

Abstract
Immune profiling of tissue through multiplex immunohistochemistry is important for the investigation of immune cell dynamics and it can contribute to disease prognosis and evaluation of treatment response in cancer patients. However, protocols for mouse formalin-fixed paraffin-embedded (FFPE) tissue have been less successful. Given that FFPE remains the most common method to fix and analyze mouse tissue, this has limited the options to study the immune system and the impact of novel therapeutics in preclinical models. In an attempt to address this, we developed an improved immunohistochemistry protocol with a more effective antigen retrieval buffer. We also validated 22 antibodies specific for mouse immune cell markers to distinguish B cells, T cells, NK cells, macrophages, dendritic cells, and neutrophils. In addition, we designed and tested novel strategies to identify immune cells for which unique antibodies are currently not available. Lastly, in the 4T1 model of breast cancer, we demonstrate the utility of our protocol and antibody panels in the quantitation and spatial distribution of immune cells.
Keywords: IHC, immune cell, FFPE, antigen retrieval, multiplex IHC
Introduction
Using immunohistochemistry to evaluate immune cell dynamics in vivo complements immunophenotyping via flow cytometry (1, 2), providing a method for validation. Further, with the advent of immunotherapy to treat cancer and other diseases, it has become increasingly important to evaluate infiltrating immune cells in the context of the whole tissue (3–5). Determining the tissue distribution of specific antigens, especially immune markers, is necessary for the diagnosis and investigation of disease. In cancer, this can assist in the understanding of immune cell behavior in the tumor microenvironment at primary and metastatic sites (6, 7).
Although multiple groups have made strides regarding multiplex IHC of human tissues for immune cell characterization (8–10), protocols using mouse formalin-fixed paraffin-embedded (FFPE) tissues are still underdeveloped. Many epitopes are formalin-fixation-sensitive and not easily detected. Historically, detection of formalin-sensitive epitopes has relied on frozen tissue (11, 12), which comes with the disadvantage of cumbersome storage and compromised tissue morphology.
Zinc-salt fixation has been proposed as an alternative to formalin-fixation as a strategy to retain tissue morphology while resulting in less epitope masking (13, 14). Zinc-salt fixation may indeed have advantages; however, there are only limited IHC studies comparing the two fixation methods. Additionally, there are concerns that zinc-based fixatives may not penetrate tissues as effectively as formalin (15). Further, to the best of our knowledge, the long-term stability of zinc-fixed tissue is unreported. Finally, formalin-fixation remains the most common fixation method for mouse tissues.
In this study, we demonstrate multiplex IHC of mouse immune cell markers with an improved antigen retrieval protocol that works efficiently for most antibodies. We used specific markers to detect populations of B cells, subtypes of T cells, natural killer (NK) cells, macrophages, dendritic cells (DCs), granulocytes and total myeloid cells. We assessed antibody specificity by utilizing tissue from wild-type, SCID, NSG, and control- or clodronate-liposome-treated mice. Lastly, we demonstrate the utility of the protocol in evaluating the immune landscape in a preclinical model of breast cancer.
Materials and Methods
Mice
Male and female BALB/c (BALB/cfC3H strain) and SCID mice were purchased from the on-campus UT Southwestern Mouse Breeding Core (Dallas, TX). Tissue from NSG mice was a kind gift from Dr. Sean Morrison (UT Southwestern Medical Center). All animals were housed in a pathogen-free environment with access to food and water ad libitum.
Macrophage Depletion
Control (‘Encapsomes’) and clodronate-liposomes (‘Clodrosomes’) were obtained from Encapsula NanoSciences LLC (Brentwood, TN, 37027 USA). 8 week-old BALB/C male mice were injected i.p. with 300 uL of Encapsomes (control) or Clodrosomes. Mice were euthanized 72 hrs post-injection and tissues were fixed in 10% neutral-buffered formalin for 48 hrs before embedding in paraffin.
Cells
4T1 breast cancer cells were obtained from the ATCC (Manassas, VA). The cells were cultured in DMEM (Sigma) supplemented with 10% fetal calf serum (RMBIO) at 37°C in a humid atmosphere with 5% CO2.
4T1 Breast Cancer Model
4T1 cells (105) were injected orthotopically into the inguinal mammary fat pat of 8-week-old female BALB/c mice. Mice were euthanized 3 weeks post injection. The established tumor was resected and fixed in 10% neutral-buffered formalin for 48 hrs before embedding in paraffin. All experiments were approved by the Institutional Animal Care and Use Committee at UT Southwestern and conducted in accordance with an approved protocol.
IHC
Tissue fixation.
All tissues were fixed in 10% neutral buffered formalin for 48 h on a shaker at room temperature. Tissues were washed and stored in PBS at 4°C until embedded. Paraffin embedding was performed by the UT Southwestern Molecular Pathology Core.
Tissue sectioning.
Before sectioning, FFPE tissue blocks were incubated in 70% ethanol with 10% glycerol for 5 minutes to soften followed by a quick rinse in distilled water. Then, 5 μm sections were cut using a Leitz 1512 rotary microtome and placed onto a water bath at 40°C for 2 minutes to reduce wrinkles. Sections were transferred to positively charged slides (#SFH1103; Biocare Medical) and allowed to dry overnight before staining.
Deparaffinization, staining, imaging.
Slides were warmed for 10 minutes in a 60°C oven before following the de-paraffinization protocol. The de-paraffinization and rehydration protocol entails incubation in xylene (3x for 5 minutes), 100% ethanol (2x for 2 minutes), 95% ethanol (2x for 2 minutes), 70% ethanol (2x for 2 minutes), 50% ethanol (2x for 2 minutes) and PBS (1× 3 minutes). To prevent sections from folding or falling off the slides during antigen retrieval and antibody stripping, the de-paraffinized slides were fixed in 10% neutral buffered formalin for 30 minutes followed by incubation in PBS (2x for 3 minutes).
