

Abstract
The introduction of solid-phase peptide synthesis in the 1960’s improved the chemical synthesis of both the A- and B-chains of insulin and insulin analogs. However, the subsequent elaboration of the synthetic peptides to generate active hormones continues to be difficult and complex due in part to the hydrophobicity of the A-chain. Over the past decade, several groups have developed different methods to enhance A-chain solubility. Two of the most popular methods are use of isoacyl dipeptides, and the attachment of an A-chain C-terminal pentalysine tag with a base labile 4-hydroxymethylbenzoic acid linker. These methods have proven effective but can be limited in scope depending on the peptide sequence of a specific insulin. Herein we describe an auxiliary approach to enhance the solubility of insulin-based peptides by incorporating a tri-lysine tag attached to a cleavable Fmoc-Ddae-OH linker. Incorporation of this linker, or “helping hand”, on the N-terminus greatly improved the solubility of chicken insulin A-chain, which is analogous to human insulin, and allowed for coupling of the insulin A- and B-chain via directed disulfide bond formation. After formation of the insulin heterodimer, the linker and tag could be easily removed using a hydrazine buffer (pH 7.5) to obtain an overall 12.6% yield based on A-chain. This strategy offers an efficient method to enhance the solubility of hydrophobic insulin-based peptides as well as other traditionally difficult peptides.
Introduction
The synthesis of insulin has had significant improvements since its discovery in 1921.1 This is particularly true since the 1960’s establishment of solid-phase peptide synthesis (SPPS), which revolutionized the capacity to synthesize the A- and B-chains, each consisting of 21 and 30 amino acids respectively.2–5 Despite this major advance, the unique properties of this endocrine hormone make its chemical synthesis complex, and over the past 50 years three major challenges have hindered the general synthesis efficiency of this molecule.6 The first challenge is formation of the correct disulfide bridges, which has been addressed and mitigated by several groups via the use of directed-disulfide bond formation.7–12 The second challenge is the low yield during the chain combination steps, which has been partially resolved by several groups via permutations of various methods and combination steps such as using silyl chloride-sulfoxide, S-sulfonation, semi-selective formation of disulfide bonds, and more recently using proinsulin as inspiration to convert a one-chain precursor into human insulin.8, 10, 13–18 The final challenge, and the focus of this work, is the hydrophobic nature of the insulin A-chain. Of the 21 amino acids that make up this chain, more than half are hydrophobic residues, and none are charged. These properties make the chain difficult to handle and purify, which further complicates forming and purifying the 51-amino acid peptide hormone consisting of an intramolecular disulfide bridge on the A-chain (CysA6-CysA11) and two intermolecular disulfide bridges (CysA7-CysB7, CysA20-CysB19).6
Due to this challenge, several groups have developed effective methods to enhance the A-chain solubility. The two most prominent methods were developed over the last decade, and they consist of using isoacyl dipeptides or a pentalysine tag on the C-terminus linked by a 4-hydroxymethylbenzoic acid linker (HMBA) (Fig. 1).19–23 The latter approach, developed by Wade et al., consists of attaching a C-terminal polycationic pentalysine tail to insulin glargine via a HMBA linker. The linker and tag are removed by treating the peptide with 0.1 M NaOH.22 Although this method effectively enhances the solubility of the A-chain, the placement of the base-labile linker is limited to the C-terminus, and the basic removal conditions can be damaging for sensitive peptides.24–26 These two items limit the scope of peptides or proteins that could benefit from this method.22, 27 An alternative approach using isoacyl dipeptides substitutes the α-nitrogen on the backbone of the peptide with the oxygen from the side chain of Thr or Ser. The native conformation of the peptide is restored via an O-to-N acyl shift after exposure to neutral or slightly basic conditions. Liu et al. demonstrated that incorporation of an isoacyl dipeptide improved solubility and facilitated purification of the A-chain. In conjunction to the isoacyl dipeptide on the A-chain, the incorporation of an isoacyl dipeptide on the B-chain led to a final synthesis yield of 24% - the highest so far reported for the total synthesis of human insulin. Although a significant advance, this method has some drawbacks such as the need to avoid neutral or basic buffers during the synthesis, deprotection, and purification steps to prevent the O-to-N acyl shift, and the potential of not having the required Ser or Thr within the peptide scheme. Additionally, for some insulin-based peptides these amino acids are inconveniently placed around disulfide bridges which play a critical role in maintaining the structural integrity of insulin.28
Fig. 1.
