

Abstract
Background
The European Society for Medical Oncology defines rare cancers as 5 or fewer cases per 100,000 persons per year. For many rare cancers, no standard of care exists, and treatment is often extrapolated. Identifying potentially targetable genomic alterations in rare tumors is a rational approach to improving treatment options. We sought to catalog these mutations in rare tumors and to assess their clinical utility.
Methods
For this retrospective analysis, we selected rare tumor patients from a dataset of patients who underwent clinical tumor genomic profiling. Sarcomas were excluded. To index potentially actionable alterations, patients’ reports were reviewed for mutations in cancer associated genes and pathways. Respective clinical records were abstracted to appraise the benefit of using a targeted therapy approach. Actionable alterations were defined as targeted by a drug available on-label, off-label, or in clinical trials.
Results
The 95 patients analyzed had 40 different tumor subtypes, most common being adenoid cystic (13%), cholangiocarcinoma (7%), and metaplastic breast (6%). At least one genomic alteration was identified in 87 patients (92%). The most common identifiable mutations were in TP53 (23%), KRAS (10%), PIK3CA (9%), CDKN2A/B (8%), BRAF (7%), MLL (7%), and ARID1A (6%). Thirty-six patients (38%) with 21 different tumors had at least one potentially actionable alteration. Thirteen patients received targeted therapy. Of these, 4 had a partial response, 6 had stable disease, and 3 had progressive disease as the best response.
Conclusion
The addition of genomic profiling to management of rare cancers adds a potential line of therapy for cancers that have little or no standard of care. In our analysis, tumors with a BRAF alteration responded well to BRAF inhibitors.
Keywords: Rare cancers, Targeted therapy, Phase I trials, Precision Oncology
Introduction
Rare cancers receive little financial support or interest from major drug developers or clinical investigators; even with adequate funding, they are by nature difficult to study even in a large academic medical center. As a result of this research quagmire, no standard of care exists for many rare cancers, and treatment is often extrapolated from case reports or small case series. Even the definition of rare cancer is not uniform. For example, the World Trade Center Health Program, a program of the US Centers for Disease Control and Prevention, defines rare diseases as having an incidence rate of less than 15 cases per 100,000 persons per year. The National Cancer Institute uses a 40,000 case cut-off but allows for more prevalent but understudied diseases such as non-Hodgkin lymphoma. The European Society for Medical Oncology (ESMO) defines rare tumors as 5 or fewer cases per 100,000 persons per year. Some investigators have estimated that as many as 25% of US cancer patients have a rare tumor type(1,2). Recognition of rare diseases’ relative collective prevalence prompted the passage of the US Rare Diseases Act of 2002 to coordinate research on the basket of rare cancers and other similarly rare diseases(3).
It is not easy to devote substantial resources to a disease that affects only a handful of people. Most would argue that capital investments in common tumor types yield substantially greater societal benefits. However, history has proven time and again that great leaps in scientific knowledge can be made by studying a rare but relatively simple disease(4). Most common cancers, such as non–small cell lung cancer, have complex genomes with many mutations and rearrangements(5). In contrast, a rare cancer such as a pediatric rhabdoid tumor may have only a single gene mutation and present a much simpler model for studying, for example, chromatin remodeling complexes(6).
There is currently a paucity of data on targetable mutations in rare tumors and a blossoming numbering of targeted therapies in development. We have previously published on the use of NGS as a diagnostic and prognostic tool in sarcomas(7), perhaps these patients can benefit from NGS as a therapeutic tool in a histology independent, mutation driven approach? The recent histology-agnostic approval of immune checkpoint inhibitor pembrolizumab across all cancers that express high levels of microsatellite instability is a landmark event. This brought the fields of genomics and immunology together to identify a genetic aberration that is a rare event (less than 5%) but occurs in a wide range of cancers. This development, along with the recent discovery of NTRK fusions across tumor types and rapid clinical translation of an effective drug targeting these fusions, are all fueling rare cancer research and the search for new drug targets(8). Some centers have even begun reporting their experience with small series of rare tumors treated successfully with NGS directed therapy(9).