For antigen retrieval, 250 ml of antigen retrieval buffer (10 mM Tris-HCl 1 mM EDTA, 10% glycerol, pH 9.0) was preheated to 80°C in a bucket inside a pressure cooker (Biocare Medical) filled with 500 ml of water. Slides were then placed inside the buffer and heated at 110°C for 18 minutes (~4–5 psi). Slides were allowed to cool at room temperature for 30 minutes followed by a PBS rinse. Tissue sections were surrounded by a hydrophobic barrier using a PAP pen and blocked for 10 minutes with 2.5% goat serum, 2.5% horse serum (Vector Laboratories) or 30 minutes Rodent Block M (Biocare Medical) if the secondary host was goat, horse, or rat, respectively.
Incubation with primary antibody was performed on a shaker for 1 h at room temperature, or at 4°C overnight. Slides were washed in PBSt (0.05% tween + 2 mM EDTA; 3x for 5 minutes) before adding the ‘polymer’ HRP- or AP-conjugated secondary antibodies (ImmPress; Vector Laboratories). Sections were incubated with secondary antibody for 30 minutes on a shaker followed by one PBSt (0.2% tween + 2 mM EDTA) and two PBSt (0.05% tween + 2 mM EDTA) washes, 5 minutes each. Sections were then incubated with chromogenic or tyramide signal amplification substrates. For multiplex staining, antibody stripping was performed in 10 mM citrate buffer (pH 6.2, 10% glycerol) in a pressure cooker at 110°C for 2 minutes before probing with the next primary antibody.
For chromogenic detection, Betazoid DAB, Warp Red, and Ferangi Blue (BDB2004L, WR806S, and FB813H respectively; Biocare Medical) were used. Substrate incubation time was determined by monitoring signal development using a microscope. After developing with chromogenic substrates, slides were counterstained with hematoxylin. Bluing was performed by running tap water over the slides for 30 seconds. Slides were cover-slipped using VectaMount (H-5501, Vector Laboratories) and scanned at 20X using the Hamamatsu NanoZoomer 2.0-HT.
For fluorescence detection, we used tyramide signal amplification (OPAL; #NEL797001KT; PerkinElmer). The following fluorophores were used: OPAL 520, OPAL 570, and OPAL 690. DAPI was used for nuclear counterstaining. To reduce the auto-fluorescence, slides were incubated at the end in TrueBlack Lipofuscin Autofluorescence Quencher (#23007; Biotium) diluted 20-fold in 70% ethanol for 30 seconds. Slides were cover-slipped using ProLong Gold (#P36931, Life Technologies). Slides were scanned at 20X using the Zeiss Axioscan.Z1 (Whole Brain Microscopy Facility, UT Southwestern). DAPI, AF488 (for OPAL520), AF555 (for OPAL570) and AF660 (for OPAL 690) channels were used to acquire images. The exposure time for image acquisition was between 5 to 100 milliseconds.
IHC Antibodies.
Primary antibodies were diluted in PBSt (0.05%) + 2 mM EDTA + 0.5% serum (goat or horse, based on the host of the secondary antibody), or Renaissance Background Reducing Diluent (PD905; Biocare Medical) when incubating sections with more than one primary antibody simultaneously. The primary antibodies and dilution factors used are displayed in Table 1. ‘Polymer’ secondary antibodies were purchased from Vector Laboratories (ImmPRESS HRP anti-rabbit IgG [#MP-7401], HRP anti-rat IgG [#MP-7404], AP anti-rat IgG [#MP-5404], and AP anti-rabbit IgG [#MP-5401]).
Table 1.
| Target/ Antibody |
Alias(es) | Company | Catalog number |
Clone | Dilution Factor, Chromoge n Staining |
Dilution Factor, Fluorescent (TSA) Staining |
|---|---|---|---|---|---|---|
| CD11b | Itgam | Abcam | ab133357 | EPR1344 | 5000 | 20000 |
| CD11c | Itgax | Cell Signaling | 97585S | D1V9Y | 100 | 200 |
| CD163 | M130 | Abcam | ab182422 | EPR19518 | 6000 | 12000 |
| CD19 | Cell Signaling | 90176 | D4V4B | 1000 | 2000 | |
| CD3e | Thermo Fisher | PA1–29547 | Polyclonal | 1000 | 3000 | |
| CD3e* | Abcam | ab11089 | CD3–12 | 250 | 750 | |
| CD4 | Sino Biological | 50134-R001 | Clone ID: 1 | 1000 | 3000 | |
| CD4 | Abcam | ab183685 | EPR19514 | 2000 | 8000 | |
| CD8a | Lyt2 | Cell Signaling | 98941 | D4W2Z | 4000 | 8000 |
| Eomes | Tbr2 | Abcam | ab183991 | EPR19012 | 500 | Not Determined |
| F4/80 | Emr1, Adgre1 | Cell Signaling | 70076 | D2S9R | 500 | 1000 |
| F4/80 | Emr1, Adgre1 | Thermo Fisher | MA5–16363 | SP115 | 500 | 800 |
| Fcgr1 | CD64 | Sino Biological | 50086-R008–50 | Clone ID: 8 | 5000 | 10000 |
| Fcgr4 | CD16–2 | Sino Biological | 50036-R011–50 | Clone ID: 11 | 2000 | 2500 |
| Foxp3 | R&D | MAB8214 | 1054C | 500 | 2000 | |
| Granzyme B | Gzmb, Ctla1 | Cell Signaling | 46890 | D6E9W | 500 | Not Determined |
| MHCII (IA/IE) | H2-Ab1 | BioLegend | 107601 | M5/114.15.2 | 200 | 500 |
| Ox40 | Tnfrsf4 | Cell Signaling | 61637 | E9U70 | 500 | Not Determined |
| Pax5 | BSAP | Abcam | ab109443 | EPR3730 | 1000 | 2000 |
| PD1 | Pdcd1 | Cell Signaling | 846S1T | D7DSW | 200 | Not Determined |
| S100a9 | MRP14, CAGB | Cell Signaling | 73425S | D3U8M | 1000 | 4000 |
| Zap70 | Srk | Cell Signaling | 2705 | 99F2 | 500 | 500 |
This antibody does not work well for combined tyramide signal amplification staining with CD4 and CD8; staining with CD3 first prevents subsequent staining for CD4 or CD8 on CD3+ cells, and vice versa.