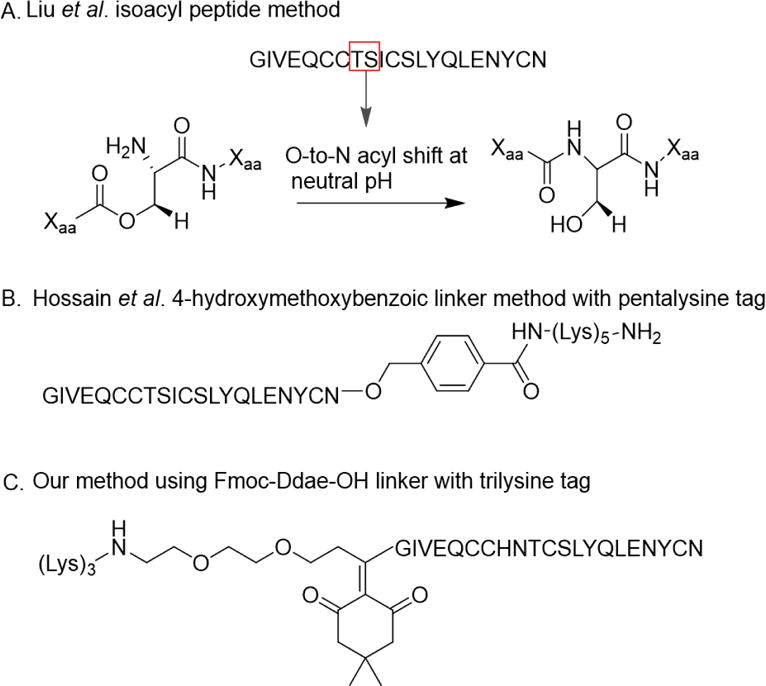
Current methods to increase solubility of insulin-based peptides. (A) Liu et al. Isoacyl peptide method using a threonine and serine to perform O-to-N acyl shift. (B) Hossain et al. a C-terminus 4-hydroxymethoxybenzoic linker with pentalysine tag. (C) Our method using a N-terminus Fmoc-Ddae-OH linker with trilysine tag.
Herein we will focus on enhancing the solubility of an insulin-based A-chain utilizing a solubility linker that can withstand the strict conditions necessary for the synthesis of insulin and can be applied to other insulin-based peptides that do not contain strategically placed Thr or Ser residues. The approach involves incorporating a tri-lysine tag linked to an N-Fmoc-1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-[2-(2-aminoethoxy) ethoxyl]-propan-1-ol (Fmoc-Ddae-OH) linker on the N-terminus of the peptide sequence (Fig. 1). The linker, better known as a “helping hand”, is easily synthesized by coupling dimedone to a Fmoc-OEG2 stretcher via carbodiimide chemistry.29 This linker is highly compatible with SPPS and has been previously introduced to the the ε-amine of lysine of challenging peptides and proteins such as the C20 peptide derived from Ebola virus and the GroES protein from E.coli, respectively.29 Furthermore, cleavage is achieved using a mild hydrazine-based phosphate buffer; limiting the potential risk for side reactions or degradation during removal.24, 25, 29, 30 In this report we introduce the helping hand to the N-terminus of the A-chain of chicken insulin, which has an 85% amino acid sequence similarity to human insulin (Fig. 2).31 Chicken insulin has a higher binding affinity to the human insulin receptor than human insulin.32 This increase in activity has been attributed to HisA8, which enhances the stability of insulin and has been used to study the activity of other insulin analogues with similar sequences.33 Furthermore, use of the helping hand to improve the solubility of the A-chain will enable future structure-activity relationship studies with chicken insulin and the synthesis of other hydrophobic insulin-based analogues.
Fig. 2.
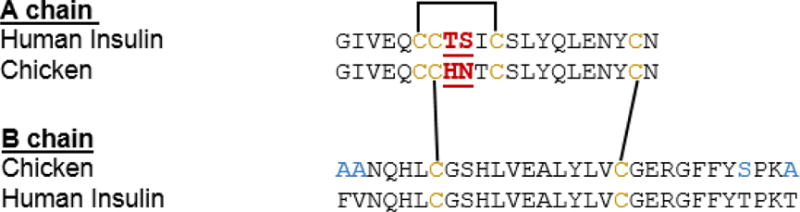
Human insulin sequence versus chicken insulin sequence. A-chain has an amino acid sequence homology of 85% and B-chain has an 87% sequence identity.