The purpose of this study was to further the understanding of molecular mechanisms underlying various rare cancers, identify potentially actionable mutations, and evaluate the response of patients to therapies chosen based on these mutations. To achieve this purpose, we retrospectively reviewed patients with rare cancers who presented to the Phase I drug development unit at our institution, underwent comprehensive genomic profiling, and received therapy through a clinical trial. We identified potentially actionable genomic alterations in with the aid of the MD Anderson Knowledge Base for Precision Medicine and defined their responses to therapy using Response Evaluation Criteria in Solid Tumors.
Patients and Methods
To fulfill our purpose of identifying potentially actionable alterations in patients with rare tumors, we first identified patients with a rare tumor from our database of 549 patients with metastatic or unresectable disease who had undergone comprehensive genomic profiling of their tumor for the purpose of precision treatment. All of these patients had been referred to the Department of Investigational Therapeutics at MD Anderson (Phase 1 clinic) where comprehensive genomic profiling was performed by Foundation Medicine using Foundation One, a CLIA-approved next-generation sequencing (NGS) platform that analyzes 236 or 315 cancer-related genes (http://www.foundationone.com). The number of genes tested was dependent on the time analysis was performed. Later tests had a larger gene panel than earlier tests. Each patient was then enrolled in a clinical trial, but clinical trial choice was not always based on NGS results and was dependent on trials open at the time. Patients with a rare tumor were identified by using the ESMO definition of rare tumors, http://www.rarecare.eu/rarecancers/Rare_Cancers_list_March2011.xls and http://www.ema.europa.eu/docs/en_GB/document_library/Other/2012/07/WC500130297.pdf. Other references used to identify rare tumors include: https://www.cdc.gov/wtc/pdfs/WTCHP_PP_RareCancers05052014.pdf2014, https://epi.grants.cancer.gov/events/rare-cancers/. For each of these cases, the pathologic diagnosis was confirmed by an MD Anderson pathologist with experience in the respective disease area.
This retrospective review was performed according to MD Anderson Institutional Review Board guidelines. Each protocol was individually approved by the institutional review board, in accordance with the declaration of Helskini, and patient informed consent was obtained prior to enrollment on respective studies. Patients did not sign separate consent for this retrospective review. Sarcomas were excluded as these had been previously analyzed in a published report (10).
Actionable mutations were defined as any mutation within either a gene or a pathway of a gene directly targeted by an approved or investigational drug. Interpretations were provided by the MD Anderson Knowledge Base for Precision Medicine (http://PCT.MDAnderson.org) as well as The Drug Gene Interaction Database (http://dgidb.genome.wustl.edu)(11). Each eligible patient’s chart was reviewed for age at first clinic visit, tumor histology, metastatic sites, number of treatment cycles, and response based on imaging (RECIST V1.1). Progression-free survival was measured from date of first dose to date of documented progression on imaging or onset of symptoms. Molecular reports were reviewed for alterations with a potentially actionable mutation.
When possible, patients were treated with a targeted agent based on genomic profiling. This was limited by available trials at time of patient presentation. Tumor mutational burden was calculated when possible. Results were grouped as low mutation burden (5 or fewer mutations per megabase), intermediate mutation burden (6–20 mutations per megabase), or high mutation burden (20 or more mutations per megabase). GENIE data were downloaded from https://www.synapse.org/#!Synapse:syn7222066/wiki/405659. Data were analyzed and visualized by using the Tableau Desktop software.
Results
From the 549 patients who underwent comprehensive genomic profiling, we identified 95 eligible patients who fit the ESMO definition of a rare tumor. Baseline demographic and clinical information for these 95 patients is shown in Table 1.
Table 1.
Baseline demographic and clinical data for 95 patients with a rare cancer
| Demographics | |
|---|---|
| Age at diagnosis, years | |
| Median | 51 |
| Range | 2–75 |
| Sex | |
| Female | 49 (52%) |
| Male | 46 (48%) |
| Patients sequenced | 95 |
| Unique tumor types | 40 |
| Metastatic site biopsy | 50 (53%) |
| Treated on clinical trial | 63 (66%) |
Rare tumor types
A variety of rare cancers was represented among the 95 patients, comprising 40 different subtypes (Table 2). The most common histologic types were adenoid cystic carcinoma (13%), cholangiocarcinoma (7%), metaplastic breast carcinoma (6%), salivary gland (5%), gallbladder carcinoma (5%), and Erdheim-Chester disease (5%).
