

Supplemental Digital Content is available in the text
Keywords: ABCD1 gene, bile acids, peroxisomal beta-oxidation, X-linked adrenoleukodystrophy
Abstract
Rationale:
X-linked adrenoleukodystrophy (X-ALD) is a rare disorder caused by mutations in the ABCD1 gene, coding for peroxisomal membrane transporter adrenoleukodystrophy (ALD) protein. The disease is characterized by accumulation of very long chain fatty acids (VLCFAs) in tissues. Adult adrenomyeloneuropathy (AMN) and the cerebral inflammatory form of ALD are the main phenotypes presenting various symptoms.
Patient concerns:
We report a case of 37-year-old patient with diagnosis of X-ALD, confirmed based on elevated VLCFA concentrations and genetic testing of ABCD1 gene. The complete clinical picture in the patient indicates AMN phenotype with cerebral involvement.
Diagnoses:
The reduced synthesis of unconjugated cholic and chenodeoxycholic acids, and the reduction to 28% to 29% of peroxisomal beta-oxidation of behenic acid and normal peroxisomal metabolism of pristanic and palmitic acid were observed in the X-ALD patient. Sanger sequencing of major genes involved in primary bile acid (BA) synthesis failed to identify pathogenic mutations of the investigated set of genes.
Interventions:
Plasma concentrations of BAs, VLCFAs, and beta-oxidation of C22:0, C16:0, and pristanic acid were studied in primary skin fibroblasts of the patient. In addition, we performed sequencing of the ABCD1, ABCD3, CYP7A1, CYP7B1, CYP27A1, HSD3B7, AKR1D1, and SLC27A5 genes in the X-ALD family.
Outcomes:
In the Polish family affected with AMN a dysregulation of the primary BA synthesis pathway was found.
Lessons:
We have demonstrated the coincidence of the adult form of X-ALD with abnormalities in BA synthesis. We suggest that decreased synthesis of BAs may be an additional dysfunction as a consequence of the ABCD1 c.659T>C, p.(Leu220Pro) mutation and may be further evidence that disturbed cholesterol metabolism is important in the pathology of ALD.
1. Introduction
X-linked adrenoleukodystrophy (X-ALD, OMIM 300100) is a rare genetic neurodegenerative disease with recessive inheritance, linked to the X chromosome. X-ALD is characterized by the accumulation of saturated, unbranched very long chain fatty acids (VLCFAs) with more than 22 carbon atoms (VLCFA) in blood and tissues. A known cause of the disease is mutations in the ABCD1 gene encoding the ALD-protein (ALDP) transporting VLCFA into peroxisomes for degradation by the enzymes of the beta-oxidation pathway. Two main phenotypes are known: adult adrenomyeloneuropathy (AMN) and the cerebral inflammatory form of adrenoleukodystrophy (CALD).[1] AMN occurs in men and with disease manifestation appearing between the ages of 20 and 40. AMN is characterized by a noninflammatory, slowly progressive neuropathy within the spinal cord and peripheral nerves.[2] The main symptoms include spastic paraparesis, sensory disturbances, and overactive bladder. Some patients with AMN have adrenal insufficiency.[2] The second phenotype CALD with inflammatory involvement is very severe, together with rapidly progressive decline in cognitive function and neurological disorders. The immune response in CALD and AMN varies and induction of the pathology may depend on different lipid classes. The incidence of X-ALD is estimated to be about 1:17 000 live births, which is considered the most common peroxisomal disease and inherited disease of the white matter of the central nervous system.[3] Within 1 family different forms of the disease with various levels of severity can occur, without any link between genotype and X-ALD phenotype, which suggests involvement of other mechanisms including environmental factors. Moreover, monozygotic twins were reported to demonstrate differing phenotypes.[4] Many studies have been done to search for phenotype modifying genes in ALD. Genes associated with fatty acid metabolism (ABCD2, ABCD3, and ABCD4), inflammation (TNFalpha, MOG, CD1, IL6, HLA, and SOD2), and vitamin B12 metabolism (ABCD4, TCN2, DHFR, CBS, MTR, MTHFR, MTRR, and RCFI) were studied.[5] Because of the lack of correlation between mutations in the ABCD1 gene and phenotype it seems to be appropriate to search for other mechanisms involved in the clinical course of the disease, the pathways and processes that may be disrupted, coexisting mutations in other genes related to lipid metabolism and peroxisomes or epigenetic regulations. Here, we present a clinical case study evaluating VLCFAs and bile acids (BAs) metabolism disorders in a Polish family with X-ALD.