Results and Discussion
Synthesis of Fmoc-Ddae-OH
An initial attempt to synthesize Fmoc-Ddae-OH following the established procedure resulted in significant formation of the O-linked dimedone ester that was slow to equilibrate to the correct C-linked product (Scheme 1). To achieve better conversion we evaluated using DCC as the activating agent however the dicyclohexylurea byproduct greatly complicated the purification. Our best results were obtained by extending the reaction time to 23h and using N, N-diisopropylethylamine (DIEA) as the base along with a catalytic amount of DMAP. This resulted in good conversion the correct C-linked product that could be isolated as described previously.29 (See ESI for details).
Scheme 1.
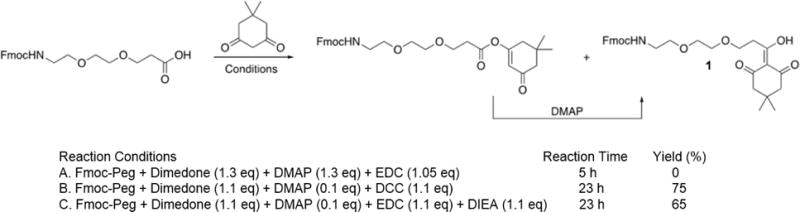
Synthesis of Fmoc-Ddae-OH linker. (A) Reaction conditions previously reported in the literature. (b) Reaction conditions using DCC produced the highest yield, however N, N’ dicyclohexylurea byproduct formation made purification difficult. (c) Reaction conditions with little byproduct formation and high yield consisted of N-Fmoc-amido dPEG-acid, 1.1 eq. dimedone, 1.1 eq. EDC-HCl, 1.1 eq. DIEA and 0.1 eq. DMAP in DCM.
Coupling of Fmoc-Ddae-OH to Chicken Insulin A-chain
The ligation of the Fmoc-Ddae-OH linker to the N-terminus of the A-chain (Scheme 2) was accomplished by first generating the A-chain using a procedure similar to that previously described.20 Briefly, we used a standard tBu/Fmoc SPPS protocol with CysA6-tert-butylthiol (StBu), CysA7-acetamidomethyl (Acm), Cysa11-p-methoxytrityl (Mmt), and CysA20-Trityl (Trt) as orthogonal protecting groups. The resulting resin-bound A-chain (2) was reacted with the Fmoc-Ddae-OH linker (1) in N-methylpyrrolidine (NMP) for 4 hours. The linker and A-chain complex (3) was subsequently washed with dimethylformamide (DMF) followed by DCM and finally treated with 20% piperidine in DMF following and standard tBu/Fmoc SPPS to add the 3 Lys residues (5).
Scheme 2.
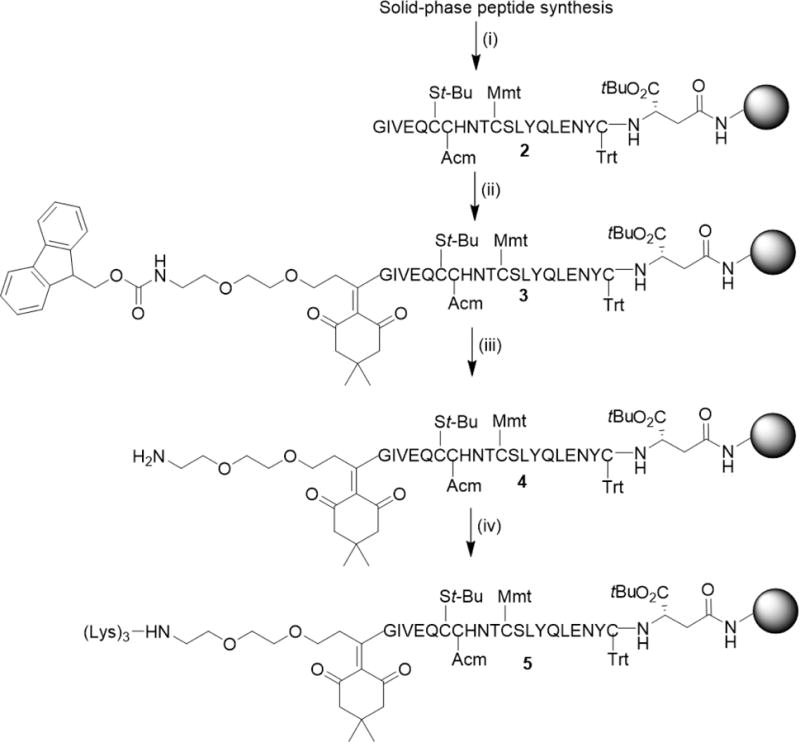
Synthesis of chicken insulin and coupling of Fmoc-Ddae-OH to the N-terminus of the A-chain. (i) Standard tBu/Fmoc SPPS protocol. (ii) 0.2 mmol Fmoc-Ddae-OH (1), 25uM resin scale in NMP, 4h RT. (iii) 20% piperidine in DMF. (iv) Standard tBu/Fmoc SPPS protocol.
Deprotection, disulfide formation, and Cleavage of A-chain with Fmoc-Ddae-OH linker and tri-lysine tag
The deprotection, disulfide formation, and cleavage of the chicken insulin A-chain containing the helping hand linker and tri-lysine tag followed a previous protocol described by Liu et al. used for the deprotection and cleavage of native insulin A-chain containing isoacyl dipeptides (Scheme 3).20 The peptide (5) was treated with 50% β-mercaptoethanol (BME) in DMF twice for 2 hours to remove the StBu protecting group on CysA6. Next the peptide was exposed to 2, 2′-dithiobis (5-nitropyridine) (DTNP) in DCM for 1 hour to activate this cysteine to SNPy [(4-nitro-pyridinyl) thiol]. Following this step, we washed the SNPy peptide (6) in a DCM cocktail consisting of 1% TFA and 5% triisopropylsilane (TIS) for 2 minutes per wash for 6 washes to remove the Mmt protecting group on CysA11. The peptide was then incubated for 5 hours in DCM to form the intramolecular disulfide bridge. Finally, peptide 7 was cleaved off resin using a TFA, TIS and H2O cocktail for a 2 hour time period and used as is. Although previous studies have detected hydrolysis Dde-derived linkers under acidic conditions over a period of 24 hours, we did not detect cleavage during our 2 hour period.30
Scheme 3.
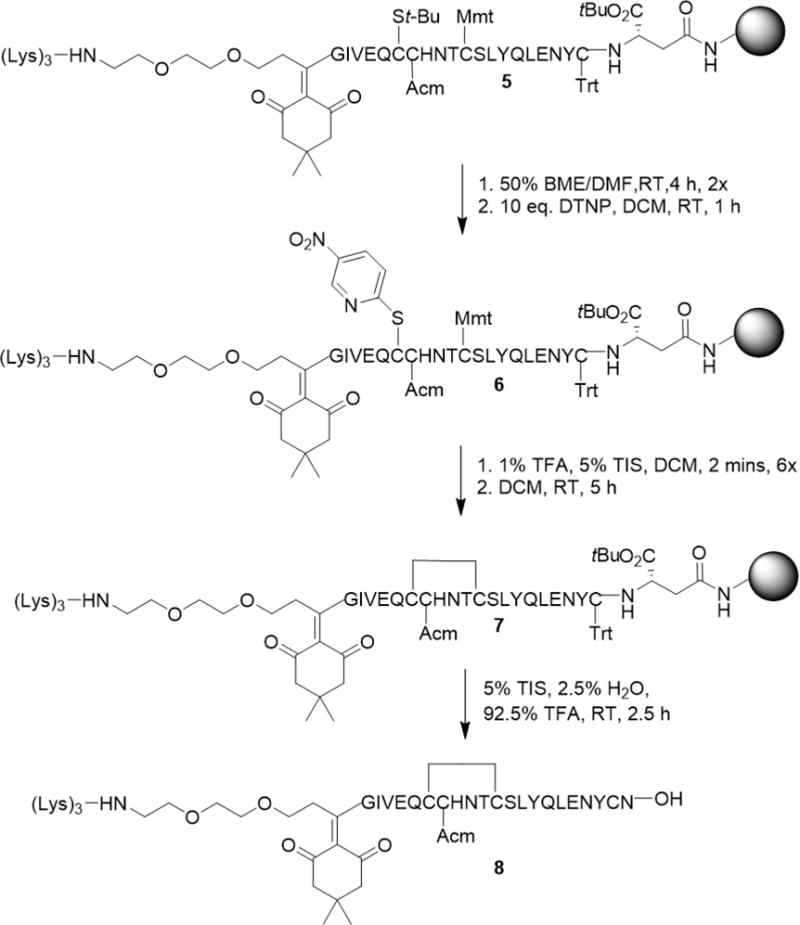
Orthogonal deprotection and cleavage of chicken insulin A-chain with Fmoc-Ddae-OH linker and trilysine tag.