Table 2.
Rare tumor histologic types and relative frequencies in our dataset along with potentially targetable genomic aberrations. Incidence rate is obtained from European Society of Medical Oncology and represents cases throughout European Union. See methods for reference.
| Disease | No. of Patients/95 | Percent | Rate/100,000 | Potential Targets | |
|---|---|---|---|---|---|
| 1 | Adenoid cystic carcinoma | 12 | 12.63% | 0.12 | KDR, KIT, PDGFRA, NOTCH, PIK3R1, PIK3CB |
| 2 | Cholangiocarcinoma | 7 | 7.37% | 0.04 | FGFR2, PIK3CA, BRAF |
| 3 | Metaplastic breast carcinoma | 6 | 6.32% | 0.06 | PIK3CA, NOTCH, PIK3R1, PTEN, MCL1 |
| 4 | Salivary gland carcinoma | 6 | 6.32% | 1.31 | AKT, CDK4, BRAF, NOTCH, RET |
| 5 | Gallbladder adenocarcinoma | 5 | 5.26% | 4.37 | FGFR2 |
| 6 | Soft-tissue histiocytosis, non-Langerhans (ECD) | 5 | 5.26% | 0.05 | BRAF, MAP2K1 |
| 7 | Small intestine carcinoid | 4 | 4.21% | 0.37 | FGFR3 |
| 8 | Thyroid anaplastic carcinoma | 3 | 3.16% | 0.17 | PIK3CA, BRAF |
| 9 | Thyroid follicular carcinoma (Hurthle cell carcinoma) | 3 | 3.16% | 0.57 | |
| 10 | Mesothelioma | 3 | 3.16% | 1.9 | |
| 11 | Appendix adenocarcinoma | 2 | 2.11% | 0.97 | FLT3 |
| 12 | Brain glioblastoma | 2 | 2.11% | 2.52 | BRAF, PIK3C2B, PTEN |
| 13 | Brain medulloblastoma | 2 | 2.11% | 0.11 | BRCA1 |
| 14 | Kidney medullary carcinoma | 2 | 2.11% | 0.01 | |
| 15 | Pancreas neuroendocrine tumor | 2 | 2.11% | 1.22 | |
| 16 | Testis germ cell tumor (non-seminoma) | 2 | 2.11% | 1.21 | PIK3CA |
| 17 | Thymus thymoma (NOS) | 2 | 2.11% | 0.13 | |
| 18 | Vagina squamous cell carcinoma | 2 | 2.11% | 1.5 | EGFR, FGF4, PTEN |
| 19 | Bladder adenocarcinoma (urachus) | 2 | 2.11% | 0.29 | PIK3CA, FLT3 |
| 20 | Malignant mixed mullerian tumor | 2 | 2.11% | 0.36 | FGFR2, PTEN |
| 21 | Kidney medullary carcinoma | 2 | 2.11% | 0.44 | |
| 22 | Adrenal gland cortical carcinoma | 1 | 1.05% | 0.18 | |
| 23 | Anus squamous cell carcinoma | 1 | 1.05% | 1.09 | |
| 24 | Brain atypical teratoid rhabdoid tumor | 1 | 1.05% | 0.01 | |
| 25 | Brain meningioma | 1 | 1.05% | 0.15 | |
| 26 | Cervix neuroendocrine carcinoma | 1 | 1.05% | 0.06 | ALK |
| 27 | Ovary/Fallopian tube serous carcinoma | 1 | 1.05% | 1 | KDR, PIK3CA, NOTCH1, FLT3, MCL1 |
| 28 | Head and neck lymphoepithelioma | 1 | 1.05% | 0.01 | MCL1 |
| 29 | Head and neck neuroendocrine carcinoma | 1 | 1.05% | 0.1 | |
| 30 | Kidney chromophobe carcinoma | 1 | 1.05% | 0.01 | |
| 31 | Lung large cell neuroendocrine carcinoma | 1 | 1.05% | 4.01 | FGFR3, PIK3CA |
| 32 | Ovary clear cell carcinoma | 1 | 1.05% | 0.32 | PIK3CA |