2. Methods
2.1. Ethical approval and consent to participate
The study, sample collection, and all procedures were approved by the Ethical Committee of the Jagiellonian University, Krakow, Poland number KBET/95/B/2012. Individual written informed consents to participate in the study were obtained from patients.
2.2. Consent to publication
Written informed consents to publication were obtained from patients.
2.3. Sample collection
Analyses of plasma BAs levels and sequencing of genes related to ALDP and peroxisomal membrane protein 70 (PMP70) transporters and genes from primary BA synthesis pathway were performed in the patient with suspicion of ALD (named X-ALD patient 1), as well as his mother and brother. The control group included 2 healthy subjects matched for age and sex. In addition screening of peroxisomes was performed on fibroblasts taken from the X-ALD patient 1. Gene expression analysis was made on fibroblasts collected from X-ALD patient 1 and a fibroblasts from a second ALD patient hemizygous to c.1661G>A, p.(Arg554His) mutation (named X-ALD 2) and 2 healthy men.
Samples of peripheral blood were collected from the X-ALD patient 1, his available family members, and the control patients to perform genetic testing and lipid screening analysis. Skin biopsies were taken from the forearm to conduct fibroblast cultures.
2.4. Studies in plasma and on DNA isolated from whole blood cells
2.4.1. Quantification of human BAs in plasma by High-performance liquid chromatography mass spectrometry
Ethylenediamine tetra-acetic acid plasma was separated from peripheral blood samples and frozen at −80°C and subsequently sent to the Institute for Clinical Chemistry and Laboratory Medicine University Hospital in the laboratory of Prof Gerd Schmitz in Regensburg (Germany) to determine BAs and sterol concentration. BA species were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described previously.[6,7] Briefly, plasma samples were spiked with a mixture of deuterated BAs as internal standard before protein precipitation. Plasma extracts were subjected to LC-MS/MS detection in negative ion mode after baseline separation of isobaric species. The LC-MS/MS system consisted of an API 4000 QTrap (AB Sciex, Darmstadt, Germany) that was coupled with electrospray ionization. Chromatographic separation was achieved by the Agilent 1200 HPLC system (Agilent, Waldbronn, Germany). Quantification was achieved using a matrix calibrator generated by standard addition.
2.4.2. Sanger sequencing
DNA was extracted from peripheral blood. Polymerase chain reaction (PCR) amplification of coding regions and intron/exon boundaries of the peroxisomal transporter ABCD1 and ABCD3 genes and CYP7A1, CYP7B1, CYP27A1, HSD3B7, AKR1D1, and SLC27A5 genes from primary BAs pathway were carried out using standard protocols. Lists of PCR primers and amplification protocols used for sequencing are added as supplementary files. Sanger amplification was performed using BigDye Terminator cycle sequencing chemistry and separated using the ABI3130XL capillary sequencer. Sequence data were compared to the appropriate reference sequences from the ENSEMBL database according to Human Genome Variation Society guidelines.
2.5. Studies in fibroblasts
Human skin fibroblasts derived from X-ALD patient 1 and X-ALD patient 2 and from the 2 control subjects were cultured in Dulbecco's Modified Eagle's Medium containing 10% fetal bovine serum and 1% antibiotic and grown at 37°C at 5% CO2 concentration.
2.5.1. VLCFAs level in fibroblasts of X-ALD patient 1
VLCFA levels were measured in fibroblasts at the Laboratory Genetic Metabolic Diseases (LGMD) in the Academic Medical Center in Amsterdam using electrospray ionization mass spectrometry as described previously by Valianpour et al.
2.5.2. Peroxisomal screening and analysis of beta-oxidation in fibroblasts of X-ALD patient 1
Peroxisomal screening included immunofluorescence microscopy measurement of the amount of catalase, detection of the ALDP and PMP70 using antibodies raised against catalase, ALD protein, and PMP70 according to the procedure described by Wanders et al.[8,9] In addition peroxisomal beta-oxidation was studied using radiolabeled C22:0, C16:0, and pristanic acid to assess peroxisomal metabolism in 2 independent experiments and compared to laboratory reference values as described.[10,11] All analyses were performed at the LGMD in the Academic Medical Center in Amsterdam.