Flow Cytometry
Single cell suspensions were prepared from C57BL/6 mouse spleens. 1×106 cells were stained with CD11b PE, Ly6C PerCP-Cy5.5, Ly6G PE-Cy7 (eBioscience), F4/80 APC (AbD Serotec) and FcγR antibodies as previously described (16, 17). Data were acquired on the FACS Canto II (BD Biosciences) and analyzed with FCS express (DeNovo Software). FcγR expression by macrophages (F4/80+), monocytes (CD11b+Ly6C+) and neutrophils (CD11b+Ly6G+) is demonstrated by mean fluorescence intensity.
Image analysis
Image analysis was conducted using Fiji software (18). During the analysis 8-bit images were thresholded at the same intensity value for a specific staining, e.g. CD3, CD11c, etc., and converted to binary format. To calculate tissue areas that are double-stained or have specific staining patterns (e.g. CD8+ and CD4-), an ‘Image Calculator Function’ was used. Then the area fraction of the signal in resulting binary images was calculated. All calculated area fractions of different staining were normalized using DAPI.
Results
We sought to develop an improved protocol for IHC on mouse FFPE sections with the following requirements: 1) reproducibility of staining across different tissues from different experiments; 2) consistent results with most antibodies used without laborious antibody-specific antigen retrieval optimization; 3) ability to perform multiplex IHC.
In accord with previous reports, we found that Tris-EDTA (pH 9.0) was optimal for most primary antibodies (19). Further, we found that the addition of 10% glycerol significantly enhanced antigen retrieval in multiple cases (Supplemental Figure 1A-L). Additionally, we found that adding a short 10% neutral buffered fixation step after deparaffinization prevented tissue folding, which can be an issue with higher pH buffers during heat-induced epitope retrieval (HIER). This did not impact epitope detection (data not shown).
For antibody stripping, some groups utilize a short HIER-like step in citrate buffer (pH 6.2), while others have reported success with incubating the slides in a 100 mM glycine-tween buffer (pH 10.0) at room temperature (14). We found that the glycine buffer-based stripping method failed to remove the primary antibody (Supplemental Figure 1-N, Q). In contrast, the short HIER-like step in citrate buffer worked effectively (Supplemental Figure 1-O, R). We suspect heat-denaturing of high affinity antibodies is particularly important in this process.
To demonstrate consistency across detection methods, we first performed single-staining on spleens using three different antibodies, starting with fluorescence (TSA) detection. Following a stripping step, we reprobed the same tissue with the same antibodies and utilized chromagen detection (Supplemental Figure 2). Cells were labeled identically with each method.
To evaluate the effect of the stripping step on antigen retrieval, we stained spleens after the primary antigen retrieval step (“Round 1,” Supplemental Figure 3). Second, we stained different spleen sections for the same markers after performing antigen retrieval and a single stripping step (“Round 2,” Supplemental Figure 3). Third, we repeated this on spleens that were subjected to antigen retrieval and two subsequent stripping steps (“Round 3,” Supplemental Figure 3). For the three antibodies tested on spleens (CD3, CD19, and Pax5), we did not observe significant differences in detection before or after the tissue was exposed to multiple stripping steps. However, this is not the case for every antibody/epitope (e.g. F4/80 [D2S9R]; Supplemental Figure 3K-L). Thus, labs should perform such testing to determine the order of staining for multiplex protocols involving antibody stripping steps.
After the primary antigen retrieval step and multiple iterations of stripping in multiplex staining, we found that the nuclear signal, via DAPI or hematoxylin, is lost if the staining is done over a longer timeframe (hours vs days; data not shown). We suspect that nuclear DNA is vulnerable to environmental DNase contamination after extensive destruction of formalin crosslinks. This issue was resolved by adding EDTA to wash buffers and antibody diluents.
Applying our revised protocol to test over 35 primary antibodies specific for mouse immune subsets, we developed a panel of lymphoid and myeloid cell markers (Table 1). These markers were chosen based on ImmGEN expression data (Figure 1, 20), previous reports (16, 17, 21–24), and antibody availability. Using chromagen and fluorescence (via tyramide signal amplification) detection methods, we observed that each marker produced expected staining patterns corresponding with known localization and expression by the respective cell types of interest (Supplemental Figures 4, 25).
Figure 1. Heatmap depicting immune cell expression of markers utilized in our immunohistochemistry panel.