| 33 | Ovary granulosa cell tumor | 1 | 1.05% | 0.12 | KDR |
| 34 | Ovary neuroendocrine carcinoma | 1 | 1.05% | 0.01 | |
| 35 | Ovary sex-cord stromal tumor | 1 | 1.05% | 0.13 | |
| 36 | Pancreas islet cell tumor | 1 | 1.05% | 0.03 | |
| 37 | Prostate neuroendocrine carcinoma | 1 | 1.05% | 0.01 | |
| 38 | Rectum squamous cell carcinoma | 1 | 1.05% | 0.07 | FGFR3, PIK3CA |
| 39 | Skin Merkel cell carcinoma | 1 | 1.05% | 0.12 | |
| 40 | Thyroid medullary carcinoma | 1 | 1.05% | 0.11 | RET |
ECD: Erdheim-Chester disease; NOS, not otherwise specified
Genomic alterations
Eighty-seven of the 95 patients (92%) had at least one genomic alteration identified. The mean number of mutations per patient was 2.6. The most frequently identified mutations were in TP53 (23%), KRAS (10%), PIK3CA (9%), CDKN2A/B (8%), BRAF (7%), MLL (7%), and ARID1A (6%) (Figure 1). Thirty-six patients (38%) had at least one potentially actionable alteration in 21 different tumors; the pathways represented by these alterations are shown in Table 2. Well-established PI3K/AKT pathway aberrations were seen in metaplastic breast carcinoma; adenoid cystic, nasopharyngeal, cholangiocarcinaom, glioblastoma; lung large cell neuroendocrine carcinoma; ovarian serous and clear cell, germ cell, and bladder adenocarcinomas; cervical squamous cell carcinoma; anaplastic thyroid carcinomas; and rectal squamous cell carcinoma. MAP kinase pathway alterations were seen in the form of BRAFV600E mutations in Erdheim-Chester disease, acinic cell tumor, cholangiocarcinoma, glioblastoma, and anaplastic thyroid carcinomas. KRAS mutations were seen predominantly in tumors of gastrointestinal or biliary origin. Amplifications dominated the landscape of FGFR alterations, which were seen in cholangiocarinoma, vaginal, metaplastic breast, rectal, ovarian serous, gallbladder, mixed mullerian, and lung large cell carcinomas and carcinoid. NOTCH pathway alterations were identified in salivary gland, adenoid cystic, and metaplastic breast carcinomas. NTRK alterations were identified in a salivary gland carcinoma. SOX2 amplification was identified in anal, lung large cell, serous ovarian, testicular, and bladder adenocarcinomas and glioblastoma. Mutations in germ-line associated genes (VHL, TSC2, NF1, NF2) were reported in anaplastic thyroid, appendiceal, lung large cell, and urachal carcinomas; carcinoid; mesothelioma.
Figure 1.
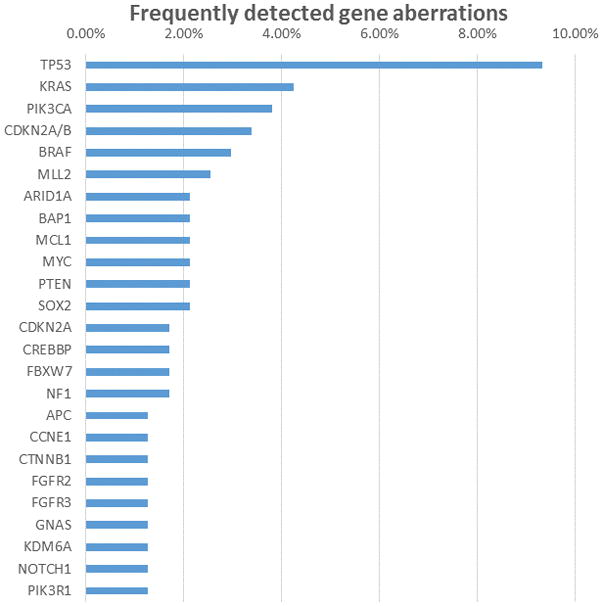
Mutations found in rare tumors
Figure 1 contains the most frequently mutated genes in 95 patients with a rare tumor.
