

Abstract
Increased expression of major histocompatibility complex (MHC) class-I genes and aberrant expression of MHC class-II genes in thyroid epithelial cells (TECs) are associated with autoimmune thyroid diseases. Previous studies have shown that methimazole (MMI) reduces MHC class-I expression and inhibits interferon-γ (IFN-γ or IFNG as listed in the MGI Database)-induced expression of the MHC class-II genes in TECs. The action of MMI on the MHC class-I genes is transcriptional, but its mechanism has not been investigated previously. In the present study, we show that in Fisher rat thyroid cell line 5 cells, the ability of MMI and its novel derivative phenylmethimazole (C10) to decrease MHC class-I promoter activity is similar to TSH/cAMP suppression of MHC class-I and TSH receptor genes, and involves a 39 bp silencer containing a cAMP response element (CRE)-like site. Furthermore, we show that C10 decreases MHC class-I gene expression to a greater extent than MMI and at 10- to 50-fold lower concentrations. C10 also reduces the IFN-γ-induced increase in the expression of MHC class-I and MHC class-II genes more effectively than MMI. Finally, we show that in comparison to MMI, C10 is a better inhibitor of specific protein–DNA complexes that are formed with a CRE-like element on the MHC class-II promoter. These data support the conclusion that the immunosuppressive mechanism by which MMI and C10 inhibit MHC gene expression mimics ‘normal’ hormonal suppression by TSH/cAMP.
Introduction
Increased expression of major histocompatibility complex (MHC) class-I molecules and aberrant expression of MHC class-II molecules on nonimmune cells are involved in the pathogenesis of autoimmune diseases. Indeed, the expression of MHC class-I and class-II molecules is increased in pancreatic β islet cells of patients with type 1 diabetes mellitus, in muscle biopsies of patients with inflammatory myopathies, and in thyroid epithelial cells (TECs) from patients with autoimmune thyroid diseases (Bottazzo et al. 1985, Kohn et al. 2000, Davies 2005, Jain et al. 2007, Gono et al. 2009). The importance of MHC class-I gene over-expression in the pathogenesis of autoimmune diseases is suggested by the observation that MHC class-I-deficient mice do not develop autoimmunity in different experimental models, such as those for systemic lupus erythematosus, type 1 diabetes mellitus, and autoimmune blepharitis (Mozes et al. 1993, Serreze et al. 1994, Chan et al. 1995, Singer et al. 1999). Down-regulation or the absence of MHC class-I gene expression is also considered a hallmark of tissue immune privilege (Ito et al. 2004, 2008).
The involvement of aberrant MHC class-II gene expression in the development of autoimmune thyroid disease has been supported by several clinical and experimental observations. For example, mice immunized with fibroblasts expressing both human TSH receptor (TSHR) and an MHC class-II molecule develop pathological features of Graves’ disease (Shimojo et al. 1996, Arima et al. 2008).
Methimazole (MMI) is widely used to treat Graves’ disease and other forms of primary hyperthyroidism, and several studies have shown that it transcriptionally down-regulates MHC class-I and class-II gene expression in Fisher rat thyroid cell line 5 (FRTL-5) cells (American Type Culture Collection, CRL-8305; Saji et al. 1992b, Singer et al. 1997, Montani et al. 1998b, Kohn et al. 2000). Furthermore, treatment of mice with MMI reduces cell-surface expression of MHC class-I molecules and prevents the development of experimental systemic lupus erythematosus, blepharitis, and experimental autoimmune uveitis (Singer et al. 1994, Chan et al. 1995, Wang et al. 2003).
In previous studies, we have characterized a tautomeric cyclic thione derivative of MMI, phenylmethimazole (compound 10, or C10; Fig. 1), as more potent than MMI in the reduction of MHC expression. In those studies, we used FRTL-5 cells as a nontransformed cell line of rat TECs, which are a widely used model of TEC functions that are relevant to human pathophysiology (Capen 1996, Fukushima et al. 2008). In these cells, we demonstrated that C10 is a powerful anti-inflammatory and immunosuppressive agent, with a greater efficacy than MMI in decreasing constitutive and interferon-γ(IFN-γ or IFNG as listed in the MGI Database)-induced increases in MHC class-I and class-II gene expression (Kohn et al. 2002, Dagia et al. 2004, Harii et al. 2005). Furthermore, in comparison to MMI, C10 reduced to a much greater extent the increases in MHC class-I and class-II gene expression that were dependent on viral infection, Toll-like receptor 3 (TLR3) stimulation, and IFN-γ (Harii et al. 2005).
Figure 1.
Chemical structures of methimazole (MMI) and 5-phenylmethimazole (C10).
In the present study, we have investigated the mechanisms of action of MMI and C10 on MHC class-I and class-II gene expression. C10 was more effective than MMI in the transcriptional down-regulation of the expression of these MHC genes and it opposed the effects of IFN-γ better, which is consistent with its suppression ofthe increased expression of these genes that results from virus- and TLR3-dependent signaling (Harii et al. 2005). We show that this C10 action is mediated by cis-acting elements that are involved in the down-regulation of MHC class-I promoter activity and of MHC class-II gene expression induced by IFN-γ. Thus, C10 is a potent immunosuppressive agent that may have clinical relevance.
Materials and Methods
Materials
C10 was a gift from Interthyr Research Corporation (Marietta, OH, USA; Kohn et al. 2002). MMI was from Sigma–Aldrich Co. Rat recombinant IFN-γ was from Life Technologies Inc., Invitrogen Corp. [α−32 P]deoxy-CTP (3000 Ci/mmol) was from MP Biomedicals Europe (Illkirch, France). The source of all other materials was Sigma–Aldrich Co., unless otherwise specified.
Cell culture
The F1 subclone of FRTL-5 cells (American Type Culture Collection, CRL-8305) was a gift from Interthyr Research Foundation (Woodinville, WA, USA). These FRTL-5 cells were grown in 6H medium: Coon’s modified Ham’s F-12 medium supplemented with 5% calf serum, 2 mM glutamine, 1 mM nonessential amino acids, and a mixture of six hormones: bovine TSH (1×10−10M), insulin (10 μg/ml), cortisol (0.4 ng/ml), transferrin (5 μg/ml), glycyl-L-histidyl-L-lysine acetate (10 ng/ml), and somatostatin (10 ng/ml). The cells were diploid between the 5th and 25th passages, and had all of the functional properties described previously (Ambesi-Impiombato 1986, Saji et al. 1992a,b, Giuliani et al. 1995, 2008, Napolitano et al. 2002). Fresh 6H medium was added every 2–3 days, and the cells were passaged every 7 days. In individual experiments, the cells were shifted to a five-hormone medium, again with 5% calf serum as described above, but without TSH (5H medium).
C10 was prepared as a 200 mM stock solution in dimelthyl-sulfoxide (DMSO) before being used, and then diluted into the medium at 37 °C to the concentrations indicated. The final DMSO concentration was identical in both control and treated samples, and did not in any case exceed 0·5% (vol/vol).
RNA isolation and northern analyses
The FRTL-5 cells were grown to 60% confluency in 6H medium, and then maintained in 5H medium (i.e. without TSH) for 6 days. They were then shifted to 6H medium for 24 h before treatments with control vehicle, C10, or MMI for 48 h. RNA was prepared using a RNeasy Mini Kit (Qiagen Inc.), with 20 μg RNA samples run on denaturing agarose gels, capillary blotted on Nytran membranes (Schleicher & Schuell-Whatman, Florham Park, NJ, USA), u.v. cross-linked, and hybridized using QuickHyb hybridization solution (Stratagene, La Jolla, CA, USA) following the manufacturer’s protocol. The probes were labeled with [α−32P]dCTP using Ladderman labeling kits (Takara Mirus Bio, Madison, WI, USA). The MHC class-I probe was a 1·0 kb HpaI fragment of the MHC class-I pH 7 clone spanning the entire cDNA insert (Saji et al. 1992b). The β-actin probe was as described previously (Saji et al. 1992a). Quantitation was performed using a BAS 1500 bioimaging analyzer (FUJI Medical Systems USA, Inc., Stamford, CT, USA).