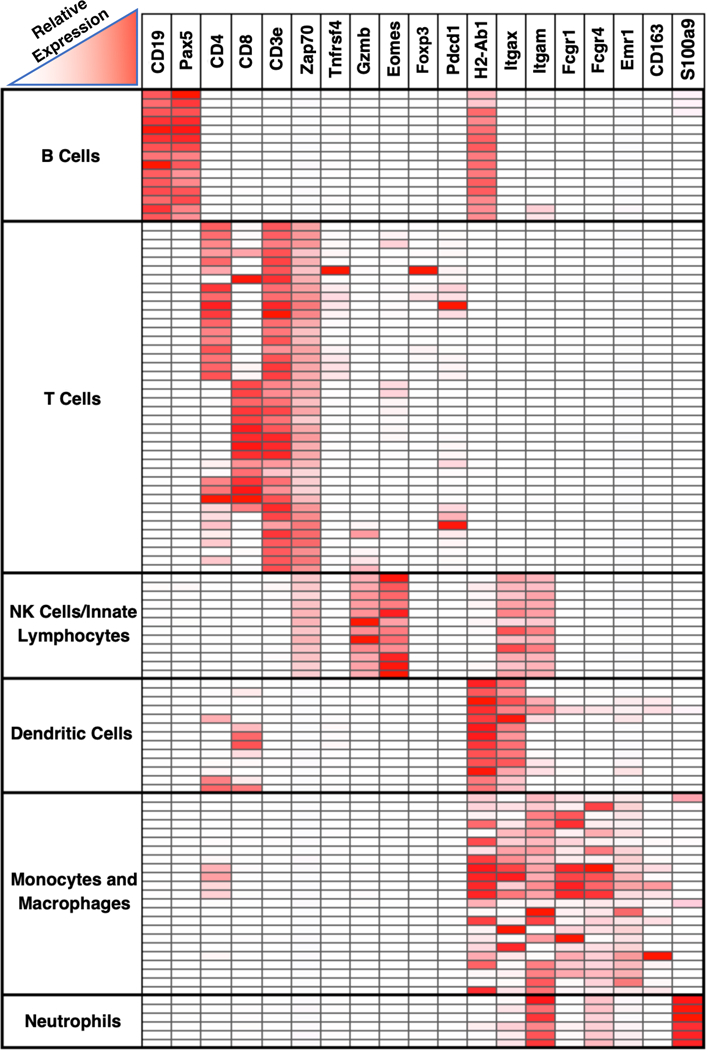
Micro-array data from ImmGen showing relative expression levels of different markers by different cell types (http://www.immgen.org ) (20). Rows represent different immune cell populations, grouped by cell type. Columns represent genes.
To validate antibody specificity, we used two strategies. First, we stained tissue in which cell types/markers of interest are expected to be decreased compared to control. For the lymphoid panel, we stained spleens from wild-type, SCID, and NSG mice (Figure 2A-G1). Compared to wild-type, we observed a decrease in lymphoid cell markers in SCID and NSG spleens. For example, CD3 and CD19 (T cell and B cell markers, respectively) are completely absent in SCID and NSG tissue. As a positive control, CD11b, mostly a myeloid-lineage marker, was consistent across all groups (Figure 2B1-D1).
Figure 2. Validation of antibody specificity.
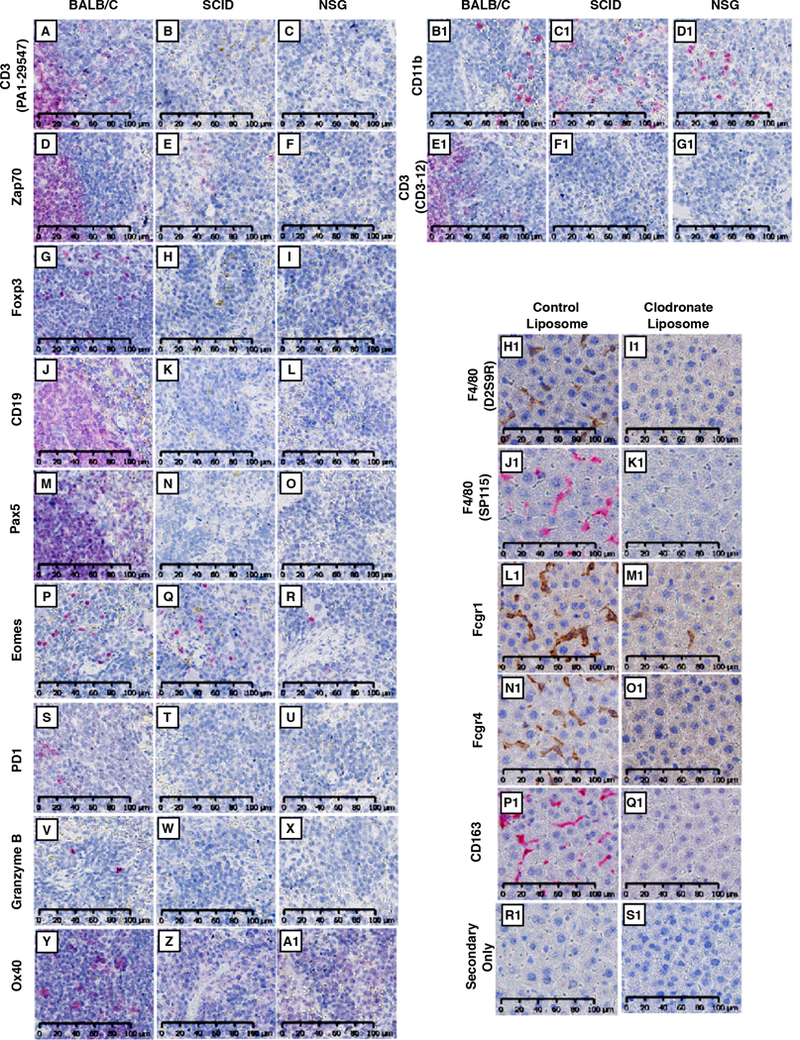
(A-G1) Wild type BALB/c, SCID, or NSG spleens were probed with antibodies specific for CD3 (PA1–29547; A, B, C), Zap70 (D, E, F), Foxp3 (G, H, I), CD19 (J, K, L), Pax5 (M, N, O), Eomes (P, Q, R), PD1(S, T, U), Granzyme B (V, W, X), Ox40 (Y, Z, A1), CD11b (B1, C1, D1), and CD3 (CD3–12; E1, F1, G1). For detection, Warp Red chromogenic substrate was used. Slides were counterstained with hematoxylin and scanned at 20X using Hamamatsu NanoZoomer 2.0-HT (H1-S1) Livers from control- or clodronate-liposome treated BALB/c mice were stained for F4/80 (D2S9R and SP115; H1, I1 and J1, K1, respectively), Fcgr1 (L1, M1), Fcgr4 (N1, O1), CD163 (P1, Q1), or secondary-only (R1, S1). Slides were scanned at 20X using the Hamamatsu NanoZoomer 2.0-HT. All images represent a 40X field-of-view. Scale bar = 100 μm for all images.