Targeted therapies
Thirteen patients received targeted therapy (Table 3). Of these, 4 had a partial response, 6 had stable disease, and 3 had progressive disease as the best response. Duration of partial response ranged from 3 months for cholangiocarcinoma to 2 years and ongoing for Erdheim-Chester disease. Some patients were unable to enroll on targeted therapy trial despite having an actionable alteration because of trial slot availability (8/21 patients).
Table 3.
Patients with a rare tumor treated on a targeted agent clinical trial and their best response.
| DISEASE | ABERRATION | THERAPY | BEST RESPONSE |
|---|---|---|---|
| CHOLANGIOCARCINOMA | BRAFV600E | BRAFi27 | PR |
| ECD | BRAFV600E | BRAFi27 | PR |
| GLIOBLASTOMA MULTIFORME | BRAFV600E | BRAFi27 | PR |
| ADENOID CYSTIC CARCINOMA | PIK3R1 E515fs*1 |
Akti28 | SD × 24 cycles |
| ADENOID CYSTIC CARCINOMA | KIT and PDGFRA amplification | KITi + PDGFRAi29 | SD × 7 cycles, continues on trial |
| SALIVARY GLAND ADENOCARCINOMA | AKT1 E17K | AKTi28 | SD × 6 cycles |
| SALIVARY GLAND ADENOCARCINOMA | BRAFV600E | BRAFi27 | PR |
| ECD | BRAFV600E | BRAFi27 | SD × 3 years |
| METAPLASTIC BREAST CARCINOMA | PIK3R1 Y580fs*19 |
Chemo + mTORi30 | SD × 7 cycles |
| METAPLASTIC BREAST CARCINOMA | PIK3CA H1047R | Chemo + mTORi30 | SD × 8 cycles |
| OVARIAN CLEAR CELL CARCINOMA | PIK3CA H1047R | Chemo + mTORi30 | PD |
| PROSTATE NEUROENDOCRINE CARCINOMA | PTEN loss | Chemo + mTORi30 | PD |
| LUNG LARGE CELL NEUROENDOCRINE CARCINOMA | PIK3CA amplification | PI3Ki31 + MEKi32 | PD |
i, inhibitor; PR, partial response; ECD, Erdheim-Chester disease; SD, stable disease; PD, progressive disease.
Tumor mutational burden
Tumor mutational burden was determined for 71 of the 95 patients. We were able to identify 2 (3%) patients (one with a nasopharyngeal carcinoma and one with a rectal squamous cell carcinoma) that had high mutational burden. Intermediate mutational burden was identified in 10 patients (14%). The rest (83%) had low mutational burden. All responders to targeted therapy had a low mutational burden.
GENIE data
We evaluated how our mutational burden data compare with those in the AACR GENIE database. We found that all tumor types in our dataset were also present in GENIE. When we plotted the average mutational burden in GENIE, we found that rare tumors had an intermediate median tumor mutation burden. This did not significantly differ from published reports on tumor mutational burden.(12) The spectrum of tumor mutation burden in GENIE ranged from very low (<1 mutation per megabase) to 20 or more, with occasional outliers having hundreds of mutations (Figure 2).
Figure 2.

Mutation burden counts from the GENIE database.
Figure 2 contains mutational burden counts form the GENIE database for tumor types in our dataset. The tumors that showed a response to a targeted agent are highlighted in red. Tumors with progressive disease on targeted therapy are highlighted in purple. Stable disease as best response is in green.
