

Abstract
During epithelial-to-mesenchymal transitions (EMTs), chick cranial neural crest cells simultaneously delaminate from the basement membrane and segregate from the epithelia, in part, via multiple protease-mediated mechanisms. Proteolytic processing of Cadherin-6B (Cad6B) in premigratory cranial neural crest cells by metalloproteinases not only disassembles cadherin-based junctions but also generates shed Cad6B ectodomains or N-terminal fragments (NTFs) that may possess additional roles. Here we report that Cad6B NTFs promote delamination by enhancing local extracellular proteolytic activity around neural crest cells undergoing EMT en masse. During EMT, Cad6B NTFs of varying molecular weights are observed, indicating that Cad6B may be cleaved at different sites by A Disintegrin and Metalloproteinases (ADAMs) 10 and 19 as well as by other matrix metalloproteinases (MMPs). To investigate Cad6B NTF function, we first generated NTF constructs that express recombinant NTFs with similar relative mobilities to those NTFs shed in vivo. Overexpression of either long or short Cad6B NTFs in premigratory neural crest cells reduces laminin and fibronectin levels within the basement membrane, which then facilitates precocious neural crest cell delamination. Zymography assays performed with supernatants of neural crest cell explants overexpressing Cad6B long NTFs demonstrate increased MMP2 activity versus controls, suggesting that Cad6B NTFs promote delamination through a mechanism involving MMP2. Interestingly, this increase in MMP2 does not involve up-regulation of MMP2 or its regulators at the transcriptional level but instead may be attributed to a physical interaction between shed Cad6B NTFs and MMP2. Taken together, these results highlight a new function for Cad6B NTFs and provide insight into how cadherins regulate cellular delamination during normal developmental EMTs as well as aberrant EMTs that underlie human disease.
INTRODUCTION
Neural crest cells are remarkably versatile and contribute substantially to the patterning of the vertebrate embryo. Upon their sorting and alignment along the neural plate border, multipotent premigratory neural crest cells become poised to undergo a highly coordinated and multi-mechanistic process called the epithelial-to-mesenchymal transition (EMT), whereby neural crest cells lose their epithelial identity and instead become mesenchymal and highly invasive. During EMT, neural crest cells undergo considerable alterations to their gene regulatory network, cytoskeleton, and intercellular adhesion system to facilitate apico-basal polarity changes and de-epithelialization, which collectively allow neural crest cells to become motile (Nieto et al., 2016). Following EMT, migratory neural crest cells travel throughout the embryo body where they differentiate and contribute to a wide array of cell types and tissues, including craniofacial bone and cartilage, the cardiac outflow tract, and neurons and glia of the peripheral nervous system. Within the cranial population of chick premigratory neural crest, EMT is triggered en masse and occurs concomitantly with delamination, a process interlinked with EMT that facilitates the separation of neural crest cells from one another, the bordering neural and non-neural ectoderm, and the basement membrane situated at the basolateral surface of the neural tube (Duband et al., 2015; Gouignard et al., 2018; Theveneau and Mayor, 2012).
Prior to EMT, chick cranial neural crest cells reside in the neural plate border as a continuous epithelia between the neural and non-neural ectoderm and originate from cell contributions from both tissues (Gouignard et al., 2018). The coordination of EMT in the bulk of the cranial neural crest cells residing in the border, which become the apices of the apposing neural folds and coincides with neural fold fusion at 5ss-6ss, is not fully understood. It is known, however, that the molecular trigger for EMT is multifactorial in nature, initiated by Wnt and BMP signaling (Stuhlmiller and Garcia-Castro, 2012), resulting in a cascade of changes in the neural crest gene regulatory network that drives cellular processes important for EMT (Simoes-Costa and Bronner, 2015). Notably, post-transcriptional regulation also positively impacts neural crest EMT, including proteolysis of membrane proteins to alter cell adhesion (Alfandari and Taneyhill, 2018; Christian et al., 2013) and changes in cell polarity achieved via cytoskeletal rearrangements that ultimately promote migration (Gouignard et al., 2018).
Changes in the adhesion protein repertoire in cranial neural crest cells are one such downstream component that facilitate both EMT and delamination to occur simultaneously (Taneyhill and Schiffmacher, 2017). As single transmembrane adhesion proteins, cadherins dimerize in cis to physically interact with dimers on neighboring cells in trans, thereby stabilizing intercellular contacts (Yap et al., 1997). In chick cranial premigratory neural crest cells, multiple cadherins, including N-cadherin, E-cadherin, and Cadherin-6B (Cad6B), are expressed simultaneously, with N-cadherin and E-cadherin also expressed in adjacent neural tube (both E-cadherin and N-cadherin) and ectoderm (E-cadherin) epithelial layers (Coles et al., 2007; Dady et al., 2012; Dady and Duband, 2017; Duband et al., 1988; Hatta et al., 1987; Lee et al., 2013; Nakagawa and Takeichi, 1995, 1998). Cad6B expression, however, is observed early on within the premigratory neural crest population, and may function to sort and amass neural crest progenitors during specification (Coles et al., 2007; Dady and Duband, 2017; Nakagawa and Takeichi, 1995). In the chick trunk, depletion of Cad6B from premigratory neural crest cells undergoing specification later reduces the number of emigrating neural crest cells, and this phenotype has been attributed to impaired de-epithelialization during EMT (Park and Gumbiner, 2010). Experimental knockdown of Cad6B expression just prior to EMT in cranial neural crest cells, however, results in premature emigration, suggesting that Cad6B still maintains a predominant function to promote cell adhesion and keep the premigratory neural crest cell domain intact, at least at this developmental stage and axial level (Coles et al., 2007).
During these pre-EMT stages, Cad6B is positively regulated by BMP signaling, and Cad6B protein achieves maximal levels within the fully apposed, fusing neural folds at the 6 somite stage (ss) (Schiffmacher et al., 2014; Sela-Donenfeld and Kalcheim, 1999; Taneyhill et al., 2007). As EMT is initiated, Cad6B is rapidly down-regulated within two hours by a core EMT transcriptional repressor complex containing Snail2 and other chromatin remodeling proteins (Strobl-Mazzulla and Bronner, 2012; Taneyhill et al., 2007). Cad6B protein is also robustly cleared within hours from the bulk of neural crest cells undergoing EMT and becomes undetectable prior to active migration. Proper levels of Cad6B in the cranial neural crest, and the rapid clearance of Cad6B at EMT, are achieved via multiple post-translational mechanisms working in concert, including Tetraspanin-mediated stabilization, clathrin-mediated endocytosis, macropinocytosis, and proteolysis (Fairchild and Gammill, 2013; Padmanabhan and Taneyhill, 2015; Schiffmacher et al., 2014). Cad6B protein is continuously cleaved by A Disintegrin and Metalloproteinases (ADAMs) 10 and 19, as well as intracellularly by the γ-secretase complex (Schiffmacher et al., 2014). Other matrix metalloproteinases (MMPs) including MMP2, MMP9, and MMP16, are active in the dorsal neural tube and may target Cad6B as well (Duong and Erickson, 2004; Monsonego-Ornan et al., 2012; Roth et al., 2017). Due to this combination of transcriptional and post-translational mechanisms, Cad6B transcription can no longer replenish membrane Cad6B protein, leading to clearance of preexisting Cad6B pools at EMT.
Cadherin proteolysis not only reduces full-length protein levels in neural crest cells but also generates both extracellular and intracellular protein fragments that possess their own novel roles (Abbruzzese et al., 2015; Mathavan et al., 2017; McCusker and Alfandari, 2009; Schiffmacher et al., 2016; Shoval et al., 2007). ADAMs and MMPs cleave cadherins in their extracellular domain to generate shed ectodomains that are often referred to as N-terminal fragments (NTFs) or soluble cadherins. The Cadherin-11 NTF has been well studied in Xenopus migratory neural crest cells, where it regulates contact inhibition of locomotion as well as migration (Abbruzzese et al., 2015; McCusker et al., 2009). Following ADAM-mediated ectodomain shedding in chick neural crest cells, the resultant membrane-bound intracellular domains (C-terminal fragment 1) of Cad6B, and potentially N-cadherin, are then subjected to γ-secretase-mediated cleavage to give rise to soluble intracellular C-terminal fragments (CTF2s) (Schiffmacher et al., 2016; Shoval et al., 2007). Both N-cadherin and Cad6B CTF2s translocate into the nucleus, and Cad6B CTF2 has been shown to up-regulate β-catenin, CyclinDI and Snail2. While both Cad6B NTFs and CTF2s are generated throughout the premigratory and EMT stages, the function of the Cad6B NTF remains unknown.
In line with our previous studies investigating Cad6B CTF2 function, the tightly regulated spatiotemporal profiles of Cad6B expression and Cad6B proteolysis within chick cranial neural crest cells also provide an opportunity to elucidate the potential roles of cadherin NTFs strictly within the context of EMT, and specifically delamination. Therefore, we investigated Cad6B NTF as a putative regulator of these cellular processes. In this study, we demonstrate the existence of multiple Cad6B NTFs generated by distinct proteases in the neural crest. In addition, our data show that Cad6B NTFs regulate the delamination process in vivo by enhancing laminin and fibronectin degradation within the dorsal neural tube basement membrane. Furthermore, in-gel zymography assays indicate that Cad6B NTFs promote delamination through a mechanism involving up-regulation of MMP2 activity that is independent of changes in MMP2 transcription but may be correlated with a direct physical interaction between Cad6B NTFs and MMP2. Collectively, these findings reveal a novel role for Cad6B NTFs and provide insight into how shed cadherin ectodomains control delamination during EMTs underscoring normal human development and disease.
MATERIALS AND METHODS
Chicken embryo culture
Fertilized chicken eggs were obtained from Centurion Poultry, Incorporated (Lexington, GA USA) and Moyer’s Chicks Hatchery (Quakertown, PA, USA) and were incubated at 38°C in humidified incubators (EggCartons.com, Manchaug, MA, USA). Embryos were staged according to the number of pairs of somites and Hamburger and Hamilton (HH) criteria (Hamburger and Hamilton, 1992).
Preparation of DNA expression constructs
An N-terminal, Hemagglutinin (HA)-tagged, full-length Cad6B coding sequence was designed by modifying the pCS2.Cad6B expression construct to include an in-frame HA coding sequence within the linker between EC2 and EC3 domain sequences as previously described (Yu, 2011). The estimated coding sequence corresponding to each Cad6B NTFs was based on both experimental and computational approaches. The primary sequence of the Cad6B short NTF (S-NTF) was based on the molecular weight of the deglycosylated NTF produced via ADAM 10 mediated Cad6B proteolysis in vitro and determined using EXPASY Compute pl/Mw tool (Artimo et al., 2012), in conjunction with C-termini predictions based on known ADAM10 cleavage sites (S-NTF/ADAM10 site: QLIQT-LSAV; MEROPS peptidase database, http://merops.sanger.ac.uk/ (Rawlings et al., 2016)). The full ectodomain (EC1-EC5) was used as the primary sequence for Cad6B long NTF (L-NTF) expression constructs. C-terminal HA-tagged coding sequences (Cad6B; accession # NP_001001758.1; S-NTF: amino acids 1–508; L-NTF: amino acids 1–615) were PCR-amplified from pCIG.Cad6B (Coles et al., 2007) and cloned into pCIG. The IRES-GFP sequence was removed from pCIG during cloning to generate pCAGGS.MMP2 HA and pCAGGs.Cad6B.S-NTF-HA and L-NTF-HA constructs. All constructs were verified for sequence accuracy (Genewiz, South Plainfield, NJ, USA).
In ovo electroporations
Expression constructs were unilaterally electroporated into premigratory midbrain neural crest cells in developing 2–3ss chick embryos using a modified version of in ovo electroporation (Itasaki et al., 1999; Schiffmacher et al., 2014). Constructs were electroporated at 3.5 pg/pl unless otherwise stated. Re-incubation times varied according to the experiment.
Cell culture and in vitro metalloproteinase inhibition assays
Chinese Hamster Ovary cells (CHO, CCL-61; American Type Culture Collection (ATCC), Manassas, VA) and chicken LMH cells (CRL-2117; ATCC) were cultured according to the provider’s instructions. Transient transfection assays were performed with Lipofectamine 2000 reagent (Thermo Fisher, Carlsbad, CA). Cells were grown to 80% confluency and transfections were performed according to the manufacturer’s protocol and published methods (Schiffmacher et al., 2014). Metalloproteinase inhibition assays were conducted using a previously characterized Flp-ln-CHO cell line expressing full-length Cad6B (tagged at the C-terminus with an HA epitope) from a single locus (Flp-ln Cad6B-HA cells) (Schiffmacher et al., 2014). A titration of working molar concentrations of GM6001 (Tocris Bioscience, Minneapolis, MN, USA) was performed to determine an effective concentration (25 μΜ) that produced statistically significant inhibition of Cad6B NTF shedding in these Flp-ln Cad6B-HA cells. Flp-ln Cad6B-HA cells were grown to 75% confluency, treated with 0.5% DMSO (control and MMP2 transfections), 25 μΜ GM6001, or 25 μΜ MMP2/MMP9 Inhibitor V (MilliporeSigma, St. Louis, MO, USA), and cultured at 5% C02 and 37°C for 18 hours. Glycoproteins, including Cad6B NTFs and MMP2, were enriched from three mis of clarified cell culture supernatants using Concanavalin A-Sepharose as previously described (Schiffmacher et al., 2014). Whole cell lysates were generated from treated cells and subjected to immunoblotting using antibodies to Cad6B (CC6DB-1, 1:80, DSHB, Iowa City, Iowa, USA); GAPDH (1:2000, sc-47778, Santa Cruz); and MMP2 (1:1000, PA1–16667, Thermo Fisher).