For macrophage markers, we stained livers from control- or clodronate-liposome-treated mice, as clodronate liposomes deplete professional phagocytic cells, primarily macrophages (26, 27). Macrophage staining was markedly reduced in clodronate liposome-treated mice (Figure 2H1-Q1).
For our second strategy to confirm antibody specificity, we utilized multiple antibodies for the same marker or cell type when possible. For example, we utilized two different antibodies for F4/80 (Figure 2H1-K1). For B cells, we utilized two different B cell-specific markers, CD19 and Pax5 (Figure 2J-O).
When developing the antibody panels, we found that for some cell types, such as DCs, neutrophils, and NK cells, antibodies for cell-specific markers were not available. To address this, we utilized multi-marker staining strategies.
For example, we developed a strategy to stain NK cells using CD3 and Zap70. CD3 is expressed by T cells, but not NK cells (Figure 1, 20). Zap70 is expressed by T cells and NK cells (Figure 1,(20). Because SCID and NSG mice lack T cells, CD3 staining is absent in SCID and NSG spleens compared to wild-type (Figure 2A-C). In contrast, some Zap70+ cells remain; presumably NK cells (Figure 2D-F). Combining these markers, we stained spleens for CD3 and Zap70 using both chromagen and fluorescence detection methods, demonstrating the presence of CD3+; Zap70+ and CD3-;Zap70+ cells, corresponding to T cells and NK cells, respectively (Figure 3A, B). In the case of chromagen staining, we first stained for CD3 using a dark brown chromagen (DAB), intentionally obscuring further staining wherever the chromagen is deposited. We subsequently performed Zap70 staining using a red chromagen (‘Warp Red’). Through this strategy, it is easy to identify Zap70 single positive cells utilizing chromagen detection (Figure 2A, inset).
Figure 3. Multiplex IHC to stain Natural Killer Cells, Dendritic cells and Neutrophils.
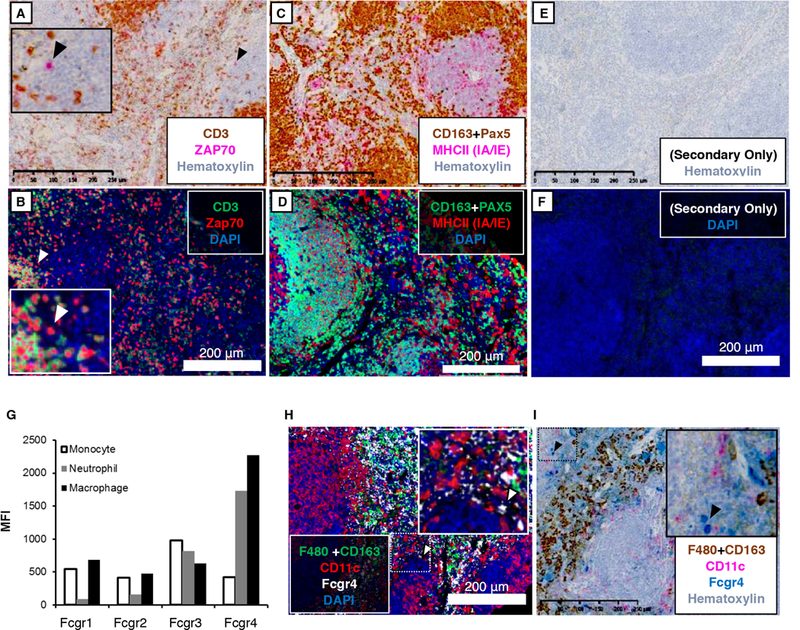
BALB/c spleens were stained for (A, B) CD3 (PA1–29547, T cells) and Zap70 (T cells, NK cells); (C, D) Pax5 (B cells), CD163 (majorly macrophages), and MHCII (IA/IE); (H, I) F4/80 (SP115), CD163, CD11c, and Fcgr4. (E, F) Secondary-antibody-only (without addition of primary antibody) was used for negative controls. Chromogenic substrates used were Betazoid DAB (brown) and Warp Red (pink). Opal 520 (green), Opal 570 (red) and Opal 690 (white) were used for fluorescence detection. Chromogen-stained sections were counterstained with hematoxylin. For fluorescence detection, sections were counterstained with DAPI (blue). Slides were scanned at 20X using the Hamamatsu NanoZoomer 2.0-HT (chromagen detection) or the Zeiss Axioscan.Z1 (fluorescence detection). Images represent a 10x field-of-view (G) Total splenocytes were isolated from C57BL/6 mice. Fcgr expression was evaluated on neutrophils (CD11b+; Ly6G+; Ly6C-), macrophages (F4/80+), and monocytes (CD11b+; Ly6c+Ly6G-). Fcgr signal is represented by mean fluorescence intensity. Scale bar = 250 μm for chromagen-stained images. Scale bar = 200 μm for fluorescent images.
We also developed a strategy to detect DCs. While CD11c has long been used as a DC marker, it is not cell-type exclusive as it is may be expressed by macrophage subsets and other immune cells (Figure 1, 20). MHCII (H2-Ab1) is also not exclusive, but it is expressed more consistently across DC subsets and is found on fewer cell types (Figure 1, 20). Because MHCII is expressed by splenic B cells and macrophages, we first stained for B cell and macrophage markers, Pax5 and CD163, respectively. By combining CD163 and Pax5 signal with the same chromogen or fluorophore, we demonstrate the effective use of a “dump channel” applied in an IHC setting (Figure 3C, D). Staining for MHCII second with a different chromagen or fluorophore revealed a distinct population of MHCII+; Pax5-; CD163- cells, which we believe to be DCs (Figure 3C, D).
