

Abstract
Necroptosis, an inflammatory form of cell death, is initiated by the activation of receptor-interacting protein kinase 3 (RIPK3), which depends on its interaction with RIPK1. Although catalytically inactive, the RIPK3 mutant D161N still stimulates RIPK1-dependent apoptosis and embryonic lethality in RIPK3 D161N homozygous mice. Whereas the absence of RIPK1 rescues RIPK3 D161N homozygous mice, we report that the absence of RIPK1 leads to embryonic lethality in RIPK3 D161N heterozygous mice. This suggested that the kinase domain of RIPK3 had a non-catalytic function that was enhanced by a conformation induced by the D161N mutation. We found that the RIPK3 kinase domain homodimerized through a surface that is structurally similar to that of the RAF family members. Mutation of residues at the dimer interface impaired dimerization and necroptosis. Kinase domain dimerization stimulated the activation of RIPK3 through cis-autophosphorylation. This noncatalytic, allosteric activity was enhanced by certain kinase-deficient mutants of RIPK3, including D161N. Furthermore, apoptosis induced by certain RIPK3 inhibitors was also dependent on the kinase dimerization interface. Our studies reveal that the RIPK3 kinase domain exhibits catalytically independent function that is important for both RIPK3-dependent necroptosis and apoptosis.
INTRODUCTION
The regulation of cell death is a key component of organismal homeostasis. Apoptosis is a form of noninflammatory programmed cell death mediated by caspases that is critical for development and to prevent autoimmune diseases. In contrast, necroptosis is an inflammatory form of regulated cell death that occurs when the caspases are inhibited and results in the release of cytosolic components and likely evolved as a response to pathogens that evade the apoptosis signaling pathway. The necroptotic pathway is initiated by the activation of receptor-interacting protein kinase 3 (RIPK3), which results in the recruitment and phosphorylation of the pseudokinase mixed lineage kinase domain–like protein (MLKL) (1). MLKL phosphorylation (p-MLKL) leads to its oligomerization and recruitment to the plasma membrane, which triggers plasma membrane leakage resulting in cell death and release of intracellular contents (2).
The activation of necroptosis by RIPK3 can be mediated by a related protein kinase, RIPK1. Activation of RIPK1 can also induce apoptosis through interactions with Fas-associated death domain protein (FADD) and is caspase-8–dependent. Although RIPK1 activation is associated with phosphorylation of the RIPK3 activation loop (AL), most studies, however, suggest that RIPK1 does not phosphorylate RIPK3 (1). Rather, it is thought that active RIPK1 uses its RIP homo-typic interaction motif (RHIM) to interact with the RIPK3 RHIM, and this interaction then promotes RIPK3 autophosphorylation and oligomerization through an ill-defined mechanism.
Eukaryotic protein kinases are generally inactive in their resting state and require a change in conformation to achieve proper activation. Activation involves repositioning of the AL to allow access of the catalytic site to adenosine triphosphate (ATP) and permitting the repositioning of the two kinase lobes for catalysis. In most cases, this change in conformation is induced by phosphorylation of the AL. The spine hypothesis posits that the active kinase conformation involves the assembly of two hydrophobic spines, the catalytic spine (C-spine) and regulatory spine (R-spine), which facilitate a conformation competent for catalysis (3). How these spines are assembled and stabilized is unique for each kinase, and illustrate key points of regulation of kinase activity (4). For certain kinases such as the epidermal growth factor receptor (EGFR) and the RAF family of kinases, kinase domain dimerization functions to induce the assembly of the spines and subsequent kinase activation. In these cases, there is asymmetry with regard to function—one of the kinases (the “activator”) induces the active conformation of the second kinase (the “receiver”) in a mechanism that is independent of catalytic activity (5, 6). This concept explains why specific inactivating mutations of BRAF are oncogenic and why certain small-molecule inhibitors to BRAF induce paradoxical downstream activation of the pathway (7–10).
An unexpected function of RIPK3 was discovered when kinase- dead, knock-in mice were generated by substitution of the aspartic acid in the conserved Asp-Phe-Gly (DFG) motif with asparagine. The DFG motif at the start of the AL coordinates divalent cations required for catalysis and forms part of the R-spine. Ripk3D161N/D161N embryos succumb to apoptosis mediated by RIPK1 and caspase-8 unlike Ripk3−/− embryos, suggesting that this mutation confers a gain-of-function activity that is kinase-independent (11). Similarly, ATP-competitive inhibitors of RIPK3 and certain infections can induce apoptosis that is RIPK1-, FADD-, and caspase-8–dependent (12, 13). Although Ripk3D161N/D161N embryonic lethality can be rescued on a RIPK1-deficient background, here, we found that Ripk3D161N/+Ripk1−/− embryos died because of necroptosis. To explain these results, we hypothesized that in the absence of RIPK1, RIPK3 may be activated by RIPK3 D161N in a manner similar to the RAF kinases involving both kinase domain dimerization and allostery.
The RAF and RIPK kinase families are members of the tyrosine kinase–like (TKL) family of serine-threonine kinases. When we compared the crystal structures of BRAF and RIPK3, we found that they both crystallize as dimers with a similar symmetry and a similar dimer interface (14). Mutation of several residues in the dimer interface prevented homodimerization of RIPK3 kinase domain alone and reduced necroptotic signaling, suggesting that dimerization of the RIPK3 kinase domain is important for its function. In addition, using certain kinase-dead mutants of RIPK3, we showed that kinase activity can be stimulated by a mechanism involving allosteric activation and cis-autophosphorylation. This suggests that the mechanism of kinase activation by allosteric dimerization reported for the RAF and EGFR families may be more broadly used than previously appreciated (15).
RESULTS
RIPK1-deficient Ripk3D161N/+ embryos display MLKL-dependent lethality
Unlike RIPK3−/− mice, kinase-dead Ripk3D161N/D161N (DFG to NFG) knock-in mice die at mid-gestation (~E11.5) from unrestrained apoptosis that is both RIPK1- and caspase-8–dependent (Fig. 1A) (11). When Ripk3D161N/+Ripk1+/− were interbred, we did not observe any Ripk3D161N/+Ripk1−/− embryos at E18.5, although all other genotypes were present at expected frequencies (Fig. 1A). These data suggested that Ripk3D161N/+Ripk1−/− embryos are lost because of mid-gestational lethality. Analysis of E12.5 Ripk3D161N/+Ripk1−/− embryos indicated that these embryos developed abnormal yolk sac vasculature compared to littermate controls (Fig. 1B). At E11.5, Ripk3D161N/+Ripk1−/− embryos showed abnormal RIPK3 auto-phosphorylation in many tissues, including placenta and liver, suggesting activation of necroptosis cell death pathways (Fig. 1C). Ripk3D161N/+Ripk1−/− embryos survived to E18.5 when necroptosis was prevented by MLKL deficiency (Fig. 1D). Lethality was dependent on the presence of the wild-type (WT) copy of Ripk3 because Ripk3D161N/−Ripk1−/− embryos survived to E18.5 (Fig. 1E). On the basis of these results, we speculated that RIPK1 loss permitted RIPK3 D161N to activate WT RIPK3 and promote necroptosis. Because RIPK1 uses its RHIM to suppress RIPK3 activation and necroptosis in the neonatal period (16–20), we hypothesized that RIPK3 D161N is a noncatalytic gain-of-function mutant. Similar DFG mutations in the BRAF kinase are oncogenic (7).
Fig. 1. RIPK3+/D161N fetuses on a RIPK1-deficient background exhibit MLKL-dependent lethality.
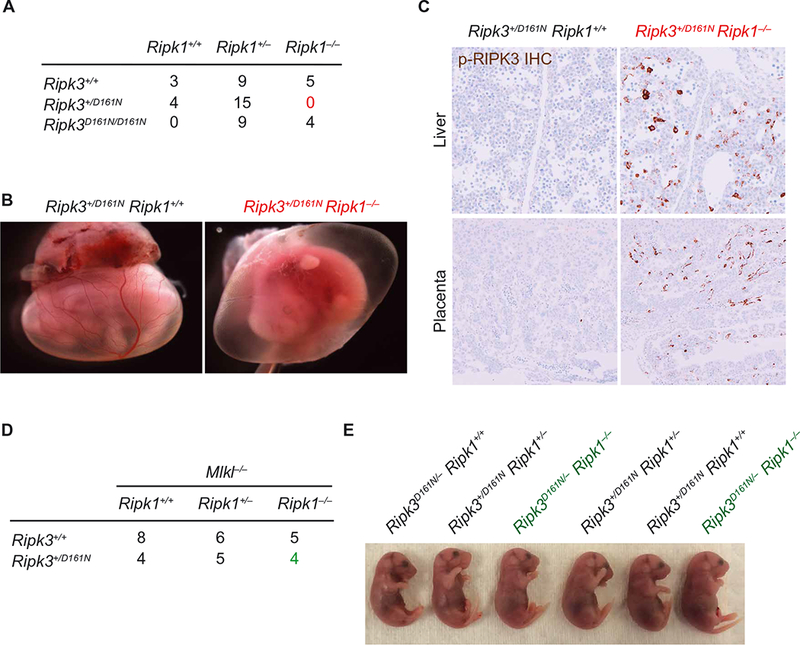
(A) Number of embryonic day 18.5 (E18.5) embryos recovered from intercrossing Ripk1+/− Ripk3+/D161N mice. Data are pooled from the analysis of 10 independent litters. (B) Whole-mount embryo imaging of E12.5 littermates. Images are representative of at least four embryos analyzed of each genotype analyzed between E12.5 and E13.5. (C) Immunohistochemical (IHC) analysis of phosphorylated RIPK3 in tissue sections from E11.5 placenta and liver. Images are representative of greater than three independent controls and three Ripk1−/−Ripk3+/D161N embryos. (D) Number of E18.5 embryos from crossing of Ripk1+/− Ripk3+/D161N Mlkl−/− and Ripk1+/− Ripk3+/+ Mlkl−/− mice. Data are pooled from the analysis of six independent litters. (E) Images of E18.5 littermates recovered from intercrossing Ripk1+/− Ripk3+/D161N and Ripk1+/− Ripk3+/− mice. Images are representative of the analysis of at least two independent litters.
The RIPK3 kinase domain interacts in a side-to-side interface similar to the RAF kinases
Catalytically inactive forms of RAF can allosterically activate WT forms of RAF through a mechanism that is dependent on kinase domain dimerization (6–8, 21). In the crystal structure of mouse RIPK3 [Protein Data Bank (PDB) ID: 4M66] (14), the kinase domain is arranged as a side-to-side molecular dimer. Strikingly, the interface of the mRIPK3 dimer is almost identical to that observed in the dimers formed by BRAF (4MNF) and CRAF (3OMV); superposition of RIPK3 against these structures yields root mean square deviation of 1.56 and 1.64 Å, respectively (Fig. 2A). This similarity suggested that the mechanism of RIPK3 activation might be similar to RAF. The dimer interface of RAF is largely composed of the loop between the αC helix and the β4 strand (6, 22). In BRAF, an arginine residue (R509) is positioned at the tip of the αC-β4 loop, adopting a “handshake pose” with the equivalent arginine on the opposite protomer to stabilize the dimer. The RIPK3 dimer interface showed a similar configuration of the αC-β4 loop with R69 of RIPK3 adopting the same position as R509 in BRAF (Fig. 2A). In addition, several other residues within the αC-β4 loop and outside the αC-β4 loop such as R96 and H156 formed part of the dimer interface of RIPK3 with E71 and E154 on the opposite protomer, respectively (Fig. 2, B to D).
Fig. 2. The RIPK3 kinase domain dimerizes using an interface similar to RAF.
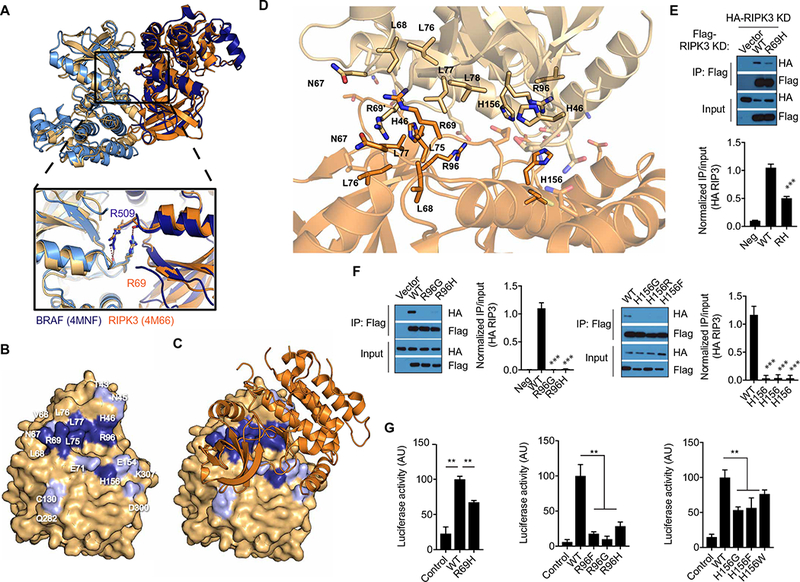
(A) Superposition of RIPK3 (orange; amber; PDB ID: 4M66) and BRAF (4MNF; navy; sky blue) molecular dimers, illustrating conserved dimer surface. Inlay panel shows a magnified view of conserved arginine in molecular handshake across dimer interface. (B) Open-book representation of the RIPK3 dimer interface surface (amber). Residues selected for mutation to validate dimer interface are colored navy blue. Additional residues that contact the opposite kinase (<3 Å) are colored sky blue. Residue numbers are given in white. (C) RIPK3 dimer interface surface with one of the RIPK3 molecules depicted with space-filling model and the second RIPK3 molecule represented as orange ribbon diagram. (D) A close-up view of the RIPK3 dimer interface (PDB ID: 4M66). Residues mutated in this study are represented by sticks and labeled in black. Transparent sticks indicate additional points of contact, which were not mutated in this study. (E and F) Co-immunoprecipitation (IP) analysis of RIPK3 kinase domain (1 to 313 amino acids) interaction in lysates of HEK293T cells cotransfected with pcDNA3-Flag RIPK3 kinase domain and the indicated pcDNA3-HA-RIPK3 kinase domain that were immunoprecipitated for Flag. Blots are representative of at least three independent experiments. Normalized band intensities are means ± SD from all experiments. (G) Split luciferase complementation assay assessment of assessment of RIPK3 dimerization in HEK293T cells transfected with constructs expressing WT RIPK3 kinase domain fused to the C-terminal domain of click beetle luciferase (CBGC-RIPK3 KD) and the indicated RIPK3 kinase domain mutations fused to the N terminus of click beetle luciferase (CBGN-RIPK3 KD). Luciferase activity data are means ± SD combined from at least two independent experiments. **P < 0.01 and ***P < 0.005 by Mann-Whitney test. AU, arbitrary units.
Although the RHIM domains of RIPK1 and RIPK3 mediate homo-and heterodimerization of the full-length proteins, whether there is an additional interaction between the kinase domains is not known. To test whether the kinase domain of RIPK3 can form dimers, we performed size exclusion chromatography using baculovirus-purified mouse RIPK3 kinase domain (S02-H303), which lacks the RHIM domain. At a concentration of 30 μM, RIPK3 migrated with an apparent molecular weight of 72 kDa, suggesting that the RIPK3 kinase domain can form dimers (fig. S1, A and B). However, protein aggregation prevented us from validating this using additional biophysical measurements. To confirm that RIPK3 can form homo-dimers in cells, we generated differentially tagged constructs using the hemagglutinin (HA) and FLAG epitopes that contained only the kinase domain (1 to 313 amino acids) of RIPK3. After their co-expression in human embryonic kidney (HEK) 293T cells, the HA epitope–tagged RIPK3 kinase domain (HA-RIPK3 KD) co- immunoprecipitated with the Flag epitope- tagged RIPK3 kinase domain (FLAG-RIPK3 KD) (Fig. 2, E and F), which indicated that RIKP3 kinase domain dimerized.
We validated that the surface interface determinants observed in the RIPK3 crystal structure were important in mediating kinase domain dimerization by testing the role of R69, R96, and H156 and found that mutating these residues prevented co- immunoprecipitation of HA-RIPK3 KD-WT (Fig. 2, E and F). To further confirm that RIPK3 forms dimers using the interface identified in the crystal structure, we performed split luciferase complementation assays in which the N-terminal (CBGN) and C-terminal (CBGC) fragments of click beetle luciferase were each fused to the RIPK3 kinase domain (23). After co-expression of RIPK3 KD-CBGN and RIPK3 KD-CBGC in HEK293T cells, we detected strong luciferase activity compared to control cells co-expressing CBGC and CBGN fragments not fused to RIPK3 (Fig. 2G and fig. S1, C to E), further confirming that the RIPK3 kinase domain homodimerizes. We then confirmed by luciferase complementation that the R69H, R96F, R96G, R96H, and H156G, H156F, and H156W had decreased luciferase activity compared to WT RIPK3-CBGN, confirming our co-immunoprecipitation results (Fig. 2G). We tested additional mutations at R69 because it is not absolutely conserved in RIPK3, and all, except R69T, exhibited decreased luciferase activity (fig. S1D). Substitution of several other residues predicted to be in the dimer interface with alanine—H46, N67, L68, L75, L76, and L77 (Fig. 2, B and D)—all impaired dimerization to varying degrees (fig. S1E). Collectively, these results indicated that the RIPK3 kinase domain formed dimers in a similar fashion to RAF.
RIPK3 kinase domain dimerization is required for necroptosis
To determine whether kinase domain dimerization is required for RIPK3 function, we reconstituted E1A-immortalized Ripk3−/− murine embryonic fibroblasts (MEFs) with WT and mutated forms of RIPK3 using lentiviral constructs. RIPK3 was co-expressed with green fluorescent protein (GFP) using a P2A peptide linker that allowed transduced cells to be distinguished from nontransduced cells. After transduction, necroptosis was induced by treatment of cells with tumor necrosis factor–α (TNF-α) and the caspase inhibitor z-VAD-fmk (combined treament abbreviated as TZ).
As expected, TZ treatment of cells transduced with WT RIPK3 stimulated cell death and loss of GFP-expressing cells within 24 hours (Fig. 3, A and B). Loss of GFP-expressing cells was not observed after reconstitution with RIPK3 K51A, confirming that RIPK3 catalytic activity is required for necroptosis (Fig. 3, A and B). To test the role of kinase domain dimerization, Ripk3−/− cells reconstituted with the R69H mutant were treated with TZ. In cells with this point mutation, the amount of cell loss was reduced as compared to cells expressing WT RIPK3 (Fig. 3, A and B). Similar results were obtained when cells were sorted for equivalent GFP expression and absolute cell death measured (fig. S2, A to C). The defect in cell death was not due to defective kinase activity, because in vitro kinase assays showed similar activities between RIPK3 R69H and WT RIPK3 (fig. S2D). The importance of kinase domain dimerization was confirmed by showing that RIPK3 H156G and RIPK3 H156R both had defects in cell loss and phosphorylation of RIPK3 and MLKL after TZ treatment (fig. S2, E and G). The in vitro kinase activity of RIPK3 H156G was not impaired in comparison to WT RIPK3, although H156R was moderately reduced (fig. S2H). These results suggest that RIPK3 dimerization is necessary for TNF-α–induced necroptosis.
Fig. 3. Necroptosis is impaired by kinase domain dimerization–defective RIPK3.
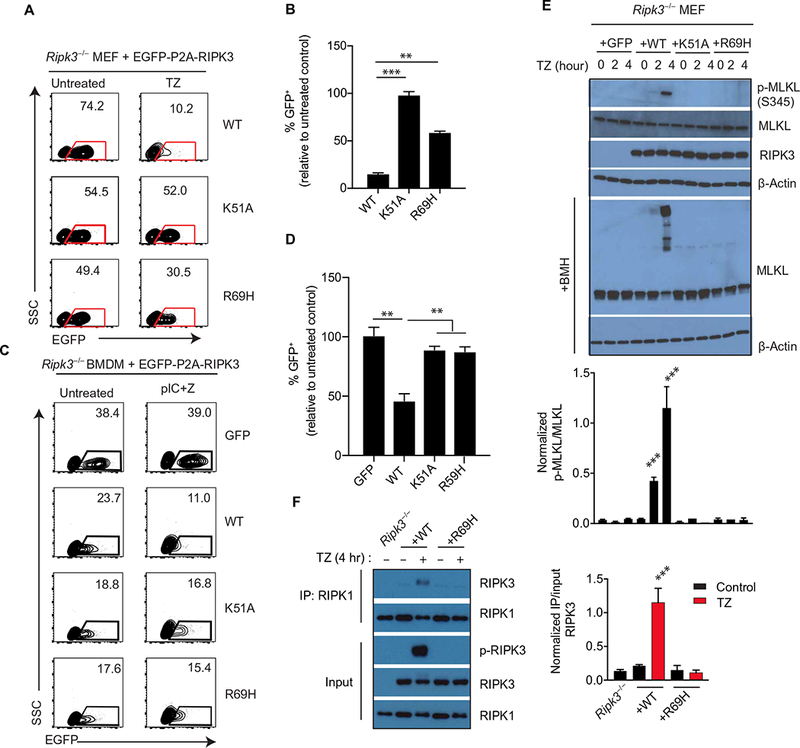
(A and B) Flow cytometry analysis of enhanced GFP (EGFP) abundance in Ripk3−/− MEFs infected with lentiviruses encoding EGFP-P2A-WT RIPK3 or the indicated mutants and treated with TNF-α + z-VAD-fmk for 24 hours. Dot plots (A) are representative of at least three independent experiments. The relative frequencies of GFP+ cells (B) are means ± SD from all experiments. SSC, side scatter. (C and D) Flow cytometry analysis of EGFP abundance in Ripk3−/− bone marrow cells transduced with lentiviral constructs encoding EGFP-P2A-WT RIPK3 or mutants and cultured for 6 days in macrophage colony- stimulating factor (M-CSF) to generate macrophages before treatment with poly(I:C)/z-VAD-fmk for 24 hours. (C) Dot plots (left) are representative of at least three independent experiments. The relative frequencies of GFP+ cells (D) are means ± SD from all experiments. (E) Western blot analysis for p-MLKL, MLKL, RIPK3, and β-actin from lysates of Ripk3−/− MEFs transduced with the indicated RIPK3 mutant construct and treated with TZ for the indicated times. Cell lysates were cross-linked with bismaleimidohexane (BMH) or directly used for immunoblotting. Blots are representative of at least three independent experiments. Normalized band intensities are means ± SD from all experiments. (F) Co-immunoprecipitation analysis of RIPK1 interactions in lysates of Ripk3−/− MEFs reconstituted with WT RIPK3 or RIPK3 R69H and treated with TZ for 4 hours that were immunoprecipitated for RIPK1. Blots are representative of at least three independent experiments. Normalized band intensities are means ± SD from all experiments. **P < 0.01 and ***P < 0.005 by Student’s t test.
In addition to TNF-α–induced necroptosis, which requires both RIPK1 and RIPK3, necroptosis can also be triggered by Toll-like receptors, such as TLR3 and TLR4, when caspase-8 is inhibited. In this setting, the RHIM-containing adaptor TIR domain–containing adapter- inducing interferon-β (TRIF) appears to engage RIPK3 independent of RIPK1 (24). To determine whether RIPK3 kinase domain dimerization is required for TLR3-induced necroptosis, we reconstituted Ripk3−/− bone marrow–derived macrophages (BMDMs) with lentiviral vectors encoding GFP alone, WT, the K51A, or the RIPK3 R69H mutations. Cells were stimulated with the TLR3 ligand poly(I:C) and z-VAD-fmk to induce necroptosis. BMDMs reconstituted with WT RIPK3 died, as indicated by the induced loss of GFP+ cells, whereas cells expressing GFP alone remained viable (Fig. 3, C and D). In comparison, BMDMs reconstituted with the K51A or R69H mutants of RIPK3 were less susceptible to TLR3-stimulated cell death (Fig. 3, C and D). Together, these results demonstrated that necroptosis induced by both TNF and TLR3 stimulation depends on RIPK3 kinase domain dimerization.
A key event in necroptosis is the phosphorylation of Thr231 and Ser232 (T231/S232) of RIPK3 (p-RIPK3) and the phosphorylation of Ser345 (S345) in p-MLKL by RIPK3. In WT RIPK3 expressing MEFs, phosphorylation of both RIPK3 and MLKL was detected maximally after 4 hours. In contrast, in both the K51A- and RIPK3 R69H- reconstituted MEFs, p-RIPK3 and p-MLKL were not detected up to the 4-hour time points (Fig. 3E). Low levels of p-MLKL were detected at later time points in RIPK3 R69H–expressing cells after TZ treatment (fig. S2F), suggesting that the ability of R69H to mediate some low levels of dimerization (Fig. 2E) can still induce some signaling (Fig. 3, A and B). Chemical cross-linking confirmed that MLKL formed oligomers, which correlated with the appearance of p-MLKL (Fig. 3E). In addition, the formation of RIPK1/RIPK3 after TZ treatment was impaired by the R69H mutation, supporting the role of the dimerization interface in the activation of RIPK3 by RIPK1 (Fig. 3F). Together, these results indicated that phosphorylation and activation of RIPK3 required kinase domain dimerization was to activate MLKL.
Allosteric activation of RIPK3 through kinase domain homodimerization
For RAF family kinases, dimerization allows one of the kinases (the activator) to induce the catalytic activity of the second kinase (the receiver). The activator kinase does not require catalytic activity but instead requires that the kinase domain be poised in a conformation that allows for “allosteric” activity (6). A specific mutation in the ATP-binding pocket in BRAF, V471F, located in a hydrophobic structure known as the C-spine (3, 25), abrogates kinase activity yet stimulates allosteric activity of BRAF by promoting the closed, active conformation of the kinase (10). Allosteric activity is not exhibited by other kinase- inactivating mutations, such as when the catalytic lysine of BRAF is mutated. To test whether kinase-inactive RIPK3 could exhibit activator function in an analogous manner, we mutated the residue analogous to BRAF V471 in the C-spine of RIPK3, V36 to phenylalanine (fig. S3A). We confirmed that RIPK3 V36F lacked catalytic activity using an in vitro kinase assay similar to the mutation of the RIPK3 catalytic lysine, K51A (Fig. 4A). Notably, Ripk3−/− MEFs reconstituted with RIPK3 V36F did not exhibit spontaneous apoptosis (fig. S3B), and consistent with its lack of kinase activity, TZ treatment did not induce cell death (fig. S3C).
Fig. 4. RIPK3 functions as an allosteric activator dependent on kinase domain homodimerization.
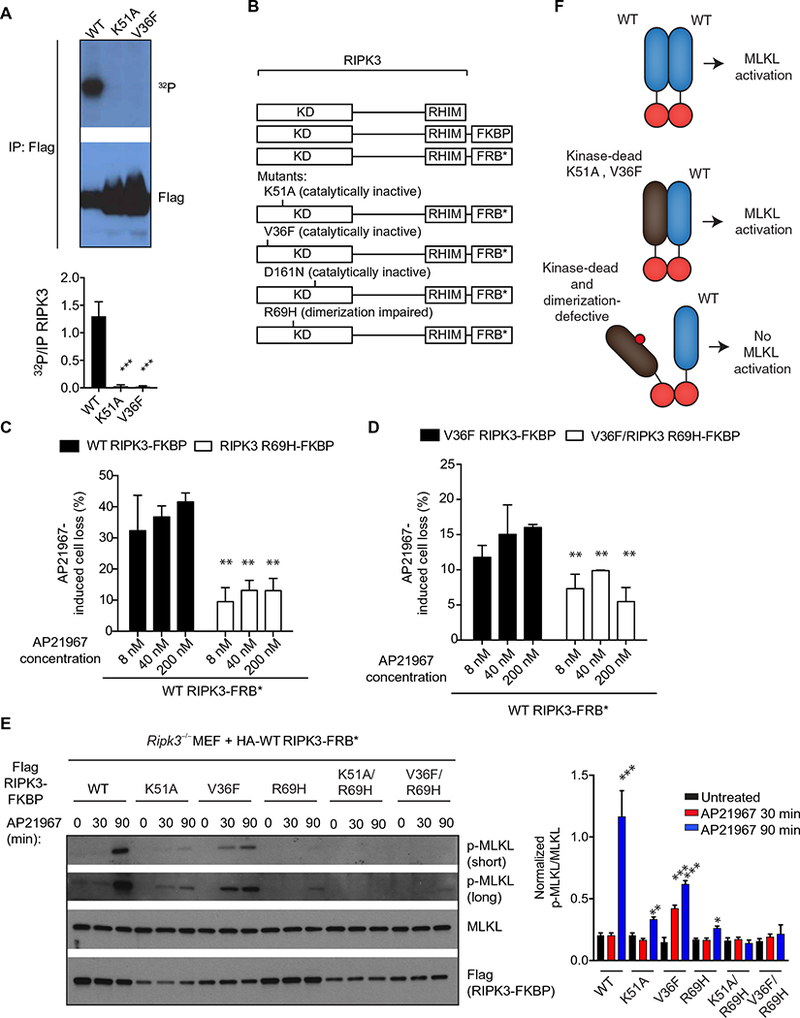
(A) Autoradiography and Western blot analysis of the in vitro kinase activity from lysates of HEK293T cells transfected with pcDNA3-Flag WT, pcDNA3-Flag K51A, or pcDNA3-Flag V36F RIPK3 constructs and immunoprecipitated for Flag 48 hours later. Blots are representative of at least three independent experiments. Normalized band intensities are means ± SD from all experiments. (B) Ripk3−/− MEFs were cotransduced with the indicated lentiviral constructs of RIPK3 fused to FRBP and FRB (B), which heterodimerize in the presence of AP21967. (C and D) CellTiter-Glo analysis of cellular viability in cells that received the indicated constructs and were pretreated with z-VAD-fmk for 1 hour before treatment with AP21967 for 6 hours. Data are means ± SD from three independent experiments. (E) Western blot analysis for p-MLKL, MLKL, and Flag in lysates from cells that received the indicated constructs and were pretreated with z-VAD-fmk before the addition of 250 nM AP21967 for the indicated time periods. Blots are representative of at least three independent experiments. Normalized band intensities are means ± SD from all experiments. (F) Model for allosteric activation of RIPK3 through kinase domain dimerization. *P < 0.05, **P < 0.01, and ***P < 0.005 by Student’s t test.
To determine whether a catalytically inactive kinase domain of RIPK3 could activate a WT RIPK3 kinase domain through dimerization, we fused full-length RIPK3 to specific domains, FK506 binding protein (FKBP) (activator) and FRB−rapamycin-binding-T2098L (FRB*) (receiver), that allowed for inducible heterodimerization by the compound, AP21967 (Fig. 4B) (26, 27). We controlled for expression of each construct in Ripk3−/− MEF cells based on co-expressed GFP and mCherry and isolated cells with similar fluorescent intensity by cell sorting and verified protein abundance by immunoblotting (fig. S4A). As expected, induced heterodimerization of WT RIPK3-FKBP with WT RIPK3-FRB* with AP21967 caused significant cell death (Fig. 4C). Forced dimerization of V36F-FKBP (activator) with WT FRB (receiver) also stimulated significant cell loss, albeit not as potently as WT RIPK3 (Fig. 4, C and D) (28, 29). Cell death was dependent on kinase domain dimerization, as R69H mutations of both constructs attenuated AP21967-induced cell death (Fig. 4, C and D).
In WT FKBP–expressing cells, p-MLKL was detected strongly only at the 90-min time point and minimally when the K51A mutant of RIPK3 was used as the activator. p-MLKL was detectable in cells that expressed the V36F mutant at both the 30- and 90-min time points but at levels lower than WT (Fig. 4E). We confirmed that RIPK3 V36F had a similar ability to WT RIPK3 to dimerize as assayed by co-immunoprecipitation (fig. S4B). As expected, the R69H mutation impaired MLKL phosphorylation by both the WT and V36F RIPK3-FKBP* constructs (Fig. 4E). These data demonstrated that certain kinase-inactive forms of RIPK3 can activate WT RIPK3 (Fig. 4F).
RIPK3 D161N acts as a strong allosteric activator
Can allosteric activation explain the biological effects of the D161N mutant of RIPK3? To test this, we mutated the RHIM domain (VQIG to AAAA) in the RIPK3-FKBP constructs (11, 12, 18) to mitigate spontaneous cell death and used the FKBP/FRB* system to control dimerization. Ripk3−/− MEFs were reconstituted with FKBP fused to RHIM-AAAA (hereafter referred to as AAAA) WT, D161N, or D161N/RIPK3 R69H and tested as activator kinases. WT RIPK3-FRB* with intact RHIM domain was co-expressed and used as the receiver kinase. When WT RIPK3 was present as both activator and receiver, we detected AP21967- induced p-MLKL and p-RIPK3 after 2 hours of stimulation [Figs. 4E and5 (A and B), and fig. S4B]. In contrast, cells expressing the D161N/AAAA mutation as the activator initiated MLKL phosphorylation earlier, with p-MLKL detectable within 30 min. This required an intact dimer interface as mutation of the R69H mutation abrogated induction of p-MLKL (Fig. 5, A and B, and fig. S4C). WT RIPK3 lacking an intact RHIM domain (WT/AAAA) was unable to function as an activator highlighting the complexity of RHIM domain interactions in regulating RIPK3 activation. Consistently, no significant cell death was observed with any of the RHIM mutants (Fig. 5C), confirming that RHIM-mediated higher- order complex formation is important for cell death (29). Similar results to RIPK3 D161N/AAAA were obtained with RIPK3 D161G/AAAA, a substitution that is also found in oncogenic BRAF (fig. S4D). A second dimer-impairing mutant, H156R, also blocked the ability of the D161N mutant of RIPK3 (RIPK3 D161N/H156R/AAAA) to function as an allosteric activator (fig. S4E).
Fig. 5. Kinase-inactive RIPK3 D161N allosterically activates WT RIPK3 and induces its cis-autophosphorylation.
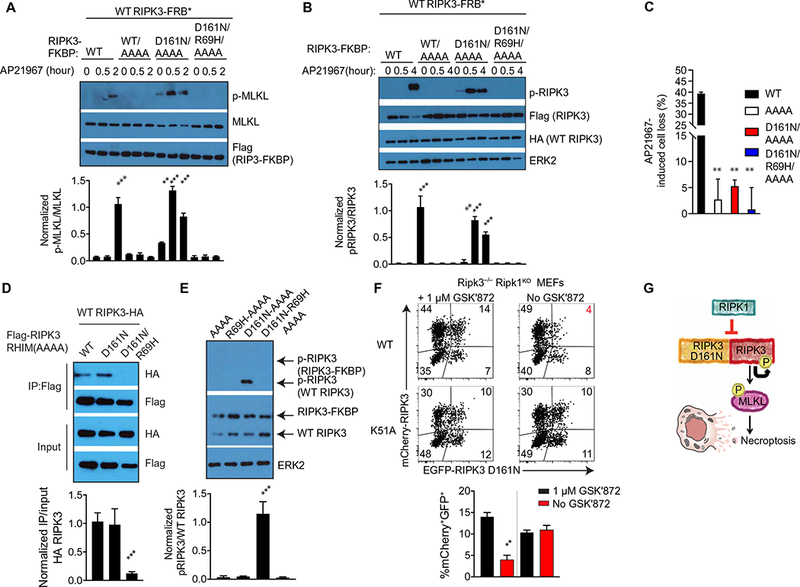
(A and B) Western blot analysis for p-MLKL (A) or pRIK3 (B) in lysates of Ripk3−/− MEFs cotransduced with the indicated constructs treated for the indicated times with 250 nM AP21967 + z-VAD-fmk. Blots are representative of three independent experiments. Normalized band intensities are means ± SD from all experiments. (C) CellTiter-Glo analysis of cell viability in Ripk3−/− MEFs cotransduced with the indicated constructs treated with 250 nM AP21967 + TZ for 6 hours. Data are means ± SD pooled from at least three independent experiments. (D) Co-immunoprecipitation analysis of RIPK3 interactions in lysates of Ripk3−/− MEFs cotransduced with the indicated constructs that were immunoprecipitated for Flag. Blots are representative of three independent experiments. Normalized band intensities are means ± SD from all experiments. (E) Western blot analysis of pRIPK3 in cell lysates from Ripk3−/− MEFs cotransduced with the indicated RIPK3-FKBP fusion constructs and WT RIPK3. Blots are representative of three independent experiments. Normalized band intensities are means ± SD from all experiments. (F) Flow cytometery analysis of EGFP and mCherry abundance in Ripk3−/−Ripk1KO MEFs cotransduced with the indicated mCherry-RIPK3 construct and EGFP-RIPK3 D161N in the presence or absence of GSK′872. Dot plots are representative of two independent experiments. The frequencies of cells expressing both constructs are means ± SD from all experiments. (G) Model for activation of WT RIPK3 by RIPK3 D161N in the absence of RIPK1. *P < 0.05, **P < 0.01, and ***P < 0.005 by Student’s t test.
We also noted that baseline RIPK3 activation seen in cells expressing RIPK3 D161N/AAAA-FKBP and WT RIPK3-FRB* (Fig. 5B) was not observed in cells expressing the dimer-defective RIPK3 D161N/R69H/AAAA. We tested whether RIPK3-D161N/AAAA had an enhanced ability to dimerize and found that D161N/AAAA associated with WT RIPK3 receiver similarly to WT RIPK3/AAAA. However, this association was reduced by the R69H mutation (Fig. 5D). Because RIPK3 D161N is kinase-inactive, it seemed unlikely that kinase transphosphorylation could explain RIPK3 autophosphorylation. Using RIPK3 D161N/AAAA fused to FKBP allowed us to distinguish WT RIPK3 from RIPK3 D161N by molecular mobility. When we co-expressed both constructs and analyzed AL phosphorylation using a RIPK3 phosphospecific antibody, we found that p-RIPK3 was only detected on WT RIPK3 in the cells that co-expressed RIPK3 D161N-FKBP (Fig. 5E). Because the D161N mutant lacks catalytic activity, these results imply that autophosphorylation of RIPK3 occurs following through cis-autophosphorylation, as was previously reported for BRAF (6).
To confirm our hypothesis that RIPK3 D161N induces allosteric activation of WT RIPK3 and necroptotic cell death in the absence of RIPK1 (fig. S6), we generated Ripk3−/− Ripk1KO MEFs using CRISPR (clustered regularly interspaced short palindromic repeats)–Cas9. We then cotransduced these cells with mCherry-WT RIPK3 or K51A along with EGFP-RIPK3-D161N in the presence of the RIPK3 inhibitor GSK′872 to prevent spontaneous necroptosis. Withdrawal of GSK′872 allowed us to quantify the fraction of GFP+ cells that were lost because of RIPK3-dependent cell death (Fig. 5, F and G). Consistent with our in vivo data, removal of GSK′872 in cells co- expressing WT and D161N RIPK3 stimulated significant cell death. However, this RIPK3-dependent cell death was not observed when GSK′872 was removed from cells co-expressing the K51A or D161N RIPK3 mutants. These data further confirmed that cell death in the absence of RIPK1 required RIPK3 kinase activity.
RIPK3-dependent apoptosis requires kinase domain dimerization with RIPK1
The rescue of Ripk3D161N/D161N (DFG to NFG) knock-in mice by Ripk1 deficiency suggests that RIPK3 D161N activates RIPK1 to induce apoptosis. To test whether this was dependent on kinase domain dimerization, we reconstituted Ripk3−/− MEFs with WT RIPK3 or RIPK3 mutants and inhibited apoptosis with z-VAD-fmk. Withdrawal of z-VAD-fmk allowed us to quantify the fraction of GFP+ cells that were lost because of apoptosis (fig. S5A). Little to no cell death occurred in Ripk3−/− MEFs reconstituted with WT, K51A, or V36F RIPK3. In contrast, there was a ~50% loss of GFP+ cells in cells reconstituted with RIPK3 D161N (Fig. 6A). This effect was abrogated in cells expressing RIPK3 D161N/R69H (Fig. 6A and fig. S5B). Because z-VAD-fmk withdrawal did not stimulate cell death in Ripk3−/−Caspase-8KO cells (Fig. 6A), cell death occurred via caspase-8–dependent apoptosis. RIPK3 mutant expression was verified by immunoblotting (fig. S5C). These data indicated that RIPK3 kinase domain dimerization was also important to stimulate apoptotic cell death.
Fig. 6. RIPK3 D161N and RIPK3 inhibitor–induced apoptosis requires Arg69 and a stable R-spine.
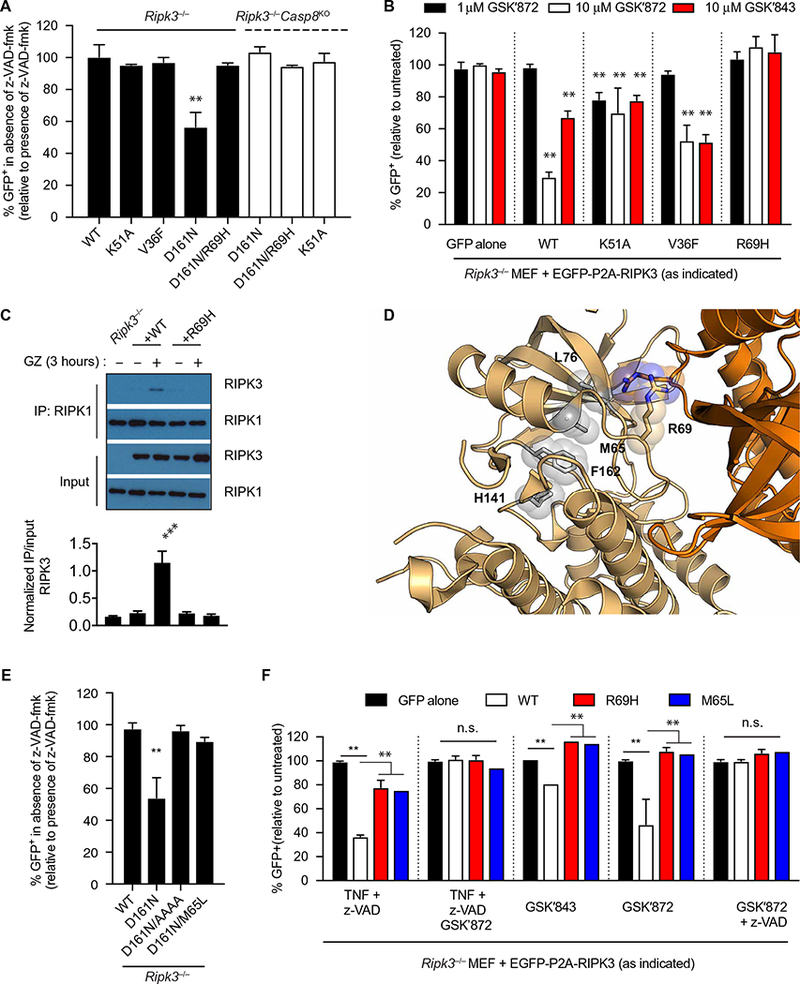
(A) Flow cytometry analysis of EGFP abundance in Ripk3−/− MEFs or Ripk3−/−Caspase-8KO MEFs transduced with the indicated EGFP-P2A-RIPK3 constructs and treated with z-VAD-fmk for 3 days. The frequencies of GFP+ cells are means ± SD pooled from at least three independent experiments. (B) Flow cytometry analysis of EGFP abundance in Ripk3−/− MEFs transduced with the indicated EGFP-P2A-RIPK3 constructs and treated with the indicated RIPK3 inhibitors. The frequencies of GFP+ cells are means ± SD pooled from at least three independent experiments. (C) Co-immunoprecipitation analysis of RIPK1 interactions in lysates of Ripk3−/− MEFs transduced with the indicated RIPK3 constructs and treated with the GSK′872 and z-VAD-fmk for 3 hours that were immunoprecipitated for RIPK1. Blots are representative of three independent experiments. Normalized band intensities are means ± SD from all experiments. (D) RIPK3 kinase domain in the active conformation (amber; PDB ID: 4M66). Features of the active conformation include the αC positioned “in” as well as a closed R-spine. R-spine residue side chains are represented by gray sticks and spheres. (E) Flow cytometry analysis of EGFP abundance in Ripk3−/− MEFs transduced with the indicated RIPK3 constructs and treated with z-VAD-fmk for 3 days. The frequencies of GFP+ cells are means ± SD pooled from at least three independent experiments. (F) Flow cytometry analysis of EGFP abundance in Ripk3−/− MEFs transduced with the RIPK3 construct and treated with the indicated inhibitors. The frequencies of GFP+ cells are means ± SD pooled from at least three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.005 by Student’s t test. n.s., not significant.
Some ATP-competitive RIPK3 inhibitors like GSK′843 and GSK′872 that inhibit necroptosis can, at certain concentrations, stimulate apoptosis in a RIPK1-, FADD-, and caspase-8–dependent manner (11, 12). Our results suggested that drug binding may induce conformational changes that induce allosteric activation similar to what was reported for certain BRAF inhibitors (7–9).To test this, we treated cells reconstituted with WT and RIPK3 R69H mutants with GSK′843 or GSK′872 and found that R69H mutations abrogated sensitivity to cell death (Fig. 6B and fig. S5D). These results suggested that apoptosis induced by these drugs required RIPK3 kinase domain dimerization. By co- immunoprecipitation, we found that WT RIPK3 formed complexes with RIPK1 after GSK′872 and z-VAD-fmk treatment, and this interaction was lost in cells expressing RIPK3 R69H (Fig. 6C). This result confirmed that these drugs induced RIPK1/RIPK3 dimers and demonstrated that allosteric activation was mediated through the dimer interface.
Destabilization of the RIPK3 R-spine impairs activator function
When kinases are in their active conformation, the C- and R-spines are assembled (3). If the active conformation of RIPK3 D161N is important for its function as an activator, destabilizing the R-spine should disrupt its ability to induce cell death (Fig. 6D). A critical R-spine residue is contributed by Met65 in the αC helix of RIPK3. Although most kinases contain leucine at this position, substitution of leucine with methionine in this position strengthens the R-spine and promotes the active conformation of the kinase (25). Thus, we hypothesized that substituting a leucine in this position could destabilize the R-spine and weaken RIPK3 D161N activator function. Accordingly, we found that apoptosis induced by D161N was completely abrogated by the M65L substitution (Fig. 6E). Cells expressing RIPK3 M65L also had a defect in TZ-induced necroptosis (Fig. 6F), and cell death was not observed after treatment with RIPK3 inhibitors GSK′843 and GSK′872 (Fig. 6F), nor were RIPK3/RIPK1 complexes formed (fig. S5E). These results indicated that disrupting the R-spine of RIPK3 prevented association with RIPK1 and cell death. Further, these data suggest that the RIPK3 D161N mutation stabilizes the closed, active conformation of RIPK3.
DISCUSSION
The activation of RIPK3 is essential to the induction of necroptosis (1, 30, 31), yet exactly how RIPK3 is activated remains elusive. Although the forced dimerization of RIPK3 is sufficient to induce necroptosis, the molecular events that explain how dimerization promotes RIPK3 kinase activity are unclear (29). On the basis of the requirement for AL phosphorylation for kinase activation, it is widely assumed that dimerization allows for kinases to cross-phosphorylate each other in trans. Although this is the mechanism of activation for kinases such as the Src family, Aurora A, IRAK4, and TBK1, how the initial kinase is activated is unclear (32–34). Allosteric activation of kinases by protein-protein interactions can potentially explain how kinase activation for some kinases is initiated. This is exemplified by cyclin binding to cyclin-dependent kinases (CDKs), which induces an active conformation of CDKs to promote catalytic activity (35). The ability of BRAF inhibitors to activate RAF kinases revealed that kinase dimerization can also function as a mechanism of allosteric activation (7, 9, 36). The ability of the pseudokinase, ERBB3, to activate EGFR family members is another example of allosteric activation (37).
Because RIPK3 and RAF are similar and belong to the TKL family, we considered that RIPK3 could be regulated similarly to RAF kinase. Accordingly, the crystal structures of RIPK3 showed that they form the same allosteric dimer as the RAF kinases (14, 38). Our alignment shows the similarity of their orientation and their respective dimer interfaces. Although interactions between RIPK3/RIPK3, RIPK1/RIPK1, and RIPK1/RIPK3 dimers are driven by RHIM domains, our work suggests that the one consequence of protein dimerization is secondary dimerization of the kinase domains and that this is the driving force for kinase activation. It is interesting that the conserved arginine in the dimer interface of murine RIPK3, R69, is not highly conserved in RIPK3 from other species. By testing almost all possible residues in this position, we found that in the mouse system, most substitutions impaired dimerization except for threonine, which appeared to promote dimerization. This is reminiscent of RAF where substitution of the conserved arginine with alanine strongly promotes dimerization (39). Supporting the rapid evolution of this signaling pathway, the mechanism of human and mouse MLKL activation is distinct likely in response to different selective forces (40). Thus, the lack of conservation of this residue may reflect the rapid evolution of RIPKs due to selective pressure exerted by pathogens (41). A comparison of kinase domains across multiple species shows lower conservation of residues in the dimer interface compared to the rest of the kinase domain, suggesting that this surface has evolved within each species.
The RIPK3 D161N knock-in mutation in mice suggests an unexpected noncatalytic function of RIPK3 (8). Our results demonstrating that WT RIPK3 can be activated by kinase-inactive forms of RIPK3 suggested that RIPK3 is allosterically activated by kinase domain dimerization. To support a mechanism of allosteric activation, we showed that the kinase domain of RIPK3 can homodimerize with itself and that RIPK3 activation was induced by dimerization with kinase domains containing the D161N or the V36F mutation. Mutations that reduced dimerization impaired the activation of WT RIPK3 and the induction of cell death.
Our model of RIPK3 dimerization can also explain the pheno-types of the RIPK3 D161N/WT RIPK3 heterozygous mice (graphically depicted in fig. S6). Here, we reported that whereas heterozygous RIPK3 D161N mice are viable when RIPK1 is present, removing RIPK1 results in embryonic lethality by constitutive necroptosis. Our data support a model in which RIPK3 D161N can allosterically activate WT RIPK3 to induce p-MLKL and necroptosis. Although the RHIM domain is required for RIPK1 inhibition of necroptosis (18), the exact mechanism is unclear. We speculate that it could be, due to competition for dimer formation from RIPK1, limiting the number of effective RIPK1/RIPK3 and RIPK3/RIPK3 complexes that could be formed.
What is the structural basis underlying the phenotypes observed with the RIPK3 D161N mutation? Mutations of the analogous aspartic acid in BRAF (D594) are relatively common in some types of tumors and can induce lung tumors in a knock-in mouse (42, 43). Although it has been reported that BRAF D594N forms constitutive dimers with CRAF, a more recent study suggests that dimers between BRAF D594N and CRAF requires prior Ras activation. Nonetheless, both studies support the idea that heterodimers formed with BRAF D594N result in activation of CRAF. Because the conformation of the kinase-inactive forms of BRAF and RIPK3 is important for allosteric activation, it seems likely that this change stabilizes the active conformation of the kinase. This conclusion was supported by the M65L mutation of RIPK3, which we predicted would destabilize the active conformation of the kinase and impaired allosteric activation. Therefore, the stability of the active conformation as defined by assembly of the R-spine may be the key to effectiveness as an allosteric activator.
On the basis of our previous work (6, 10), we predicted that the V36F mutant of RIPK3 would impair the kinase activity of RIPK3 and also act as an allosteric activator. V36 is positioned in the back of the ATP pocket, and a substitution with phenylalanine blocks ATP binding (10) by mimicking the adenine ring of ATP and closes the two kinase lobes by stabilizing the C-spine (10). We found that V36F functioned as a moderate allosteric activator, to a lesser extent than D161N, but stronger than K51A. We suspect that changes in the dynamics of the kinase domain lobes mediated by the V36F and D161N mutations underlie their different efficiencies in allosteric activation.
RIPK3-dependent caspase activation has been reported downstream of lipopolysaccharide treatment in the setting of inhibitor of apoptosis protein (IAP) deficiency and influenza A infection (40, 44). Because the kinase activity of RIPK1 is not required for its function in RIPK3-mediated apoptosis, interaction of an “active” RIPK3 with RIPK1 may function to induce conformational changes that allow for FADD and caspase-8 binding. An “activating” interaction between RIPK3 D161N and RIPK1 can explain embryonic lethality and constitutive apoptosis in the D161N/D161N RIPK3 mice when RIPK1 is present (45).
Given that two families of TKL kinases (RAF and RIPK) now appear to use an allosteric activation mechanism of activation driven by dimerization, it will be interesting to determine whether this is a general phenomenon of other TKL kinases and/or whether it occurs more broadly in the kinase family. A better understanding of this mechanism can potentially transform our understanding of kinase activation and inform the better development of kinase inhibitors.
MATERIALS AND METHODS
Mice
All animal experiments were approved by the Genentech Institutional Animal Care and Use Committee. Ripk3−/−, Ripk3D161N/D161N, and Ripk1−/− (11) were generated at Genentech as previously described. Mlkl−/− (46) were obtained from the Walter and Eliza Hall Research Institute. Mice were maintained in a specific pathogen–free facility. The experiments were not randomized, and the investigators were not blinded to allocations during experiments and outcome assessment. No statistical methods were used to predetermine sample size. For timed pregnancies, mice were designated E0.5 on the morning a vaginal plug was detected. Embryos were harvested at the indicated times and genotyped by polymerase chain reaction (PCR) using primers listed below. 5′-GATGCCAGACATGAAGCATCC-3′, 5′-GGAGGATTAAAGGTCTGAGGC-3′, and 5′-TCATTGCCTCCCTTACTACCC-3′ amplified 265–base pair (bp) WT and 231-bp Ripk1-DNA fragments. Ripk3 genotyping primers: 5′-AGAAGATGCAGCAGCCTCAGCT-3′, 5′-ACGGACCCAGGCTGACTTATCTC-3′, and 5′-GGCACGTGCACAGGAAATAGC-3′ amplified 130-bp WT and 298-bp Ripk3-DNA fragments; 5′-CGCTTTAGAAGCCTTCAGGTTGAC-3′ and 5′-ACGGACCCAGGCTGACTTATCTC-3′ amplified 345-bp WT and 379-bp Ripk3D161N-DNA fragments; and 5′-TATGACCATGGCAACTCACG-3′, 5′-ACCATCTCCCCAAACTGTGA-3′, and 5′-TCCTTCCAGCACCTCGTAAT-3′ amplified 498-bp WT and 158-bp Mlkl-DNA fragments.
Immunohistochemistry
After collection, embryos and placentas were immersion-fixed in formalin and transferred to phosphate-buffered saline (PBS) before tissue processing. Tissues were then dehydrated in 70% ethanol followed by Flex 95 (Richard-Allan Scientific) and Flex 100 solutions, cleared in xylene, and routinely infiltrated and embedded in paraffin. Embryos and placentas were embedded in separate blocks.
Formalin-fixed, paraffin-embedded tissue sections were stained with rabbit anti-mouse phospho-RIPK3 antibody (5 μg/ml; GEN135–35-9, Genentech) recognizing phosphorylated residues Thr231, as previously described (18). Immunohistochemistry was performed on the Ventana Discovery XT platform with Cell Conditioning 1 standard antigen retrieval. The reaction was detected with the HQ amplification system (p-RIPK3) using 3,3′ diaminobenzidine as the chromogen and hematoxylin counterstain.
Constructs and cell culture
Mouse Ripk3 complementary DNA (cDNA) was obtained as a GeneBlock Fragment from Integrated DNA Technologies and cloned into pcDNA3 (Invitrogen) using Gibson Assembly. Lentiviral constructs were generated using the pUltra-EGFP-P2A-T2A and pUltra-mCherry-P2A-T2A vectors (gifts from A. Brunger; Addgene) with RIPK3 cloned in-frame with EGFP-P2A. All mutations were generated using PCR and verified by conventional Sanger sequencing. FKBP and FRB constructs were originally obtained from Addgene, and FRB was modified with T2098L mutation and is designated FRB*. The following vectors were then constructed using Gibson Assembly pUltra-EGFP-P2A-RIPK3- GSG-FRB*-HA and pUltra-mCherry-2xFlag-RIPK3-GSG- FKBP.
E1A-immortalized Ripk3−/− MEFs (11) and HEK293T cells were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM). To prepare lentivirus, HEK293T cells were transfected with the pUltra- based third-generation lentiviral vectors along with psPAX2 and pVSG-G using a Mirus LT-1 transfection reagent. Viral supernatant was collected at 48 hours after transfection and used to infect target cells with the addition of protamine.
To generate Ripk3−/−Caspase-8KO and Ripk3−/−Ripk1KO MEFs, Ripk3−/− MEFs were transfected with pcDNA3-GFP and pX330 at a ratio of 1:20 using the Amaxa Nucleofector MEF1 kit (Lonza). Forty-eight hours later, ~100,000 GFP+ cells were sorted, and the polyclonal population was screened for lack of Caspase-8 expression by immunoblotting. The single-guide RNA sequences used were 5′-TGAGATCCCCAAATGTAAGC-3′ (Casp8–1), 5′-AATCCTCGATCTTCCCCAGC-3′ (Casp8–2), and 5′-GGGTCTTTAGCACGTGCATC-3′ (Ripk1–1).
Generation of BMDMs
Femurs and tibias from Ripk3−/− C57BL6/J mice were manually flushed to harvest bone marrow cells, and red blood cells were lysed in ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA). Cells were cultured in complete DMEM containing 10% of CMG cell-conditioned medium (containing M-CSF) for 6 days to obtain BMDMs. On days 1 and 2 of culture, cells were spin-fected with lentiviral supernatant produced by HEK293T cells at 2000 rpm for 1 hour. Cells were harvested on day 6 for stimulation.
Chemicals
rmTNF-a (50 ng/ml; Peprotech) was used unless otherwise indicated. z-VAD-fmk (Enzo) was used at 20 μM. Low–molecular weight poly(I:C) (50 μg/ml; Invivogen) was used. AP21967 (A/C heterodimerizer; Clontech) was used at the indicated concentrations. RIPK3 inhibitors GSK′842 (Aobious) and GSK′872 (Millipore) were used at the indicated concentrations.
In vitro kinase assay
For in vitro kinase assays, immunoprecipitates were washed once with a kinase reaction buffer [25 mM Hepes, 10 mM MgCl2, 0.5 mM Na3VO4, 0.5 mM dithiothreitol (DTT) (pH 7.4)], then resuspended in 25 μl of kinase reaction buffer, and then incubated with 5 μCi of [γ−32P]-ATP (PerkinElmer), and reactions were incubated at 30°C for 30 min. Reactions were terminated by the addition of a 4× SDS sample buffer and subject to immunoblotting and autoradiography.
Immunoprecipitation and Western blotting
For Flag immunoprecipitation, cells were washed with ice-cold PBS and lysed in the following lysis buffer: 20 mM tris-HCl (pH 7.5), 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM EGTA, 1% NP-40, 50 mM NaF, 1 mM Na3VO4, and protease inhibitors (Sigma-Aldrich). The cell lysates were centrifuged at 20,000g for 10 min, and the supernatants were subjected to immunoprecipitation at 4°C for 1 hour using Flag M2 Agarose Beads (A2220, Sigma-Aldrich). After the immunoprecipitation, the beads were washed three times in lysis buffer, and the immuno-precipitated proteins were subsequently analyzed by immunoblotting.
For RIPK1 immunoprecipitation, cells were lysed in the following buffer: 20 mM tris-HCl (pH 7.5), 137 mM NaCl, 1 mM EDTA, 1.5 mM MgCl2, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM NaF, 1 mM Na3VO4, and protease inhibitors (Sigma-Aldrich), followed by centrifugation at 20,000g for 10 min. Immunoprecipitation was carried out using anti-RIPK1 (BD Biosciences) and incubated at 4°C overnight. Antibody complexes were recovered after 4-hour incubation with Magnetic Protein G Dynabeads (Thermo Fisher Scientific). Beads were washed three times with lysis buffer before elution with 1× SDS sample buffer.
Proteins were separated by 8 or 10% SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Bio-Rad). Membranes were incubated with primary antibodies, as indicated, followed by detection with horseradish peroxidase (HRP)–conjugated species-specific antibody to immunoglobulin light chain (115–035-174, 211–032-171; Jackson ImmunoResearch) and a Luminata HRP substrate (Millipore).
Expression and purification of MmRIPK3KD
The expression of baculovirus construct MmRIPK3 (S02-H303) N- terminal histidine, which is the same construct used for PDB deposition 4M69, carrying the same point mutations (distal to dimer interface), was expressed in T.ni cells. The cell paste was homogenized in 50 mM tris (pH 8.0), 0.5 M NaCl, 5% glycerol, 10 mM imidazole, 10 mM BME, 0.25% CHAPS, and protease inhibitors (Roche), and the lysate was incubated with Ni-NTA beads (Qiagen) for 1 hour at 4°C. The protein bound to Ni-NTA resin was pelleted by centrifugation at 1900 rpm. The resin was then packed into a gravity column and washed with the lysis buffer supplemented with 30 mM imidazole. The bound protein was then eluted using lysis buffer + 250 mM imidazole and immediately injected onto S200 10/300 Increase column (GE Healthcare) that had been preequilibrated with 50 mM tris (pH 8.0), 500 mM NaCl, 5% glycerol, 1 mM tris(2-carboxyethyl)phosphine. Gel filtration standards (Bio-Rad) were then injected over the same column. Ve/Vo was plotted against the log of molecular weight to generate a standard curve. The red triangle indicates the position of MmRIPK3, indicating that the construct is a dimer at the RIPK3 concentration injected (1 mg/ml).
Split luciferase assay
HEK293T cells were transfected with pRK-based CBGN and CBGC constructs (23). Six hours after transfection, cells were lysed, and lucif-erase activity was measured using the Bright-Glo Luciferase Assay System (Promega) in a plate reader.
BMH cross-linking
Lysates were prepared as previously described (47). Briefly, 50 μl of cell lysates was incubated with 125 μM BMH (Thermo Fisher Scientific) for 5 min, and reactions were stopped with the addition of DTT at a final concentration of 50 mM. Samples were analyzed by immunoblot as described above.
Antibodies
The following antibodies were used in this study: anti-Flag (M2, Sigma-Aldrich), anti-HA (H6908, Sigma-Aldrich), anti-RIPK3 (ADI-905–242, Enzo), anti-RIPK1 (38/RIP, BD), anti-MLKL (AP14272b, Abgent), anti–p-MLKL(S345, Abcam), anti–p-RIPK3 (18), anti- β-actin (AC-74, Sigma-Aldrich), and anti-ERK2 (C-14, Santa Cruz Biotechnology).
Cell death assays
Cell death was analyzed using flow cytomery or a CellTiter-Glo Luminescent Cell Viability Assay kit (Promega). For flow cytometry, after the indicated treatment, plated cells were washed with 1× PBS to remove dead cells, trypsinized, and resuspended in DMEM/10% fetal bovine serum (FBS). An equal volume of 2% FBS/1× PBS/2 mM EDTA containing 4′,6-diamidino-2-phenylindole (DAPI) was added to exclude dead cells. For certain experiments, relative number of cells was quantified by the addition of CountBright Absolute Counting Beads to wells (Thermo Fisher Scientific). Samples were analyzed on LSR II or LSRFortessa (BD Biosciences). Data were analyzed by FlowJo (Tree Star), and cells in the displayed plots were pregated on DAPI− singlets.
CellTiter-Glo assays were performed according to the manufacturer’s instructions. In brief, 2 to 3 × 103 cells were seeded in a 96-well plate. After treatment, equal volume of CellTiter-Glo reagent was added to the cell culture medium, which had been equilibrated to room temperature for 30 min, and cells were lysed with shaking for at least 15 min at room temperature. Luminescence recording was performed with Cytation 5 (Biotek). The percentage of cell loss was determined by the percentage of ATP loss relative to untreated controls.
Supplementary Material
Acknowledgments:
We thank J. Hu and H. Yu for insightful discussions. We thank T. Egawa and D. Verbaro for technical support.
Funding:
This work was supported by grants from the NIH and by Genentech. S.R. was supported by the NIH Training Grant 5T32HL007317.
Footnotes
Competing interests:
K.N., M.M., J.D.W., J.G.Q., C.S., D.M.W., and A.S.S. are employees of Genentech.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
www.sciencesignaling.org/cgi/content/full/11/544/eaar2188/DC1
Fig. S1. Biochemical analysis of the RIPK3 kinase domain.
Fig. S2. Characterization of Ripk3−/− MEFs after reconstitution with WT RIPK3 and dimerization-defective RIPK3 mutations.
Fig. S3. Expression of RIPK3 V36F does not cause apoptosis or TZ-dependent necroptosis.
Fig. S4. Characterization and kinetics of FKBP-FRB RIPK3 fusion system for induced heterodimerization.
Fig. S5. RIPK3-induced apoptosis by RIPK3 D161N and the RIPK3 inhibitor GSK′872.
Fig. S6. Model of the mechanism of RIPK3 D161N function.
REFERENCES AND NOTES
- 1.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK-M, Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang H, Sun L, Su L, Rizo J, Liu L, Wang L-F, Wang F-S, Wang X, Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Kornev AP, Taylor SS, Ten Eyck LF, A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kornev AP, Taylor SS, Dynamics-driven allostery in protein kinases. Trends Biochem. Sci 40, 628–647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J, An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS, Shaw AS, Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R, Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N, RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S, RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, Shaw AS, Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. U.S.A 108, 6067–6072 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM, Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ, RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moriwaki K, Bertin J, Gough PJ, Chan FK-M, A RIPK3–caspase 8 complex mediates atypical pro–IL-1β processing. J. Immunol 194, 1938–1944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y, Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 5, 70–78 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Kung JE, Jura N, Structural basis for the non-catalytic functions of protein kinases. Structure 24, 7–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong Y-N, Janke LJ, Kelliher MA, Kanneganti T-D, Green DR, RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, Bertin J, Gough PJ, Balachandran S, Mocarski ES, RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. U.S.A 111, 7753–7758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose-Girma M, Warming S, Solon M, Ngu H, Webster JD, Dixit VM, RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Lin J, Kumari S, Kim C, Van T-M, Wachsmuth L, Polykratis A, Pasparakis M, RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 540, 124–128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers TW, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JFM, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, Luoh S-M, Anderson DJ, Ludlam MJC, Wiesmann C, Ultsch M, Friedman LS, Malek S, Belvin M, Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 501, 232–236 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M, Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol 22, 37–43 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Villalobos V, Naik S, Bruinsma M, Dothager RS, Pan M-H, Samrakandi M, Moss B, Elhammali A, Piwnica-Worms D, Dual-color click beetle luciferase heteroprotein fragment complementation assays. Chem. Biol 17, 1018–1029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES, Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem 288, 31268–31279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu J, Ahuja LG, Meharena HS, Kannan N, Kornev AP, Taylor SS, Shaw AS, Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol 35, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollock R, Giel M, Linher K, Clackson T, Regulation of endogenous gene expression with a small-molecule dimerizer. Nat. Biotechnol 20, 729–733 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T, An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu X-N, Yang Z-H, Wang X-K, Zhang Y, Wan H, Song Y, Chen X, Shao J, Han J, Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SWG, Albert ML, Green DR, Oberst A, RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X, Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137, 1100–1111 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C, Dong M-Q, Han J, RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC, Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. U.S.A 109, 9378–9383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zorba A, Buosi V, Kutter S, Kern N, Pontiggia F, Cho Y-J, Kern D, Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. eLife 3, e02667 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferrao R, Zhou H, Shan Y, Liu Q, Li Q, Shaw DE, Li X, Wu H, IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly. Mol. Cell 55, 891–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massagué J, Pavletich NP, Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, Misteli T, Chapman PB, Sosman JA, Graeber TG, Ribas A, Lo RS, Rosen N, Solit DB, RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, Jura N, Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci. Signal 7, ra114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 38.Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, Morales T, Smith D, Brandhuber BJ, Hymowitz SG, Malek S, Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26, 402–413 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Baljuls A, Mahr R, Schwarzenau I, Müller T, Polzien L, Hekman M, Rapp UR, Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J. Biol. Chem 286, 16491–16503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Spall SK, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, Benetatos CA, Condon SM, Wong WW-L, Silke J, Vaux DL, Vince JE, RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun 6, 6282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newton K, Manning G, Necroptosis and inflammation. Annu. Rev. Biochem 85, 743–763 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R; Cancer Genome Project, Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Nieto P, Ambrogio C, Esteban-Burgos L, Gómez-López G, Blasco MT, Yao Z, Marais R, Rosen N, Chiarle R, Pisano DG, Barbacid M, Santamaría D, A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature 548, 239–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin III TH, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, Verbist K, Gough PJ, Bertin J, Hartmann BM, Sealfon SC, Kaiser WJ, Mocarski ES, López CB, Thomas PG, Oberst A, Green DR, Balachandran S, RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, Dai W, Young SN, Tanzer MC, Wardak A, Liang L-Y, Cowan AD, Hildebrand JM, Kersten WJA, Lessene G, Silke J, Czabotar PE, Webb AI, Murphy JM, Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun 9, 2422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J-G, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RCJ, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS, The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR, Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.