2.6. Gene expression
Total RNA was isolated from the cultured fibroblasts using the RiboPure Kit (Ambion). Samples with RIN above 8.0 were selected for further analysis. Sample preparations for Agilent microarray hybridization (SurePrint G3 Unrestricted Human GE 8x 60K Microarray -AMADID-G4858A_028004) were performed according to the manufacturer's instruction.
Data analysis was performed based on the limma[12] library in R environment [R Core Team (2016)]. Normalization and data analysis were performed for 1-color spotted microarrays. A linear model was used for analyzing designed experiments and the assessment of differential expression by comparison of selected groups of data X-ALD versus control patients. A linear model (lmFit and contrasts.fit functions) was used to compute fold changes and t-statistics for the contrasts of interest. Adjustment for multiple testing, using Benjamini and Hochberg method to control the false discovery rate at 5% across all genes and all contrasts was calculated.
3. Case report
3.1. Case description of the X-ALD patient
A 37-year-old man presented with a 7-year history of progressive behavioral abnormality, difficulty in walking due to axonal and demyelination injury, and disorder of micturition with disturbances of the storage function of the bladder. His medical history includes information about thinning eyebrows and hair starting at the age of 4 years. The patient experienced his first epileptic attack at the age of 37 years, with subsequent reoccurrence. Endocrine testing showed high level of serum adrenocorticotropic hormone, lack of adrenal reserve in the short Synacthen test, and low level of dehydroepiandrosterone sulfate allowing the suspicion of ALD. Magnetic resonance imaging of the brain showed abnormal signal intensities in the white matter typical for ALD, with increased signal on T2 and decreased signal on the T1 sequence located in the parieto-occipital and corpus callosum. Electromyography showed axonal and demyelinating injury in the right peroneal nerve. The diagnosis of X-ALD was confirmed based on elevated VLCFA concentrations and genetic testing of ABCD1 gene. The complete clinical picture in the patient indicates AMN phenotype with cerebral involvement. The parameters such as aminotransferases and bilirubin levels were within normal limits, whereas liver ultrasonography showed no abnormalities.
Family history revealed that the patient's mother, father, and younger brother were ALD asymptomatic. Carrier testing documented the ABCD1 mutation in the mother.
3.2. ABCD1 mutation analysis
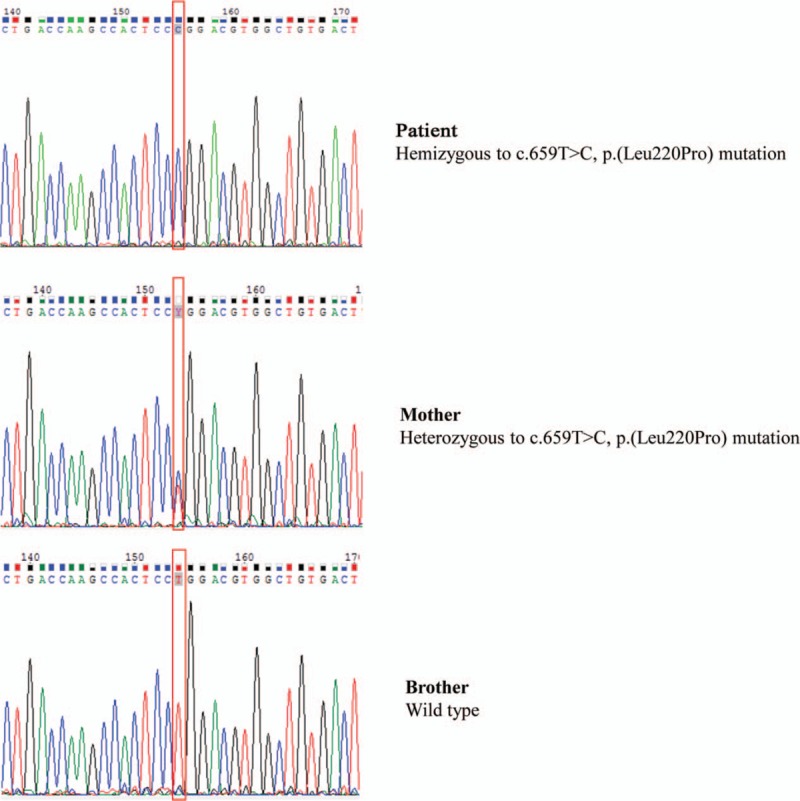
ABCD1 gene sequencing revealed a hemizygous c.659T>C, p.(Leu220Pro) mutation in exon 1 in peripheral blood DNA of the X-ALD patient 1. Carrier testing revealed the presence of the same heterozygous ABCD1 mutation in the mother. At DNA of X-ALD 2 patient was detected as hemizygous c.1661G>A, p.(Arg554His) mutation in 7 exon of ABCD1 gene. The results of sequencing of the gene ABCD1 in X-ALD patient 1 and family is shown in Figure 1.
Figure 1.
The electropherograms of exon 1 sequencing in ABCD1 gene in family members showing position 659 of the coding DNA sequence. X-ALD = X-linked adrenoleukodystrophy.
3.3. Determination of VLCFA concentrations and beta-oxidation activity of C22:0 and C16:0 fatty acid in X-ALD patient 1 fibroblasts
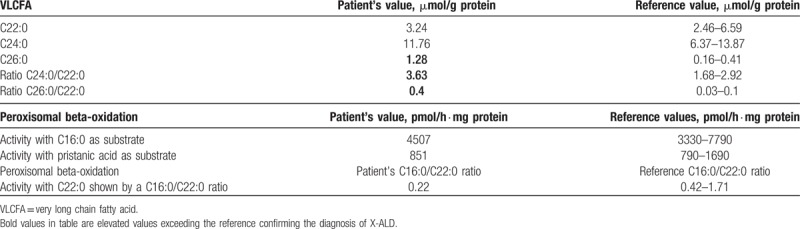
X-ALD patient 1 had an abnormal VLCFAs profile with increased C26:0 concentration and C24:0/C22:0 and C26:0/C22:0 ratio (Table 1). The peroxisomal beta-oxidation activity of the C22:0 was reduced to 28% to 29% as shown by a reduced C16:0/C22:0 ratio in 2 independent experiments compared to laboratory reference values (Table 1).
Table 1.
Results of very long chain fatty acid levels and activity of beta-oxidation with C22:0, C16:0, and pristanic acids as substrates in fibroblasts of X-linked adrenoleukodystrophy patient 1.
3.4. Analysis of plasma bile acids and sterols
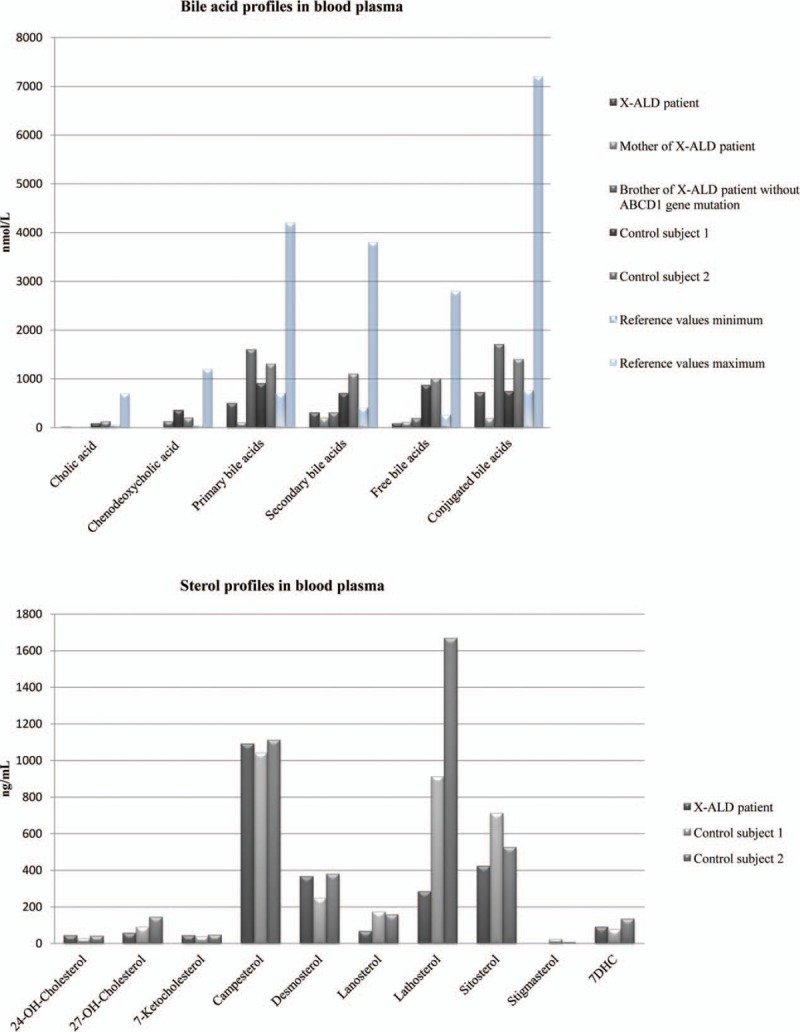
We found a significant decrease in plasma BAs levels in the X-ALD patient 1 as shown in Table 2. In general, total plasma BAs and the primary BA species were significantly lower compared to the healthy controls. Among all species-free BA species revealed the most significantly reduced plasma levels, with very low levels of unconjugated cholic acid (CA) and chenodeoxycholic acid (CDCA). The level of BAs was more reduced in the patient's mother. In addition, analysis of the sterol levels in the plasma showed a significant reduction of lathosterol in the X-ALD patient 1 relative to control subjects (Fig. 2).
Table 2.
Plasma bile acids in the family members.

Figure 2.
Plasma bile acids and sterol profiles in blood plasma in the family members.
3.5. Beta-oxidation with pristanic acid as substrates in X-ALD patient 1 fibroblasts
We found a normal beta-oxidation of pristanic acid in peroxisomes of fibroblasts from X-ALD patient 1 (Table 1). Pristanic acid was used as a representative marker of proper beta-oxidation of branched chain fatty acids because BAs intermediate are handled by the same enzymes: alpha-methylacyl-CoA racemase, acyl-CoA oxidase 2, hydroxysteroid 17-beta dehydrogenase 4, and sterol carrier protein 2.
3.6. Sequencing of genes ABCD3, CYP7A1, CYP7B1, CYP27A1, HSD3B7, AKR1D1, and SLC27A5
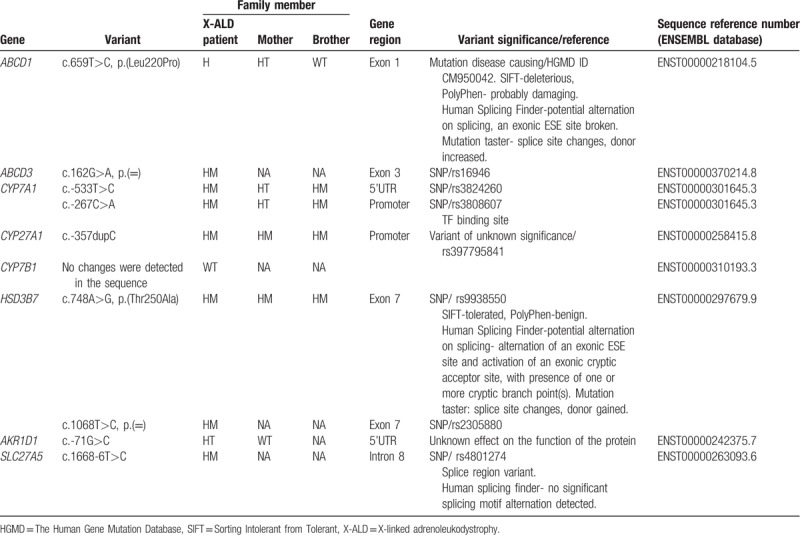
No genetic mutations were found with high probability to affect function of constituents of the pathway analyzed potentially causing an abnormal BA profile (Table 3).
Table 3.
Genotypes of family members in the genes related to the synthesis of bile acids and ABC transporters.
3.7. Gene expression in fibroblasts of AMN patients
Gene expression analysis was performed on Agilent microarrays (see methods for detail) to search for genes and related pathways involved in the pathogenesis of the disease, related especially to peroxisomes and cholesterol synthesis. We demonstrated a statistically significant 4.4-fold decrease in expression of the pleckstrin homology domain containing A6 (PLEKHA6) gene and 5.7-fold increase of LXN (latexin) gene (adj.P < .05) in X-ALD versus control patients. The third gene, lysosomal protein transmembrane 5 (LAPTM5) (fold of change [FC] = 5.1 upregulated) showed a moderate trend toward significance (adj.P = .062), which may also suggest the involvement of this protein in the pathogenesis of X-ALD; results presented in Table 4).
Table 4.
Gene expression results in adrenomyeloneuropathy and control fibroblasts.

4. Discussion
In our patient (X-ALD 1) we detected a previously described homozygous c.659T>C, p.(Leu220Pro) mutation in the ABCD1 gene.[13] Analysis of the VLCFAs level and beta-oxidation of C22:0 in the patient's fibroblasts confirmed the diagnosis of the X-ALD in the patient.[14]
Furthermore, for the first time we have demonstrated the coincidence of the adult form of X-ALD with abnormalities in BA synthesis. The reduced synthesis of primary BAs especially CA and CDCA was observed in the X-ALD patient 1 and his mother.
There are many descriptions of clinical cases related to disorders in peroxisome metabolism; however, there are no reports on simultaneous increase in VLCFAs and reduction of CA and CDCA. Patients with Zellweger syndrome have a decreased number of hepatic peroxisomes, impaired plasmalogen synthesis, and increased levels of VLCFA, BAs precursors, pipecolic and phytanic acids.[15] This group described 19 adult Zellweger syndrome patients, presenting a wide range of symptoms, but none of them had both CA and CDCA levels below reference value and at the same time increased C26:0 levels.[15] The used in our study catalase immunofluorescence method documented presence of the not impaired catalase importing peroxisomes in patient fibroblasts. In addition, the absence symptoms of hepatic injury exclude peroxisomal biogenesis disorder.
The abnormal BA profile suggests that the disorder could be a result of a coexistence of a mutation in another gene, probably influencing the metabolism of BAs. X-linked ALD is a peroxisomal disorder and BAs are metabolized in peroxisomes; therefore, we suspected disturbances in these organelles. Normal beta-oxidation of branched chain fatty acids was observed in the X-ALD patient 1 fibroblasts, suggesting that BAs beta-oxidation inside the peroxisome was not disturbed. The BA profile suggests that there may be either an impaired transport of 3α,7α-dihydroxycholestanoic acid (DHCA) and 3α,7α,12α-trihydroxycholestanoic acid (THCA) into the peroxisomes due to lack of acyl-CoA synthetase producing DHCA-CoA and THCA-CoA or absence of its transporter. It has been demonstrated that the transport of DHCA-CoA and THCA-CoA into peroxisomes is mediated by the PMP70 protein encoded by the ABCD3 gene.[11] Therefore we checked for the presence of the PMP70 protein in the peroxisomal membrane by microscopy with antibodies against this protein, which revealed normal peroxisomal staining. Sanger sequencing of the ABCD3 gene demonstrated no mutation. Furthermore we excluded mutations in the patient's main genes related to the synthesis of BAs outside the peroxisomes. We sequenced genes known from earlier studies on congenital BA synthesis deficiencies or catalyzing key steps in synthesis of CA and CDCA.
Gan-Schreier et al suggested that transport of the VLCFA and BAs is mediated by a common translocator—ALDP, for which they compete. It has been demonstrated in studies on mouse and rat peroxisomes that incubation at high concentrations of VLCFAs inhibited the conjugation of BAs with taurine.[16] These studies suggest that BAs and VLCFAs transport can be activated by the same enzymes or transported into peroxisomes with the same protein and may compete with each other.
ALDP and PMP70 form homo-oligomers in peroxisomal membranes; however, in vivo experiments showed their capacity to form ALDP-PMP70 heterodimers.[17] Braiterman et al[18] demonstrated that overexpression of PMP70 corrects the VLCFA β-oxidation defect in skin fibroblasts from X-ALD patients. It has been suggested that PMP70 partially constitutes the function of ALDP, which may explain the residual 10% to 30% VLCFA β-oxidation activity observed in fibroblasts from X-ALD patients and in our patient at the level of 28%.[17,18] Decreased synthesis of BAs presented in our results could confirm the hypothesis that PMP70 partially takes over the function of the ALDP protein. Decreased concentrations of BAs were found only in family members with the c.659T>C, p.(Leu220Pro) mutation, which may suggest that it is associated with a mutation in the ABCD1 gene.
In 2 articles published in 2016[19,20] it was demonstrated that metabolites of cholesterol conversion may play a significant role in the pathology of X-ALD. Cholesterol biosynthesis occurs partly in peroxisomes, transformation starts from acetyl-CoA (presumably derived from peroxisomal VLCFA beta-oxidation) to farnesyl diphosphate in these organelles with engagement of 8 enzymes.[21] Nury et al[19] showed that plasma 7-ketocholesterol (7KC), a lipid peroxidation product, is increased in X-ALD patients. 7KC was shown to be formed during oxidative stress through nonenzymatic chemical oxidation of cholesterol and induced 3 main symptoms of ALD: inflammation, cell death, and peroxisomal dysfunction. In another experiment in induced pluripotent stem cells, derived from fibroblasts of patients with childhood cerebral ALD, upregulation of cholesterol 25-hydroxylase (CH25H) with a high production of 25-hydroxycholesterol (25-HC) and subsequent activation of the NLRP3 inflammasome leading to cerebral inflammation were found.[20] The results of previous studies show accumulation of cholesterol in the lysosomes from patient fibroblasts with peroxisomal disorders and from mouse models with silenced ABCD1, ACOT8, BAAT, TMEM135, PEX1, PEX3, PEX6, PEX10, PEX26, and NPC1 genes.[22] Furthermore it has been proven that cholesterol can be transported to peroxisomes from lysosomes through lysosome-peroxisome membrane contact with the binding of peroxisomal lipid phosphatidylinositol-4,5-bisphosphate [PI (4,5)P2] by the lysosomal protein synaptotagmin VII (Syt7) and requires functionally active of lysosomes and peroxisomes.[22]
In our study we showed decreased expression of PLEKHA6 and increased LAPTM5 in fibroblasts of X-ALD patients. PLEKHA6 is expressed in many tissues, with the highest level in the adult brain, heart, and kidney.[23] It is involved in intracellular signaling and phosphatidylinositol lipid binding.[24] There are no published results showing which phospholipids may be bound to the PLEKHA6 protein. Based on the sequence similarities between PEPP1 and its homologs, PEPP2 and PLEKHA6 binding with phosphatidylinositol-3-phosphate was suggested.[25] PLEKHA6 contains a PH domain, which can bind phospholipids—phosphatidylinositol-3,4,5-trisphosphate and PI (4,5)P2.[25] The observed significantly reduced expression of the PLEKHA6 gene may relate this protein to the pathology of X-ALD. One of the possible mechanisms is that insufficient expression of PLEKHA6 may enhance the problem with cholesterol transport between lysosomes and peroxisomes in X-ALD patients. Taking into account the process of cholesterol transport between lysosomes and peroxisomes with the participation of phospholipid PI (4,5)P2 anchored in the peroxisome membrane it is possible that the PLEKHA6 gene may be engaged in this process probably by the transport or binding of this PI (4,5)P2 lipid or other phosphoinositides. However, confirmation of this hypothesis requires further studies.
To summarize in our X-ALD family with AMN we showed a new disturbance with abnormal C24-BAs (CDCA and CA) levels, which are the final metabolites of cholesterol conversion suggesting an additional phenotype related to cholesterol metabolism. In the studied family no cause of the disorder in the synthesis of BAs was found. Exome sequencing is required to find the gene responsible for this disorder, which would allow to confirm or rule out the hypothesis that the disorder is associated with the presence of the ABCD1 gene mutation leading to abnormal function of peroxisomes.
In conclusion, the presented results may be further evidence that disturbed cholesterol metabolism is important in the pathology of ALD.
Acknowledgments
The authors thank the patients and their families for their cooperation. The authors are grateful to the Jagiellonian University Małopolska Centre of Biotechnology for granting usage of equipment to perform hybridization and scanning of slides for gene expression experiments.
Author contributions
Conceptualization: Teresa Płatek, Beata Kieć-Wilk, Gerd Schmitz, Aldona Dembińska-Kieć.
Data curation: Teresa Płatek, Evelyn Orso, Barbara Zapała, Anna Polus, Monika Chojnacka, Urszula Ciałowicz, Gerd Schmitz.
Formal analysis: Teresa Płatek, Barbara Zapała, Anna Polus, Monika Chojnacka, Urszula Ciałowicz, Małgorzata Malczewska-Malec.
Funding acquisition: Teresa Płatek.
Investigation: Teresa Płatek, Anna Polus, Bogdan Solnica, Aldona Dembińska-Kieć.
Methodology: Teresa Płatek, Evelyn Orso, Barbara Zapała, Anna Polus, Monika Chojnacka.
Project administration: Teresa Płatek.
Resources: Teresa Płatek.
Software: Monika Piwowar.
Validation: Teresa Płatek, Monika Piwowar, Gerd Schmitz.
Visualization: Teresa Płatek, Monika Piwowar, Evelyn Orso.
Writing – original draft: Teresa Płatek.
Writing – review and editing: Evelyn Orso, Barbara Zapała, Anna Polus, Beata Kieć-Wilk, Monika Piwowar, Monika Chojnacka, Urszula Ciałowicz, Małgorzata Malczewska-Malec, Gerd Schmitz, Bogdan Solnica, Aldona Dembińska-Kieć.
Supplementary Material
Footnotes
Abbreviations: 7KC = 7-ketocholesterol, ALD = adrenoleukodystrophy, ALDP = ALD-protein, AMN = adrenomyeloneuropathy, BA = bile acid, CA = cholic acid, CALD = cerebral inflammatory form of adrenoleukodystrophy, CDCA = chenodeoxycholic acid, DHCA = 3α,7α-dihydroxycholestanoic acid, LGMD = Laboratory Genetic Metabolic Diseases, PI (4,5)P2 = phosphatidylinositol-4,5-bisphosphate, PMP70 = peroxisomal membrane protein 70, THCA = 3α,7α,12α-trihydroxycholestanoic acid, VLCFA = very long chain fatty acid, X-ALD = X-linked adrenoleukodystrophy.
This work was supported by grants for young scientists from the Polish Ministry of Science and Higher Education: “Genetics and lipidomics of adrenoleukodystrophy” under agreement No: K/DSC/001368 and by the FP7-Program of the European Community under grant agreement No 202272, IP-Project LipidomicNet.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 2012;1822:1465–74. [DOI] [PubMed] [Google Scholar]
- [2].Weber FD, Wiesinger C, Forss-Petter S, et al. X-linked adrenoleukodystrophy: very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum Mol Genet 2014;23:2542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vogel BH, Bradley SE, Adams DJ, et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: Diagnostic protocol, surveillance protocol and treatment guidelines. Mol Genet Metab 2015;114:599–603. [DOI] [PubMed] [Google Scholar]
- [4].Korenke GC, Krasemann E, Doerr HG, et al. Cerebral adrenoleukodystrophy (ALD) in only one of monozygotic twins with an identical ALD genotype. Ann Neurol 1996;40:254–7. [DOI] [PubMed] [Google Scholar]
- [5].Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet 2015;8:109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Scherer M, Gnewuch C, Schmitz G, et al. Rapid quantification of bile acids and their conjugates in serum by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2009;877:3920–5. [DOI] [PubMed] [Google Scholar]
- [7].Gnewuch C, Liebisch G, Langmann T, et al. Serum bile acid profiling reflects enterohepatic detoxification state and intestinal barrier function in inflammatory bowel disease. World J Gastroenterol 2009;15:3134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wanders RJ, Wiemer EA, Brul S, et al. Prenatal diagnosis of Zellweger syndrome by direct visualization of peroxisomes in chorionic villus fibroblasts by immunofluorescence microscopy. J Inherit Metab Dis 1989;12suppl 2:301–4. [DOI] [PubMed] [Google Scholar]
- [9].Ferdinandusse S, Jimenez-Sanchez G, Koster J, et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum Mol Genet 2015;24:361–70. [DOI] [PubMed] [Google Scholar]
- [10].Dacremont G, Cocquyt G, Vincent G. Measurement of very long-chain fatty acids, phytanic and pristanic acid in plasma and cultured fibroblasts by gas chromatography. J Inherit Metab Dis 1995;18suppl 1:76–83. [DOI] [PubMed] [Google Scholar]
- [11].Wanders RJ, Denis S, Ruiter JP, et al. Measurement of peroxisomal fatty acid beta-oxidation in cultured human skin fibroblasts. J Inherit Metab Dis 1995;18suppl 1:113–24. [DOI] [PubMed] [Google Scholar]
- [12].Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ligtenberg MJ, Kemp S, Sarde CO, et al. Spectrum of mutations in the gene encoding the adrenoleukodystrophy protein. Am J Hum Genet 1995;56:44–50. [PMC free article] [PubMed] [Google Scholar]
- [14].Valianpour F, Selhorst JJM, Van Lint LEM, et al. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 2003;79:189–96. [DOI] [PubMed] [Google Scholar]
- [15].Berendse K, Engelen M, