation (400 rpm for 5 min). The supernatant was centrifuged further (400 rpm for 5 min and two cycles of 1500 rpm for 5 min) to obtain NPCs. The NPCs did not contain hepatocytes, as assessed by light microscopy. Cells were treated with (CD16/CD32) FC block (BD Biosciences) for 30 mins at room temperature. Cell viability was determined by staining APC-Cy7 – fixable viability dye. To obtain the total number of neutrophils NPCs were treated with antibodies to CD45, CD11b, and Ly6G (BD Biosciences). Data were analyzed using FlowJo software.
Statistical and Computational Analyses
Results are expressed as the mean ± standard error of mean (SEM). Group comparisons were performed using ANOVA and Student’s t-test. A p<0.05 was considered statistically significant. For the cytokine analysis, data were analyzed using SigmaPlot™ 11 software (Systat Software, Inc., San Jose, CA). We carried out Dynamic Network Analysis (DyNA) inferences as previously described by our group (9, 17, 18). Dynamic network analysis (DyNA) was carried out to define, in a granular fashion, the central inflammatory network nodes as a function of time.
Using inflammatory mediator measurements of at least three time points, dynamic networks were created over four consecutive time periods (0–1 h, 1–3 h, 3–6 h, and 6–24 h) using Matlab® software. Connections, defined as the numbers of trajectories of serum inflammatory mediators that move in parallel, were created if the Pearson correlation coefficient between any two nodes (inflammatory mediators) at the same time interval was greater or equal to a threshold of 0.7 (a correlation value commonly used to characterize trajectories that move in parallel either up or down). The network complexity for each time-interval was calculated using the following formula: Sum (N1 + N2 +…+ Nn)/n-1, where N represents the number of connections for each mediator and n is the total number of mediators analyzed. The total number of network connections represents the sum of the number of connections across all time-intervals for all mice in a given subgroup.
RESULTS
Liver Ischemia-Reperfusion induces and maintains a pro-inflammatory microenvironment associated with injury.
We first sought to evaluate a time course of the liver I/R injury after 1, 3, 6 and 24 hours of reperfusion. Serum ALT, AST, and LDH levels peaked at 6 hours after reperfusion (Figure 1A, 1B and 1C) confirming acute liver injury and then decline by 24 hours. The hepatic histological changes were consistent with the elevated serum ALT and AST levels, with the presence of severe sinusoidal dilation and confluent pericentral hepatocellular necrosis which continued to evolve and expand at 24 hours (Figure 1D and Figure 1E). The levels of the 20 inflammatory mediators present in the reperfused lobe of the ischemic liver during the first 24 hours after reperfusion are shown in Supplementary Figure 1. Nineteen of the 20 examined inflammatory mediators were found to be significantly elevated at one time or another, with most peaking at approximately 6 hours and returning toward normal by 24 hours. These results are consistent with other observations suggestive of a complex interactive chain of mediators secreted by liver parenchymal and non-parenchymal cells interacting with infiltrates of circulating leukocytes. In addition to studying individual inflammatory mediators, recent work acknowledges the importance of delineating the interactions of these mediators with each other to create this inflammatory milieu(11). Therefore, we next sought to define, in a more granular fashion, the time evolution of dynamic networks of local inflammation in the livers of the mice after liver I/R. We employed data-driven computation, specifically dynamic network analysis (DyNA) (17–19), to discern and compare the interconnections among inflammatory mediators in the liver environment of mice receiving liver I/R over defined ranges of time. DyNA showed an increase in the total number of network connections over all the time intervals (Figure 1F), with an increasing complexity rising over time (Figure 1G). Supplementary Table 1 indicates the number of interconnections for each cytokine.
Figure 1.
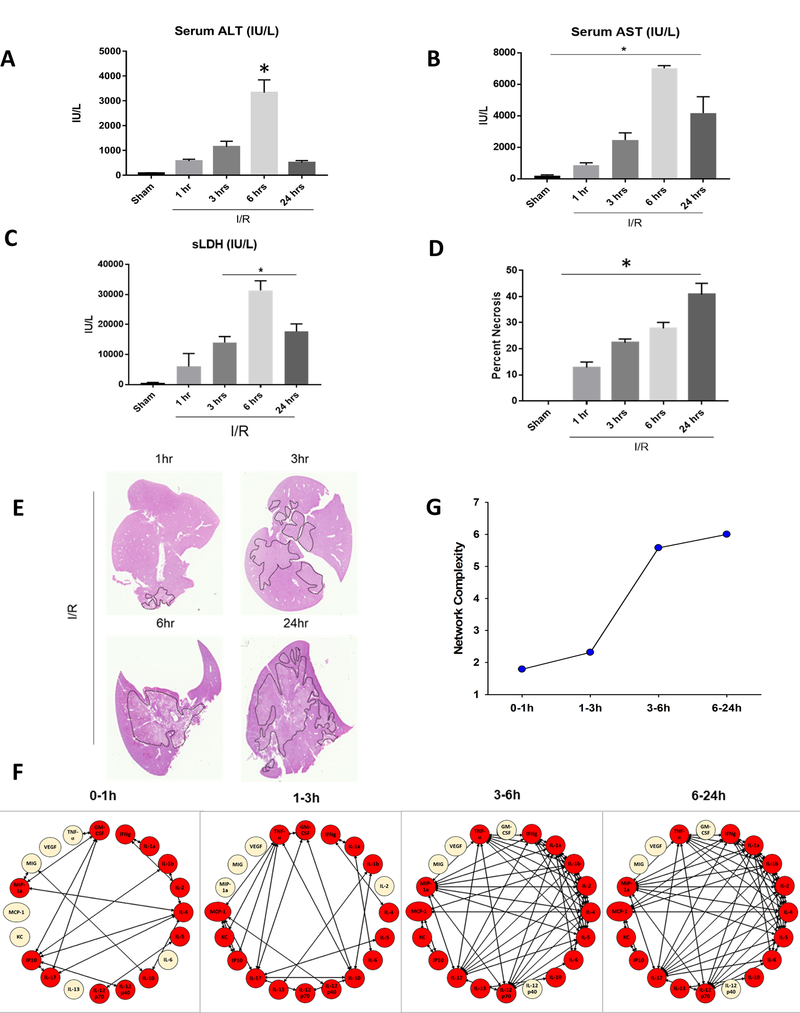
Liver Ischemia-Reperfusion induces and maintains a pro-inflammatory microenvironment to promote injury. (A-C) Serum ALT, Serum AST and Serum LDH levels were assessed after 1 hour of ischemia and 1, 3, 6 or 24 hours of reperfusion. Data represent the mean ± SE (n = 6 mice/group). (D) Quantification of necrotic hepatocytes in H&E stained liver sections after a time course of liver I/R. (E) Representative H&E stained liver sections showing areas of hepatic necrosis in I/R-treated mice reperfused at different time points. Liver tissue from the ischemic lobes was obtained at multiple time points after reperfusion and analyzed for the presence of inflammatory mediators followed by Dynamic Network Analysis (DyNA) as described in the Materials and Methods. (F) and (G) Networks were characterized by both innate and lymphoid mediators that evolved into a large complex network with time.
Differential Networks of inflammation implicate IL-17A as an early central driver of inflammation after I/R followed by a cytokine storm
Following liver I/R, a complex chain reaction develops between inflammatory cells and their cytokine products as shown in Figure 1. Figure 2 shows individual mediators and their connectivities at the different time intervals extrapolated from Figure 1F. Inferred dynamic networks in the livers of mice after I/R were characterized by interconnections among both innate immune mediators (MIP-1α, IL-1β, TNF-α, IFN-γ) and lymphoid mediators (IL-2, IL-4, IL-13, IL-17A), starting within the first hour post-injury (Figure 2B). Between 1 and 3 hours, DyNA inferred a central role for IL-17A, which developed the most connections with an array of both innate and lymphoid cytokines (Figure 2C). By 6 hours, the networks evolved into a much larger, reverberating mediator complex, a phenomenon often labelled a “cytokine storm” (Figure 2D and 2E). By 6 hours, the central cytokine with regards to connectivity remained IL-17A, with more than 10 connections, but this cytokine had been joined in the interactive mix with 10 other cytokines with 8 or more connections each. Only IL-10 and IL-13 remained unconnected with the components of the cytokine storm after 6 hours.
Figure 2.
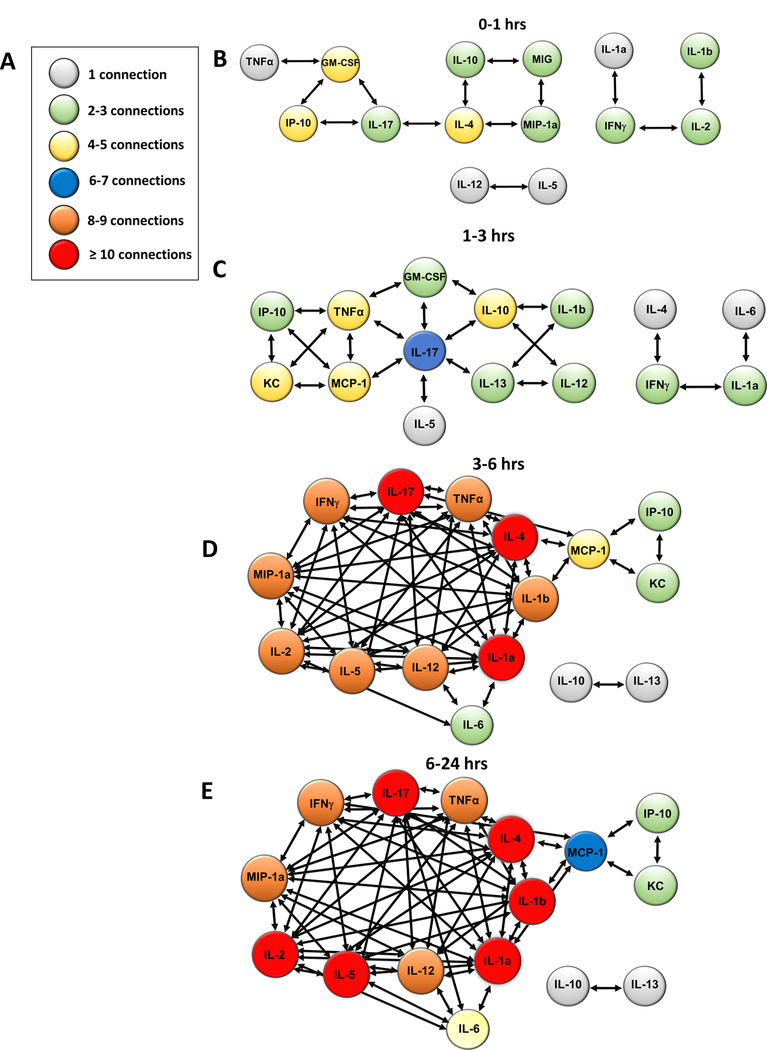
Differential Networks of inflammation implicate IL-17A as an early [1–3 hours] central driver of inflammation after I/R followed by a cytokine storm. (A-E) Inferred dynamic networks in the livers of mice after I/R were characterized by interconnections among both innate immune mediators and lymphoid mediators at the various timepoints of reperfusion.
IL-17A promotes liver injury after I/R.
The analysis in Figure 2 is consistent with the idea that IL-17A plays an early central role in the organization of a self-propagating cytokine storm in response to liver I/R. To test the DyNA-generated hypothesis associating IL-17A with the pro-inflammatory environment after liver I/R, recombinant mouse IL-17A (rIL-17) or IL-17A neutralizing antibodies (anti-IL-17) were administered intraperitoneally to mice immediately after reperfusion was initiated. rIL-17 increased liver damage significantly in mice subjected to I/R, whereas anti-IL-17 reduced liver damage as evidenced serum ALT levels compared to control mice receiving PBS (mean ALT levels IU/L: Control: 2797±361 , Anti-IL-17: 1093±512, rIL-17: 5399±760, P<0.001) (Figure 3A). Of note, rIL-17 injected intraperitoneally caused no elevation of serum ALT in sham treated mice (data not shown). Histopathologic examination revealed that the extent of liver necrosis paralleled serum ALT levels; anti-IL-17 administration reduced liver necrosis and rIL-17 administration augmented it (Figure 3B and Figure 3C). DyNA was performed on the ischemic liver lobe to compare mice treated with anti-IL-17 to control mice (Figure 3D). When IL-17 was blocked immediately after reperfusion was initiated, there was a marked decrease in the number of significant mediators and connections in the treated mice, with a decreased network complexity as compared to controls in Figure 2D. Figure 3D and 3E illustrate that the cytokine network became disassembled by early administration of anti-IL-17, with a marked decrease in the number of connections between the various mediators. As a consequence, TNFα with 7 connections, IL-13 with 6 connections, and IL-4 with 5 connections were inferred to have replaced IL-17A as central mediators. IL-13 and IL-4 have long been considered prototypic anti-inflammatory cytokines and are virtually absent from networks of unmodified I/R. These two cytokines have assumed a highly connected role after anti-IL-17 administration. Figure 3E compares the effect of administration of anti-IL-17 at time of unclamping (time 0 of reperfusion), at 2 hours after unclamping, and at 4 hours after unclamping. Whereas administration of anti-IL-17 at time of reperfusion disassembled the subsequent cytokine storm, administration at later time points had virtually no effect on the cytokine network. Supplementary Table 1 indicates the number of connections made for each cytokine depending on the time of administration of anti-IL-17.
Figure 3.
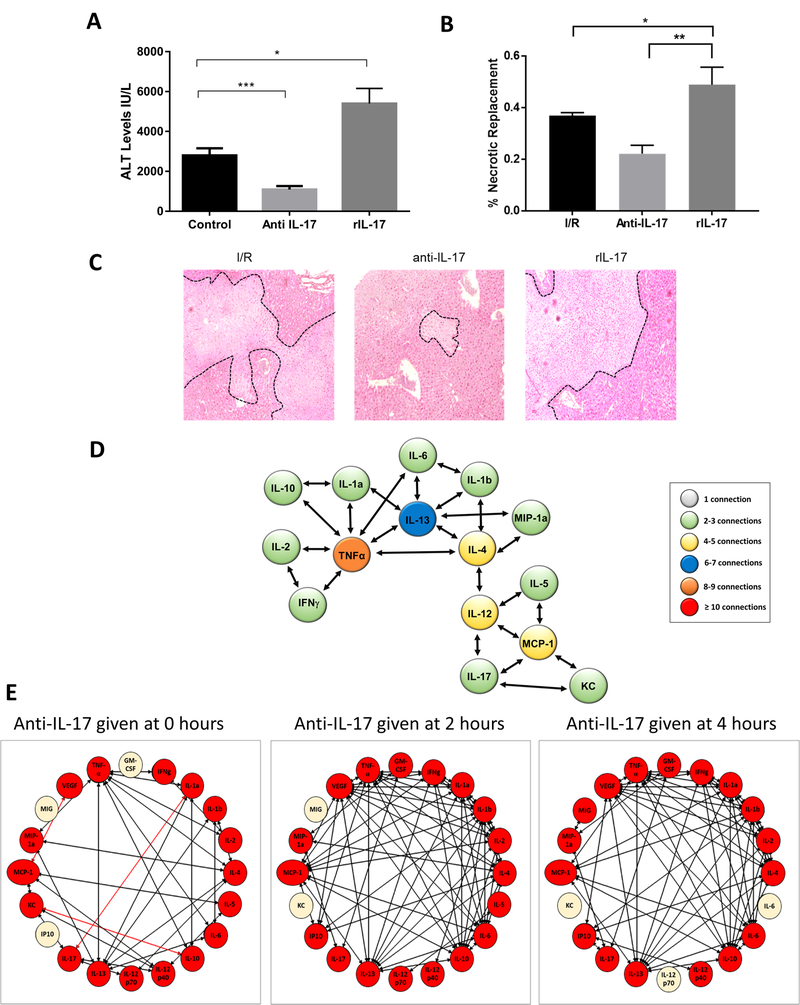
IL-17 promotes liver injury after I/R. (A) I/R-treated mice were given monoclonal neutralizing Anti-IL-17 or recombinant IL-17 (rIL-17) at the time of reperfusion. Serum ALT levels were assessed after 1 hour of ischemia and 6 hours of reperfusion. Data represent the mean ± SE (n = 6 mice/group). (B) Quantification of areas of hepatic areas of necrosis in H&E stained liver sections from control, anti-IL-17 or rIL-17-treated mice 6 hours after reperfusion. (C) Representative H&E stained liver sections showing decreased areas of hepatic necrosis in I/R-treated mice receiving anti-IL-17 and increased areas of necrosis in mice treated with rIL-17. (D) Inferred dynamic networks in the livers of mice treated with anti-IL-17 at the time of reperfusion. Disassembly of the inflammatory network is seen with reduced complexity of mediator interactions. (E) DyNA analysis of I/R livers of mice treated with anti-IL-17 at the time of unclamping (time 0 hours reperfusion) reveals a decrease in the number and complexity of connections between inflammatory mediators compared to control mice from Figure 1 and 2. In contrast, the complexity of the network remains high when anti-IL-17 was given two or four hours after reperfusion. *P < 0.05, **P<0.01, ***P<0.001. The red lines indicate negative correlation between the inflammatory mediators.
IL-17A increases neutrophil infiltration and neutrophil extracellular trap (NET) formation after I/R.
We next sought to determine a mechanism by which a putative, IL-17A mediated cytokine storm could exacerbate liver I/R injury. Neutrophils play a crucial role in the response to sterile inflammation induced by liver I/R (5). Previous studies have implicated IL-17A in sustaining neutrophil recruitment to areas of inflammation,(20). In addition, we have shown previously that liver I/R induced increases in ALT and liver necrosis are accompanied by increases in hepatic neutrophil infiltration and intrahepatic NET formation (4–6). We therefore examined whether the damaging effects of IL-17A in the setting of liver I/R were related to an increase in neutrophil influx and NET formation in the livers. Figure 4A and 4B show that there was a significant decrease in infiltrating neutrophils within the ischemic liver lobes in the mice treated early with anti-IL-17 compared to liver lobes from control mice. We found that the serum level of MPO-DNA complexes, a marker of circulating nucleosomes that are derived from NET formation(4), was decreased significantly in mice undergoing liver I/R with early anti-IL-17 treatment compared to controls (Figure 4C). Furthermore, ischemic lobes also exhibited significantly higher levels of citrullinated-histone H3, a specific marker of NET formation(4), after liver I/R, which was significantly reduced when mice were treated with anti-IL-17 (Figure 4B and 4D). On the other hand, treatment of mice with rIL-17 significantly increased neutrophil influx and NET formation (Figure 4B to 4D and Supplementary Figure 2B). We then examined whether IL-17A can induce neutrophils directly to form NETs. In vitro, rIL-17 was incubated with murine peripheral-derived neutrophils. Similar to A23-treated neutrophils (positive control), significantly increased citrullinated-H3 expression was observed in neutrophils and neutrophil media, respectively, after stimulation with rIL-17, compared to levels in control unstimulated neutrophils (Figure 4E, Figure 4F). rIL-17 induced significant NET formation in vitro consistently, as shown by immunofluorescence imaging compared to control. Increased NET formation was also confirmed with flow cytometry analysis showing a time dependent increase in the formation of NETs (Sytox green) and neutrophil ROS levels (DHR staining) compared to untreated neutrophils (Supplementary Figures 2C and 2D). Of note, similar effects of IL-17 in inducing NET formation were observed using mouse bone marrow derived neutrophils and neutrophils derived from human peripheral blood (Supplementary Figure 3A and 3B).
Figure 4.
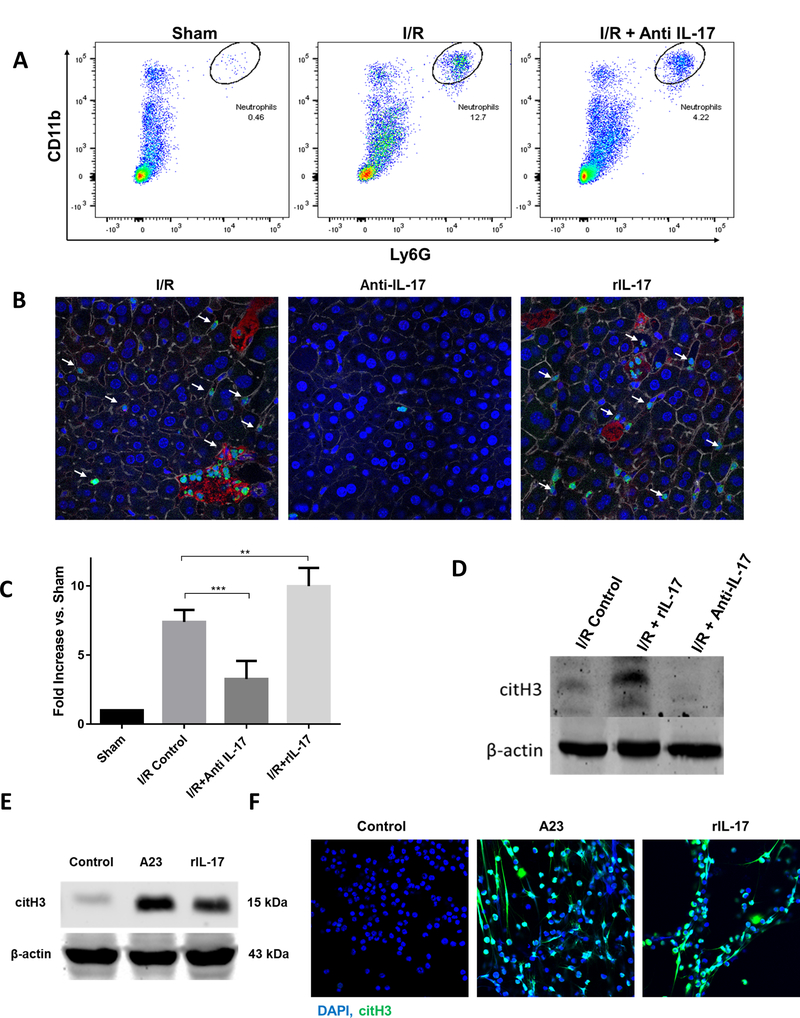
IL-17 increases neutrophil infiltration and neutrophil extracellular trap (NET) formation after I/R. (A) Flow cytometer analysis of liver after ischemia and 6 hour of perfusion reveals increased neutrophil infiltration compared to sham. Neutrophil influx in significantly decreased in mice treated with anti-IL-17. (B) Using confocal microscopy, there is a significant decrease in hepatic infiltrating neutrophils 6 hours hepatic I/R in anti-il-17 treated mice, and an increase in rIL-17 treated mice compared to control mice (mean 2.7 μm2 Ly6G+ area/total cells for control versus 0.7 μm2 Ly6G+ area/total cells for anti-IL-17 mice versus 3.8 μm2 Ly6G+ area/total cells for rIL-17 treated mice, p < 0.001). White arrows indicate neutrophils; Ly6G (green), nuclei (blue), citrullinated histone [citH3] (red), actin (white). Scale Bars 50μm. (C) NETs acutely form in liver tissue 6 hours after liver I/R as assessed by serum levels of MPO-DNA compared to sham. MPO-DNA levels are significantly reduced in mice treated with anti-IL-17 at the time of reperfusion; MPO-DNA levels are increase in livers of mice treated with rIL-17A (D) Cit-H3 protein levels in the liver were determined by Western blot in I/R, I/R + rIL-17, and I/R + anti-IL-17 mice groups (6 per group) 6 hours after liver I/R. Levels of citH3 were elevated when rIL-17 had been given and minimized when anti-IL-17 had been given. (E) In vitro, mouse peripheral derived-neutrophils were treated with A23 (5uM) to induce citrullinated histone [NET formation]. Similarly, citH3 levels were increased when neutrophils were treated with rIL-17 (F) Confocal images show in vitro neutrophils forming NETs when treated with rIL-17 or A23. Nuclei (blue), citH3 (green). Scale Bars 100μm. *P < 0.05, **P<0.01, ***P<0.001.
IL-17A and NETs initiate inflammatory responses and exacerbate liver injury in vivo
Thus far, we have shown that in the presence of rIL-17 there is an increase in neutrophil infiltration, NET formation, and liver injury after I/R. In addition, in vitro, rIL-17 can stimulate NET formation directly. Therefore, we next sought to test our hypothesis that rIL-17 drives the inflammatory cascade and contributes to sterile injury through its role in inducing NET formation. Therefore, we subjected mice to liver I/R and administered rIL-17 or a combination of rIL-17 and DNAse (a potent inhibitor of NET effect (21)). Plasma MPO-DNA levels were used as markers of NET formation. As seen in Figure 5A and 5B and Supplementary Figure 3C, rIL-17 increased blood levels of MPO-DNA as well as liver injury; both effects were ameliorated significantly, but not obliterated totally, in the presence of DNAse. Furthermore, DyNA analysis suggested that blocking NET formation with DNAse disassembled the cytokine networks by radically reducing the number of participating cytokines and their mutual interconnections. The reformed network revealed a less robust central organizing function now played by IL-4 and IL-12. Of importance, the central role that IL-17A played in the cytokine network induced by liver I/R (Figure 2) was minimized by DNAse, suggesting that NET formation has a positive feedback function on the IL-17A driven dynamic inflammation networks that elicit NET formation (Figure 5D).
Figure 5.
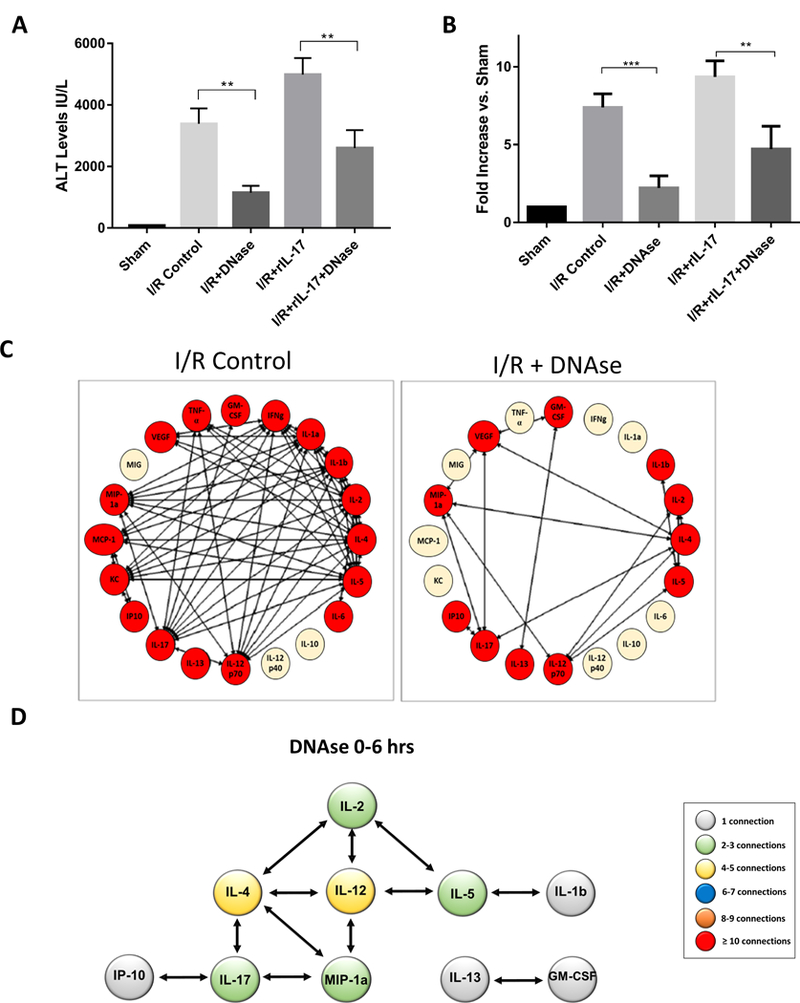
IL-17 and NETs initiate liver injury exacerbated by administration of rIL-17 administration and modulated by administration of DNAse. (A) Serum ALT levels of mice after liver I/R in various treatment groups. In the presence of DNase, a NET inhibitor, the effects of rIL-17 were significantly ameliorated as evident by serum ALT levels. This correlated with decreased NET formation as seen in (B). (C and D) DyNA analysis of I/R livers of mice treated with DNAse reveal disassembly of the inflammatory network with a significant decrease in the number and complexity of inflammatory mediator connections compared to control mice.
DISCUSSION
Our principal finding in this study is that surgical stress, in the form of liver I/R, is associated with a rising complexity of dynamic inflammation networks within the liver, and that there is an early critical role for IL-17A in mediating and orchestrating the immune response to liver I/R to result in the post-I/R injury. We also establish in a mouse model a critical role for both IL-17A and NETs in the pathophysiology of liver I/R (Figure 6). Inflammation is an essential host response for healing of damaged tissues(1). Trauma, surgical manipulation, shock, and organ transplantation are clear examples in which hepatic ischemia followed by reperfusion occurs and the liver I/R model has often been employed to study the inflammatory changes of acute sterile injury(22). In this model, the response to hepatic lobar I/R is dysregulated in that hepatic necrosis evolves in the ischemic portions of the liver. Previous experiments by ourselves and others have shown convincingly that neutrophilic infiltration of the reperfused lobe and the development of NETs are critical steps to liver damage (4, 5). NETs arise from the enzymatic citrullination of nuclear histones, leading to the ultimate expulsion from the cell of the nuclear chromatin with its embedded proteins together with the granules and other cytoplasmic contents(23). DNA and the toxic DAMPs embedded within it act as alarmins, augmenting inflammation and promoting microvascular thrombosis, thereby destroying hepatic tissue (4). The effects of NETosis can be prevented or reversed in several ways including genetic elimination of the enzymes responsible, or by the administration of enzyme inhibiting drugs, or the systemic administration of DNAse, which dissolves the extracellular chromatin(4). It has been recently shown that neutrophilia in the genetic absence of endogenous DNAses spontaneously leads to sinusoidal thrombosis, hepatic necrosis, and death(21).
Figure 6.
Schematic drawing depicting the interplay between IL-17 and NETs to orchestrate the injury after liver I/R.
NETs take hours to evolve when generated in vitro, and there may be therapeutic insights to be gained by understanding how the inflammatory milieu generated by I/R serves to generate NETs in vivo. The complexity of the inflammatory milieu defies simple analysis; nineteen of the twenty inflammatory mediators assessed became significantly elevated within the first six hours following initiation of reperfusion. In order to determine the critical cytokines within ischemic tissue that play important organizing roles in the induction of NETosis, we employed data-driven dynamic network analysis of twenty cytokines and chemokines in the I/R liver lobe. During the period of reperfusion following an ischemic event in the liver, a complex, non-linear cellular and humoral response is set in motion dependent not only on the individual measurable components but also in which positive and negative feedbacks between these components occur over time (3). The complexity of these events has made it difficult to identify potential therapeutic targets which could effectively modulate the adverse pathophysiological outcomes. Computational modeling of these responses has now been found to be of use in analyzing the multimodal interactions of biological processes. Dynamic network analysis [DyNA] is one computerized algorithm that can assess how the multiple inflammatory mediators which appear within the blood or tissue in response to injury interact over time. Indeed, it has identified the possible early drivers and secondary inflammatory mediators of inflammation in a variety of experimental and clinical responses to injury. Relying on reductionist experimental results alone, one cannot effectively detect dynamic and complex interactive effects of the various mediators in inflammation. Many studies have focused attention on the role of single or several selected cytokines found to be elevated in inflamed tissue and blood after injury and thus hypothesized to play important roles as mediators of tissue injury or death. Indeed, the therapeutic targeting of suspect inflammatory mediators has been moderately effective in certain chronic autoimmune diseases (24). However, acute inflammatory insults such as hemorrhagic shock or liver I/R have been resistant to modulation (22). Focusing on a single inflammatory mediator as a causal factor may not take account of the fact that the environment which follows acute insults evolves with such speed and is soon composed of such a large network of diverse proteins engaged in a complex and difficult to stop chain reaction among inflammatory cells and their cellular products(11). Such cytokine storms, however, may be analyzed computationally over short periods of time, revealing networks and interactions among mediators. Such network analysis has been found useful in trauma, hemorrhage, wound healing, allograft rejection, and other chronic inflammatory events which follow acute insults (7–11).
The present study was designed to define the dynamic evolution of the cytokine networks which rapidly emerge within a few hours of liver I/R. We hypothesized that a few inflammatory mediators would be secreted early in the process and serve as organizational centers (nodes) of complexity subject to rational therapeutic modulation. In fact, IL-17A, proved to serve as such an organizational center. Administration of recombinant IL-17A, though harmless in itself, exacerbated the degree of liver damage induced by liver I/R. Furthermore, the early, but not delayed, administration of neutralizing antibodies to IL-17A markedly reduced the degree of neutrophil infiltration and liver damage induced by I/R. As expected, the administration of DNAse significantly reduced the hepatic damage even if recombinant IL-17A had been administered. Taken together, these data give support to the hypothesis that IL-17A plays a central organizing role in the inflammatory cascade responsible for the induction of NETosis in this sterile injury model. Notably, recombinant IL-17A was capable of inducing NETs from murine-derived neutrophils in vitro. However, it cannot be concluded that IL-17A is solely responsible for NETosis in this in vivo model because many of the mediators in the evolving cytokine storm have been shown capable of inducing NETosis alone or in combination.
Dynamic network analysis not only helped to identify IL-17A as a critical inflammatory node in the early evolving cytokine cascades which induced NETosis, but also demonstrated that the inflammatory network leading to NETosis became disassembled by the early administration of anti-IL-17A. In the absence of IL-17A activity in the early hours post liver I/R, the cytokine networks appeared to be re-assembled with central roles for TNFα, IL-13, and IL-4, the latter pair being generally regarded as anti-inflammatory/pro-reparative cytokines. Concurrently neutrophil infiltration, intra hepatic NETosis and hepatic necrosis were reduced. These changes in the network after early anti-IL-17A administration showed decreases in cytokine network connectivity. Administration of DNAse also showed decreases in cytokine connectivity and network disassembly coincident with reduction in liver necrosis, but the reformed networks were not identical to that induced by anti-IL-17 – perhaps highlighting the complexity of the reverberating self-reinforcing mediator milieu once disturbed.
Ineterleukin-17A-related pathways have been best described in mediating chronic inflammatory diseases such as rheumatoid arthritis, asthma, systemic lupus erythematosus, multiple sclerosis, and allograft rejection; however, little is known regarding role of IL-17A in innate immunity or its role regarding acute liver I/R injury (25, 26). In inflammation resulting from infection, activation of dendritic cells is thought to produce IL-23 with subsequent activation of IFN-α/β and IFN-γ/δ and natural killer T cell production of IL-17A. The cascade leads to the recruitment of neutrophils to sites of infection (25). Pro-inflammatory cytokines and chemokines are produced by stromal, epithelial, and endothelial cells in response to IL-17A to recruit neutrophils rapidly to sites of infection (27–30). In our liver I/R model, a model of sterile inflammation, we demonstrated a similar role of IL-17A in recruiting neutrophils to the site of inflammation. In addition, computational analysis showed an early role of IL-17A in coordinating the inflammatory response and orchestrating the cytokine/chemokine response, suggesting a critical role in the innate immune response to sterile inflammation.
Much attention in previous studies has focused on the production of IL-17A by a subset of T cells called Th17 cells (31). While IL-17A production by T cells is widely studied especially in chronic inflammatory processes, it is increasingly appreciated that widely diverse types of innate immune cells can also produce IL-17A(32). Recent studies demonstrate that IL-17A+ cells can be found in atherosclerotic plaques (33). Similarly, in renal ischemia reperfusion injury, neutrophils were found to be the major source of IL-17A production that in turn positively regulate further neutrophil transmigration to the injured organ (34). Interestingly, in psoriasis, neutrophils were found to be the predominant cell types that contain IL-17A in human skin. Furthermore, IL-17A is released through the formation of neutrophil extracellular traps(35). In our model of liver I/R, IL-17A stimulated neutrophil recruitment to the liver and induced NET formation that contributed the injury after liver ischemia. As NETs can also release IL-17A(35), this sets the stage to a self-sustaining inflammatory process to be set in motion as depicted in Figure 6. We hypothesize that the relationship among IL-17A, neutrophils and NETs is a feedback-feedforward communication that leads to the vicious cycle that results in the injury after liver I/R. Inhibiting NETs or IL-17A breaks the vicious cycle and ameliorates the injury after liver I/R.
As mentioned previously, I/R injuries result in the production and local release of DAMPs and downstream pro-and anti-inflammatory chemokines, cytokines, free radicals that enter the circulation affecting distant organs and tissues. This leads to a highly complex inflammatory network as depicted in our study. Inflammation in general leads to resolution, re-establishment of homeostasis, and tissue repair. However, a self-sustaining, seemingly irreversible inflammatory process can also be set in motion, driving a systemic, malignant pathobiology across multiple organ systems which further exacerbate an acute injury or lead to a chronic state of inflammation (1). In our experiments, when IL-17A was neutralized, there was a significant decrease in the complexity of the inflammatory networks and a decrease in the injury. Of interest, the DyNA analysis, after anti-IL-17A was given, revealed the integration of both IL-10 and IL-13 into the main inflammatory network. Both IL-10 and IL-13 have been shown to protect against hepatic I/R and promote resolution of inflammation (36, 37),(38). A better understanding of the natural processes that lead to resolution and reestablishment of homeostasis and tissue repair are warranted.
There are a number of potential limitations to the use of DyNA to draw conclusions as to the causal events in the response to any biological event. The first limitation involves the choice of the specific panel of inflammatory mediators. There are likely a number of mediators in our inflammatory mediators that could have been added, including virtually all of the proteins released initially by liver cells exposed to liver I/R. In the present study, we sought to assess mediators which have be implicated in responses to acute injury and that we have shown previously to be released by stressed liver cells (39). A second limitation involves the choice of tissue to sample: for this reason, we chose the sample the tissue subjected to I/R rather than the serum. A third limitation involves the choice of time interval to study. One of our most important findings was that in liver I/R, the importance of IL17A could only be discerned in the first few hours, when this cytokine made the most connections with the developing cytokine storm. Once discovered by network analysis, validation was provided by the inhibition of the development of the cytokine storm by neutralization of IL-17A. Conversely, the administration of rIL-17A recapitulated many of the responses seen. Nonetheless, the search for other initiating or driving mediators must be undertaken in subsequent work among which are DAMPs released form hypoxic tissue itself.
In conclusion, identifying inflammatory networks that reflect dynamic changes in the inflammatory response stemming from sterile inflammation, such as liver I/R, could lead to opportunities for directed therapeutic intervention, enhance outcomes after liver ischemia reperfusion and improve patient outcomes after liver surgery. In this regard, our in vivo and in silico studies identified biomarkers such as IL-17A and NETs as potential therapeutic targets in liver I/R injury. More generally, the use of novel computational methodology could streamline studies of liver I/R in humans or other inflammatory diseases.
Supplementary Material
List of abbreviations:
- I/R
ischemia reperfusion
- DyNA
dynamic network analysis
- IL
interleukin
- NET
neutrophil extracellular trap
- MPO
myeloperoxidase
- DNA
deoxyribose nucleic acid
- DAMPs
damage associated molecular patterns
- ALT
alanine aminotransferase
- MCP-1
monocyte chemotactic protein
- MIP-1α
macrophage inflammatory protein
- MIG
monokine induced by gamma interferon
- IFN- γ
interferon-γ
- VEGF
vascular endothelial growth factor
- GM CSF
granulocyte-macrophage colony stimulating factor
- TNF-α
tumor necrosis factor alpha-α
- ELISA
enzyme linked immunosorbent assay
- WT
wild type
- NPC
nonparenchymal cell
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Chen GY, and Nuñez G. 2010. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol 10: 826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Golen RF, Reiniers MJ, Olthof PB, van Gulik TM, and Heger M. 2013. Sterile inflammation in hepatic ischemia/reperfusion injury: present concepts and potential therapeutics. J. Gastroenterol. Hepatol 28: 394–400. [DOI] [PubMed] [Google Scholar]
- 3.Klune JR, and Tsung A. 2010. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg. Clin. North Am 90: 665–77. [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, and Tsung A. 2015. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62: 600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaeschke H 2006. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol. Gastrointest. Liver Physiol 290: G1083–8. [DOI] [PubMed] [Google Scholar]
- 6.Al-Khafaji AB, Tohme S, Yazdani HO, Miller D, Huang H, and Tsung A. 2016. Superoxide induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-dependent mechanism. Mol. Med 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vodovotz Y, and Billiar TR. 2013. In silico modeling: methods and applications to trauma and sepsis. Crit. Care Med 41: 2008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azhar N, Ziraldo C, Barclay D, Rudnick DA, Squires RH, Vodovotz Y, and Pediatric Acute Liver Failure Study Group. 2013. Analysis of serum inflammatory mediators identifies unique dynamic networks associated with death and spontaneous survival in pediatric acute liver failure. PLoS One 8: e78202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, Mi Q, Barclay D, Zuckerbraun B, Peitzman AB, Sperry J, and Billiar TR. 2016. Temporal Patterns of Circulating Inflammation Biomarker Networks Differentiate Susceptibility to Nosocomial Infection Following Blunt Trauma in Humans. Ann. Surg 263: 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziraldo C, Mi Q, An G, and Vodovotz Y. 2013. Computational Modeling of Inflammation and Wound Healing. Adv. wound care 2: 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vodovotz Y, Csete M, Bartels J, Chang S, and An G. 2008. Translational systems biology of inflammation. PLoS Comput. Biol 4: e1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, Barthwal MK, and Dikshit M. 2012. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McInnes IB, and Schett G. 2007. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol 7: 429–442. [DOI] [PubMed] [Google Scholar]
- 14.Brennan FM, and McInnes IB. 2008. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Invest 118: 3537–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y. Isolation of human and mouse neutrophils ex vivo and in vitro. Methods Mol Biol . 2012. [DOI] [PubMed]
- 16.Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, Brinkmann V, von Bernuth H, and Zychlinsky A. 2017. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife [DOI] [PMC free article] [PubMed]
- 17.Abboud A, Namas RA, Ramadan M, Mi Q, Almahmoud K, Abdul-Malak O, Azhar N, Zaaqoq A, Namas R, Barclay DA, Yin J, Sperry J, Peitzman A, Zamora R, Simmons RL, Billiar TR, and Vodovotz Y. 2016. Computational Analysis Supports an Early, Type 17 Cell-Associated Divergence of Blunt Trauma Survival and Mortality. Crit. Care Med 44: e1074–e1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mi Q, Constantine G, Ziraldo C, Solovyev A, Torres A, Namas R, Bentley T, Billiar TR, Zamora R, Puyana JC, and Vodovotz Y. 2011. A dynamic view of trauma/hemorrhage-induced inflammation in mice: Principal drivers and networks. PLoS One 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Namas RA, Vodovotz Y, Almahmoud K, Abdul-Malak O, Zaaqoq A, Namas R, Mi Q, Barclay D, Zuckerbraun B, Peitzman AB, Sperry J, and Billiar TR. 2016. Temporal patterns of circulating inflammation biomarker networks differentiate susceptibility to nosocomial infection following blunt trauma in humans. Ann. Surg 263: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laan M, Cui ZH, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh BE, and Lindén A. 1999. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol 162: 2347–52. [PubMed] [Google Scholar]
- 21.Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, Bilyy R, Krenn V, Renné C, Renné T, Kluge S, Panzer U, Mizuta R, Mannherz HG, Kitamura D, Herrmann M, Napirei M, and Fuchs TA. 2017. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science (80-. ) 358: 1202–1206. [DOI] [PubMed] [Google Scholar]
- 22.Serracino-Inglott F, Habib NA, and Mathie RT. 2001. Hepatic ischemia-reperfusion injury. Am. J. Surg 181: 160–6. [DOI] [PubMed] [Google Scholar]
- 23.Brinkmann V, and Zychlinsky A. 2012. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol 198: 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nickoloff BJ, and Nestle FO. 2004. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest 113: 1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwakura Y, and Ishigame H. 2006. The IL-23/IL-17 axis in inflammation. J. Clin. Invest 116: 1218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miossec P, and Kolls JK. 2012. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov 11: 763–76. [DOI] [PubMed] [Google Scholar]
- 27.Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, and Armitage RJ. 1995. Human IL-17: A Novel Cytokine Derived from T Cells. J. Immunol 155: 5483–5486. [PubMed] [Google Scholar]
- 28.Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, Khatcherian A, Gonzalez J, Pierson KC, White TR, Pensabene C, Coats I, Novitskaya I, Lowes MA, and Krueger JG. 2008. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol 159: 1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, and Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med 203: 2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peric M, Koglin S, Kim S-M, Morizane S, Besch R, Prinz JC, Ruzicka T, Gallo RL, and Schauber J. 2008. IL-17A enhances vitamin D3-induced expression of cathelicidin antimicrobial peptide in human keratinocytes. J Immunol 181: 8504–8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tesmer LA, Lundy SK, Sarkar S, and Fox DA. 2008. Th17 cells in human disease. Immunol. Rev 223: 87–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cua DJ, and Tato CM. 2010. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol 10: 479–89. [DOI] [PubMed] [Google Scholar]
- 33.De Boer OJ, Van Der Meer JJ, Teeling P, Van Der Loos CM, Idu MM, Van Maldegem F, Aten J, and Van Der Wal AC. 2010. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J. Pathol 220: 499–508. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Huang L, Vergis AL, Ye H, Bajwa A, Narayan V, Strieter RM, Rosin DL, and Okusa MD. 2010. IL-17 produced by neutrophils regulates IFN-??-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Invest 120: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, and Bruce AT. 2011. Mast Cells and Neutrophils Release IL-17 through Extracellular Trap Formation in Psoriasis. J. Immunol 187: 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshidome H, Kato A, Miyazaki M, Edwards MJ, and Lentsch AB. 1999. IL-13 activates STAT6 and inhibits liver injury induced by ischemia/reperfusion. Am. J. Pathol 155: 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lentsch AB, Kato A, Yoshidome H, McMasters KM, and Edwards MJ. 2000. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology 32: 169–173. [DOI] [PubMed] [Google Scholar]
- 38.Yoshidome H, Kato A, Edwards MJ, and Lentsch AB. 1999. Interleukin-10 suppresses hepatic ischemia/reperfusion injury in mice: implications of a central role for nuclear factor kappaB. Hepatology 30: 203–8. [DOI] [PubMed] [Google Scholar]
- 39.Ziraldo C, Vodovotz Y, Namas RA, Almahmoud K, Tapias V, Mi Q, Barclay D, Jefferson BS, Chen G, Billiar TR, and Zamora R. 2013. Central role for MCP-1/CCL2 in injury-induced inflammation revealed by in vitro, in silico, and clinical studies. PLoS One [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.