Discussion
In this study we sought to uncover the mutational landscape of rare tumors and to identify potentially actionable aberrations. We found that a variety of such targets exist including PI3K, FGFR, EGFR, NOTCH, NTRK, and MAPK pathways. Clinically, the patients with robust responses had BRAF V600E or PI3K pathway mutations. Additionally, we found that rare tumors tend to have a low to intermediate mutational burden and this is confirmed by data not only from our patients, but from the AACR GENIE database.
Rare tumors continue to suffer from relative research neglect. This is unfortunate because they potentially have a distinct advantage from a genomic perspective. They can have simple genomes with relatively few mutations. Studying a genome with low mutational burden allows for more straightforward hypothesis testing regarding a particular driver mutation or targeted therapy. In seeking to define a new way of studying and perhaps treating rare tumors, in a “genome first, histology second” approach, we reviewed NGS data from patients enrolled on various phase I trials at our institution. Naturally, the sample was small, and less than 20% of the tumors qualified as rare. We could have enlarged this sample by expanding beyond the strict ESMO definition of rare cancer, but we deliberately chose to restrict our analysis to truly the most underrepresented tumors. As expected, our dataset was diverse, with 40 different tumor types, and may not accurately reflect the (true) relative incidences of these diseases. Adenoid cystic carcinoma should not be more incident than metaplastic breast cancer, but it is likely over-represented in our dataset because patients with this cancer tend to live longer and be more fit to participate in a clinical trial than those with metaplastic breast cancer. The diversity of tumors makes any kind of outcomes conclusions impossible. This may be acceptable, as drugs with rare tumor indications hardly ever achieve regulatory approval through a placebo-controlled randomized phase III trial. Instead, regulatory agencies will accept phase II data as sufficient evidence of drug activity(13).
Our experience was colored by a preponderance of patients with BRAF V600E mutations, some of whom had remarkable responses to BRAF-directed therapy. This was likely due to a selection bias because at least one clinical trial available specifically sought to identify BRAF-mutant cancers for treatment with targeted agent vemurafenib(14). This selection bias is also likely the reason that BRAF inhibition led to the strongest clinical responses and is consistent with several prior reports of BRAF inhibitor activity(15–18). If our data were enriched with, for example, PI3K or KIT pathway mutations, then the respective small molecule inhibitors of those mutations would likely have resulted in the best clinical responses. Most patients in this review did not receive a matched therapy. As our clinic and the list of available trials continues to evolve with the landscape of targeted therapies, we may find more patients being enrolled on matched therapies. This is especially evident as we continue to enlist patients to trials such as TAPUR, NCI-MATCH, and My Pathway which aim to definitely answer the question of targeted therapy as best approach. We note that dramatic clinical responses in patients with PI3K pathway mutations were not observed. This is consistent with prior publications.
One question that arises is the meaning of stable disease. Oncologists frequently reassure anxious patients that a stable scan is a good scan. Perhaps this is less meaningful to those with a potentially indolent disease such as adenoid cystic carcinoma, where stable disease for many years may become a normal part of life. Or it may be good news for a patient with metaplastic breast carcinoma or cholangiocarcinoma. Investigators have shown that patients with stable disease may benefit just as much as patients with a partial response(19). Others have shown that even a marginal response that falls under the purview of stable disease can have a survival advantage(20). In gastrointestinal stromal tumor, for example, any decrease in size of more than 10% or decrease in density greater than 15% indicates a response to therapy(21). From a scientific or regulatory approval perspective, stable disease is not a response and needs stronger supporting data such as a survival advantage to be clinically meaningful. Showing a survival advantage in a rare tumor with an even rarer mutation is an almost unrealistic expectation as the numbers required to show benefit may be unattainable in a single institution or in a timeframe meaningful to current patients.
An important hypothesis that arises from these data is whether low mutational burden increases probability of response to targeted therapy. Intuitively, finding an actionable mutation on a backdrop of many other mutations makes it unlikely that this mutation is driving the cancer. Alternatively, a single potentially actionable mutation on a backdrop of a quiescent tumor genome is more likely to yield therapeutic success. An analysis of a larger dataset for targeted therapy response in patients with low TMB versus intermediate or high TMB may further elucidate this question.