Synthesis, Deprotection, and Cleavage of Chicken Insulin B-chain
Due to the sequence homology of chicken insulin to human insulin the synthesis, deprotection, and cleavage of the B-chain followed a similar procedure as previously described (Scheme 4).20 The B-chain was synthesized using 2-chlorotrityl chloride resin (See ESI for attachment of first amino acid to resin), and CysB7 and CysB19 were protected using Acm and Trt, respectively. Cleavage was completed using a TFA, TIS, H2O cocktail with 2,2′-dithiodipyridine (DTDP) to activate CysB19 to 2-pyridinethiol, and the B-chain was used without further purification.
Scheme 4.
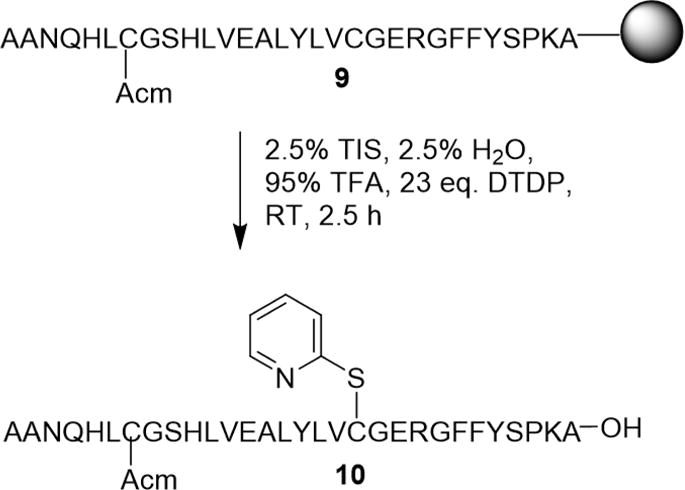
Orthogonal deprotection and cleavage of chicken insulin B-chain
Ligation of Chicken A and B-chain and Removal of Helping Hand
To form the first intermolecular disulfide bridge (CysA20 and CysB19), the A- and B-chains were dissolved and mixed in 6 M urea containing 0.2 M NH4HCO3 (pH 8) buffer solution for 30 minutes. The heterodimer (11) was obtained in a 33.1% yield after purification using HPLC. At this point the helping hand with the tri-lysine tag was removed from the A-chain using a hydrazine-based buffer (pH 7.5) similar to that previously described without the use of dithiothreitol (DTT)29 to afford peptide 12. The tag was removed at this point since the formation of the heterodimer lessens the need for enhanced solubility. To form the second intermolecular bridge (CysA7 and CysB7) the heterodimer (12) was treated with 25 equivalents of I2 in 77% aqueous AcOH for 15 minutes to obtain a yield of 42.7% after HPLC purification. Furthermore, we hypothesized that removing the solubility tag after formation of the second intermolecular disulfide bridge could increase the reaction yield (Scheme 5), but this procedure generated a similar yield as removing the tag after formation of the first intermolecular bridge. We hypothesize that perhaps the length and bulkiness of the linker and solubility tag may hinder the facile formation of the last intermolecular bridge between the two chains as we observed longer reaction times for the formation of the last bridge.
Scheme 5.
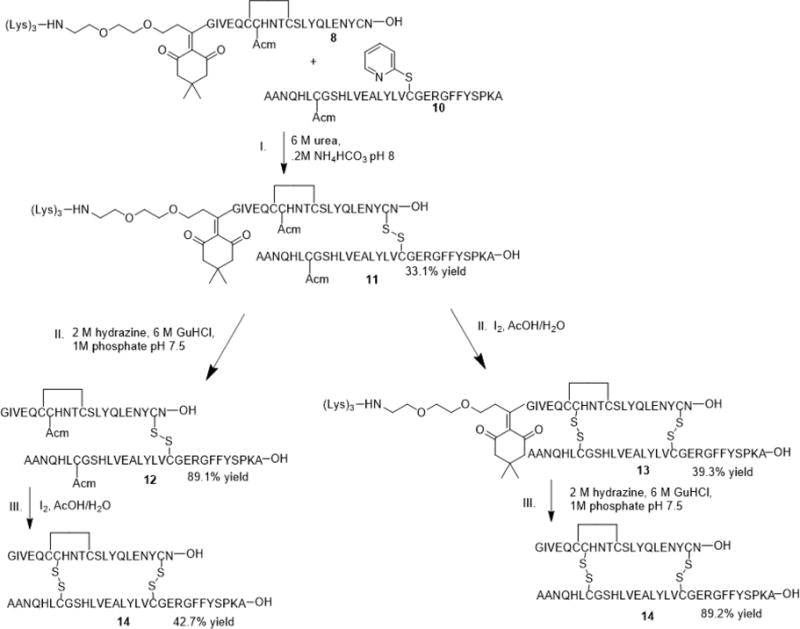
Three-step combination protocol for removal of solubility tag and linker after formation of the first intermolecular bridge (12) and removal of solubility tag and linker after formation of the second intermolecular bridge (13).
In Vitro Cell-Based Assays of Chicken Insulin
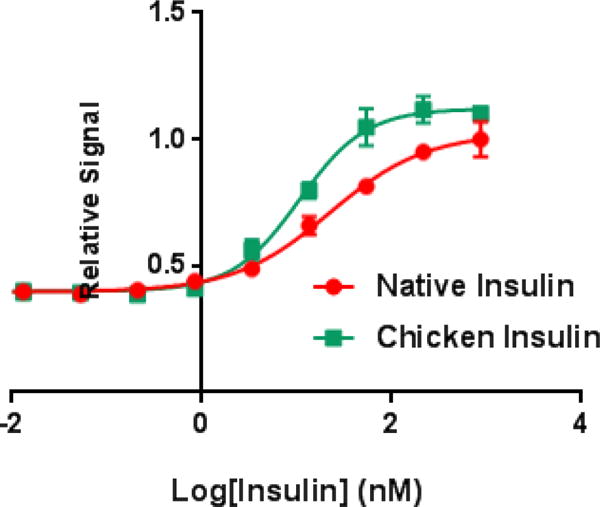
The resulting chicken insulin was tested for its ability to stimulate the insulin signaling pathway via a phospho-AKT (Ser 473) cell-based assay. In brief, NIH 3T3 cells overexpressing the insulin receptor isoform B were incubated with human insulin or synthetic chicken insulin in a range of concentrations for 30 minutes. After incubation, we lysed the cells and measured the levels of p-AKT at Ser 473. We observed chicken insulin had similar bioactivity to native insulin although previous studies indicate that chicken insulin has a ~5 fold higher in binding affinity to the insulin receptor (Fig. 3).32 This, however, is not surprising as previous studies indicate the potency of human insulin binding to the insulin receptor does not necessarily correlate to the bioactivity.34 Furthermore, we performed a p-IR beta assay to evaluate the ability of chicken insulin to induce phosphorylation of the insulin receptor only when the receptor is phosphorylated at Tyr 1150/1151. We observed chicken insulin could stimulate the phosphorylation of the insulin receptor with two 2-fold greater potency than native insulin, which is more consistent with the receptor binding data previously reported.
Fig. 3.
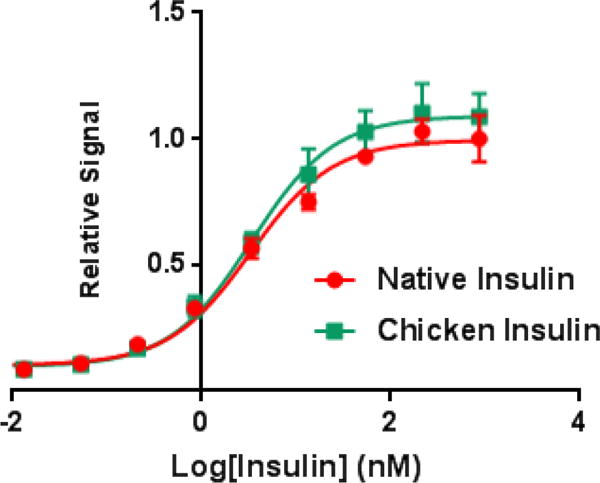
pAKT cell-based assay measuring the bioactivity of chicken insulin versus human insulin in NIH 3T3 cells overexpressing the human insulin receptor. (Native Insulin EC50 = 3.27 nM, Chicken Insulin EC50 = 3.33 nM).
Conclusions
In this study, an efficient alternative procedure is employed to enhance the solubility of insulin-based peptides. This method uses a tri-lysine solubility tag linked to a PEG-dimedone based linker that can be easily synthesized and coupled to the N-terminus of insulin-based peptides. This linker only requires an N-terminus or a lysine side chain, and its removal does not require harsh basic conditions that can be detrimental to sensitive peptides. Our studies further show that this linker can withstand harsh acidic conditions required for peptide cleavages, as well as reducing/oxidizing and basic conditions required for the deprotection of various cysteine-protecting groups. Our method shows that removing the linker and solubility complex after the formation of the first intermolecular bridge between CysA20-CysB19 does not improve the overall yield during the formation of the second intermolecular bridge (yield of 38.0% vs. 35.0% respectively, over two steps starting from peptide 11).
Moreover, using this method we were able to chemically synthesize chicken insulin, and to our knowledge this is the first reported chemical synthesis of this vertebrate insulin. This approach further demonstrates the helping hand is a suitable traceless linker that can overcome the hydrophobicity of insulin-based peptides without leaving a mark on the N-terminus which is critical for insulin binding to its receptor. This method can be used to facilitate the synthesis of new insulin analogues and could further be used to synthesize other peptides fragments with low solubility; in particular this method could be used in library-based applications where peptide sequences vary significantly. Finally, this method could be combined with the isoacyl dipeptide strategy to further enhance solubility and improve the overall yield of difficult peptides.
Supplementary Material
Fig. 4.
Phospho-IR beta cell-based assay measuring the endogenous levels of insulin receptor beta only when phosphorylated at Tyr 1150/1151 when stimulated by human insulin versus chicken insulin in NIH 3T3 cells overexpressing the human insulin receptor. (Native Insulin EC50 = 22.20 nM, Chicken Insulin EC50 = 10.54 nM).
Acknowledgments
We thank Utah Science Technology and Research Initiative (USTAR), JDRF (to D. H. C.) and NIH Grant AI076168 (to M.S.K.) for funding. J.M.N. is supported by NIDDK T32 Metabolism Training Grant and M. E. P. was supported by NIHLB Cardiovascular Training Grant. We thank Dr. Michael Jacobsen for helpful discussion.
Footnotes
Electronic Information (ESI) available: Detailed synthesis procedure and mass spectrometry data; along with cell assay protocol.
Conflicts of Interest
There are no conflicts to declare.
References
- 1.Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. CMAJ. 1991;145:1281–1286. [PMC free article] [PubMed] [Google Scholar]
- 2.Merrifield RB. Journal of the American Chemical Society. 1963;85:2149–2154. [Google Scholar]
- 3.Kung YT, Du YC, Huang WT, Chen CC, Ke LT. Sci Sin. 1965;14:1710–1716. [PubMed] [Google Scholar]
- 4.Katsoyannis PG, Tometsko A, Zalut C. J Am Chem Soc. 1966;88:166–167. doi: 10.1021/ja00953a033. [DOI] [PubMed] [Google Scholar]
- 5.Marglin A, Merrifield RB. J Am Chem Soc. 1966;88:5051–5052. doi: 10.1021/ja00973a068. [DOI] [PubMed] [Google Scholar]
- 6.Mayer JP, Zhang F, DiMarchi RD. Biopolymers. 2007;88:687–713. doi: 10.1002/bip.20734. [DOI] [PubMed] [Google Scholar]
- 7.Sieber P, Kamber B, Hartmann A, Johl A, Riniker B, Rittel W. Helv Chim Acta. 1977;60:27–37. doi: 10.1002/hlca.19770600105. [DOI] [PubMed] [Google Scholar]
- 8.Akaji K, Fujino K, Tatsumi T, Kiso Y. Journal of the American Chemical Society. 1993;115:11384–11392. [Google Scholar]
- 9.Annis I, Hargittai B, Barany G. Methods in Enzymology. Vol. 289. Academic Press; 1997. pp. 198–221. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Luo EY, Flora DB, Mayer JP. Organic Letters. 2013;15:960–963. doi: 10.1021/ol400149j. [DOI] [PubMed] [Google Scholar]
- 11.Liu F, Li P, Gelfanov V, Mayer J, DiMarchi R. Accounts of Chemical Research. 2017;50:1855–1865. doi: 10.1021/acs.accounts.7b00227. [DOI] [PubMed] [Google Scholar]
- 12.Wu F, Mayer JP, Gelfanov VM, Liu F, DiMarchi RD. The Journal of Organic Chemistry. 2017;82:3506–3512. doi: 10.1021/acs.joc.6b03078. [DOI] [PubMed] [Google Scholar]
- 13.Dixon GH, Wardlaw AC. Nature. 1960;188:721. doi: 10.1038/188721a0. [DOI] [PubMed] [Google Scholar]
- 14.Sohma Y, Kent SB. J Am Chem Soc. 2009;131:16313–16318. doi: 10.1021/ja9052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tofteng AP, Jensen KJ, Schaffer L, Hoeg-Jensen T. Chembiochem. 2008;9:2989–2996. doi: 10.1002/cbic.200800430. [DOI] [PubMed] [Google Scholar]
- 16.Du YC, Zhang YS, Lu ZX, Tsou CL. Sci Sin. 1961;10:84–104. [PubMed] [Google Scholar]
- 17.Sohma Y, Hua QX, Whittaker J, Weiss MA, Kent SB. Angew Chem Int Ed Engl. 2010;49:5489–5493. doi: 10.1002/anie.201001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patil NA, Tailhades J, Karas JA, Separovic F, Wade JD, Hossain MA. Angew Chem Int Ed Engl. 2016;55:14552–14556. doi: 10.1002/anie.201604733. [DOI] [PubMed] [Google Scholar]
- 19.Sohma Y, Taniguchi A, Skwarczynski M, Yoshiya T, Fukao F, Kimura T, Hayashi Y, Kiso Y. Tetrahedron Letters. 2006;47:3013–3017. [Google Scholar]
- 20.Liu F, Luo EY, Flora DB, Mezo AR. Angew Chem Int Ed Engl. 2014;53:3983–3987. doi: 10.1002/anie.201310735. [DOI] [PubMed] [Google Scholar]
- 21.Thalluri K, Kou B, Gelfanov V, Mayer JP, Liu F, DiMarchi RD. Organic Letters. 2017;19:706–709. doi: 10.1021/acs.orglett.6b03876. [DOI] [PubMed] [Google Scholar]
- 22.Hossain MA, Belgi A, Lin F, Zhang S, Shabanpoor F, Chan L, Belyea C, Truong HT, Blair AR, Andrikopoulos S, Tregear GW, Wade JD. Bioconjugate Chemistry. 2009;20:1390–1396. doi: 10.1021/bc900181a. [DOI] [PubMed] [Google Scholar]
- 23.Williams GM, Cooper GJ, Lee K, Whiting L, Brimble MA. Org Biomol Chem. 2013;11:3145–3150. doi: 10.1039/c3ob40322j. [DOI] [PubMed] [Google Scholar]
- 24.Nicolás E, Pedroso E, Girald E. Tetrahedron Letters. 1989;30:497–500. [Google Scholar]
- 25.Dolling R, Beyermann M, Haenel J, Kernchen F, Krause E, Franke P, Brudel M, Bienert M. Journal of the Chemical Society, Chemical Communications. 1994:853–854. doi: 10.1039/C39940000853. [DOI] [Google Scholar]
- 26.Shabanpoor F, Separovic F, Wade JD. Vitamins & Hormones. Vol. 80. Academic Press; 2009. pp. 1–31. [DOI] [PubMed] [Google Scholar]
- 27.Liu F, Zaykov AN, Levy JJ, DiMarchi RD, Mayer JP. J Pept Sci. 2016;22:260–270. doi: 10.1002/psc.2863. [DOI] [PubMed] [Google Scholar]
- 28.Chang SG, Choi KD, Jang SH, Shin HC. Mol Cells. 2003;16:323–330. [PubMed] [Google Scholar]
- 29.Jacobsen MT, Petersen ME, Ye X, Galibert M, Lorimer GH, Aucagne V, Kay MS. J Am Chem Soc. 2016;138:11775–11782. doi: 10.1021/jacs.6b05719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galibert M, Piller V, Piller F, Aucagne V, Delmas AF. Chemical Science. 2015;6:3617–3623. doi: 10.1039/c5sc00773a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conlon JM. Peptides. 2001;22:1183–1193. doi: 10.1016/s0196-9781(01)00423-5. [DOI] [PubMed] [Google Scholar]
- 32.Conlon JM, Cavanaugh ES, Mynarcik DC, Whittaker J. Biochem J. 1996;313(Pt 1):283–287. doi: 10.1042/bj3130283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss MA, Hua QX, Jia W, Nakagawa SH, Chu YC, Hu SQ, Katsoyannis PG. J Biol Chem. 2001;276:40018–40024. doi: 10.1074/jbc.M104634200. [DOI] [PubMed] [Google Scholar]
- 34.Menting JG, Gajewiak J, MacRaild CA, Chou DH, Disotuar MM, Smith NA, Miller C, Erchegyi J, Rivier JE, Olivera BM, Forbes BE, Smith BJ, Norton RS, Safavi-Hemami H, Lawrence MC. Nat Struct Mol Biol. 2016;23:916–920. doi: 10.1038/nsmb.3292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.