Flow-cytometric analysis
The FRTL-5 cells were grown to 60% confluency in 6H medium, and then maintained in 5H medium (i.e. without TSH) for 6 days. They were then shifted to 6H medium for 24 h before treatments with control vehicle, C10, MMI, or IFN-γ for 48 h.
For MHC class-I-molecule analysis, single cell suspensions were prepared and stained as described previously (Saji et al. 1992a, Shimojo et al. 1996, Giuliani et al. 2000, Napolitano et al. 2002); here, a directly FITC monoclonal antibody against the rat MHC class-I molecule was used: FITC-anti-rat RT1A (clone OX-18) (BD Pharmingen, San Jose, CA, USA). The same procedure was followed to evaluate MHC class-II molecules, except using a FITC mouse anti-rat monoclonal antibody to MHC class-II: FITC anti-rat RT1B (clone OX-6) (AbD Serotec, Kidlington, UK).
An isotype-matched control was used for the detection of background fluorescence: FITC mouse IgG1 isotype control (BD Pharmingen).
Plasmids
The luciferase chimeras of the swine MHC class-I (PD1) 5′-flanking region, p(–1100)Luc, p(–203)Luc, and p(–127)Luc, have been described previously (Napolitano et al. 2002). Briefly, different lengths of the 5′-flanking region of the PD1 (Pdcd1 as listed in the MGI Database) gene were inserted into a pGL2 basic vector (Promega Corp). The flanking regions and the corresponding constructs were named after the number of the base pair at the 5′ end of the + 1 bp, with the latter indicating the start of transcription. The –137 bp region of the HLA-DRα gene promoter (Montani et al. 1998a,b) was inserted into the BglII–HindIII site of the pGL2 basic vector. The sequences of all of the constructs used were confirmed by standard methods (Sanger et al. 1977).
Transfection and luciferase assay
Stable transfectant cells that contained the PD1–luciferase chimeras were obtained using a diethylaminoethyl (DEAE)-dextran procedure (Lopata et al. 1984, Giuliani et al. 1995, Napolitano et al. 2002). Near confluent cells in 6H medium were co-transfected with 20 μg plasmid DNA and 2 μg pMAMneo (Clontech BD Biosciences). After 2 days, G418 (Invitrogen) was added to the medium to a final concentration of 400 μg/ml, and after 3 weeks, the G418-resistant colonies were pooled and used.
For the luciferase assay, cells were grown to 60% confluency in 6H medium, and then maintained in 5H medium (i.e. without TSH) for 6 days. They were then shifted to 6H medium for 24 h before treatments with control vehicle, C10, MMI, or IFN-γ for 36 h as indicated.
The –137 bp HLA-DRα–Luc chimera was transiently transfected into FRTL-5 cells using the DEAE-dextran method indicated above. The cells were grown to 60% confluency, maintained in 5H medium (i.e. no TSH) for 6 days, and then returned to 6H medium 12 h before transfection. The cells were co-transfected with 20 μg –137 bp HLA-DRα–Luc chimera plasmid and 2 μg pRL-TK vector (Promega) as an internal control for efficiency of transfection. Twelve hours after transfection, the cells were treated with vehicle, IFN-γ, C10, or MMI for 36 h as indicated. The transfection efficiency did not deviate by more than 3% across all conditions, with corrections made as appropriate.
Luciferase assays were performed using a luciferase assay system (Promega), following the manufacturer’s instructions, and a LUMAT LB 9507 luminometer (EG & G Berthold, Bundoora, Australia). All values were normalized according to total cell protein.
Cell extracts
FRTL-5 cells were grown in the presence of complete 6H medium until 60% confluency, shifted into 5H medium (i.e. no TSH) for 6 days, and then treated as indicated. Cell extracts were prepared using a modification of a previously described protocol (Dignam et al. 1983). Briefly, the cells were washed twice in cold PBS, pH 7·4, scraped, and centrifuged (500g; 5 min; 4 °C). The cell pellet was resuspended in two volumes of Dignam buffer C (25% glycerol, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES–KOH), pH 7·9, 1·5 mM MgCl2, 420 mM NaCl, 0·5 mM dithiothreitol (DTT), 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 0·5 mM phenylmethylsulfonyl fluoride). The final NaCl concentration was adjusted to 420 mM on the basis of the cell pellet volume. The cells were lysed by repeated cycles of freezing and thawing, and then the cell extracts were centrifuged at 100 000 g at 4 °C for 20 min. Finally, the supernatants were recovered, aliquoted, and stored at –70 °C.
Electrophoretic mobility shift assays
DNA probes were created by restriction enzyme treatments of the chimeric luciferase constructs indicated above, and purified from 2% agarose gels using QIAEX (Qiagen Inc). These were labeled with [α−32P]dCTP using Klenow fragment (New England Biolabs Inc., Ipswich, MA, USA), and then purified on 8% native polyacrylamide gels.
The electrophoretic mobility shift assays (EMSAs) were performed as described previously (Giuliani et al. 1995, Montani et al. 1998a,b). The binding reactions in low salt and with no detergent included 1·5fmol [32P]DNA, 3 μg cell extract, and 3 μg poly(dI-dC) in 10 mM Tris–Cl, pH 7·9, 1 mM MgCl2, 1 mM DTT, 1 mM EDTA, and 5% glycerol, in a total volume of 20 μl, with incubations at room temperature for 30 min. Where indicated, unlabeled oligo-nucleotide competitors or antibodies were added to the binding reactions, followed by incubation with the cell extracts for 20 min before addition of the labeled DNA. The oligonucleotide competitors used were CRE1, spanning base pairs from –127 to –90 of the PD1 gene and containing the CRE-like site (TGACGCGA); nonpalindromic CRE (NPCRE), a derivative oligonucleotide spanning base pairs from –127 to –90 in which the CRE-like element had been mutated to a nonpalindromic substitution (CGACACGA); 5′-CRE, a derivative oligonucleotide spanning base pairs from –127 to –108 and lacking the CRE-like site and the 3′-flanking region; TSHR suppressor element binding protein-1 (TSEP-1), a single-strand oligonucleotide containing one of the TSEP-1 binding sites of the Tshr gene (5′-AAACTACCTCTCAACGCATCCG-3′). The antibodies used were activator transcription factor (ATF)-1 (F1–1, sc-241), ATF2 (C19, sc-187), and CREB binding protein (CBP or CREBBP as listed in the MGI Database) (A-22, sc-369) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Following the incubations, the reaction mixtures were electrophoresed on 4 or 5% native polyacrylamide gels at 160 V in 0·5 × or 1 × Tris-borate-EDTA buffer at room temperature. The gels were dried and autoradiographed.
Protein determination
Protein concentrations were determined using BCA protein assay kits (Pierce Biotechnology Inc., Rockford, IL, USA), with crystalline BSA as standard.
Statistical analysis
All experiments were repeated at least three times with independent batches of cells. The data are given as means ± S.D. The significance between experimental values was determined by two-way ANOVA, with P<0·05 or better when the data from all of the experiments were considered.