Co-lmmunoprecipitation, N-linked deglycosylation, and immunoblot analysis of Cad6B NTFs
Chicken LMH cells grown in 60 mm plates were either co-transfected with pCS2.HA-Cad6B and pCIG.MMP2 together or singly with pCS2 using Lipofectamine 2000 as described above. The next day, cells were re-incubated in three mis of fresh media containing low serum levels (2% FBS). Following a 24 hour incubation, supernatants were collected and processed as previously described (Schiffmacher et al., 2014). HA-tagged Cad6B NTFs generated via HA-Cad6B proteolysis were immunoprecipitated using an HA antibody (3F10; MilliporeSigma) and magnetic Pierce protein A/G beads (Thermo Fisher). For N-linked deglycosylation of full-length Cad6B and Cad6B NTFs, Concanavalin A-Sepharose-mediated enrichments were first performed as previously described (Schiffmacher et al., 2014). Following bead/protein complexing and washing with Tris-buffered Saline/1% TX-100 (TBST), glycoproteins were eluted by boiling at 100°C for 10 minutes in 1X Glycoprotein Denaturing Buffer (New England Biolabs, Ipswich, MA, USA). Eluates were split into two equal volumes, with one sample further diluted in glycobuffer containing PNGase F. Untreated samples were also diluted with glycobuffer but did not receive PNGase F. All samples were incubated for seven hours at 37°C according to the manufacturer’s protocol. Relative mobility shifts between samples were analyzed by immunoblotting as previously described (Schiffmacher et al., 2014). Primary antibodies used for immunoblotting were: Cad6B (CC6DB-1, 1:80, DSHB, Iowa City, Iowa, USA); GAPDH (1:2000, sc-47778, Santa Cruz); and MMP2 (1:2000, PA1–16667, Thermo Fisher). Immunoblots were serially probed between antibody stripping with Restore Western Blot Stripping Buffer according to the manufacturer’s instructions (Thermo Scientific). Immunoblot images for figures were gamma-modified and processed using Photoshop CS6 (Adobe Systems, San Jose, CA, USA). Immunoblot band volumes (intensities) were calculated from unmodified immunoblot images using Image Lab software (Bio-Rad). Fold changes in shed Cad6B NTF levels were presented as means and standard error of the means (SEMs) of band volumes (n = 5), and fold changes in full-length Cad6B were presented as means and SEMs of Cad6B/GAPDH ratios (n = 3). Immunoblots were analyzed by ANOVA using the PROC MIXED model in SAS statistical software (SAS Institute, Cary, NC), and levels were deemed significantly different (p < 0.05) using the PDIFF procedure.
Immunohistochemistry
Immunohistochemical detection of various proteins was performed on whole embryos or transverse sections following 4% paraformaldehyde fixation and cryostat-sectioning as in (Schiffmacher et al., 2014). The following primary antibodies and concentrations were used: GFP (1:750, clone 3E6, Thermo Fisher) HA (1:1000, 3F10, MilliporeSigma; or 1:250, 12CA5, Thermo Scientific); Snail2 (1:100, C19G7, Cell Signaling Technologies, Danvers, MA); laminin (1:100, 3H11, DSHB; or 1:500, PA1–84171, Thermo Fisher); fibronectin (1:500, B3/D6, DSHB); ZO-1 (1:50, 402300, Thermo Fisher); and Cad6B (1:80, CCD6B-1, DSHB). Appropriate fluorescently-conjugated secondary antibodies (Alexa Fluors 488, 594, 647, Thermo Fisher) were used at a concentration of 4 pg/ml. Slides were mounted using Fluoromount G containing DAPI to mark cell nuclei. Unless otherwise noted, images from four to seven serial symmetrical transverse sections through the midbrain of a minimum of three embryos were collected using an LSM Zeiss 800 microscope with Airyscan and a 63X oil objective (NA 1.4) at room temperature (Carl Zeiss Microscopy, Thornwood, NY) and analyzed with the Zen Blue software (Zeiss). For each individual section, laser power and gain were set to capture images just below saturation limits for all channels requiring pixel intensity quantification. For sections used in laminin and fibronectin fluorescent intensity quantifications, transverse symmetry was determined by comparing the migratory neural crest cell domain length (as marked by Snail2 immunostaining) between electroporated and unelectroporated sides. Only sections with distance ratios of 1.0 +/− 0.15 were used as replicates. All exported files were processed in Photoshop CS6 (cropping, minor gamma adjustments), and a representative image was chosen for applicable figures. Neural tube laminin and fibronectin fluorescent intensity was determined on original Zeiss CZI files using the profile line intensity tool in the Zen Blue software (Zeiss). Line profiles were drawn over laminin- or fibronectin-immunostained basement membranes at high magnification, from their start at the dorsal tube midline up to the first hinge point (a maximum of 75 μm). Equidistant line profiles were then drawn along the unelectroporated basement membrane. Exported pixel intensity data sets were rendered into graphs using Excel software. Total laminin or fibronectin levels within quantified basement membranes were calculated as the sum of the areas under the curve between all consecutive data points, with each pixel intensity measurement taken at every 1.01 μm (20X). Total areas under the curve were then presented as ratios of the electroporated side over the unelectroporated side. Ratios were compared between treatments and were deemed significantly different (p < 0.05) using the PDIFF procedure with Tukey’s adjustment in SAS statistical software.
Neural crest cell explant gelatin zymography
For gelatin zymography, embryos at 3ss were electroporated with plasmids and re-incubated for three hours post-electroporation before midbrain dorsal neural folds (containing premigratory neural crest cells) were dissected out in PB-1 standard medium and placed into chamber slides (Nunc Lab-tek II, Thermo Scientific) coated with 16.6 pg/ml poly-L-lysine (P5899, MilliporeSigma, Burlington, MA, USA) and 10 pg/ml fibronectin (356008, Corning, NY, USA). Equal numbers of tissue (12–15 per experiment) were explanted and incubated overnight in serum-free Dulbecco’s Modified Eagle’s Medium (DMEM, 10–013-CV, Mediatech) containing 1X N2 supplement (Thermo Fisher) at 5% C02 and 37°C. Explants were counted to confirm equal explant numbers between treatments following attachment. Media was replaced with 500 pi DMEM/N2 media and re-incubated for 24 hours. Supernatants were collected and centrifuged at 300 x g for five minutes at 4°C. The top 400 pi was concentrated to 100 pi using 3K Amicon Ultra size exclusion columns (MilliporeSigma). Gelatin zymography on supernatants was performed as previously described (Hu and Beeton, 2010) on Novex Zymogram Plus 10% gelatin gels (Thermo Fisher). Gels were developed overnight at 37°C and stained with Pierce Coomassie Plus Bradford reagent (Thermo Fisher.) Gels were imaged with the ChemiDoc XRS system (Bio-Rad, Hercules, CA), and inverse band densitometries at 72 kDa (the molecular weight of pro-MMP2) were calculated from unmodified immunoblot images (0.1 to 0.125 second exposures) using Image Lab software (Bio-Rad). Relative enzymatic activities were generated by normalizing inverse band volumes of each treatment replicate at 72 kDa to the mean GFP treatment band volume. Fold changes are presented as means and SEMs of these ratios. Immunoblots were analyzed by ANOVA using the PROC MIXED model in SAS statistical software (SAS Institute, Cary, NC), and levels were deemed significantly different (p < 0.05) using Tukey’s adjustment. Following two 1X PBS washes, explant lysates were collected by adding 50 pi of 1% NP-40 lysis buffer directly to explants, and processed for immunoblotting, as described above.
QPCR
Changes in gene expression following overexpression of L-NTF-HA or GFP were measured by SYBR Green-based QPCR as previously described (Schiffmacher et al., 2016). For MMP2, MMP9, MT2-MMP/MMP15, MT3-MMP/MMP16, and TIMP2, relative levels of expression were back-calculated from a standard curve generated from a reference cDNA originating from pooled 6–10ss midbrain RNA and normalized to chick 18S rRNA levels. Normalizations to chick HPRT1 and GUSB, both transcribed by RNA polymerase II, were also carried out and yielded identical results (Fig. S4). All 78S-normalized replicate levels were then normalized again to the GFP control mean to arbitrarily set the GFP control mean at 1. To account for potential genomic DNA contamination and amplification during reverse transcription, QPCR was also performed on identical sample cDNA synthesis reactions lacking reverse transcriptase. Recombinant L-NTF-HA transcripts were amplified using an antisense primer that only hybridizes to the 3’ HA tag sequence. L-NTF-HA levels were assessed using a standard curve consisting of serial dilutions of pCAGGs.L-NTF-HA plasmid. All primers were designed using Primer-BLAST software (Ye et al., 2012) and data were analyzed by analysis of variance (ANOVA) using the PROC MIXED model (SAS). Levels were deemed significantly different (p < 0.05) using the PDIFF procedure. Data were transformed to meet assumptions of normality and homogeneity of variance.
RESULTS
Cranial neural crest cells dismantle tight junction complexes and degrade basement membranes prior to Cad6B protein down-regulation
The spatiotemporal expression pattern of Cad6B protein during early chick embryo development has been well documented along the entire anterior-posterior axis. Thus, Cad6B is often utilized as a nonnuclear protein marker for designating the specified premigratory neural crest domain residing in the neural plate border (Coles et al., 2007; Nakagawa and Takeichi, 1998; Schiffmacher et al., 2014). In cranial neural crest cells, Cad6B is also co-expressed with E-cadherin prior to, and throughout early, EMT. As EMT progresses, however, migratory neural crest cells rapidly deplete their Cad6B membrane pools but still maintain E-cadherin (Dady et al., 2012; Lee et al., 2013). The sharp repression of Cad6B expression between 6ss and 7ss (1–2 hours), along with the concomitant turnover of Cad6B protein (6ss-8ss, 3 hours) suggest that Cad6B may play exclusive roles in regulating neural crest cell EMT and delamination (Dady and Duband, 2017; Schiffmacher et al., 2014; Taneyhill et al., 2007). Indeed, our previous investigation into Cad6B CTF2 function supports this hypothesis, as Cad6B CTF2s complex with β-catenin, translocate into the nucleus, and induce transcription of genes important for EMT (Schiffmacher et al., 2016). How Cad6B protein levels in the cranial neural crest fluctuate in accordance with other aspects of EMT, such as alterations in tight junctions and the basement membrane of the neural folds, however, has yet to be examined. To this end, we performed Cad6B immunohistochemistry during somite stages when EMT is occurring through the chick midbrain region, in combination with antibodies that mark tight junctions (Zonula Occludens-1, ZO-1) and the basement membrane (fibronectin), which are also compatible for triple-label immunohistochemistry (Fig. 1). While ZO-1 may not always exclusively mark tight junctions, we found that ZO-1 spatiotemporal protein expression at the midbrain level overlaps with that observed for the tight junction core protein Claudin-1 (Fishwick et al., 2012). It is important to note that Duband and Thiery thoroughly characterized fibronectin expression during cranial neural crest cell EMT over 35 years ago, and our fibronectin immunolabeling results reflect the same spatiotemporal expression pattern (Duband and Thiery, 1982). With recent technical advances, though, we can now document the presence of these different proteins relative to one another using triple-labeling and high-resolution confocal microscopy (Fig. 1), thereby gaining additional insight into the levels and distribution of Cad6B, ZO-1-possessing tight junctions, and fibronectin within the basement membrane throughout EMT.
Figure 1. Cad6B levels in cranial neural crest cells undergoing EMT down-regulated dismantling of apical tight junctions and basement membrane degradation.
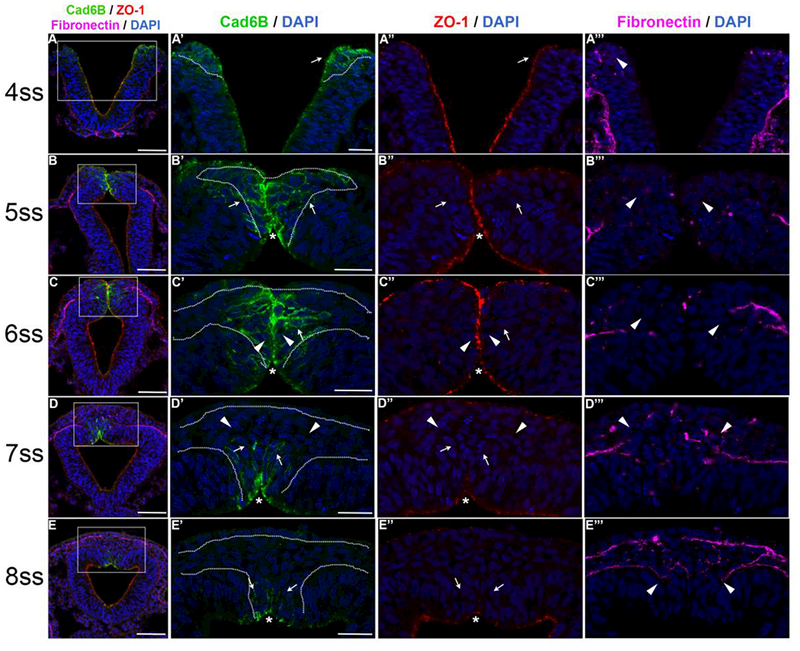
Representative transverse midbrain sections taken through embryos that have been immunostained for Cad6B (green), ZO-1 (red), and fibronectin (violet). DAPI stain labels cell nuclei (blue). (A-E, left column) Lower magnification images depicting triple-label immunohistochemistry (Cad6B/ZO-1/fibronectin) merged with DAPI. (A’-E’) Higher magnification images of dorsal neural tubes from section images in (A-E, highlighted by the white boxes) demonstrating Cad6B and DAPI. (A’’-E’’) The same images in (A’-E’) but showing ZO-1. (A’’’-E’’’) The same images in (A’’-E’’) but demonstrating fibronectin. All asterisks in (A’-E’) demarcate the population of Cad6B-positive cells that remain polarized and exhibit ZO-1 expression. (A-A’’’) Cad6B-positive midbrain neural crest cells at the 4ss. Epithelial premigratory neural crest cells (A’, A’’, arrows) express both Cad6B and ZO-1 at apical membranes and adhere to the basement membrane (A’’’, arrowhead). (B-B’’’) The midbrain neural crest cell domain expands at the 5ss, with most of the neural crest cells maintaining expression of Cad6B- and likely ZO-1 (B’, B’’, arrows; B’’’, arrowheads). (C-C’’’) Midbrain neural crest cells at the 6ss, when EMT is occurring en masse. Some neural crest cells at the midline have not yet initiated EMT, as they still express ZO-1 (similar to Claudin-1 at these axial levels and somite stages (Fishwick et al., 2012)) and appear polarized (O’, arrowheads). Delaminating Cad6B-positive neural crest cells (O’, C’’, arrows, and C’’’, arrowheads) appear to be oriented laterally towards the fused basement membranes. (D-D’’’) Midbrain neural crest cells at the 7ss. Leading neural crest cells exhibiting significantly diminished Cad6B levels (D’-D’’’, arrowheads) begin to emigrate through the fused basement membrane, which are followed by newly depolarized Cad6B-positive/ZO-1-negative neural crest cells (D’, D’’, arrows) that have begun to emigrate from the midline. (E-E’’’) Remaining premigratory Cad6B/ZO-1 double-positive neural crest cells (E’, E’’, arrows), and the bulk of Cad6B-negative migratory neural crest cells that have completed EMT (E’’’, arrowheads), at the 8ss. The duration between somite stages is about 1.5 hours. Scale bars in (A-E) are 50 pm. Scale bars in (A’-E’) are 20 pm and also apply to corresponding images in (A’’’-E’’’).
Prior to EMT, chick neural crest cells (labeled throughout the figure with the white dashed lines) within the prospective midbrain at the 4ss exist as an epithelium at the apex of the rising neural folds, and are continuous with the pseudostratified neuroepithelium and cuboidal ectoderm epithelium. As the neural folds rise and appose, thus forcing the basement membranes of the neural tube and ectoderm to come into contact, premigratory neural crest cells express Cad6B, exhibit apical polarity, and possess associated tight junctions (depicted by ZO-1, Fig, 1A’’, arrow). In addition, Cad6B-positive premigratory neural crest cells remain columnar in shape and maintain adherence to the basement membrane (depicted by fibronectin immunolabeling, Fig. 1A’’’, arrowhead). At the 5ss, the neural fold apices are in direct apposition to each other and the Cad6B-positive domains are now in contact. At the apical membranes, ZO-1 and robust Cad6B expression are maintained, indicating that premigratory neural crest still possess zonula adherens and have not de-epithelialized (Fig. 1B’, B’’, asterisk). A small population of Cad6B-positive premigratory neural crest cells are rounded in shape, although it cannot be determined if they have already depolarized and completed apical constriction (Fig. 1B’ 1B’’, and also at the 6ss, 1C’, 1C’’, arrows). These round cells may have detached from the luminal side of the neural tube or could still possess protrusions anchoring them to the midline (Fig. 1B’’’ and at the 6ss, C’’’, arrowheads).
By the 6ss, many neural crest cells along the midline exhibit strong Cad6B and ZO-1 labeling on their apposed apical membranes, suggesting that their underlying zonula adherens are still evident and that they have not yet initiated EMT. This subpopulation (Fig. 1C’, arrowheads) maintains the distinct midline of the dorsal neural tube observed at the 5ss (Fig. 1C’, asterisk), and their apical junctions would restrict movement of any of the fully delaminated neural crest cells now observed dorsally (Fig. 1C’, arrows). Nevertheless, at the 6ss, the Cad6B-positive neural crest domain is many cell layers thick, with numerous neural crest cells residing a cell layer beyond the midline (Fig. 1C’, arrows) and appearing depolarized without contacts to the midline or basement membrane. The majority of the Cad6B-positive lateral membranes possessed by neural crest cells are also still oriented towards the discontinuous basement membrane (Fig 1C’’’, arrowheads).
As EMT ensues en masse for the entire domain at the 7ss, the leading neural crest cells, which have already migrated through the fused ectoderm/neural tube basement membrane, exhibit decreased Cad6B levels (Fig. 1D’, D’’, D’’’ arrowheads). This population of neural crest cells corresponds to de-epithelialized, Snail2-positive cells adjacent to the nonneural ectoderm to which Lee et al. refer as ectomesenchymal cells, and they are the first to fully turnover their Cad6B membrane pools (Figs. 1D’, D’’, arrowheads) (Lee et al., 2013). On the other hand, neural crest cells residing along the midline at the 6ss (Fig. 1C’, C’’, arrowheads) have now fully disassembled their apical tight junctions and formed a dense cluster of newly depolarized neural crest cells, but still maintain Cad6B expression as they begin to emigrate through the basement membrane (Fig. 1D’, arrows). At the 8ss, a Cad6B-positive neural crest domain still remains (Fig. 1E’, arrows), and these neural crest cells have yet to de-epithelialize as evidenced by the presence of strong apical Cad6B and ZO-1 labeling (Fig. 1E’, E’’, asterisk; also observed in Fig. 1C’, D’, asterisks). These cells (Fig. 1E’, E’’, arrows) will also soon down-regulate Cad6B protein levels, complete EMT, and emigrate out of the neural tube between the ends of the discontinuous neural tube basement membrane (Fig. 1E’’’, arrowheads). We also ascertained the localization of the basement membrane protein laminin, relative to fibronectin and Cad6B, by performing triple-labeling using antibodies to each of these proteins across the 4–8ss. We find that fibronectin and laminin show comparable spatiotemporal distribution throughout the basement membrane of the dorsal neural tube with respect to Cad6B (Fig. S1). As such, ZO-1, and fibronectin or laminin, can be used to accurately represent how tight junctions and the basement membrane, respectively, change during EMT.
In summary, our spatiotemporal expression analysis from the 4ss-8ss (6 hours) highlights the dynamic changes in Cad6B expression that occur within individual cranial neural crest cells collectively undergoing EMT and demonstrates that Cad6B protein is progressively down-regulated across these developmental stages after tight junctions and basement membranes are lost. These results highlight the importance of rapid, post-translational mechanisms, such as proteolysis, to remove Cad6B from the neural crest population during EMT.
Cad6B proteolysis generates multiple NTFs of various molecular weights during EMT
Our previous studies demonstrated that proteolytic processing of Cad6B in cranial neural crest cells generates multiple NTFs of different molecular weights, including a predominant ~91 kDa NTF that accumulates during EMT (Schiffmacher et al., 2014). Due to the decreased resolution of higher molecular weight proteins on the 12% SDS-PAGE gel used in that analysis, an accurate evaluation of the relative mobilities of fainter NTF bands, in comparison to full-length Cad6B, could not be accomplished. Therefore, a more detailed analysis of Cad6B NTF numbers and molecular weights was performed by increasing Cad6B input and SDS-PAGE resolution (7–8% polyacrylamide). In addition, treatment with Peptide-N-Glycosidase F (PNGase F), which removes asparagine-linked glycans from at least two predicted sites within the Cad6B ectodomain (NetNGLyc 1.0 server, http://www.cbs.dtu.dk/services/NetNGlyc), was used to provide a more accurate prediction of NTF molecular weight based on the peptide itself.
Our first objective was to assess endogenous NTF molecular weights generated during midbrain EMT using our enhanced protocols. Dorsal neural folds containing premigratory neural crest cells, and dorsal neural tubes containing neural crest cells undergoing EMT en masse, were excised from the midbrains of 5ss and 6ss embryos, respectively. Lysates were subjected to Concanavalin A enrichment and then equally divided into a control sample or further treated with PNGase F. Immunoblot analysis reveals that in both 5ss and 6ss lysates, full-length Cad6B protein at 122 kDa is reduced by 16 kDa to 106 kDa following N-linked deglycosylation (denoted by #, Fig. 2A, lanes 1–4; Table 1). In addition to full-length Cad6B, at least two NTFs are detected in both the 5ss and 6ss lysates, with the larger NTF at 95 kDa (denoted by +, Fig. 2A, lanes 1 and 3, long NTF (L-NTF)) corresponding to the observed NTF (~91 kDa) in our previous study (Schiffmacher et al., 2014). In this new experiment, however, we identified a second Cad6B NTF (denoted by *, Fig. 2A, lanes 1 and 3, short NTF (S-NTF)) with a molecular weight at 83 kDa, which was faintly detectable in the 5ss lysate but more evident in the 6ss lysate. Following PNGase F treatment, L-NTF (95 kDa) and S-NTF (83 kDa) were reduced to 80 kDa and 70 kDa, respectively (Fig. 2A, lanes 2 and 4, + and *). The 15 kDa and 13 kDa reductions in relative mobilities for L-NTF and S-NTF were not significantly different from the 16 kDa reduction observed with full-length Cad6B, suggesting that the glycosylated asparagines are all located within the Cad6B N-terminus that is shared among full-length Cad6B, Cad6B L-NTF, and Cad6B S-NTF. Interestingly, a third Cad6B NTF at 89 kDa was also detected in the PNGase F-treated 6ss lysate (Fig. 2A, lane 4), and this band does not have any corresponding equivalent in the untreated 6ss sample that is 13–16 kDa greater in size. Complete N-linked deglycosylation of the more abundant full-length Cad6B indicates that PNGase F treatment was sufficient to fully remove these post-translational modifications. More importantly, PNGase F treatment reveals that the various NTFs evident in the untreated lysate are not N-linked glycosylation variants of a single shed Cad6B ectodomain. Altogether, our analysis reveals that multiple Cad6B NTFs of varying molecular weight are shed during the pre-EMT and EMT stages.
Figure 2. Multiple Cad6B NTFs of various lengths are shed from cranial neural crest cells during EMT.

(A) Cad6B proteolysis in cranial neural crest cells generates multiple shed ectodomains or NTFs during pre-EMT (5ss) and EMT (6ss) stages. Neural crest domains were excised, pooled, and converted to lysates for Concanavalin A enrichment. Glycoprotein eluates were equally divided with one half serving as a control to the other half that was treated with Peptide-N-Glycosidase F (PNGase F) to perform N-linked deglycosylation. In untreated samples (lane 1, 5ss, and lane 3, 6ss), a 122 kDa full-length Cad6B protein (#), 95 kDa NTF (L-NTF, +), and smaller 83 kDa NTF (S-NTF, *) were observed. Full removal of N-linked glycosylations through treatment with PNGase F in both the 5ss sample (lane 2) and the 6ss sample (lane 4) decreased molecular weights of full-length Cad6B (#) by 16 kDa to 106 kDa, L-NTF (+) by 15 kDa to 80 kDa, and S-NTF (*) by 13 kDa to 70 kDa. These comparable shifts (13–16 kDa) indicate that the NTFs are not post-translational modification variants of a single NTF but two NTFs of different length maintaining the original N-linked moieties existing on full-length Cad6B. (B) ADAM10 and an endogenous CHO cell protease differentially cleave Cad6B in vitro to generate two distinct NTFs. CHO cells transiently transfected with a Cad6B expression construct endogenously shed 88 kDa Cad6B L-NTFs into the supernatant (+, lanes 1 and 3). Treatment with PNGase F reduces its molecular weight by 17 kDa to produce a 71 kDa fragment (+, lane 2). In CHO cells transfected with both Cad6B and ADAM10 expression constructs, an additional ADAM10-specific 70 kDa S-NTF is shed (*, lane 1) that is reduced by 14kDa to 56 kDa following N-linked deglycosylation. Similar molecular weight shifts between L-NTFs (+) and S-NTFs (*) indicate that both NTFs exhibit the original N-linked glycosylation status as the full-length Cad6B. (C) Broad-spectrum protease inhibitor (GM6001) and MMP2/MMP9-specific Inhibitor V both significantly abrogate CHO cell protease shedding of Cad6B in vitro. 18 hours treatment of a Flp-In CHO cell line stably expressing Cad6B-HA with 25 μM GM6001 inhibits NTF shedding by 58.4% (black bars: GM6001 vs. CTRL, p < 0.05, n = 5), which subsequently increases Cad6B retention on CHO cell membranes by 72.8% (gray bars: GM6001 vs. CTRL, p < 0.05, n = 3). 18 hours treatment with 25 μM MMP2/MMP9 Inhibitor V also significantly inhibited NTF shedding by 52.1% (black bars: INH V vs. CTRL, p < 0.05, n = 5), which led to a 30.3% increase in Cad6B membrane retention (gray bars: INH V vs. CTRL, p = 0.065, n = 3). As a positive control, MMP2 was transiently transfected into Flp-In CHO Cad6B-HA cells to increase Cad6B shedding. Increasing supernatant MMP2 levels resulted in a 30.5% increase in 88 kDa L-NTF levels (gray bars: MMP2 vs. CTRL, p < 0.05, n = 5) and a significant reduction in CHO cell membrane Cad6B pools (45.9%, black bars: MMP2 vs. CTRL, p < 0.05, n = 3). No other bands were observed upon MMP2 overexpression, indicating that recombinant MMP2 and CHO cell MMP2/MMP9 cleave Cad6B at the same site in vitro. Treatment levels were analyzed by ANOVA, and asterisks denote treatments that are statistically different (p < 0.05). Immunoblots are representatives of independent experiments, and GAPDH served as a loading control for cell lysates. (D) HA-tagged versions of L-NTF and S-NTF constructs in Fig. S2 express secreted, N-linked glycosylated NTFS at similar molecular weights between neural crest cells in vivo and CHO cells in vitro. The HA-tagged long NTF or L-NTF-HA is comprised of the full Cad6B ectodomain including all 5 EC domains, and the fully N-linked deglycosylated forms are comparable in molecular weight when expressed in vivo (74 kDa, bottom left blot) or in vitro (72 kDa, top blot). The HA-tagged short NTF or S-NTF-HA construct, consisting of EC1-EC4.5 domains, expresses a 61 kDa deglycosylated fragment in vivo (bottom right blot), which is comparable in molecular weight to the deglycosylated 62 kDa NTF (top blot) expressed in CHO cells. The differences observed in glycosylated versions of L-NTF-HA expressed in vivo and in vitro (84 kDa versus 93 kDa, respectively) and S-NTF-HA expressed in vivo and in vitro (70 kDa versus 72 kDa, respectively) may be due to minor differences in N-linked glycosylation moieties between chicken neural crest cells and mammalian CHO cells.
Table 1.
Molecular weight analysis of full-length Cad6B and Cad6B NTFs generated in vitro and in vivo*
| Molecular Weight in kDa (Mean) | |||||
|---|---|---|---|---|---|
| Experiments (n) | PNGase F − | PNGase F + | |||
| Protein | Source | (untreated) | (treated) | Difference | |
| Full-length Cad6B ( # in Fig. 2) |
In vivo | 2 | 122 | 106 | 16 |
| In vitro | 1 | 135 | 113 | 22 | |
| In vivo | 2 | 95 | 80 | 15 | |
| Cad6B L-NTF ( + in Fig. 2) |
Rec. L-NTF-HA (In vivo) | 1 | 84 | 74 | 10 |
| In vitro | 4 | 88 | 71 | 17 | |
| Rec. L-NTF (In vitro) | 1 | 92 | 73 | 19 | |
| Rec. L-NTF-HA (In vitro) | 1 | 93 | 72 | 21 | |
| In vivo | 1 | 83 | 70 | 13 | |
| Cad6B S-NTF ( * in Fig. 2) |
Rec. S-NTF-HA (In vivo) | 1 | 70 | 61 | 9 |
| In vitro | 3 | 70 | 56 | 14 | |
| Rec. S-NTF (In vitro) | 1 | 71 | 59 | 12 | |
| Rec. S-NTF-HA (In vitro) | 1 | 72 | 62 | 10 | |
All analyses were performed on 7−10% SDS-PAGE gels.
The presence of numerous Cad6B NTFs during cranial neural crest EMT suggests that full-length Cad6B could be regulated by several proteases cutting at unique sites within the Cad6B N-terminus. Our previous work identified ADAM10 and ADAM19 as two key transmembrane metalloproteinases that limit Cad6B expression within the premigratory neural crest domain in vivo and generate a 70 kDa Cad6B NTF in vitro (Schiffmacher et al., 2014). To ascertain the molecular weight of the N-linked deglycosylated 70 kDa NTF (which could correspond to the in vivo S-NTF), serial Concanavalin A pulldown and PNGase F treatment was performed on conditioned media collected from CHO cells co-transfected with Cad6B-HA and ADAM10 expression constructs. CHO cells lack endogenous cadherins and are thus a cell line of choice for cadherin research (Niessen and Gumbiner, 2002; Paterson et al., 2003). Comparison of untreated and PNGase F treated supernatants reveals that the ADAM10-generated 70 kDa NTF (denoted by *, Fig. 2B, lane 1) is reduced by 14 kDa to 56 kDa (denoted by *, Fig. 2B, lane 2), indicating that the contribution of the N-linked glycosylations to the relative mobility of the ADAM10-shed NTF are comparable to those in NTFs generated by Cad6B proteolysis in vivo. While both ADAM10 and ADAM19 produce NTFs of similar size in vitro, we also noted that, in the absence of recombinant ADAM expression, full-length Cad6B is cleaved by an endogenous CHO cell protease that sheds an 88 kDa NTF (denoted by +, Fig. 2B, lane 3), which is also observed when ADAM10 is co-expressed with Cad6B (denoted by +, Fig. 2B, lane 1). PNGase F treatment reduces the molecular weight of this fragment to approximately 71 kDa (denoted by +, Fig. 2B, lane 2), similar to the Cad6B L-NTF observed via immunoblot in vivo (Fig. 2A, 95 kDa, lanes 1 and 3, +, versus 80 kDa, PNGase F treated in lanes 2 and 4, +). These in vitro results suggest that Cad6B is also likely cleaved by two proteases in vivo before and during EMT, generating the S-NTF through ADAM-mediated proteolysis and the L-NTF through another unidentified protease.
MMPs and ADAMs differentially cleave Cad6B to generate two distinct NTFs in vitro
The Cad6B NTFs generated in vivo and in vitro by recombinant ADAM10 and an unknown endogenous CHO cell protease are highly similar, but not identical, in relative mobilities. We hypothesized that the smallest in vivo 83 kDa Cad6B NTF (Fig. 2A, lanes 1 and 3, *, S-NTF) corresponds to the 70 kDa NTF generated by ADAM10 in vitro (Fig. 2B, lane 1, *). In line with this, the larger in vivo-produced 95 kDa Cad6B NTF (Fig. 2A, lanes 1 and 3, +, L-NTF) may in fact represent the proteolytic product of another protease that is also responsible for shedding the 88 kDa Cad6B NTF in CHO cells (Fig. 2B, lanes 1 and 3, +). Several metzincin proteases have been shown to cleave cadherins to give rise to larger NTFs (~90 kDa) including MMPs (Marambaud et al., 2002; Noe et al., 2001). CHO cells have been previously shown to express the gelatinase MMP9 (Elliott et al., 2003), which may aid in the processing of N-cadherin in neural crest cells (Monsonego-Ornan et al., 2012). MMP2 is also expressed in CHO cells (RT-PCR analysis, our unpublished data) and in chick neural crest cells undergoing EMT (Duong and Erickson, 2004). To identify putative protease candidates responsible for generating Cad6B L-NTF (Fig. 2, +), we performed metalloproteinase inhibition assays in Flp-In Cad6B-HA stable CHO cells, which constitutively express Cad6B from a single genomic integration site (Fig. 2C) (Schiffmacher et al., 2014). With 100% of cells expressing Cad6B from a single allele, we were able to increase ectodomain shedding and improve our ability to detect changes within full-length Cad6B protein that otherwise were not previously observed in high copy transient cotransfections (Schiffmacher et al., 2014). Flp-In Cad6B-HA stable CHO cells were treated for 18 hours with 25 μΜ GM6001, a broad spectrum metalloproteinase inhibitor, or 25 μΜ Inhibitor V, which selectively and irreversibly inhibits the gelatinases MMP2 and MMP9 by binding to MMP active sites (Ikejiri et al., 2005). Following chemical treatment, both conditioned media and cell lysates were collected to analyze changes in Cad6B NTF shedding and full-length Cad6B levels using a Cad6B antibody. As a putative positive control, Flp-In Cad6B stable CHO cells were transiently transfected with an MMP2 expression construct, re-incubated for 24 hours, and replenished with 3 mls fresh media (+ 0.5% DMSO) at the same time when other wells were being treated with inhibitors (time point = 0). Flp-In Cad6B-HA stable CHO cells treated with 0.5% DMSO served as a vehicle control for metalloproteinase inhibition.
GM6001 treatment (25 μM) resulted in a significant 58.4% reduction in Cad6B L-NTF shedding versus the control group (Fig. 2C, p < 0.05, n = 5), and a significant 72.8% increase in relative full-length Cad6B levels versus the control (p < 0.05, n = 3). At the same molar concentration, Inhibitor V treatment yielded a significant 52.1% reduction in Cad6B L-NTF levels at 88 kDa (Fig. 2C, p < 0.05, n = 5), and a modest but statistically insignificant 30.3% increase in relative full-length Cad6B levels (p = 0.065, n = 3). Conversely, MMP2 overexpression decreased full-length Cad6B levels between treatment and control lysates by 45.9% (Fig. 2C, p < 0.05, n = 3), which translated into a 30.5% increase in Cad6B L-NTF shedding (p < 0.05, n = 5). Immunoblot analysis of supernatants following control and chemical treatment confirmed our initial RT-PCR results that MMP2 is expressed in CHO cells and demonstrates the high levels of secreted MMP2 in the MMP2 overexpression control. More importantly, we were unable to detect any additional NTF bands via immunoblot that would indicate that recombinant MMP2 was cleaving Cad6B at another site. Together, these in vitro assays demonstrate that gelatinase MMPs cleave Cad6B at an alternate site that is C-terminal to where ADAMs 10 and 19 proteolytically process Cad6B in vitro (Fig. 2D). Therefore, the in vitro generation of both an 88 kDa NTF by CHO MMPs, and a 70 kDa NTF by recombinant ADAMs 10 and 19, serve as a model to explain rapid proteolysis of Cad6B and generation of 95 kDa L-NTF and 83 kDa S-NTF in vivo in cranial neural crest cells undergoing EMT en masse in the chick midbrain (6ss). Since both S-NTF and L-NTFs are simultaneously generated during EMT, we next investigated their putative functions in vivo.
Constructs expressing Cad6B NTF fragments mimic NTFs generated by ADAM10 and MMPs
Given that we are technically unable to perform Cad6B NTF loss-of-function assays in our model system, we focused our efforts on elucidating EMT stage-specific roles for Cad6B NTFs by employing in vivo gain-of-function assays. To accomplish this, we designed expression constructs that express either untagged or HA-tagged NTFs that would mimic NTFs shed by ADAMs and MMPs in vivo. Untagged L-NTF and S-NTF expression constructs were first designed and analyzed for their ability to express secreted NTFs comparable in molecular weight to NTFs generated via full-length Cad6B proteolysis. The Cad6B L-NTF construct expresses a 92 kDa fragment containing all five extracellular repeat (EC) domains (Fig. S2). An expression construct was also designed to generate S-NTF that corresponds to the cleavage product generated via ADAM10-mediated proteolysis in vitro (Fig. S2 and Fig. 2B, D). A prediction of the S-NTF primary sequence was calculated by comparing the molecular weight of the N-linked deglycosylated S-NTF observed in vitro (55 kDa) with theoretical molecular weights of unmodified mature ectodomain fragments (49 kDa, EC1-EC4; 62 kDa, EC1-EC5) using the Expasy Compute pI/Mw tool (Wilkins et al., 1999). In addition, we employed the MEROPS database to hone in on potential ADAM10 cleavage sites within the EC4 domain by searching for primary sequence enriched with specific amino acids at key positions within probable ADAM10 cleavage sites (see Methods and Materials).
Constructs were first transfected into CHO cells, and collected media possessing these NTFs were analyzed in the presence or absence of PNGase F. L-NTF expression is comparable in relative mobility to the NTF generated via CHO cell MMP mediated-proteolysis in vitro (denoted by +, Fig. S2, 92 kDa band in lane 3 versus 88 kDa band in lane 1). Furthermore, the N-linked deglycosylated L-NTF peptide is similar in relative mobility to the corresponding MMP-shed NTF, which indicates that the recombinant NTF possesses the same degree of N-linked glycosylation as full-length Cad6B in CHO cells (Fig. S2, 73 kDa band in lane 4 versus 71 kDa band in lane 2, +). When expressed in vitro, the recombinant S-NTF (denoted by *, Fig. S2, 71 kDa band in lane 5) was very similar in relative mobility to the ADAM10-cleaved S-NTF (Fig. S2, 70 kDa band in lane 1, *), and PNGase F treatment resulted in comparable reductions in molecular weight (Fig. S2, 59 kDa band in lane 6 versus 56 kDa band in lane 2, respectively, *).
DNA constructs with C-terminal HA tags were also designed to express NTF-HA variants mimicking the ADAM10- and CHO cell MMP-produced NTFs observed in vitro (Figs. 2B, D). In addition, the same constructs were electroporated independently into chick premigratory neural crest cells at the 3ss and electroporated neural folds were collected, pooled, and lysed six hours later at 7ss. Interestingly, HA-tagged NTFs expressed in vivo were very similar in molecular weight to NTFs generated in CHO cells (Fig. 2D, Table 1). Both L- and S-NTFs, however, did not recapitulate the molecular weights of the endogenous NTFs detected in Fig. 2A (L-NTF: 84 kDa versus 95 kDa, respectively; S-NTF: 70 kDa versus 83 kDa, respectively). These differences in molecular weight suggest that the Cad6B ectodomain may possess additional post-translational modifications that are not imparted on NTFs during recombinant NTF expression in vivo. Together, these in vitro data demonstrate that each construct expresses NTFs that are secreted and N-linked glycosylated to an extent shared by the full-length Cad6B ectodomain, and, notably, sufficiently mimic the S-NTF and L-NTF generated via Cad6B proteolysis.
Cad6B NTF overexpression in vivo reduces laminin and fibronectin levels in the dorsal neural tube basement membrane
Our previous studies demonstrated that Cad6B protein is continuously processed by ADAM10 and ADAM19 prior to, and during, EMT, and that the intracellular Cad6B CTF2 plays a distinct role in modulating EMT effector gene expression as long as Cad6B levels are maintained. We hypothesized that Cad6B NTFs also play a unique role in regulating EMT, but unlike Cad6B CTF2, NTF function may not be entirely cell-autonomous given that the Cad6B NTFs are shed peptides. As such, Cad6B NTFs may contribute to the coordination of EMT that occurs within the entire population of Cad6B-positive cranial neural crest cells. To ascertain possible functions for Cad6B NTFs, we performed gain-of-function assays in vivo using HA tagged NTF plasmids and assessed changes in the basement membrane by examining laminin (Fig. 3) and fibronectin (Figs. 3, S3) during EMT stages.
Figure 3. Overexpression of either L-NTF or S-NTF in cranial neural crest cells results in precocious laminin and fibronectin breakdown and premature delamination.

(A-C’’) Representative transverse sections taken through the midbrain region of 8–9ss embryos expressing L-NTF-HA (A–A’’), S-NTF-HA (B-B’’), or GFP (C-C’’), followed by immunostaining for HA or GFP (green), laminin (red), and Snail2 (violet). (A-C, left column) Low magnification (20×) images display merged triple-label immunohistochemistry with nuclear DAPI stain (blue). (A’-C’, middle column) Higher magnification images of dorsal neural tubes from section images in (A-C) demonstrating HA (or GFP) and laminin co-localization. (A’’-C’’, right column) The same images in (A’-C’) but showing laminin and Snail2 co-localization. Dotted lines in (A’, A’’, B’, B’’, C’, C’’) mark dorsal neural tube midlines created by fusing neural folds. Arrowheads in (A’, B’) indicate marked reductions in laminin immunoreactivity as well as laminin “patchiness.” The scale bar in (A) is 50 μm and applicable to (B, C), while the scale bar in (A’) is 20 μm and applicable to (A’’, B’, B’’, C’, C’’). (D) Representative line scans of laminin immunostaining intensity along the basement membranes of the L-NTF-HA-expressing side in (A-A’’, red) and the unelectroporated contralateral side in (A-A’’, blue) in a medial (0 μm) to lateral (45 μm) direction. (E) Basement membrane levels of laminin and fibronectin (for fibronectin images, see Fig. S3) for each section (treatment and control sides) were quantified by generating ratios of the areas under the curve between treatment (L-NTF-HA, S-NTF-HA, GFP, or wildtype (WT)) line scans versus the unelectroporated, contralateral side (see D). Ratios lower than one indicate a reduction in laminin or fibronectin. At least 15 symmetric sections (see Section 2) originating from at least three embryo midbrains were analyzed per treatment group. Both S-NTF-HA and L-NTF-HA overexpression caused significant reductions in both laminin (33% and 32% reductions, respectively, p < 0.05) and fibronectin (23% and 21% reductions, respectively, p < 0.05) levels versus GFP controls. GFP ratios were not significantly different from wildtype controls. Treatment levels were analyzed by ANOVA, and asterisks denote treatments that are significantly different to GFP controls (p < 0.05).
The rapid dynamics of basement membrane degradation within the midbrain dorsal neural tube (3 hours, 6ss to 8ss) results in unique fibronectin and laminin patterning at each successive somite stage (Fig. 1, Fig. S1). Furthermore, basement membrane distribution changes significantly along the anterior-posterior axis of the head within a single embryo during EMT between the 6ss-8ss. In the context of delamination, these two observations confound any interpretation of basement membrane phenotypes when making embryo-to-embryo comparisons between treated and control embryos. To overcome these limitations, we employed a unilateral in ovo electroporation approach to target one side of the premigratory neural crest domain for L-NTF-HA, S-NTF-HA, and GFP overexpression while contralateral, unelectroporated sides served as stage- and axial level-matched controls (Schiffmacher et al., 2014). Embryos were electroporated at the 3ss to sufficiently target the premigratory neural crest domain prior to neural fold fusion and were collected eight hours later at the 8ss to determine the effects of NTF overexpression on basement membrane integrity. Performing our analysis at the 8ss allows electroporated neural crest cells sufficient time to express NTFs, undergo EMT, and migrate along the neural tube basement membrane (Fig. 1E). Furthermore, the medial basement membrane region is less patchy at the 8ss than at prior somite stages (Duband and Thiery, 1982, 1987), which allows for better assessment of extracellular matrix protein levels along the continuous neural tube basement membrane.
We performed laminin or fibronectin immunostaining on transverse tissue sections of electroporated embryos and observed a wide range of phenotypes involving changes in basement membrane protein levels and delamination (Fig. 3 and Fig. S3). The most consistent phenotypes observed within dorsal neural tubes containing S-NTF- or L-NTF-expressing cells, as compared to the contralateral, unelectroporated control side, were a decrease in laminin and fibronectin immunoreactivity and increased instances of discontinuous protein (“patchiness”) (Figs. 3A’, 3B’, S3A’, S3B’ arrowheads). Within a few transverse sections, NTF-positive neural tube cells were observed precociously delaminating through the overlying basement membrane (data not shown). Interestingly, we observed no change in the number of neural crest cells migrating from the DNA-electroporated side versus the contralateral unelectroporated side for all S-NTF-HA, L-NTF-HA, or GFP treatments (mean ratios of cell counts (electroporated/unelectroporated); GFP: 0.99; S-NTF-HA: 1.05; L-NTF-HA: 1.01).
To quantify laminin and fibronectin loss, we performed line intensity scans of laminin or fibronectin immunostaining within midbrain transverse sections along the basement membranes from the midline laterally to where neural tube curvature begins (see Fig. 3D showing line scans for laminin from the L-NTF-HA transverse section electroporated side (red line) and contralateral control side (blue line) in Fig. 3A’). The sums of pixel intensities along each equidistant line intensity scan were compared between unelectroporated contralateral sides and electroporated sides to obtain a ratio for each section (electroporated/unelectroporated) (Fig. 3E). Transverse section symmetry was also assessed to ensure asymmetrically-sectioned embryos were not included in the analysis (see Methods and Materials). For unelectroporated wildtype embryo and GFP-overexpressing controls, ratio means were close to 1 (Fig. 3E, laminin: 0.94 and 1.07, respectively, p = 0.6; fibronectin: 1.03 and 0.99, respectively, p = 0.7) and were not significantly different from each other, signifying that basement membrane protein levels were not altered between sides of the midbrain neural tube. For both S-NTF-HA and L-NTF-HA overexpression, however, mean ratios for both laminin and fibronectin were significantly decreased from both wildtype and GFP-overexpressing controls, indicating that laminin and fibronectin levels were markedly reduced in the basement membrane in contact with S-NTF- or L-NTFpositive neural crest cells (Fig. 3E, laminin: 33% and 32% reduction from GFP, respectively, p < 0.05; fibronectin: 23% and 21% reduction from GFP, respectively, p < 0.05). Interestingly, mean ratios for S-NTF-HA and L-NTF-HA treatment groups (for both laminin and fibronectin) were not statistically different from each other (Fig. 3E, p = 0.95), indicating that both NTF variants function to regulate both basement membrane proteins. Collectively, these results suggest that in cranial neural crest cells, Cad6B cleavage by multiple proteases during EMT generates functional NTFs that play roles in reducing laminin and fibronectin, promoting subsequent delamination of neural crest cells from the overlying neural tube basement membrane.
Neural crest cells overexpressing Cad6B L-NTFs possess increased MMP2 activity
An ectopic reduction in laminin and fibronectin within the basement membranes of electroporated neural crest cells secreting elevated levels of Cad6B NTFs suggests that Cad6B NTFs play a role in increasing proteolytic breakdown of extracellular matrix components. Chick cranial neural crest cells express a range of proteases that may process laminin and fibronectin during EMT to promote basement membrane degradation, including ADAM10, MMP16, and MMP subfamily gelatinases MMP2 and MMP9 (Christian et al., 2013; Duong and Erickson, 2004; Monsonego-Ornan et al., 2012; Roth et al., 2017; Schiffmacher et al., 2014). Due to the high degree of efficiency with which MMPs process laminin, we hypothesized that elevated Cad6B L-NTF levels accumulating in the extracellular space induce localized MMP activity. To this end, we performed gelatin zymography with supernatants collected from electroporated neural crest explants to assess gelatin degradation. Explants were cultured in DMEM/N2 buffer, which does not possess any inherent gelatinase activity, for 24 hours to allow proteases to accumulate to detectable levels (Duong and Erickson, 2004). Gelatin zymography analysis indicated significant proteolytic activity at 72 and 62 kDa, which correspond to the pro- and active form of MMP2, respectively (Fig. 4). Lower molecular weight bands observed on the zymogram have been reported previously with respect to MMP2 in the trunk neural crest (Duong and Erickson, 2004) and likely represent MMP2 breakdown products that still retain proteolytic activity (see (Fridman et al., 1992; Toth et al., 2012)). Furthermore, the gel proteolysis pattern of control GFP- and L-NTF overexpressing explant cultures aligned at equal relative mobilities with degradation patterns elicited by recombinant MMP2-overexpressing explants, which were included as positive zymography controls, further confirming the identity of MMP2 as the protease. By analyzing the inverse pixel intensity of the bands corresponding to MMP2 activity at 72 kDa, we found that cumulative (recombinant plus endogenous) MMP2 secretion resulted in significantly increased gelatin degradation over endogenous MMP2 secretion observed in the GFP explant culture (9.8 +/− 1.9-fold increase, p < 0.05, n = 5). Importantly, in explant cultures containing neural crest cells secreting elevated levels of Cad6B L-NTFs, we also detected an increase of endogenous MMP2 activity over GFP controls (2.0 +/− 0.26-fold increase, p < 0.05, n = 5). Interestingly, while we detected a faint band at 92 kDa in our zymogram, which could correspond to the pro-form of MMP9, we did not note any activity at 82–84 kDa, the molecular weight for the mature form of MMP9 (Shapiro et al., 1995), which reportedly plays a role in processing N-cadherin and laminin during hindbrain and trunk neural crest cell migration (Monsonego-Ornan et al., 2012). Altogether, these data indicate that elevated levels of Cad6B NTF secretion increase endogenous MMP2 activity in the cranial neural crest.
Figure 4. Cad6B L-NTF-HA overexpression increases MMP2 activity ex vivo.
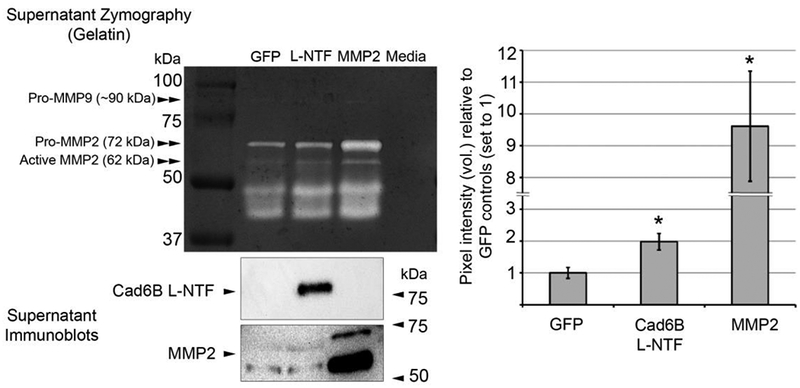
3–4ss embryo neural folds containing premigratory neural crest cells electroporated unilaterally with L-NTF-HA or GFP expression plasmids were excised, pooled, and incubated ex vivo for 48 hours. Conditioned supernatants were collected from the last 24 hours of incubation and tested for recombinant L-NTF and MMP2 expression (immunoblot) and protease activity (gelatin zymography). Immunoblots were performed to ensure that explants were sufficiently electroporated and provide a qualitative, not quantitative, assessment of MMP2 levels after L-NTF or MMP2 overexpression in this assay. Elevated recombinant L-NTF levels in the supernatant resulted in a 2.0 (+/− 0.26)-fold increase in gel degradation at 72 kDa vs. GFP control supernatants, which corresponds to MMP2-mediated degradation. Conditioned supernatants collected from positive control MMP2-overexpressing explants led to a 9.8 (+/− 1.9)-fold increase vs. GFP control supernatants. Asterisks denote a significant difference from GFP (p < 0.05, n = 5). In addition to pro- and active forms of MMP2, faint gelatinase activity was also noted at ~ 90 kDa, indicating possible activity by MMP9. Degraded MMP2 with intact catalytic sites, and possibly other lower molecular weight gelatinases, account for the activity observed between 37 and 50 kDa.
Cad6B NTFs do not increase localized MMP2 activity through upstream transcriptional regulation
The mechanism(s) by which cadherin NTFs increase MMP2 activity has not been well studied in neural crest cells or in other model systems. One report demonstrated that treatment of a human bronchial epithelial cell line with conditioned media containing soluble E-cadherin (E-cadherin NTFs) transcriptionally up-regulates MMP2, MMP9, and MT1-MMP/MMP14 (Nawrocki-Raby et al., 2003). In line with this study, we hypothesized that secreted Cad6B NTFs function as a co-receptor/ligand that initiates signal transduction and subsequent downstream transactivation of MMP2. To explore this possibility, we performed quantitative PCR (QPCR) for MMP9, MT2-MMP/MMP15, MT3-MMP/MMP16, and TIMP2 on cDNA prepared from pooled cranial neural crest cells excised from electroporated embryos (Fig. 5). MMP9, MT2-MMP/MMP15, MT3-MMP/MMP16, and TIMP2 were included in our analysis as their protein products posttranslationally regulate MMP2 levels in neural crest cells and their transcriptional up-regulation could indirectly explain our observed increases in MMP2 activity (Fig. 4) (Cantemir et al., 2004; Christian et al., 2013; Monsonego-Ornan et al., 2012; Patterson et al., 2013; Roth et al., 2017).
Figure 5. Cad6B NTFs do not induce MMP2 levels through transcriptional regulation in cranial neural crest cells in vivo.
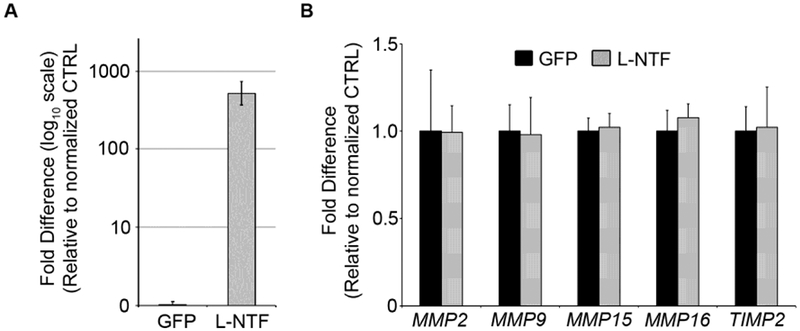
(A) Embryos were unilaterally electroporated with GFP (black) or L-NTF (gray) DNA expression constructs at the 3ss and incubated for an additional 5 hours when neural crest cells are undergoing EMT en masse and dorsal neural tube basement membrane is being degraded (6–7ss). Excised neural crest cells (five embryos per replicate) were pooled and lysed for RNA extraction, followed by generation of cDNA. (A) Electroporation efficiency and relative expression levels of L-NTF were assessed using primers designed to amplify L-NTF. (B) Gene levels were normalized to 18S ribosomal RNA, and the graph shows differences in gene expression as determined by calculating the fold-change from the control group (GFP; arbitrarily set to 1; n = 3).
Embryos were electroporated with either L-NTF-HA or GFP (control) at the 3ss and re-incubated for 5 hours to the 6ss stage when EMT is occurring en masse. We have previously validated the short 5 hour incubation as sufficient time to allow recombinant protein expression and downstream induction of transcriptional target genes (Schiffmacher et al., 2016). cDNAs were generated from pooled electroporated neural folds, and samples were evaluated for L-NTF-HA overexpression via QPCR prior to being included as a replicate (Fig. 5A). At the level of L-NTF-HA expression achieved, transient expression of L-NTF-HA did not induce transcript levels for any of the genes tested (Fig. 5B). Of note, our MMP QPCR analysis is consistent with expression analyses previously performed by other groups who have demonstrated the protein products of MMP9 and MMP2 upstream regulators in neural crest or in tissues with which neural crest cells make contact (Cai et al., 2000; Cantemir et al., 2004; Monsonego-Ornan et al., 2012; Patterson et al., 2013; Roth et al., 2017). Collectively, these findings suggest that MMP2 activity in our explants was not augmented due to direct MMP2 transcription or indirectly via an increase in various MMPs that function upstream to regulate MMP2.
Shed Cad6B NTFs physically interact with MMP2
Our current data reveal that Cad6B NTFs post-transcriptionally control MMP2 activity. To gain further insight into this mechanism, we performed co-immunoprecipitation experiments to assess whether MMP2 and shed Cad6B NTFs could biochemically interact. In order to overcome the inability of the Cad6B antibody (CCD6B-1) to sufficiently immunoprecipitate Cad6B protein in vitro, we generated a modified Cad6B expression construct containing an HA tag in the N-terminal ectodomain of Cad6B (HA-Cad6B). The insertion of the HA tag into the linker region between EC2 and EC3 does not alter N-linked glycosylations (unpublished) or expression (see Fig. 6 input panel), but does disrupt the CCD6B-1 epitope, rendering this antibody unable to bind Cad6B in immunoassays. The HA-Cad6B construct was cotransfected into chick LMH cells with recombinant chick MMP2. LMH cells were chosen over CHO cells as the model system so that recombinant protein interactions were occurring in a chick proteomic background (in case other chick-specific proteins are required to facilitate an NTF/MMP2 interaction). Furthermore, the antibody used to detect chick MMP2 binds to it with low affinity and can only detect high levels of recombinant chick MMP2 overexpression. This strategy eliminates any misinterpretation of potential MMP2 co-immunoprecipitations as either pulldown contamination or co-immunoprecipitations of endogenously expressed MMP2 (as in the case with CHO cell MMP2, which binds to the MMP2 antibody with greater affinity). Interestingly, LMH cells express both MMP2 and MMP9, as assessed by RT-PCR (not shown), but our MMP2 antibody does not bind sufficiently to allow for endogenous MMP2 detection. Our data indicated that LMH cells proteolytically process HA-Cad6B to liberate HA-tagged NTFs that can bind recombinant MMP2 (lane 1, anti-HA IP). This interaction was only observed in the presence of both recombinant MMP2 and HA-tagged full-length Cad6B, and not detected in the ‘HA-Cad6B only’ transfections, although cleavage of HA-tagged full-length Cad6B occurred in the absence of exogenous MMP2 (lane 2, anti-HA IP). Taken together, these results reveal that Cad6B NTFs can interact with MMP2 in vitro and suggest that MMP2 binding to Cad6B NTFs shed in vivo could be critical to increasing localized MMP2 activity.
Figure 6. L-NTFs generated via LMH cell-mediated proteolysis of full-length HA-Cad6B physically associate with recombinant MMP2.
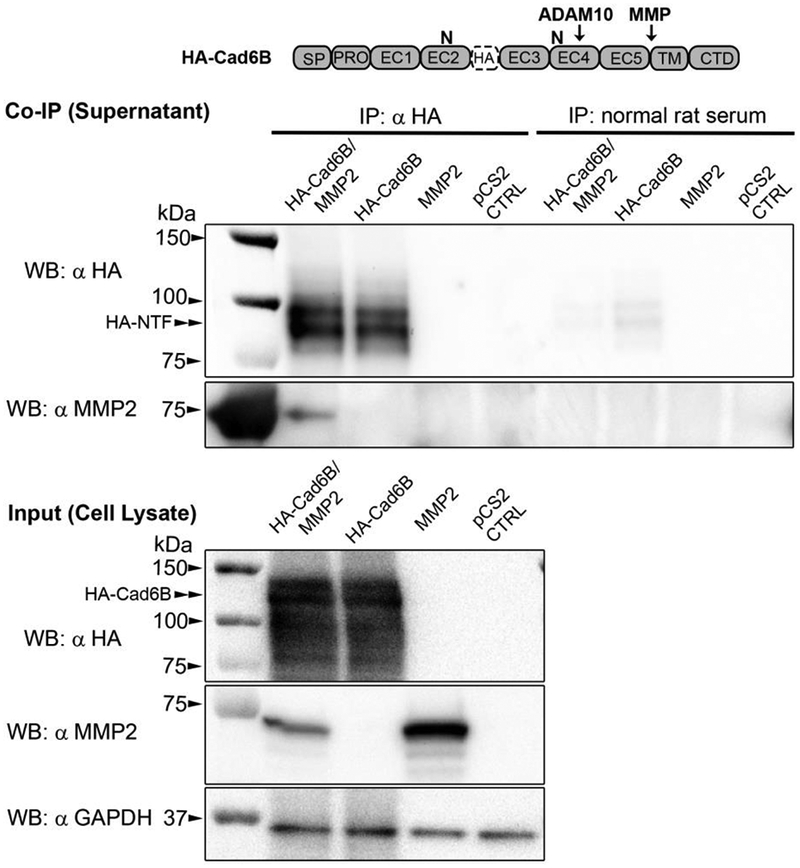
LMH cells were co-transfected with HA-tagged full-length Cad6B and chick MMP2 expression plasmids. Single transfections of HA-Cad6B or MMP2 were also included as co-immunoprecipitation controls. Following overnight transfections, cells were grown in low serum media for an additional 24 hours. Conditioned medias were then subjected to HA antibody pulldown and immunoblotting for HA and MMP2 (serially probed on the same immunoblot). HA immunoblots indicate that cleaved HA-tagged L-NTFs accumulate in the media and are sufficiently immunoprecipitated only in supernatants of cells transfected with HA-Cad6B. No full-length HA-Cad6B was detected in any respective supernatants. Pulldown of HA-tagged L-NTFs from ‘HA-CAD6B only’ control demonstrate that most of the L-NTFs are generated by endogenous LMH proteases (LMH cells express both MMP2 and MMP9, unpublished). Serial immunoblotting with an MMP2 antibody reveals that recombinant 72 kDa MMP2 is co-immunoprecipitated with HA-tagged NTFs. Normal rat serum immunoprecipitations were performed as negative controls for each treatment. Immunoblotting of treatment cell lysates serve as treatment inputs as well as qualitative transfection controls for HA-Cad6B and MMP2. GAPDH immunoblotting was also performed as a loading control between treatments.
DISCUSSION
The coordinated EMTs that occur in cranial neural crest cells of the chick are unique from EMTs occurring in chick trunk neural crest cells and other animal models. A significant distinction is that chick cranial neural crest cell EMT is initiated at a very precise moment in embryonic development that takes place after neural crest specification, neural fold elevation and fusion, and, on the whole embryo level, at a specific somite stage (~ 6ss). Following specification, the premigratory neural crest gene regulatory network implements a transcriptional program that directs EMT by modulating hundreds of downstream effector genes (Bronner and Simoes-Costa, 2016). This core of EMT regulators, in concert with factors contributing to epigenetic modifications, is thought to control the range of cellular mechanisms that contribute towards the successful completion of EMT (Hu et al., 2014; Nieto et al., 2016). As the transcriptional and epigenetic networks initiate the EMT program, additional layers of regulatory input from outside the nucleus contribute to the robustness and temporal coordination of EMT, including the delamination of neural crest cells. These inputs create feedback loops and can affect the EMT regulatory network.
Our collective studies illustrate how the presence and activity of different proteases, along with expression of specific substrates (Cad6B) at the neural crest cell membrane, can generate multifaceted, regulatory feedback into more than one mechanistic component that is part of the EMT program (Fig. 7). Full-length Cad6B is a known substrate of ADAM 10 and ADAM 19 within the premigratory neural crest domain, and knockdown of either ADAM in vivo results in increased apicolateral retention of Cad6B protein (Schiffmacher et al., 2014). Morpholino-mediated knockdown of Cad6B just prior to EMT increases the proportion of the cranial neural crest cell pool becoming emigratory at once, indicating that Cad6B functions to maintain neural crest cell adhesion (Coles et al., 2007). In this regard, experimental depletion of Cad6B levels at EMT stages mimics the endogenous mechanism of ADAM-mediated Cad6B cleavage. Proteolytic processing of Cad6B imparts a second regulatory input into EMT by generating a soluble intracellular Cad6B CTF2 peptide that complexes with β-catenin to transcriptionally control genes involved in directing EMT, including Snail2 (Schiffmacher et al., 2016). Now we provide evidence for a third downstream regulatory input that contributes to the regulation of neural crest cell delamination. Shed Cad6B NTFs function to increase localized MMP2 activity to process extracellular substrates such as basement membrane components (laminin, Fig. 3; fibronectin, Fig. S3) and potentially full-length Cad6B as well (Fig. 7). Moreover, multiple Cad6B NTFs of varying molecular weights are present during EMT stages, indicating differential cleavage by both ADAMs and MMPs (Fig. 2).
Figure 7. Cad6B proteolysis and its impact on EMT.
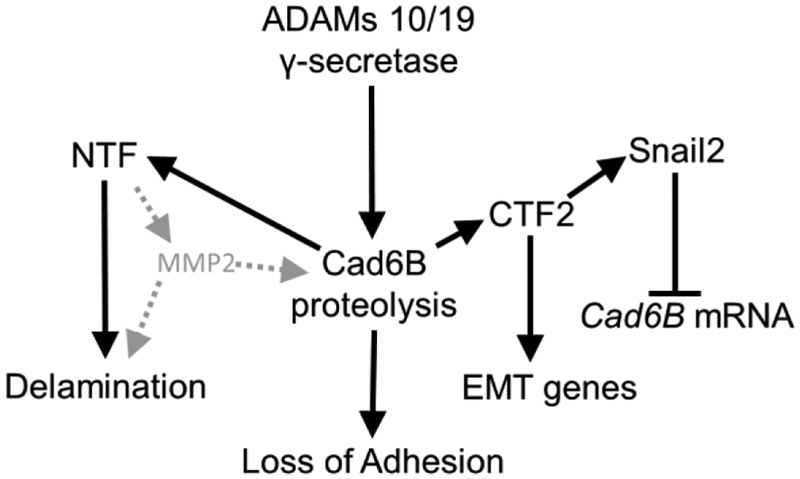
Cad6B proteolysis from multiple proteases during EMT contributes several downstream regulatory inputs into the neural crest EMT program including loss of cell adhesion (downregulation of full-length Cad6B, (Schiffmacher et al., 2014)), transcriptional regulation of EMT genes including Snail2 (increased levels of Cad6B CTF2, (Schiffmacher et al., 2016), and breakdown of basement membranes contributing to delamination (increased levels of shed Cad6B NTFs).
Our data do not exclude the possibility that Cad6B is processed by additional proteases expressed in the chick midbrain (MMP9, MT2-MMP/MMP15, MT3-MMP/MMP16; Fig. 5). Given the rapid morphological and cellular changes in the dorsal neural tube at EMT, multiple proteases may regulate EMT, with each one processing specific substrates at defined times. For example, ADAM10 protein is restricted to the apical region of premigratory neural crest cells and therefore is likely not the primary protease that cleaves Cad6B within depolarized neural crest cells located away from the midline (Fig. 1C’, arrows, 6ss). At early EMT stages, however, ADAM19 is highly expressed within newly emigratory neural crest cells exiting the dorsal neural tube, and is likely processing any remaining membrane Cad6B to generate SNTFs as neural crest cells penetrate through the fused basement membranes (Fig. 1D’, D’’, arrowheads). Within this neural crest subdomain, ADAM19 and MMP2 may both process Cad6B as a shared substrate, yet generate two distinct Cad6B NTFs. Thus, the proteases acting on Cad6B to yield NTFs may depend upon the developmental stage of the embryo and specifically where neural crest cells are in the EMT process (e.g., polarized neural crest cells still attached to the basement membrane versus depolarized neural crest cells detached from the basement membrane; Fig. 1). Interestingly, our immunohistochemistry experiments for Cad6B use the only commercially available Cad6B antibody, which was raised to the Cad6B extracellular domain (Nakagawa and Takeichi, 1998). Based on our initial experiments (Fig. 6), this epitope resides within EC2 and EC3 (our unpublished data); therefore, full-length Cad6B, along with NTFs generated by ADAMs and MMPs, would be recognized by the antibody. As such, the Cad6B protein distribution documented at the developmental stages shown herein represents the combination of full-length Cad6B and shed Cad6B NTFs (Fig. 1). Nevertheless, our immunohistochemistry data are in good agreement with what we have observed in immunoblots of chick neural crest at specific developmental stages (examining both full-length Cad6B and shed NTFs; herein, (Schiffmacher et al., 2014), and (Schiffmacher et al., 2016)) and at the transcript level for Cad6B by in situ hybridization (Dady and Duband, 2017; Fairchild and Gammill, 2013; Schiffmacher et al., 2014).
Our in-gel zymography results on neural crest explant supernatants indicate that MMP2 activity is positively regulated by the presence of Cad6B NTFs (Fig. 4). Lower molecular weight bands (37–50 kDa) on the zymogram likely correspond to MMP2, but not MMP9, breakdown products. This is due to their size, their increased intensity after NTF and MMP2 overexpression, and the fact that we do not detect the active form of MMP9 on the zymogram (82–84 kDa; (Shapiro et al., 1995)), unlike what is noted in the chick hindbrain and trunk, where MMP9 regulates N-cadherin and laminin during neural crest migration (Monsonego-Ornan et al., 2012). Alternatively, these lower molecular weight bands could represent the activity of other gelatinases that have yet to be identified in neural crest cells (e.g., MMP3; pro-form: 54–59 kDa, active form: 47.5k Da; (O’Brien et al., 2007)). We were unable to identify MMP3 through QPCR conducted on chick midbrain neural crest-derived cDNA, however, suggesting that it may not be present, although further confirmation of this is necessary through the use of MMP3-expressing tissue as a template.
Prior immunohistochemical analysis revealed MMP2 protein within the extracellular space between fusing ectoderm and neural tube basement membranes from the 4ss (HH8) and the 8ss (HH9+) stages (Cai et al., 2000). It is possible that those leading emigratory neural crest cells still maintaining Cad6B expression (Fig. 1C’’, arrowheads) and able to shed NTFs come into contact with localized sources of MMP2, whereby these shed Cad6B NTFs (generated by either ADAMs or MMP2) augment the ability of MMP2 to degrade basement membrane proteins including laminin, fibronectin, many collagen types, aggrecan, and vitronectin (Christian et al., 2013). Cad6B NTFs may also be interacting with MMP2 in a cell autonomous fashion, as MMP2 has been shown to be secreted by trunk and cardiac neural crest cells in vitro, in accordance with our own midbrain neural crest explant data (Fig. 4; (Cantemir et al., 2004; Duong and Erickson, 2004)). We are currently limited in our ability to assess MMP2 and Cad6B protein colocalization within chick dorsal neural tubes at the 6–8ss due to technical issues with commercially available MMP2 antibodies that do not work in chick tissue by immunohistochemistry. Nonetheless, our current data point to a role for secreted Cad6B NTFs in enhancing localized MMP2 activity to degrade fibronectin and laminin.
Our QPCR data rule out changes in MMP2, or genes encoding upstream regulators of MMP2, at the level of transcription, strongly suggesting that post-transcriptional mechanism(s) are employed instead to increase MMP2 activity in the presence of Cad6B NTFs. We hypothesize that cleavage of full-length Cad6B by an MMP, such as MMP2, generates an L-NTF that enhances MMP2 activity through a biochemical interaction between L-NTF and MMP2. To test this hypothesis, we performed co-immunoprecipitation experiments that revealed a biochemical interaction between shed Cad6B L-NTFs and MMP2 in vitro. Intriguingly, LMH cells express both MMP2 and MMP9 transcripts, the protein products of which could function to cleave full-length Cad6B in this experiment to liberate NTFs. Levels of endogenous pro-MMP2 are not detectable in this system with our current antibody (which recognizes both the pro- and active forms of MMP2), likely due to a reduced binding affinity to the endogenous MMP2 protein and/or the general presence of low levels of MMP2 in these cells, as noted in all lanes of the input blot (Fig. 6). Taken together, we now propose that binding of NTFs to MMP2 could lead to increased localization, extracellular retention or recruitment, as well as stabilization (or decreased turnover) of pro- or active MMP2 protein and/or increased conversion of pro-MMP2 to active MMP2 by its upstream regulators. Future studies will be geared towards dissecting the molecular basis by which Cad6B NTFs control MMP2 and potentially other proteases in the neural crest.
As discussed previously, other MMPs are expressed in the dorsal neural tube and thus could be responsible for processing extracellular matrix components. Our QPCR data indicated that midbrain neural crest-derived cDNAs possess low levels of MMP9 transcripts, although it is important to note that detection of protease transcripts or protein do not automatically correlate with protease activity. In line with our QPCR data, we detected a faint band corresponding to MMP9 activity by gelatin zymography within our neural crest explant supernatants, indicating MMP9 may also contribute to the loss of laminin we observed following overexpression of Cad6B NTFs in midbrain neural crest cells in vivo. Our current data does not discern if MMP2 and MMP9 have differential enzyme activities for Cad6B. In our protease inhibitor assay, MMP2/MMP9 Inhibitor V treatment inhibited the generation of the 88 kDa Cad6B L-NTF (Fig. 2B, lanes 1 and 3, +; C). This selective MMP2/MMP9 Inhibitor V (Ki = 16 nM for MMP2, 180 nM for MMP9; (Ikejiri et al., 2005)) was able to significantly inhibit Cad6B proteolysis at the same molar concentration used for GM6001, even though GM6001 is a more potent inhibitor of MMP2 and MMP9 (Ki = 0.5 nM for MMP2, 0.2 nM for MMP9; (McNulty et al., 2009)). Our future experiments will aim to further investigate the link between the observed NTF-mediated increase in MMP2 activity ex vivo with the noted loss of laminin and fibronectin following NTF overexpression in vivo, as well as identify additional proteases that mediate other aspects of EMT.
The identification of more than one Cad6B NTF generated in vivo not only suggests that multiple proteases can process Cad6B at separate cleavage sites, but also calls into question whether the NTFs possess different functions. Our N-linked deglycosylation efforts improved our accuracy in determining the true length of each NTF primary sequence as well as allow for better comparison of molecular weights between in vitro- and in vivo-derived NTFs. Remaining differences in molecular weight between N-linked deglycosylated NTFs and their theoretical molecular weights suggest that Cad6B ectodomains possess other major post-translational modifications such as O-linked mannosylations and O-linked GlcNAcylations (Carvalho et al., 2016). Furthermore, these additional post-translational modifications likely account for the differences in molecular weights between endogenous NTFs generated via proteolysis in vivo (Fig. 2A) and recombinant NTFs overexpressed in vivo (Fig. 2D). Our future experiments will decipher these other in vivo modifications using available endoglycosidases and to determine the amino acid sequence of NTF C-termini.
In our overexpression assays, both S-NTF-HA and L-NTF-HA elicited the same phenotype of diminished laminin and fibronectin levels, indicating that the protein subdomains required for their function are within the first 4 EC domains (Fig. 3). The functions of type II Cadherin-11 NTFs in Xenopus have been extensively studied in the context of neural crest cell migration (Abbruzzese et al., 2015; Mathavan et al., 2017; McCusker and Alfandari, 2009). Xenopus ADAM13 cleaves Cadherin-11 ectodomains C-terminal to the EC3 domain, and this EC1–3 fragment plays a major role in promoting neural crest migration independent of its ability to bind other cadherins. A recent report reveals that Cadherin-11 EC1–3 can bind to receptors from several growth factor families, including ErbB2 (EGF Receptor 2), which leads to AKT phosphorylation and persistence of neural crest cell migration (Mathavan et al., 2017). These results were validated through in vitro studies, which showed that Cadherin-11 EC1–3 enhances the ability of a heterodimer consisting of ErbB2 and ErbB3 to interact with PI3K, a kinase that resides upstream of AKT (Mathavan et al., 2017). Importantly, inhibition of PI3K abrogates neural crest migration, suggesting a critical role for this pathway during migration and providing additional support for the importance of AKT in the Xenopus neural crest, as was shown in another recent study (Bahm et al., 2017). In light of these data, it is intriguing to speculate that Cad6B NTFs could function similarly in chick cranial neural crest to mediate activation of a PI3K/AKT pathway, but instead leading to expression of genes required for neural crest cell EMT as opposed to migration. This is not without precedence, as PI3K/AKT signaling has been well documented to induce EMT through multiple mechanisms, including increases in MMPs, as has been observed in cancer cells (Wu et al., 2011; Xu et al., 2015; Zuo et al., 2011)
CONCLUSIONS
Our data provide strong evidence for another unique role for a cadherin proteolytic fragment in promoting neural crest cell EMT. While Cad6B CTF2s activate EMT effector genes including Snail2, a repressor of Cad6B transcription, to facilitate EMT, our findings reveal that Cad6B NTFs can function post-transcriptionally. Distinct Cad6B NTFs are generated in the cranial neural crest through both ADAM- and MMP-mediated proteolysis. Cad6B NTF- overexpressing neural crest cells exhibit a reduction in laminin and fibronectin within the neural tube basement membrane, leading to their precocious delamination. Moreover, Cad6B NTF overexpression results in enhanced activity of MMP2, a known protease that can process laminin, fibronectin, and other components of the extracellular matrix. Notably, Cad6B NTFs can physically interact with MMP2, pointing to a possible mechanism by which MMP2 activity could be enhanced. These findings imply, yet again, that a Cad6B proteolytic product (NTFs) can exert feedback regulation on full-length Cad6B, this time at the level of the protein itself, through increases in the activity of a protease (MMP2) that can process Cad6B protein (Fig. 7). Altogether, these results shed additional light on the role of cadherin proteolytic fragments in neural crest cell EMT and further reveal the importance of post-translational regulation within the EMT regulatory program.
Supplementary Material
Representative transverse midbrain sections taken through embryos that have been immunolabeled for Cad6B (green), laminin (red), and fibronectin (violet). DAPI stain labels cell nuclei (blue). (A-D, left column) Lower magnification images depicting triple-label immunohistochemistry (Cad6B/laminin/fibronectin) merged with DAPI. (A’-D’) Higher magnification images of dorsal neural tubes from section images in (A-D), demonstrating laminin and DAPI. (A’’-D’’) The same images in (A’-D’) but showing fibronectin. “Laminin antibody only” immunohistochemistry controls (E, E’) were performed in the presence of its respective secondary antibody (red channel, E) as well as the secondary antibody used for fibronectin (E’) in (A-D). Reciprocal controls for fibronectin immunolabeling were also performed (F, F’). The lack of immunolabeling observed in E’ and F’ demonstrate that neither the laminin antibody nor the fibronectin antibody exhibit any bleed-through activity during imaging or cross reactivity with the unassociated secondary antibody.
NTF expression plasmids designed to express NTFs mimicking NTFs observed in vivo (Fig. 2A) and in vitro (Fig. 2B) produce secreted, N-linked glycosylated NTFs (lanes 3–6) of comparable molecular weight to the L-NTFs (+) and S-NTFs (*) generated via CHO cell MMP and recombinant ADAM 10- mediated Cad6B proteolysis in vitro (lanes 1 and 2). The L-NTF construct, consisting of EC1-EC5 domains, expresses a 92 kDa fragment (+, lane 3) similar in molecular weight to the 88 kDa NTF (+, lane 1) observed after cleavage by a CHO MMP. The S-NTF construct, consisting of EC1-EC4.5 domains, expresses a 71 kDa fragment (*, lane 5) comparable in molecular weight to the 70 kDa NTF (*, lane 1) observed after cleavage by ADAM10. Removal of N-linked glycosylations results in similar band shifts between the cleaved L-NTF (+, lane 2) and recombinant L-NTF (+, lane 4) (17 kDa versus 19 kDa, respectively). Comparable shifts were also observed between cleaved S-NTF (*, lane 2) and recombinant S-NTF (*, lane 6) (12 kDa versus 14 kDa, respectively).
(A-C’’) Representative transverse sections taken through the midbrain region of 8–9ss embryos expressing L-NTF-HA (A-A’’), S-NTF-HA (B-B’’), or GFP (C-C’’), followed by immunostaining for HA or GFP (green), fibronectin (red), and Snail2 (violet). (A-C, left column) Low magnification (20x) images display merged triple-label immunohistochemistry with nuclear DAPI stain (blue). (A’-C’, middle column) Higher magnification images of dorsal neural tubes from section images in (A-C) demonstrating HA (or GFP) and fibronectin co-localization. (A’’-C’’, right column) The same images in (A’-C’) but showing fibronectin and Snail2 co-localization. Dotted lines in (A’, A’’, B’, B’’, C’, C’’) mark dorsal neural tube midlines created by fusing neural folds. Arrowheads in (A’, B’) indicate marked reductions in fibronectin immunoreactivity. The scale bar in (A) is 50 pm and applicable to (B, C), while the scale bar in (A’) is 20 pm and applicable to (A’’, B’, B’’, C’, C’’).
Normalization of candidate gene levels to other reference genes including HPRT1 and GUSB do not result in altered changes in gene expression between GFP and L-NTF overexpression. QPCR results displayed in Fig. 5, which are normalized to 18S RNA levels, were reassessed using reference genes transcribed by RNA polymerase II. These results are in agreement with our findings in Fig. 5.
HIGHLIGHTS.
Neural crest cells reduce tight junctions and basement membrane prior to Cad6B loss.
Cad6B proteolysis generates distinct NTFs of various molecular weights during EMT.
Cad6B NTFs promote delamination through a reduction in laminin and fibronectin.
MMP2 activity is increased in Cad6B NTF-expressing neural crest cells.
Shed Cad6B NTFs and MMP2 physically interact.
ACKNOWLEDGEMENTS
We thank Ms. Karishma Sharma for technical assistance. The authors declare no competing financial interests. This work was supported by grants to L.A.T. (R01DE024217; Research Scholar Grant, RSG-15-023-01-CSM, American Cancer Society) and A.T.S (F32DE022990).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbruzzese G, Becker SF, Kashef J, Alfandari D, 2015. ADAM13 cleavage of cadherin-11 promotes CNC migration independently of the homophilic binding site. Dev Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfandari D, Taneyhill LA, 2018. Cut loose and run: The complex role of ADAM proteases during neural crest cell development. Genesis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H, 2012. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40, W597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahm I, Barriga EH, Frolov A, Theveneau E, Frankel P, Mayor R, 2017. PDGF controls contact inhibition of locomotion by regulating N-cadherin during neural crest migration. Development 144, 2456–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner ME, Simoes-Costa M, 2016. The Neural Crest Migrating into the Twenty-First Century. Curr Top Dev Biol 116, 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai DH, Vollberg TM, Hahn-Dantona E, Quigley JP, Brauer PR, 2000. MMP-2 expression during early avian cardiac and neural crest morphogenesis. Anat Rec 259, 168–179. [DOI] [PubMed] [Google Scholar]
- Cantemir V, Cai DH, Reedy MV, Brauer PR, 2004. Tissue inhibitor of metalloproteinase-2 (TIMP-2) expression during cardiac neural crest cell migration and its role in proMMP-2 activation. Dev Dyn 231, 709–719. [DOI] [PubMed] [Google Scholar]
- Carvalho S, Reis CA, Pinho SS, 2016. Cadherins Glycans in Cancer: Sweet Players in a Bitter Process. Trends Cancer 2, 519–531. [DOI] [PubMed] [Google Scholar]
- Christian L, Bahudhanapati H, Wei S, 2013. Extracellular metalloproteinases in neural crest development and craniofacial morphogenesis. Crit Rev Biochem Mol Biol 48, 544–560. [DOI] [PubMed] [Google Scholar]
- Coles EG, Taneyhill LA, Bronner-Fraser M, 2007. A critical role for Cadherin6B in regulating avian neural crest emigration. Dev Biol 312, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dady A, Blavet C, Duband JL, 2012. Timing and kinetics of E- to N-cadherin switch during neurulation in the avian embryo. Dev Dyn 241, 1333–1349. [DOI] [PubMed] [Google Scholar]
- Dady A, Duband JL, 2017. Cadherin interplay during neural crest segregation from the non-neural ectoderm and neural tube in the early chick embryo. Dev Dyn 246, 550–565. [DOI] [PubMed] [Google Scholar]
- Duband JL, Dady A, Fleury V, 2015. Resolving time and space constraints during neural crest formation and delamination. Curr Top Dev Biol 111, 27–67. [DOI] [PubMed] [Google Scholar]
- Duband JL, Thiery JP, 1982. Distribution of fibronectin in the early phase of avian cephalic neural crest cell migration. Dev Biol 93, 308–323. [DOI] [PubMed] [Google Scholar]
- Duband JL, Thiery JP, 1987. Distribution of laminin and collagens during avian neural crest development. Development 101, 461–478. [DOI] [PubMed] [Google Scholar]
- Duband JL, Volberg T, Sabanay I, Thiery JP, Geiger B, 1988. Spatial and temporal distribution of the adherens-junction-associated adhesion molecule A-CAM during avian embryogenesis. Development 103, 325–344. [DOI] [PubMed] [Google Scholar]
- Duong TD, Erickson CA, 2004. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn 229, 42–53. [DOI] [PubMed] [Google Scholar]
- Elliott P, Hohmann A, Spanos J, 2003. Protease expression in the supernatant of Chinese hamster ovary cells grown in serum-free culture. Biotechnol Lett 25, 1949–1952. [DOI] [PubMed] [Google Scholar]
- Fairchild CL, Gammill LS, 2013. Tetraspanin18 is a FoxD3-responsive antagonist of cranial neural crest epithelial-to-mesenchymal transition that maintains cadherin-6B protein. Journal of cell science 126, 1464–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishwick KJ, Neiderer TE, Jhingory S, Bronner ME, Taneyhill LA, 2012. The tight junction protein claudin-1 influences cranial neural crest cell emigration. Mechanisms of development 129, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman R, Fuerst TR, Bird RE, Hoyhtya M, Oelkuct M, Kraus S, Komarek D, Liotta LA, Berman ML, Stetler-Stevenson WG, 1992. Domain structure of human 72-kDa gelatinase/type IV collagenase. Characterization of proteolytic activity and identification of the tissue inhibitor of metalloproteinase-2 (TIMP-2) binding regions. J Biol Chem 267, 15398–15405. [PubMed] [Google Scholar]
- Gouignard N, Andrieu C, Theveneau E, 2018. Neural crest delamination and migration: Looking forward to the next 150 years. Genesis, e23107. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL, 1992. A series of normal stages in the development of the chick embryo. 1951. Dev Dyn 195, 231–272. [DOI] [PubMed] [Google Scholar]
- Hatta K, Takagi S, Fujisawa H, Takeichi M, 1987. Spatial and temporal expression pattern of N-cadherin cell adhesion molecules correlated with morphogenetic processes of chicken embryos. Dev Biol 120, 215–227. [DOI] [PubMed] [Google Scholar]
- Hu N, Strobl-Mazzulla PH, Bronner ME, 2014. Epigenetic regulation in neural crest development. Dev Biol 396, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Beeton C, 2010. Detection of functional matrix metalloproteinases by zymography. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S, 2005. Potent mechanism-based inhibitors for matrix metalloproteinases. J Biol Chem 280, 33992–34002. [DOI] [PubMed] [Google Scholar]
- Itasaki N, Bel-Vialar S, Krumlauf R, 1999. ‘Shocking’ developments in chick embryology: electroporation and in ovo gene expression. Nat Cell Biol 1, E203–207. [DOI] [PubMed] [Google Scholar]
- Lee RT, Nagai H, Nakaya Y, Sheng G, Trainor PA, Weston JA, Thiery JP, 2013. Cell delamination in the mesencephalic neural fold and its implication for the origin of ectomesenchyme. Development 140, 4890–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK, 2002. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21, 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathavan K, Khedgikar V, Bartolo V, Alfandari D, 2017. The ectodomain of cadherin-11 binds to erbB2 and stimulates Akt phosphorylation to promote cranial neural crest cell migration. PLoS ONE 12, e0188963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker C, Cousin H, Neuner R, Alfandari D, 2009. Extracellular cleavage of cadherin-11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol Biol Cell 20, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker CD, Alfandari D, 2009. Life after proteolysis: Exploring the signaling capabilities of classical cadherin cleavage fragments. Commun Integr Biol 2, 155–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty AL, Weinberg JB, Guilak F, 2009. Inhibition of matrix metalloproteinases enhances in vitro repair of the meniscus. Clin Orthop Relat Res 467, 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsonego-Ornan E, Kosonovsky J, Bar A, Roth L, Fraggi-Rankis V, Simsa S, Kohl A, Sela-Donenfeld D, 2012. Matrix metalloproteinase 9/gelatinase B is required for neural crest cell migration. Dev Biol 364, 162–177. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M, 1995. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development 121, 1321–1332. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M, 1998. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development 125, 2963–2971. [DOI] [PubMed] [Google Scholar]
- Nawrocki-Raby B, Gilles C, Polette M, Bruyneel E, Laronze JY, Bonnet N, Foidart JM, Mareel M, Birembaut P, 2003. Upregulation of MMPs by soluble E-cadherin in human lung tumor cells. Int J Cancer 105, 790–795. [DOI] [PubMed] [Google Scholar]
- Niessen CM, Gumbiner BM, 2002. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J Cell Biol 156, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP, 2016. EMT: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Noe V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M, 2001. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. Journal of cell science 114, 111–118. [DOI] [PubMed] [Google Scholar]
- O’Brien M, O’Shaughnessy D, Ahamide E, Morrison JJ, Smith TJ, 2007. Differential expression of the metalloproteinase MMP3 and the alpha5 integrin subunit in human myometrium at labour. Mol Hum Reprod 13, 655–661. [DOI] [PubMed] [Google Scholar]
- Padmanabhan R, Taneyhill LA, 2015. Cadherin-6B undergoes macropinocytosis and clathrin-mediated endocytosis during cranial neural crest cell EMT. Journal of cell science 128, 1773–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Gumbiner BM, 2010. Cadherin 6B induces BMP signaling and de-epithelialization during the epithelial mesenchymal transition of the neural crest. Development 137, 2691–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AD, Parton RG, Ferguson C, Stow JL, Yap AS, 2003. Characterization of E-cadherin endocytosis in isolated MCF-7 and chinese hamster ovary cells: the initial fate of unbound E-cadherin. J Biol Chem 278, 21050–21057. [DOI] [PubMed] [Google Scholar]
- Patterson RA, Cavanaugh AM, Cantemir V, Brauer PR, Reedy MV, 2013. MT2-MMP expression during early avian morphogenesis. Anat Rec (Hoboken) 296, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Finn R, 2016. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 44, D343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth L, Kalev-Altman R, Monsonego-Ornan E, Sela-Donenfeld D, 2017. A new role of the membrane-type matrix metalloproteinase 16 (MMP16/MT3-MMP) in neural crest cell migration. The International journal of developmental biology 61, 245–256. [DOI] [PubMed] [Google Scholar]
- Schiffmacher AT, Padmanabhan R, Jhingory S, Taneyhill LA, 2014. Cadherin-6B is proteolytically processed during epithelial-to-mesenchymal transitions of the cranial neural crest. Molecular biology of the cell 25, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmacher AT, Xie V, Taneyhill LA, 2016. Cadherin-6B proteolysis promotes the neural crest cell epithelial-to-mesenchymal transition through transcriptional regulation. J Cell Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela-Donenfeld D, Kalcheim C, 1999. Regulation of the onset of neural crest migration by coordinated activity of BMP4 and Noggin in the dorsal neural tube. Development 126, 4749–4762. [DOI] [PubMed] [Google Scholar]
- Shapiro SD, Fliszar CJ, Broekelmann TJ, Mecham RP, Senior RM, Welgus HG, 1995. Activation of the 92-kDa gelatinase by stromelysin and 4-aminophenylmercuric acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP). J Biol Chem 270, 6351–6356. [DOI] [PubMed] [Google Scholar]
- Shoval I, Ludwig A, Kalcheim C, 2007. Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development 134, 491–501. [DOI] [PubMed] [Google Scholar]
- Simoes-Costa M, Bronner ME, 2015. Establishing neural crest identity: a gene regulatory recipe. Development 142, 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl-Mazzulla PH, Bronner ME, 2012. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. The Journal of cell biology 198, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhlmiller TJ, Garcia-Castro MI, 2012. Current perspectives of the signaling pathways directing neural crest induction. Cell Mol Life Sci 69, 3715–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill LA, Coles EG, Bronner-Fraser M, 2007. Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development 134, 1481–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneyhill LA, Schiffmacher AT, 2017. Should I stay or should I go? Cadherin function and regulation in the neural crest. Genesis 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, Mayor R, 2012. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev Biol [DOI] [PubMed] [Google Scholar]
- Toth M, Sohail A, Fridman R, 2012. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods in molecular biology (Clifton, N.J 878, 121–135. [DOI] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF, 1999. Protein identification and analysis tools in the ExPASy server. Methods in molecular biology (Clifton, N.J 112, 531–552. [DOI] [PubMed] [Google Scholar]
- Wu ST, Sun GH, Hsu CY, Huang CS, Wu YH, Wang HH, Sun KH, 2011. Tumor necrosis factor α induces epithelial-mesenchymal transition of renal cell carcinoma cells via a nuclear factor kappa B independent mechanism. Exp Biol Med (Maywood) 236, 1022–1029. [DOI] [PubMed] [Google Scholar]
- Xu W, Yang Z, Lu N, 2015. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr 9, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Gumbiner BM, 1997. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol 13, 119–146. [DOI] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL, 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, 2011. Tools for studying the role of N-cadherin mediated extracellular interaction in neuronal development and function. Cell adhesion & migration 5, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo JH, Zhu W, Li MY, Li XH, Yi H, Zeng GQ, Wan XX, He QY, Li JH, Qu JQ, Chen Y, Xiao ZQ, 2011. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem 112, 2508–2517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative transverse midbrain sections taken through embryos that have been immunolabeled for Cad6B (green), laminin (red), and fibronectin (violet). DAPI stain labels cell nuclei (blue). (A-D, left column) Lower magnification images depicting triple-label immunohistochemistry (Cad6B/laminin/fibronectin) merged with DAPI. (A’-D’) Higher magnification images of dorsal neural tubes from section images in (A-D), demonstrating laminin and DAPI. (A’’-D’’) The same images in (A’-D’) but showing fibronectin. “Laminin antibody only” immunohistochemistry controls (E, E’) were performed in the presence of its respective secondary antibody (red channel, E) as well as the secondary antibody used for fibronectin (E’) in (A-D). Reciprocal controls for fibronectin immunolabeling were also performed (F, F’). The lack of immunolabeling observed in E’ and F’ demonstrate that neither the laminin antibody nor the fibronectin antibody exhibit any bleed-through activity during imaging or cross reactivity with the unassociated secondary antibody.
NTF expression plasmids designed to express NTFs mimicking NTFs observed in vivo (Fig. 2A) and in vitro (Fig. 2B) produce secreted, N-linked glycosylated NTFs (lanes 3–6) of comparable molecular weight to the L-NTFs (+) and S-NTFs (*) generated via CHO cell MMP and recombinant ADAM 10- mediated Cad6B proteolysis in vitro (lanes 1 and 2). The L-NTF construct, consisting of EC1-EC5 domains, expresses a 92 kDa fragment (+, lane 3) similar in molecular weight to the 88 kDa NTF (+, lane 1) observed after cleavage by a CHO MMP. The S-NTF construct, consisting of EC1-EC4.5 domains, expresses a 71 kDa fragment (*, lane 5) comparable in molecular weight to the 70 kDa NTF (*, lane 1) observed after cleavage by ADAM10. Removal of N-linked glycosylations results in similar band shifts between the cleaved L-NTF (+, lane 2) and recombinant L-NTF (+, lane 4) (17 kDa versus 19 kDa, respectively). Comparable shifts were also observed between cleaved S-NTF (*, lane 2) and recombinant S-NTF (*, lane 6) (12 kDa versus 14 kDa, respectively).
(A-C’’) Representative transverse sections taken through the midbrain region of 8–9ss embryos expressing L-NTF-HA (A-A’’), S-NTF-HA (B-B’’), or GFP (C-C’’), followed by immunostaining for HA or GFP (green), fibronectin (red), and Snail2 (violet). (A-C, left column) Low magnification (20x) images display merged triple-label immunohistochemistry with nuclear DAPI stain (blue). (A’-C’, middle column) Higher magnification images of dorsal neural tubes from section images in (A-C) demonstrating HA (or GFP) and fibronectin co-localization. (A’’-C’’, right column) The same images in (A’-C’) but showing fibronectin and Snail2 co-localization. Dotted lines in (A’, A’’, B’, B’’, C’, C’’) mark dorsal neural tube midlines created by fusing neural folds. Arrowheads in (A’, B’) indicate marked reductions in fibronectin immunoreactivity. The scale bar in (A) is 50 pm and applicable to (B, C), while the scale bar in (A’) is 20 pm and applicable to (A’’, B’, B’’, C’, C’’).
Normalization of candidate gene levels to other reference genes including HPRT1 and GUSB do not result in altered changes in gene expression between GFP and L-NTF overexpression. QPCR results displayed in Fig. 5, which are normalized to 18S RNA levels, were reassessed using reference genes transcribed by RNA polymerase II. These results are in agreement with our findings in Fig. 5.