Next, we developed a strategy to distinguish neutrophils from macrophages. Using knowledge from the literature and considering the available antibodies for mouse targets, we stained for CD163 and Fcgr4. According to ImmGen (Figure 1, 20) and previous reports (16, 17), Fcgr4 is expressed by macrophages and neutrophils. This is corroborated by flow cytometric analysis (Figure 3G). Based on this information, we expect that splenic macrophages are CD163+; Fcgr4+ and neutrophils are CD163-; Fcgr4+. Performing this multiplex staining reveals cells that follow these staining patterns (Figure 3H, I).
Finally, we tested our immune panels and multiplex staining on 4T1 tumors, which is an immune cell-heavy model of breast cancer. We did multiplex IHC staining using chromogen and fluorescent detection methods (Figure 4). Figure 4AB shows CD3+; CD4+ and CD3+; CD8+ T cells. We also detected CD3-; CD4+ and CD3-; CD8+ cells which may be NK or DCs. Notably, all CD3+ T cells were labeled with either CD4 or CD8, demonstrating the specificity and sensitivity of the staining, as the presence of double-negative or double-positive T-lineage cells are unreported in this model.
Figure 4. Multiplex IHC staining of 4T1 tumor tissues.
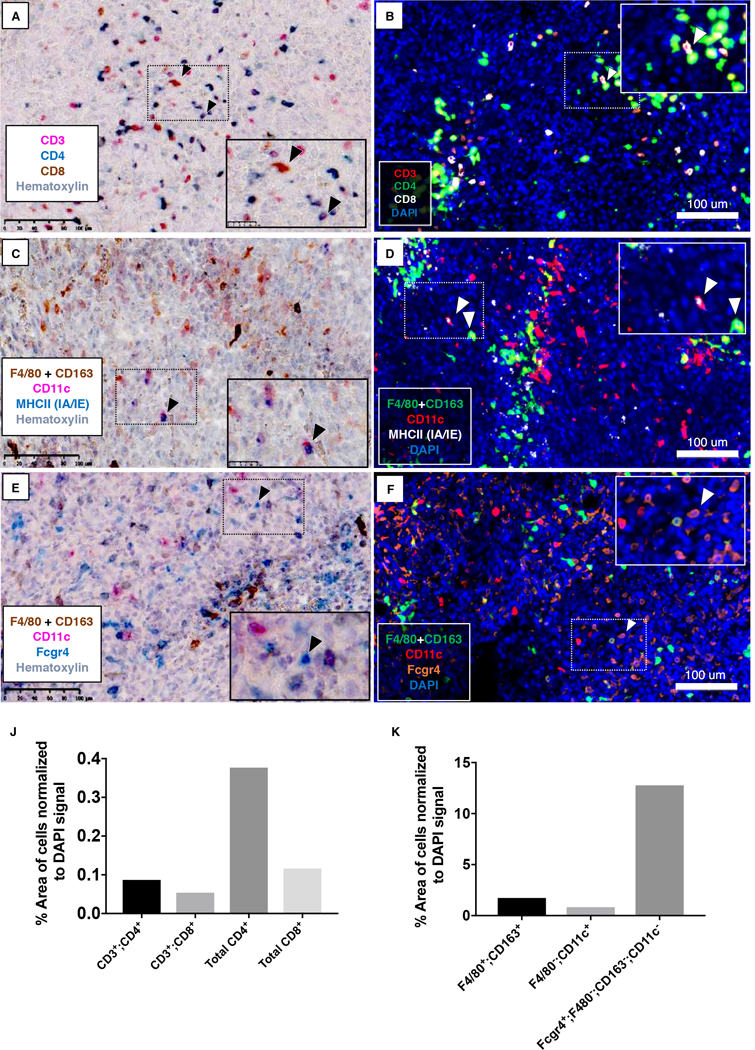
4T1 primary tumors were stained for (A, B) CD3 (PA1–29547; T cells), CD4 (EPR19514; T cells, NK cells, DCs), and CD8 (T cells, NK cells, DCs); (C, D), CD11c, Fcgr4, and CD163+F4/80(SP115); (E, F) CD11c, MHCII (IA/IE), and CD163+F4/80(SP115). Chromogenic substrates used were Betazoid DAB (brown), Warp Red (pink), and Feranji Blue (blue) (A, C, E). Opal 520 (green), Opal 570 (red), and Opal 690 (white or orange) were used for fluorescent staining (B, D, F). Chromogen sections were counterstained with hematoxylin, whereas DAPI staining was used for fluorescence detetction. Slides were scanned at 20X using the Hamamatsu NanoZoomer 2.0-HT (chromagen detection) or the Zeiss Axioscan.Z1 (fluorescence detection). Images represent a 20x field-of-view. Inset: Higher magnification. (B, C, D) Arrows point to CD3+;CD8+ (B), CD11c+;MHCII+ (C; D, left arrow), or CD163&F4/80+;CD11c+ cells (D, right arrow). (E, F) Arrows point to Fcgr4+; CD11c- CD163&F4/80- cells. (J, K) Cell types in 4T1 tumor sections were quantitated as % area of positive cells normalized to DAPI signal. Scale bar = 100 μm for all images.
Figure 4D (inset, right arrow) shows the presence of CD11c+ macrophages. This corresponds to tumor-associated macrophages, as resident tissue macrophages in the mammary fat pad are CD11c- (28). We also can detect F4/80-; CD163-; CD11c+; MHCII+ DCs (Figure 4D, inset, left arrow).
Figure 4JK shows quantitation of different immune cell types in 4T1 tumors normalized to the DAPI signal. 4T1 tumors have been reported to have high neutrophil infiltration (29). Consistent with this, our analysis shows a high frequency of F4/80-; CD163-; Fcgr4+ cells which are presumably neutrophils (Figure 4E-F).
Comparatively, we also found modest numbers of tissue macrophages (F4/80+; CD163+; CD11c-), DCs (F4/80-; CD163-; CD11c+; MHCII+) and T cells (CD3+; CD4+ and CD3+; CD8+), data which is consistent the findings of other groups (30–32). In sum, this data illustrates how an effective multiplex IHC strategy can be exploited to investigate the immune dynamics of the tumor microenvironment.
Discussion
In this study, we produced a modified FFPE IHC protocol for mouse tissue that provides a more effective antigen retrieval method. Antigen retrieval has been a longstanding challenge in the field, often requiring different retrieval protocols for different epitopes (14, 33, 34). Our protocol works consistently well for all of the antibodies tested. The use of glycerol in the Tris-EDTA antigen retrieval buffer greatly increases the detection of multiple antigens and may be a better standard for mouse FFPE tissues. Also, the use of EDTA in the wash buffers prevents degradation of DNA by nucleases preserving nuclear morphology; a consideration that is absent in other multiplex protocols. Lastly, we found that treatment with 10% formalin immediately prior to antigen retrieval helps to maintain tissue integrity and reduces loss of tissue during multiple rounds of stripping and re-probing.
We also propose a novel strategy of using multiple markers to stain for cell types for which unique markers/antibodies have not been identified or are not available commercially. This strategy can be particularly helpful to identify DCs, neutrophils, and NK cells in the tumor microenvironment. With new emerging immune regulators (35) and therapies, this strategy provides an excellent tool to investigate the effect of drug treatment and prognosis of disease.
Overall, our protocol of multiplex IHC staining on mouse tissue provides an improved strategy for reproducible staining. Using our strategy and by properly planning the sequence of target antibodies, visualization of multiple epitopes is only limited by the number of filter channels in the microscope. This protocol has the potential to improve the utility of multiplex IHC to investigate the immune system in vivo, including the analysis of the tumor immune microenvironment and mechanisms of action of immune-related drugs in preclinical models.
Supplementary Material
Acknowledgments:
The authors thank D. Primm for editorial assistance, Drs. E. Koay and A. Zijlstra, and members of the Brekken lab for advice and helpful discussion.
Financial support: The work was supported by NIH grants R01 CA192381 to R.A. Brekken, and U54 CA210181 (PI: M. Ferrari) to R.A. Brekken and A. Zeimys; and F31 CA19603301 to N. Sorrelle; the Effie Marie Cain Scholarship in Angiogenesis Research to R.A. Brekken; and L.N. Dahal was funded by the Bloodwise Programme Grant (#12050).
Abbreviations
- DC
dendritic cell
- NK
natural killer cell
- FFPE
formalin-fixed paraffin-embedded
- HIER
heat-induced epitope retrieval
- IHC
immunohistochemistry
- MHC
major histocompatibility complex
- NSG
NOD SCID gamma
- SCID
severe combined immunodeficiency
- TSA
tyramide signal amplification
Footnotes
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data and materials availability: All materials are available from commercial sources or can be derived using methods described in this study. Allrelevant data are reported in the article
Conflict of Interests: none
References
- 1.Szantho E, Karai B, Ivady G, Bedekovics J, Szegedi I, Petras M, Ujj G, Ujfalusi A, Kiss C, Kappelmayer J, and Hevessy Z 2018. Comparative Analysis of Multicolor Flow Cytometry and Immunohistochemistry for the Detection of Disseminated Tumor Cells. Appl Immunohistochem Mol Morphol 26: 305–315. [DOI] [PubMed] [Google Scholar]
- 2.Saravanan L, and Juneja S 2010. Immunohistochemistry is a more sensitive marker for the detection of myeloperoxidase in acute myeloid leukemia compared with flow cytometry and cytochemistry. Int J Lab Hematol 32: e132–136. [DOI] [PubMed] [Google Scholar]
- 3.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, Diao L, Wang J, Parra-Cuentas ER, Wistuba II, Soucheray M, Thai T, Asahina H, Kitajima S, Altabef A, Cavanaugh JD, Rhee K, Gao P, Zhang H, Fecci PE, Shimamura T, Hellmann MD, Heymach JV, Hodi FS, Freeman GJ, Barbie DA, Dranoff G, Hammerman PS, and Wong KK 2016. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res 76: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sridharan V, Gjini E, Liao X, Chau NG, Haddad RI, Severgnini M, Hammerman P, El-Naggar A, Freeman GJ, Hodi FS, Rodig SJ, Dranoff G, and Schoenfeld JD 2016. Immune Profiling of Adenoid Cystic Carcinoma: PD-L2 Expression and Associations with Tumor-Infiltrating Lymphocytes. Cancer Immunol Res 4: 679–687. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig KF, Du W, Sorrelle NB, Wnuk-Lipinska K, Topalovski M, Toombs JE, Cruz VH, Yabuuchi S, Rajeshkumar NV, Maitra A, Lorens JB, and Brekken RA 2018. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res 78: 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, and Slingluff CL Jr. 2012. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 72: 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, and Krummel MF 2016. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parra ER, Uraoka N, Jiang M, Cook P, Gibbons D, Forget MA, Bernatchez C, Haymaker C, Wistuba II, and Rodriguez-Canales J 2017. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci Rep 7: 13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stack EC, Wang C, Roman KA, and Hoyt CC 2014. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 70: 46–58. [DOI] [PubMed] [Google Scholar]
- 10.Dixon AR, Bathany C, Tsuei M, White J, Barald KF, and Takayama S 2015. Recent developments in multiplexing techniques for immunohistochemistry. Expert Rev Mol Diagn 15: 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofman FM, and Taylor CR 2013. Immunohistochemistry. Curr Protoc Immunol 103: Unit 21 24. [DOI] [PubMed] [Google Scholar]
- 12.Wagrowska-Danilewicz M, and Danilewicz M 2009. Immunofluorescence on paraffin-embedded sections in evaluation of immune complex deposits in renal biopsy specimens. Pol J Pathol 60: 3–9. [PubMed] [Google Scholar]
- 13.Beckstead JH 1994. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. J Histochem Cytochem 42: 1127–1134. [DOI] [PubMed] [Google Scholar]
- 14.Feng Z, Jensen SM, Messenheimer DJ, Farhad M, Neuberger M, Bifulco CB, and Fox BA 2016. Multispectral Imaging of T and B Cells in Murine Spleen and Tumor. J Immunol 196: 3943–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lykidis D, Van Noorden S, Armstrong A, Spencer-Dene B, Li J, Zhuang Z, and Stamp GW 2007. Novel zinc-based fixative for high quality DNA, RNA and protein analysis. Nucleic Acids Res 35: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tutt AL, James S, Laversin SA, Tipton TR, Ashton-Key M, French RR, Hussain K, Vaughan AT, Dou L, Earley A, Dahal LN, Lu C, Dunscombe M, Chan HT, Penfold CA, Kim JH, Potter EA, Mockridge CI, Roghanian A, Oldham RJ, Cox KL, Lim SH, Teige I, Frendeus B, Glennie MJ, Beers SA, and Cragg MS 2015. Development and Characterization of Monoclonal Antibodies Specific for Mouse and Human Fcgamma Receptors. J Immunol 195: 5503–5516. [DOI] [PubMed] [Google Scholar]
- 17.Dahal LN, Gadd A, Edwards AD, Cragg MS, and Beers SA 2018. UC-1V150, a potent TLR7 agonist capable of activating macrophages and potentiating mAb-mediated target cell deletion. Scand J Immunol 87: e12666. [DOI] [PubMed] [Google Scholar]
- 18.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, and Schmid B 2012. Fiji: an open-source platform for biological-image analysis. Nat. Meth. 9: 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pileri SA, Roncador G, Ceccarelli C, Piccioli M, Briskomatis A, Sabattini E, Ascani S, Santini D, Piccaluga PP, Leone O, Damiani S, Ercolessi C, Sandri F, Pieri F, Leoncini L, and Falini B 1997. Antigen retrieval techniques in immunohistochemistry: comparison of different methods. J Pathol 183: 116–123. [DOI] [PubMed] [Google Scholar]
- 20.Heng TS, Painter MW, and Immunological Genome Project C 2008. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9: 1091–1094. [DOI] [PubMed] [Google Scholar]
- 21.Au-Yeung BB, Deindl S, Hsu LY, Palacios EH, Levin SE, Kuriyan J, and Weiss A 2009. The structure, regulation, and function of ZAP-70. Immunol Rev 228: 41–57. [DOI] [PubMed] [Google Scholar]
- 22.Shiraishi D, Fujiwara Y, Horlad H, Saito Y, Iriki T, Tsuboki J, Cheng P, Nakagata N, Mizuta H, Bekki H, Nakashima Y, Oda Y, Takeya M, and Komohara Y 2018. CD163 Is Required for Protumoral Activation of Macrophages in Human and Murine Sarcoma. Cancer Res 78: 3255–3266. [DOI] [PubMed] [Google Scholar]
- 23.Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V 2012. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cobaleda C, Schebesta A, Delogu A, and Busslinger M 2007. Pax5: the guardian of B cell identity and function. Nat Immunol 8: 463–470. [DOI] [PubMed] [Google Scholar]
- 25.Bronte V, and Pittet MJ 2013. The spleen in local and systemic regulation of immunity. Immunity 39: 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claassen E, and Van Rooijen N 1984. The effect of elimination of macrophages on the tissue distribution of liposomes containing [3H]methotrexate. Biochim Biophys Acta 802: 428–434. [DOI] [PubMed] [Google Scholar]
- 27.Seiler P, Aichele P, Odermatt B, Hengartner H, Zinkernagel RM, and Schwendener RA 1997. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol 27: 2626–2633. [DOI] [PubMed] [Google Scholar]
- 28.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, and Li MO 2014. The cellular and molecular origin of tumor-associated macrophages. Science 344: 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, Nakasone ES, Hearn SA, Kuttner V, Qiu J, Almeida AS, Perurena N, Kessenbrock K, Goldberg MS, and Egeblad M 2016. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 8: 361ra–138.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Hunborg P, Varvares MA, Hoft DF, Hsueh EC, and Peng G 2015. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 6: 17462–17478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song H, Jung JI, Cho HJ, Her S, Kwon SH, Yu R, Kang YH, Lee KW, and Park JH 2015. Inhibition of tumor progression by oral piceatannol in mouse 4T1 mammary cancer is associated with decreased angiogenesis and macrophage infiltration. J Nutr Biochem 26: 1368–1378. [DOI] [PubMed] [Google Scholar]
- 32.Roland CL, Lynn KD, Toombs JE, Dineen SP, Udugamasooriya DG, and Brekken RA 2009. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One 4: e7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi SR, Key ME, and Kalra KL 1991. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem 39: 741–748. [DOI] [PubMed] [Google Scholar]
- 34.Shi SR, Liu C, and Taylor CR 2007. Standardization of immunohistochemistry for formalin-fixed, paraffin-embedded tissue sections based on the antigen-retrieval technique: from experiments to hypothesis. J Histochem Cytochem 55: 105–109. [DOI] [PubMed] [Google Scholar]
- 35.Sorrelle N, Dominguez ATA, and Brekken RA 2017. From top to bottom: midkine and pleiotrophin as emerging players in immune regulation. J Leukoc Biol 102: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.