Results
C10 decreases constitutive MHC class-I gene expression in FRTL-5 cells
In a previous report, we showed that at a maximally effective concentration of 5 mM, MMI transcriptionally decreased MHC class-I expression in TECs (Saji et al. 1992b).
In the present study, we have analyzed the effects of the MMI derivative C10 on MHC class-I gene expression. The treatment of FRTL-5 cells with C10 resulted in a 66 ± 7% decrease in MHC class-I RNA levels, a greater decrease than that obtained with MMI (45 ± 5%) (Fig. 2A). Importantly, C10 was maximally effective at a concentration 10-fold lower than MMI (0·5 vs 5·0 mM); no further effects were seen at higher C10 concentrations (Fig. 2A). Treatment with solvent alone (0·5% DMSO) did not show any significant effects relative to nontreated cells (Fig. 2A, Cont DMSO versus Cont). Therefore, under all of the following conditions, the control cells were treated with 0·5% DMSO, a concentration equivalent to or lower than that used for the vehicle in treated cells.
Figure 2.
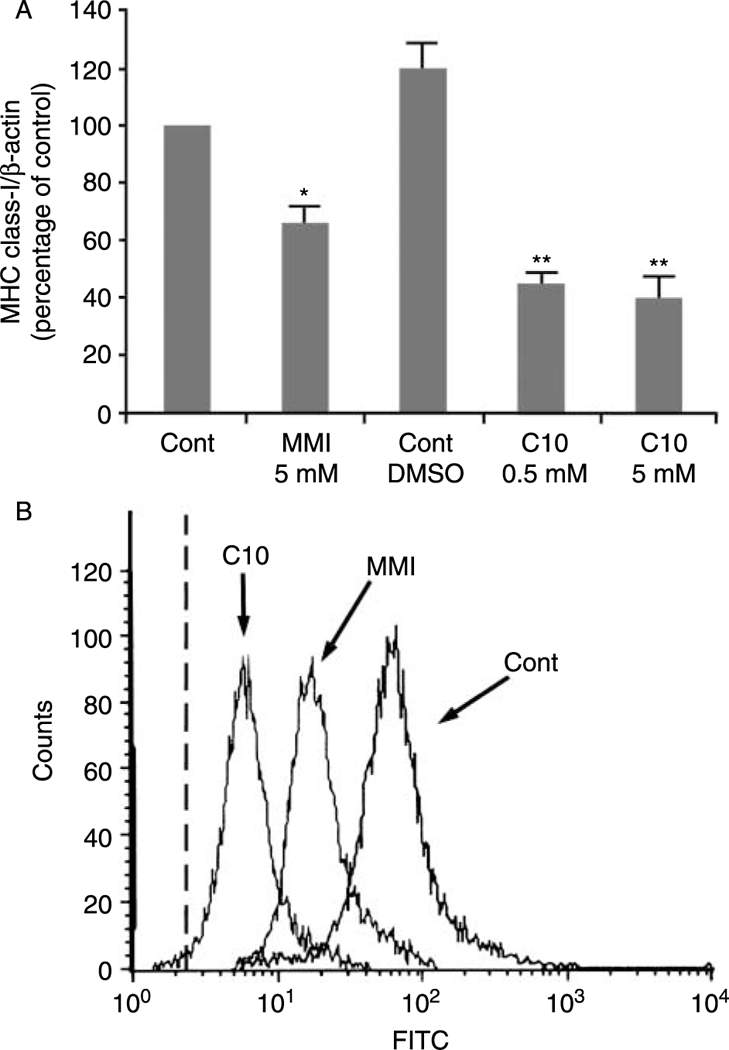
Effects of MMI and C10 on MHC class-I gene expression. (A) Effects of MMI and C10 on MHC class-I RNA levels. FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments as indicated. Data are mean MHC class-I/β-actin ±S.D. expressed as percentages of control from three separate experiments. *P<0·05 compared to control;**P<0·05 compared to control and to MMI-treated cells. (B) Flow-cytometric analysis of the expression of surface MHC class-I molecules. FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium with control vehicle for C10 (0·5% DMSO;Cont) without and with 5 mM MMI, or 0·5 mM C10 for 48 h as indicated. The cells were stained as detailed in Methods. The dashed line represents the isotype-matched control.
The decrease in MHC class-I RNA levels following C10 treatment was accompanied by a decrease in class-I surface molecule expression, as evaluated by flow-cytometric analysis (Fig. 2B). Furthermore, at a concentration of 0·5 mM, C10 significantly decreased cell-surface expression of MHC class-I molecules to a greater degree than that achieved with 5 mM MMI (Fig. 2B).
MMI and C10 decrease MHC class-I gene promoter activity through a DNA sequence located within 127 bp of the 5′ end of the transcription start site
The addition of either 5 mM MMI or 0·5 mM C10 to FRTL-5 cells stably transfected with a construct containing the –1100 bp of the extended MHC class-I promoter region ligated to the luciferase reporter gene [p(–1100)Luc] significantly reduced the luciferase activity (Fig. 3A). To map the MMI/C10-responsive element, FRTL-5 cells stably transfected with the 5′ deletion mutants p(–203)Luc and p(–127)Luc (Fig. 3B) were treated with 5 mM MMI or 0·5 mM C10. Both compounds decreased MHC class-I promoter activity in cells transfected with p(–203)Luc and p(–127)Luc, whereas the activity of the control pGL2 promoter was not affected (Fig. 3A).
Figure 3.
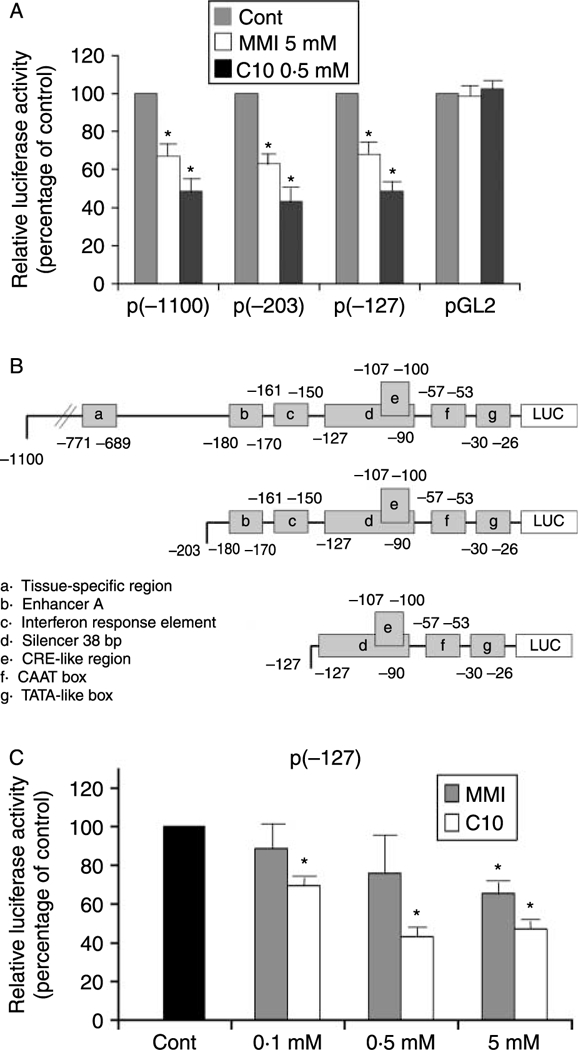
Effects of MMI and C10 on MHC class-I promoter activity in FRTL-5 cells stably transfected with PD1 promoter-luciferase chimeras. (A) G418-resistant FRTL-5 cells stably transfected with p(−1100)Luc, p(−203)Luc, p(−127)Luc, and the control vector pGL2 were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium as indicated. Luciferase activity is expressed as percentages of control activity. Data are means of four separate experiments. *P<0·05. (B) Diagrammatic representation of the different class-I-Luc chimeras. Some of the different regulatory cis-elements are noted. (C) Dose-response effects of MMI and C10 on the promoter activity of the p(−127)Luc chimera. G418-resistant FRTL-5 cells were stably transfected with p(−127)Luc, and FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium as indicated. Luciferase activity as in (A), with data as means of three separate experiments. *P<0·05.
The effects of both C10 and MMI on all three of the constructs were maximal after 36 h of treatment. Expression levels of p(–1100)Luc were 67·0 ± 7·2 and 48·5 ± 8·7% of control after treatment with MMI and C10 respectively, whereas those of p(–203)Luc were 63·0 ± 5·0 and43·0 ± 8·0% of control respectively and those of p(–127)Luc were 68·2 ± 9·2 and 48·6 ± 4·5% of control respectively. No further significant decreases were observed after 48 and 72 h of treatment (data not shown). The effects of C10 were concentration dependent, reaching a maximum at 0·5 mM, with no increases in the effects to 5 mM (Fig. 3C). In contrast, the effects of MMI were only significant at 5 mM (Fig. 3C).
These data indicate that the action of MMI and C10 is mediated by a DNA sequence element located between –127 bp and the start of transcription. This region encompasses a previously described silencer element that spans base pairs from –127 to –90 (Fig. 3B, element d), which contains a CRE-like site (Fig. 3B, element e) (Saji et al. 1997, Kirshner et al. 2000).
To identify the DNA–protein complexes associated with the C10/MMI-responsive region, EMSAs were performed using a [32P]-labeled probe corresponding to the 5′-flanking region of the PD1 gene between –168 and +1 bp. The EMSAs with extracts derived from nontreated FRTL-5 cells generated a series of complexes, A–E (Fig. 4A, lane 2). As shown previously, TSH treatment of the FRTL-5 cells induced the appearance of two additional complexes, F and G (Fig. 4A, lane 4). Interestingly, treatment of FRTL-5 cells with both 0·5 mM C10 and 5 mM MMI also induced the appearance of bands F and G (Fig. 4A, lanes 5 and 3). Thus, TSH/cAMP, C10, and MMI all induced the same pattern of bands in EMSAs, suggesting that common complexes are induced by all of these treatments.
Figure 4.
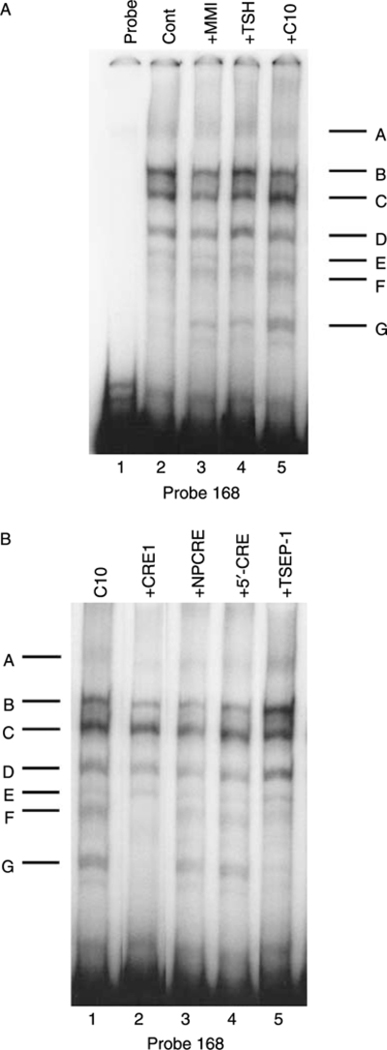
Effects of MMI and C10 on binding of proteins to the −168 bp probe. Representative EMSAs performed using cell extracts, as detailed in Materials and Methods, and the −168 bp probe, which spans the region between −168 and +1 bp from the transcription start site of the PD1 gene. (A) FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments in 5H medium as indicated with 0·5% DMSO (Control), 5 mM MMI, 1×10−10 M TSH, or 0·5 mM C10 for 36 h. Protein-DNA complexes are labeled as A-G (see text). Lane 1, probe alone. (B) FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with the probe incubated in 5H medium with 0·5 mM C10 alone or with 100-fold excess of unlabeled competitors as indicated for 36 h. Protein-DNA complexes are labeled as A-G (see text). Similar results were obtained using cell extracts from cells treated with 5 mM MMI (data not shown).
The EMSA bands F and G induced by TSH/cAMP treatment of FRTL-5 cells required the CRE sequence contained within the 168 bp segment (Saji et al. 1997, Kirshner et al. 2000). To determine whether the C10-induced complexes similarly depended on the CRE, competition studies were performed using a nonlabeled 38 bp oligonucleotide that spans the MHC class-I CRE. Indeed, the intact CRE (CRE1), but neither a mutant CRE (NPCRE) nor a sequence flanking CRE (5′-CRE), inhibited the formation of the C10-induced bands F and G (Fig. 4B). Thus, the C10-induced complexes, as with the TSH/cAMP-induced complexes, required an intact CRE.
The formation of the F and G complexes was also inhibited by a single-strand oligonucleotide containing one of the TSEP-1 binding sites of the Tshr gene (Fig. 4B, lane 5 versus lane 1) (Napolitano et al. 2000). The same results were obtained using extracts from cells treated with MMI (data not shown).
C10 and MMI reduce the effects of IFN-γ on MHC class-I and class-II gene expression
Previous studies from our laboratory have demonstrated that MMI opposes IFN-γ-mediated induction of the MHC class-I molecule expression in FRTL-5 cells (Saji et al. 1992b). Therefore, we next examined whether C10 has a similar effect.
FRTL-5 cells stably transfected with the p(–203)Luc class-I promoter, which contains an IFN response element (Fig. 3B), were treated for 36 h in 6H medium containing 100 U/ml IFN-γ without or with 0·5 mM C10 or 5 mM MMI. As shown in Fig. 5A, both C10 and MMI dramatically blocked IFN-γ-dependent activation of the MHC class-I gene promoter. The effect of C10 was greater than that of MMI (33·8 ± 6·2 vs 50·0 ± 4·3% inhibition respectively) at a 10-fold lower concentration.
Figure 5.
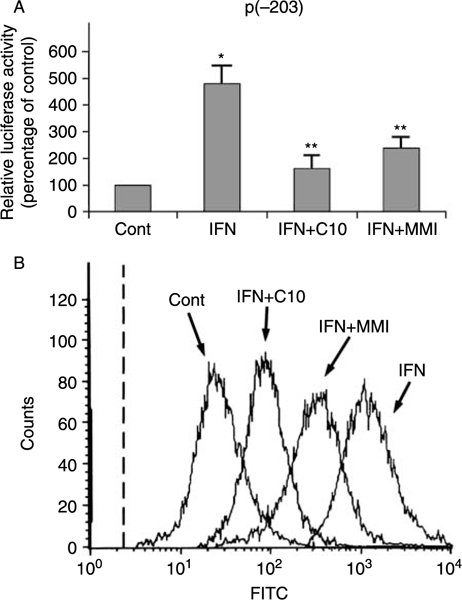
Effects of C10 and MMI on IFN-γ-induced increase in MHC class-I gene expression. (A) G418-resistant FRTL-5 cells stably transfected with p(−203)Luc were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium with 0·5% DMSO as vehicle (Cont) or 100 U/ml IFN-γ without or with 0·5 mM C10 or 5 mM MMI as indicated for 36 h. Luciferase activity is expressed as percentages of control activity, with data as means of three separate experiments. *P<0·05 compared to control; **P<0·05 compared to IFN. (B) Flow-cytometric analysis of the expression of surface MHC class-I molecules. FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium as in (A) as indicated for 48 h. The cells were stained with a fluoresceinated monoclonal antibody that specifically reacts with epitope S of the rat MHC class-I molecule. The dashed line represents the isotype-matched control.