Our results also highlight the need for basket studies for the study of patients with a rare cancer. The availability of the VE-Basket trial(14) had an important effect on patients harboring the BRAF alteration. The availability of trials such as TAPUR from American Society of Clinical Oncology or the National Cancer Institute-Match will likely help patients with rare cancers be enrolled on clinical trials. We have questioned whether whole genome or whole exome sequencing would add valuable information. Keeping in mind the available therapies even in our very large center, all of the targets of these drugs are covered in a limited gene panel. The added cost and complexity of more extensive panels does not warrant the likely limited benefit over a cancer gene directed panel.
Studying these rare tumors to further human knowledge is an admirable and worthwhile goal. As physicians we must remember that with each of those repositories of hidden knowledge comes a patient desperately seeking hope for a cure. In this golden age of DNA sequencing, it is no longer acceptable to simply study a patient. We must use this robust new technology to benefit patients, not just the scientific endeavor. It is with this patient-centered viewpoint that we recognized a paucity of data on mutations and potential targets in rare tumors with the full recognition that arguments exist for and against universal genomic testing.(22)
Several studies have shown benefit to precision oncology which were nicely compiled in a meta-analysis of 32 phase II trials showing that a precision strategy was an independent predictor of success.(23) However, one randomized study – SHIVA trial – did not show a survival advantage for molecularly targeted agents. While this study is important in showing feasibility of precision oncology in a randomized setting, it should be noted that more than 80% of patients received single agent mTOR inhibitor or hormonal therapy. The inference from this study should be that single agent mTOR inhibitors or hormonal therapies may be futile in the refractory setting.(24) Recently, the MyPathway trial used four currently approved targeted therapies to treat patients with advanced solid tumors harboring alterations in HER-2, EGFR, BRAF, or Hedgehog pathway. The study produced meaningful objective responses in almost a quarter of patients.(25)
At our center the type of rare tumor and the department they present in drives decisions on sending comprehensive genomic profiling. Some cancers such as cholangiocarcinoma are increasingly being molecularly profiled and frequently referred to our phase 1 clinic for access to targeted therapies. Other departments and practitioners have been slower to catch on, but we are hoping that our analysis will entice other providers who see rare tumors to send molecular profiling more consistently. As this practice becomes more common, we will find more and more patients who can benefit from targeted therapy. If we do not seek, we do not find.
We conclude that the addition of comprehensive genomic profiling adds a possible line of therapy for rare cancers that have little or no standard of care. Potentially targetable genes/pathways in these cancers include EGFR, VEGF, PTEN/AKT, MAPK, ALK, NOTCH, FGF, RET, FLT3, KIT, and PDGFRA. Tumors with the BRAFV600E alteration responded well to BRAF inhibitors in our patients. mTOR inhibitors are effective in cancers with a PTEN/PI3K alteration. KIT inhibitors have activity in caners with KIT and PDGFRA alterations other than gastrointestinal stromal tumors. Most tumors will not have an actionable mutation. Tumor mutational burden may be inversely proportional to response to targeted therapy.
Acknowledgments
Financial Support: The University of Texas MD Anderson Cancer Center is supported by the National Institutes of Health Cancer Center Support Grant CA016672. V. Subbiah acknowledges the Shannon Wilkes Sarcoma Research funds. This work was supported in part by Cancer Prevention Research Institute of Texas Grant RP110584 and National Center for Advancing Translational Sciences (Grant UL1 TR000371, Principal Investigator D. D. McPherson) (Center for Clinical and Translational Sciences).
The funding sources had no input into the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication.
The authors would like to thank Katherine L Hale for her assistance in editing this manuscript.
Footnotes
Conflict of Interest: Apostolia-Maria Tsimberidou receives research funding from FMI.
References
- 1.Greenlee RT, Goodman MT, Lynch CF, Platz CE, Havener LA, Howe HL. The Occurrence of Rare Cancers in U.S. Adults, 1995–2004. Public Health Rep. 2010;125(1):28–43. doi: 10.1177/003335491012500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeSantis CE, Kramer JL, Jemal A. The burden of rare cancers in the United States: The Burden of Rare Cancers in the United States. CA: A Cancer Journal for Clinicians. 2017;67(4):261–72. doi: 10.3322/caac.21400. [DOI] [PubMed] [Google Scholar]
- 3.RARE DISEASES ACT OF 2002. Volume 107-2802002. p 116.
- 4.Jamshidi F, Nielsen TO, Huntsman DG. Cancer genomics: why rare is valuable. J Mol Med (Berl) 2015;93(4):369–81. doi: 10.1007/s00109-015-1260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. GENOMIC LANDSCAPE OF NON-SMALL CELL LUNG CANCER IN SMOKERS AND NEVER SMOKERS. Cell. 2012;150(6):1121–34. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012;122(8):2983–8. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groisberg R, Roszik J, Conley A, Patel SR, Subbiah V. The Role of Next-Generation Sequencing in Sarcomas: Evolution From Light Microscope to Molecular Microscope. [DOI] [PubMed] [Google Scholar]
- 8.Subbiah V, Roszik J. Towards precision oncology in RET-aberrant cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato S, Kurasaki K, Ikeda S, Kurzrock R. Rare Tumor Clinic: The University of California San Diego Moores Cancer Center Experience with a Precision Therapy Approach. LID - theoncologist.2017-0199 [pii] LID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groisberg R, Hong DS, Holla V, Janku F, Piha-Paul S, Ravi V, et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget. 2017;5(0) doi: 10.18632/oncotarget.16845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner AH, Coffman AC, Ainscough BJ, Spies NC, Skidmore ZL, Campbell KM, et al. DGIdb 2. 0: mining clinically relevant drug–gene interactions. Nucleic Acids Research. 2016;44(D1):D1036–D44. doi: 10.1093/nar/gkv1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Medicine. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chabner B. Approval of New Agents after Phase II Trials. Am Soc Clin Oncol Educ Book. 2012:e1–3. doi: 10.14694/EdBook_AM.2012.32.e1. [DOI] [PubMed] [Google Scholar]
- 14.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. New England Journal of Medicine. 2015;373(8):726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2018;36(1):7–13. doi: 10.1200/JCO.2017.73.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planchard D, Besse B, Groen HJM, Souquet P-J, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. The Lancet Oncology. 17(7):984–93. doi: 10.1016/S1470-2045(16)30146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2017.5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen S, Meric-Bernstam F, Hong DS, Hess KR, Subbiah V. Co-occurring Genomic Alterations and Association With Progression-Free Survival in BRAFV600-Mutated Nonmelanoma Tumors. Journal of the National Cancer Institute. 2017;109(10) doi: 10.1093/jnci/djx094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, Teng Y, Jin B, Zhao M, Yu P, Hu X, et al. Initial partial response and stable disease according to RECIST indicate similar survival for chemotherapeutical patients with advanced non-small cell lung cancer. BMC Cancer. 2010;10:681. doi: 10.1186/1471-2407-10-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19(3):508–15. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 21.Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25(13):1753–9. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 22.Subbiah V, Kurzrock R. Universal Genomic Testing Needed to Win the War Against Cancer: Genomics IS the Diagnosis. JAMA Oncol. 2016;2(6):719–20. doi: 10.1001/jamaoncol.2016.0078. [DOI] [PubMed] [Google Scholar]
- 23.Schwaederle M, Zhao M, Lee JJ, Eggermont AM, Schilsky RL, Mendelsohn J, et al. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(32):3817–25. doi: 10.1200/JCO.2015.61.5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Tourneau C, Delord J-P, Gonçalves A, Gavoille C, Dubot C, Isambert N, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. The Lancet Oncology. 16(13):1324–34. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 25.Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2018 doi: 10.1200/JCO.2017.75.3780. JCO2017753780. [DOI] [PubMed] [Google Scholar]
- 26.Pubchem. Vemurafenib.
- 27.Pubchem. AKT Kinase Inhibitor.
- 28.Pubchem. DCC-2618.
- 29.Pubchem. Temsirolimus.
- 30.Pubchem. 944396-07-0.
- 31.Trametinib. Pubchem.