These results for the class-I promoter activity were confirmed by flow-cytometric analysis of the surface molecule expression (Fig. 5B).
Since IFN-γ induces de novo MHC class-II gene expression that is blocked by MMI (Montani et al 1998b), we next evaluated the effects of C10 on IFN-γ induction. FRTL-5 cells were treated with 100 U/ml IFN-γ without or with 0·5 mM C10 or 5 mM MMI for 48 h. As shown by flow-cytometric analysis in Fig. 6A, FRTL-5 cells do not normally express MHC class-II molecules on their surface (Fig. 6A, Cont), while IFN-γ treatment induces their appearance (Fig. 6A, IFN). As with MMI, C10 significantly inhibited this IFN-γ effect (Fig. 6A, IFN + C10 and IFN + MMI respectively).
Figure 6.
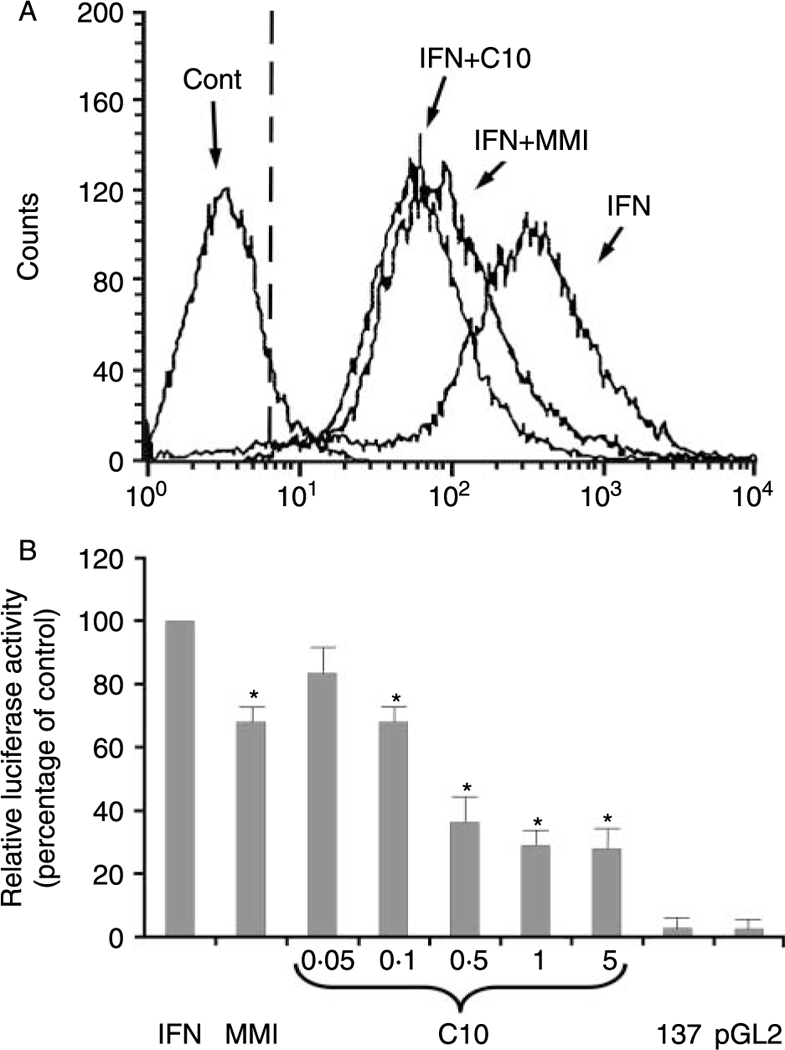
Effects of C10 and MMI on IFN-γ-induced MHC class-II molecule expression. (A) Flow-cytometric analysis of the expression of surface MHC class-II molecules. FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium with 0·5% DMSO as vehicle (Cont) or with 100 U/ml IFN-γ without or with 0·5 mM C10 or 5 mM MMI as indicated for 48 h. The cells were stained with a fluoresceinated monoclonal antibody that specifically reacts with RT1B (clone OX-6) of the rat MHC class-II molecule. The dashed line represents the isotype-matched control. (B) Effects of C10 on MHC class-II promoter activity. Transient transfections of FRTL-5 cells with the −137 bp HLA-DRα-Luc chimera were performed as described in Materials and Methods. Twelve hours after transfection, the cells were treated for 48 h with 100 U/ml INF-γ plus 0·5% DMSO as the IFN-induction control (IFN) or with 5 mM MMI or C10 at indicated concentrations. 137: cells transfected with−137 bp HLA-DRα-Luc chimera without 100 U/ml IFN-γ treatment; pGL2: cells transfected with plasmid control vector alone (pGL2 basic) with no further treatment. Luciferase activity is expressed as percentages of IFN-induced activity, with data as means of four separate experiments. *P<0·05 compared to IFN.
C10 action on the MHC class-II promoter is similar to that of MMI, but its effects are seen at 10- to 50-fold lower concentrations
To determine whether C10 and MMI target MHC class-II gene transcription, FRTL-5 cells were transiently transfected with a construct containing 137 bp of a minimal HLA-DRα promoter (HLA-DRα–Luc) (Balducci-Silano et al. 1998, Montani et al. 1998a,b). The cells were treated for 48 h with 100 U/ml IFN-γ without or with 5 mM MMI or C10 at the concentrations shown in Fig. 6B. In the absence of IFN-γ treatment, the –137 bp HLA-DRα promoter was not active in transiently transfected FRTL-5 cells, relative to the vector control (Fig. 6B, 137 versus pGL2). Treatment of FRTL-5 cells with rat recombinant IFN-γ induced –137 bp HLA-DRα–Luc chimera activity (Fig. 6B, IFN). However, addition of C10 suppressed this IFN-γ effect in a dose-dependent fashion (Fig. 6B, C10). The maximal effect of C10 was seen at 0·5 mM (36·0±9·8% of control), the same concentration that had maximal activity in decreasing MHC class-I gene expression. There were no significant increases in the inhibitory action of C10 at higher concentrations (Fig. 6B, C10, 5 mM).
As consistently seen, C10 was more effective than MMI and its effects were present at 10- to 50-fold lower concentrations (Fig. 6B, C10 versus MMI). The C10 effects were significant after 24 h of treatment (with MHC class-II gene expression being 66·0±4·0% of control cells treated with IFN-γ) and maximal after 48 h (with MHC class-II gene expression being 36·0 ± 9·8% of control cells).
To further investigate the mechanism of action of C10 on MHC class-II gene expression, we performed EMSAs using the –137 bp fragment of the HLA-DRα minimal promoter as a radiolabeled probe. As described previously (Montani et al. 1998b), cell extracts from IFN-γ-treated cells (100 U/ml) increased the abundance of a constitutive protein–DNA complex (Fig. 7A, lane 3, band A) and induced the appearance of a new faster migrating complex, (Fig. 7A, lane 3, band B). Treatment of cells with 0·5 mM C10 suppressed the effects of IFN-γ on the formation of both of these complexes (Fig. 7A, lane 5 versus lane 3). C10 was more effective than MMI at a concentration that is 10-fold lower (Fig. 7A, lane 5 versus lane 4).
Figure 7.
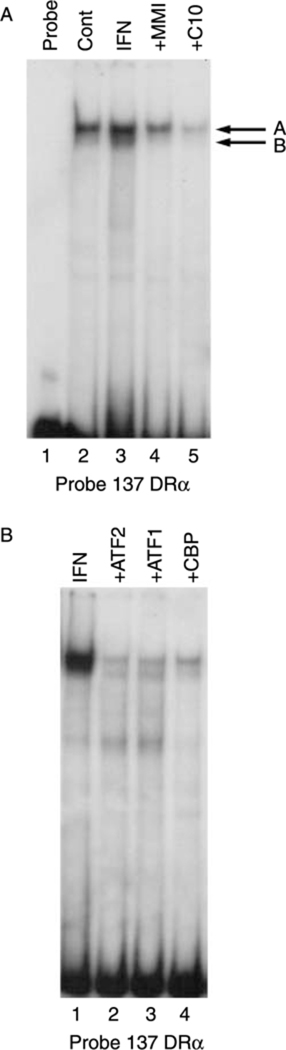
Effects of MMI and C10 on the binding of proteins to the −137 bp HLA-DRα probe. Representative EMSAs performed using cell extracts, as detailed in Materials and Methods, and the −137 bp HLA-DRα probe. (A) Effects of C10 on the IFN-γ-induced protein-DNA complexes. FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with final treatments in 6H medium as indicated with 0·5% DMSO as vehicle (Cont), or with 100 U/ml IFN-γ without or with 5 mM MMI or 0·5 mM C10, for 48 h. Protein-DNA complexes are labeled as A and B. Lane 1, probe alone. (B) Effects of antisera specific for ATF1, ATF2, and CBP on the formation of the IFN-γ-induced complexes. FRTL-5 cells were grown and prepared as detailed in Materials and Methods, with the probe incubated in 6H medium with 100 U/ml IFN-γ and with antibodies as indicated for 48 h.
Pre-incubation of FRTL-5 cell extracts with antibodies against the transcription factors ATF-1, ATF-2, and CBP, as described in Montani et al. (1998b), reduced the abundance of these two protein–DNA complexes (Fig. 7B, lanes 3, 2, and 4 respectively versus lane 1), suggesting a potential involvement of these transcription factors in the regulation of the MHC class-II gene. Further studies are in progress to evaluate this hypothesis.
Discussion
MMI is a thionamide drug that is widely used to treat hyperthyroidism. Its primary effect is to inhibit thyroid hormonogenesis by interfering with the action of the thyroid peroxidase (Cooper 2005a). As well as antithyroid activity, MMI has anti-inflammatory and immunosuppressive effects (Mitsiades et al. 2001, Cooper 2005a, Davies 2005). Although the latter effects have been viewed in the context of MMI action on thyroid hormone synthesis, several studies have suggested direct anti-inflammatory and immunomodulatory effects of MMI. Among other data, it has been shown that MMI inhibits a) the increase of CD69 and Fas ligand expression in T cells and TECs respectively from patients with Graves’ disease; and b) the constitutive and/or IFN-induced increased expression of the MHC class-I and class-II genes and ICAM-1 gene in TECs (Weetman et al. 1984, Saji et al. 1992b, Gessl & Waldhaus 1998, Montani et al. 1998b, Kim et al. 2001, Mitsiades et al. 2001, Zantutt-Wittmann et al. 2001, Cooper 2005a, Davies 2005).
In the present study, we have shown that a phenyl derivative of MMI, C10 (Fig. 1), is 10- to 50-fold more potent than MMI at suppressing abnormally increased MHC molecules in TECs. Specifically, using FRTL-5 cells, we have shown that C10 decreases constitutive and IFN-γ-induced MHC class-I gene expression. Both C10 and MMI affect MHC class-I promoter activity by targeting a region within 168 bp of transcription initiation. Moreover, we have shown here that these actions of MMI and C10 mimic the actions of TSH/cAMP on MHC class-I gene expression. Indeed, MMI and C10 act on a region that includes the constitutive 38 bp silencer of the MHC class-I promoter, which contains a CRE-like sequence (Saji et al. 1997, Kirshner et al. 2000). Both MMI and C10 induced the formation of two protein-DNA complexes that were not distinguishable in EMSAs from those induced by TSH/cAMP, the formation of which is associated with the down-regulation of the MHC class-I gene induced by TSH/cAMP
The competition for these two complexes by an oligonucleotide containing a binding site for the Y-box protein TSEP-1 suggests that TSEP-1 is involved in their formation. One hypothesis is that TSEP-1 can interact with other transcription factors, such as CREB, ATF1, and TTF1, which are known regulators ofthe TSHR and MHC genes in TECs (Saji et al. 1997, Kirshner et al. 2000, Kohn et al. 2000, Napolitano et al. 2000). Further studies are in progress to evaluate this hypothesis and to determine the transcription factors that constitute these complexes.
As already noted above, C10 is more effective than MMI in the regulation of MHC class-I and class-II genes at a concentration that is 10- to 50-fold lower. This is important because MMI anti-inflammatory and immunosuppressive effects are seen at concentrations that exceed those seen in vivo by 10- to 50-fold (Saji et al. 1992b, Cooper 2005b).
These observations are of particular interest given the role that the overexpression of HLA molecules on nonimmune cells has in the pathogenesis of thyroid autoimmune diseases (Bottazzo et al. 1985, Kohn et al. 2000, Davies 2005, Creus et al. 2009). It has been hypothesized (Harii et al. 2005) that several insults to nonimmune cells of target tissues (such as viral infections, dsRNA, or tissue injury) can activate TLR3 overexpression and signaling, and then induce an innate immune gene response, including secretion of both type 1 (IFN-α or IFN-β) and type 2 (IFN-γ) IFNs. This would result in an overexpression of TLR, MHC molecules, and cytokines by the target cells, which would then recruit and activate lymphocytes and hence initiate an autoimmune response (Kohn et al. 2000, Harii et al. 2005, Gianoukakis et al. 2008, Pisetsky 2008). Given this hypothesis, it is important to stress that both MMI and, to a greater extent, C10 inhibit overexpression of TLR3 molecules and the signaling induced by dsRNA and viruses in TECs (Harii et al. 2005). Indeed, C10 and MMI are strong inhibitors of STAT-1 and STAT-3 phosphorylation (Kim et al. 2001, Harii et al. 2005). Activation of the STAT proteins is one of the mechanisms by which dsDNA and dsRNA activate MHC, IFNs, TLRs, and other related genes (Kohn et al. 2000, Harii et al. 2005).
The mechanisms of action of these compounds explain their broader anti-inflammatory effects and suggest a therapeutic role for C10 beyond thyroid autoimmune diseases (Dagia et al. 2004). We are now carrying out animal studies to investigate these potential therapeutic properties of C10 in vivo. Preliminary results indicate that C10 has potential for autoimmune diseases regardless of thyroid function. Indeed, using Shimojo’s mouse model of Graves’-like disease (Arima et al. 2008), we have found that the administration of C10 can prevent the onset of the disease in the majority of the mice without causing hypothyroidism (Cerrone et al. 2008). Further studies are in progress to confirm these preliminary observations.
In summary, our data show that C10 inhibits constitutive and IFN-γ-induced MHC gene expression in TECs to a greater extent than MMI. Although the effects of MMI on MHC class-I gene expression were reported several years ago, the present study is the first to investigate its action at the promoter level. We have identified some of the cis-acting elements that are involved in the MMI/C10 effects, and we speculate that some of the transcription factors involved in TSH/cAMP regulation of the MHC and TSHR genes are also implicated in the C10/MMI effects on the MHC genes. These C10 effects on the MHC genes, together with its action on TLRs and IFN-regulated genes, suggest that it should have a role in the treatment of diseases involving dysfunction of innate immunity. Thus, C10 holds promise as a therapeutic agent for the treatment of autoimmune/inflammatory and other diseases, e.g. cancer, in which the TLR and STAT proteins have pathogenic roles.
Acknowledgements
The authors are grateful to Massimo Ruzzi and Christian Mazzocco for their technical assistance, and to Christopher P Berrie, MA, MPhil, PhD, for linguistic revision of the manuscript. Additionally, the authors would like to acknowledge the anonymous reviewers who have helped them to improve the presentation of their study.
Funding
Funding for this study was in part through a grant from the nonprofit Interthyr Research Foundation.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicial to the impartiality of the research reported herein.
References
- Ambesi-Impiombato FS 1986. Living, fast-growing thyroid cell strain, FRTL-5. US Patent 4,608,341. Interthyr Research Foundation Inc., MD, USA. 26 Aug 1986.
- Arima T, Shimojo N, Yamaguchi K, Tomiita M, Kohn LD & Kohno Y 2008. Enhancement of experimental Graves’disease by intranasal administration of a T-cell epitope of the thyrotropin receptor. Clinical Immunology 127 7–13. [DOI] [PubMed] [Google Scholar]
- Balducci-Silano PL, Suzuki K, Ohta M, Saito J, Ohmori M, Montani V, Napolitano G, Shong M, Taniguchi SI, Pietrarelli M et al. 1998. Regulation of major histocompatibility (MHC) class-II humanleukocyte antigen-DRα gene expression in thyrocytes by single-strand-binding protein-1, a transcription factor that also regulates thyrotropin receptor and MHC class-I gene expression. Endocrinology 139 2300–2313. [DOI] [PubMed] [Google Scholar]
- Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG & Gamble DR 1985. In-situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas of diabetic patients. New England Journal of Medicine 313 353–360. [DOI] [PubMed] [Google Scholar]
- Capen CC 1996. Anatomy: comparative anatomy and physiology In Werner and Ingbar’s the Thyroid, edn 7, pp 19–38. Eds Braverman LE & Utiger RD. Philadelphia: Lippincott-Raven Publishers. [Google Scholar]
- Cerrone D, Giuliani C, Rossi C, Bucci I, Arima T, Shimojo N, Kohno Y, Kohn LD, Napolitano G & Monaco F 2008. Phenylmethimazole prevents Graves’ disease onset in an animal model. Thyroid 18 S-39. [Google Scholar]
- Chan C, Gery I, Kohn LD, Nussenblatt R, Mozes E & Singer DS 1995. Periocular inflammation in mice with experimental systemic lupus erythematosus (SLE): a new experimental blepharitis and its modulation. Journal of Immunology 154 4830–4835. [PubMed] [Google Scholar]
- Cooper DS 2005a. Antithyroid drugs. New England Journal of Medicine 352 905–917. [DOI] [PubMed] [Google Scholar]
- Cooper DS 2005b. Treatment of thyrotoxicosis In Werner and Ingbar’s the Thyroid, edn 9, pp 665–694. Eds Braverman LE & Utiger RD. Philadelphia: Lippincott-Raven Publishers. [Google Scholar]
- Creus KK, De Paepe B & De Bleecker JL 2009. Idiopathic inflammatory myopathies and the classical NF-kB complex: current insights and implications for therapy. Autoimmunity Reviews 8 627–631. [DOI] [PubMed] [Google Scholar]
- Dagia NM, Harii N, Meli AE, Sun X, Lewis CJ, Kohn LD & Goetz DJ 2004. Phenyl methimazole inhibits TNF-α-induced VCAM-1 expression in an IFN regulatory factor-1-dependent manner and reduces monocytic cell adhesion to endothelial cells. Journal ofImmunology 173 2041–2049. [DOI] [PubMed] [Google Scholar]
- Davies TF 2005. The pathogenesis of Graves’ disease In Werner and Ingbar’s the Thyroid, edn 9, pp 457–473. Eds Braverman LE & Utiger RD. Philadelphia: Lippincott-Raven Publishers. [Google Scholar]
- Dignam J, Lebovitz R & Roeder R 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research 11 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T, Nedachi T, Akizawa H, Akahori M, Hakuno F & Takahashi S-I 2008. Distinct modes of activation of phosphatidylinositol 3-kinase in response to cyclic adenosine 3, 5-monophosphate or insulin-like growth factor I play different roles in regulation of cyclin D1 and p27Kip1 in FRTL-5 cells. Endocrinology 149 3729–3742. [DOI] [PubMed] [Google Scholar]
- Gessl A & Waldhaus W 1998. Elevated CD69 expression on naive peripheral blood T cells in hyperthyroid Graves’ disease and autoimmune thyroiditis: discordant effect of methimazole on HLA-DR and CD69. Clinical Immunology and Immunopathology 87 168–175. [DOI] [PubMed] [Google Scholar]
- Gianoukakis AG, Khadavi N & Smith TJ 2008. Cytokines, Graves’ disease, and thyroid-associated ophthalmopathy. Thyroid 18 953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani C, Saji M, Napolitano G, Palmer LA, Taniguchi SI, Shong M, Singer DS & Kohn LD 1995. Hormonal modulation of major histocompatibility complex class-I gene expression involves an enhancer-A-binding complex consisting of fra-2 and the p-50 subunit of NF-kB. Journal of Biological Chemistry 270 11453–11462. [DOI] [PubMed] [Google Scholar]
- Giuliani C, Napolitano G, Mastino AS, Di Vincenzo S, D’Agostini C, Grelli S, Bucci I, Singer DS, Kohn LD, Monaco F et al. 2000. Thymosin-α1 regulates MHC class-I expression in FRTL-5 cells at a transcriptional level. European Journal of Immunology 30 778–786. [DOI] [PubMed] [Google Scholar]
- Giuliani C, Noguchi Y, Harii N, Napolitano G, Tatone D, Bucci I, Piantelli M, Monaco F & Kohn LD 2008. The flavonoid quercetin regulates growth and gene expression in rat FRTL-5 thyroid cells. Endocrinology 149 84–92. [DOI] [PubMed] [Google Scholar]
- Gono T, Katsumata Y, Kawaguchi Y, Soejima M, Wakasugi D, Miyawaki M, Tsukahara S, Yamanaka H & Hara M 2009. Selective expression ofMHC class I in the affected muscle of a patient with idiopathic inflammatory myopathy. Clinical Rheumatology 28 873–876. [DOI] [PubMed] [Google Scholar]
- Harii N, Lewis CJ, Vasko V, Benavides-Peralta U, Sun X, Ringel MD, Saji M, Giuliani C, Napolitano G, Goetz DJ et al. 2005. Thyrocytes express a functional Toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. MolecularEndocrinology 19 1231–1250. [DOI] [PubMed] [Google Scholar]
- Ito T, Ito N, Bettermann A, Tokura Y, Takigawa M & Paus R 2004. Collapse and restoration of MHC class-I-dependent immune privilege: exploiting the human hair follicle as a model. American Journal of Pathology 164 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Meyer KC, Ito N & Paus R 2008. Immune privilege and the skin. Current Directions in Autoimmunity 10 27–52. [DOI] [PubMed] [Google Scholar]
- Jain A, Sharma MC, Sarkar C, Bhatia R, Singh S & Handa R 2007. Major histocompatibility complex class-I and -II detection as a diagnostic tool in idiopathic inflammatory myopathies. Archives of Pathology & Laboratory Medicine 131 1070–1076. [DOI] [PubMed] [Google Scholar]
- Kim H, Lee T-H, Hwang YS, Bang MA, Kim KH, Suh JM, Chung HK, Yu D- Y, Lee K- K, Kwon O- Y et al. 2001. Methimazole as an antioxidant and immunomodulator in thyroid cells: mechanisms involving interferon-g signaling and H2O2 scavenging. Molecular Pharmacology 60 972–980. [DOI] [PubMed] [Google Scholar]
- Kirshner S, Palmer L, Bodor J, Saji M, Kohn LD & Singer DS 2000. Major hystocompatibility class-I gene transcription in thyrocytes: a series of interacting regulatory DNA sequence elements mediate thyrotropin/cyclic adenosine3′,5′-monophosphaterepression. MolecularEndocrinology 14 82–89. [DOI] [PubMed] [Google Scholar]
- Kohn LD, Napolitano G, Singer DS, Molteni M, Scorza R, Shimojo N, Kohno Y, Mozes E, Nakazato M, Ulianich L et al. 2000. Graves’ disease: a host defence mechanism gone awry. International Reviews of Immunology 19 633–664. [DOI] [PubMed] [Google Scholar]
- Kohn LD, Curley RWJ & Rice JM 2002. Methimazole derivatives and tautomeric cyclic thiones to treat autoimmune diseases. US Patent 6,365,616. Sentron Medical Inc., MD, USA, Department of Health and Human Services, DC, USA. 02 Apr 2002.
- Lopata MA, Cleveland DW & Sollner-Webb B 1984. High level expression of a chloramphenicol acetyl transferase gene by DEAE-dextran-mediated DNA transfections coupled with a dimethylsulfoxide or glycerol shock treatment. Nucleic Acids Research 12 5707–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N, Poulaki V, Mitsiades CS, Koutras DA & Chrousos GP 2001. Apoptosis induced by FasL and TRAIL/Apo2L in the pathogenesis of thyroid diseases. Trends in Endocrinology and Metabolism 12 384–390. [DOI] [PubMed] [Google Scholar]
- Montani V, Taniguchi SI, Shong M, Suzuki K, Ohmori M, Giuliani C, Napolitano G, Saji M, Fiorentino B, Reimold AM et al. 1998a. Major histocompatibility class-II HLA-DRα gene expression in thyrocytes: counter regulation by the class-II transactivator and the thyroid Y-box protein. Endocrinology 139 280–289. [DOI] [PubMed] [Google Scholar]
- Montani V, Shong M, Taniguchi SI, Suzuki K, Giuliani C, Napolitano G, Saito J, Saji M, Fiorentino B, Reimold AM et al. 1998b. Regulation of major histocompatibility class-II gene expression in FRTL-5 thyrocytes: opposite effects of interferon and methimazole. Endocrinology 139 290–302. [DOI] [PubMed] [Google Scholar]
- Mozes E, Kohn LD, Hakim F & Singer DS 1993. Resistance ofMHC class-I-deficient mice to experimental systemic lupus erythematosus. Science 261 91–93. [DOI] [PubMed] [Google Scholar]
- Napolitano G, Montani V, Giuliani C, Di Vincenzo S, Bucci I, Todisco V, Laglia G, Coppa A, Singer DS, Nakazato M et al. 2000. Transforming growth factor-β1 down-regulation of major histocompatibility complex class-I in thyrocytes: coordinate regulation of two separate elements by thyroid-specific as well as ubiquitous transcription factors. Molecular Endocrinology 14 486–505. [DOI] [PubMed] [Google Scholar]
- Napolitano G, Bucci I, Giuliani C, Massafra C, Di Petta C, Devangelio E, Singer DS, Monaco F & Kohn LD 2002. High glucose levels increase major histocompatibility complex class-I gene expression in thyroid cells and amplify interferon-g action. Endocrinology 143 1008–1017. [DOI] [PubMed] [Google Scholar]
- Pisetsky DS 2008. The role of innate immunity in the induction of autoimmunity. Autoimmunity Reviews 8 69–72. [DOI] [PubMed] [Google Scholar]
- Saji M, Moriarty J, Ban T, Kohn LD & Singer DS 1992a. Hormonal regulation of major histocompatibility complex class-I genes in rat thyroid FRTL-5 cells: thyroid-stimulating hormone induces a cAMP-mediated decrease in class-I expression. PNAS 89 1944–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saji M, Moriarty J, Ban T, Singer DS & Kohn LD 1992b. Major histocompatibility complex class-I gene expression in rat thyroid cells is regulated by hormones, methimazole and iodide, as well as interferon. Journal of Clinical Endocrinology and Metabolism 75 871–878. [DOI] [PubMed] [Google Scholar]
- Saji M, Shong M, Napolitano G, Palmer LA, Taniguchi SI, Ohmori M, Ohta M, Suzuki K, Kirshner LS, Giuliani C et al. 1997. Regulation of major histocompatibility complex class-I gene expression in thyroid cells: role of the cAMP response element-like sequence. Journal of Biological Chemistry 272 20096–20107. [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S & Coulson AR 1977. DNA sequencing with chain-terminating inhibitors. PNAS 74 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serreze DV, Leiter EH, Christianson G, Greiner D & Roopenian DC 1994. Major histocompatibility complex class-I-deficient NOD-B2mnull mice are diabetes and insulinitis resistant. Diabetes 43 505–509. [DOI] [PubMed] [Google Scholar]
- Shimojo N, Kohno Y, Yamaguchi K, Kikuoka S, Hoshioka A, Niimi H, Hirai A, Tamura Y, Saito Y, Kohn LD et al. 1996. Induction of Graves’-like disease in mice by immunization with fibroblasts transfected with the thyrotropin receptor and a class-II molecule. PNAS 93 11074–11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer DS, Kohn LD, Zinger H & Mozes E 1994. Methimazole prevents induction of experimental systemic lupus erythematosus in mice. Journal of Immunology 153 873–880. [PubMed] [Google Scholar]
- Singer DS, Mozes E, Kirshner S & Kohn LD 1997. Role of MHC class-I molecules in autoimmune disease. Critical Reviews in Immunology 17 463–468. [PubMed] [Google Scholar]
- Singer DS, Zinger H, Kohn LD & Mozes E 1999. Differing MHC class-I requirements for induction and propagation ofexperimental systemic lupus erythematosus. European Journal of Immunology 29 2259–2268. [DOI] [PubMed] [Google Scholar]
- Wang P, Sun SH, Silver PB, Chan CC, Agarwal RK, Wiggert B, Kohn LD, Jamieson GA Jr & Caspi RR 2003. Methimazole protects from experimental autoimmune uveitis (EAU) by inhibiting antigen-presenting-cell function and reducing antigen priming. Journal of Leukocyte Biology 73 57–64. [DOI] [PubMed] [Google Scholar]
- Weetman AP, Holt ME, Campbell AK, Hall R & McGregor AM 1984. Methimazole and generation of oxygen radicals by monocytes: potential role in immunosuppression. BMJ 288 518–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zantutt-Wittmann DE, Tambascia MA, da Silva Trevisan MA, Pinto GA & Vassallo J 2001. Antithyroid drugs inhibit in-vivo HLA-DR expression in thyroid follicular cells in Graves’ disease. Thyroid 11 575–580. [DOI] [PubMed] [Google Scholar]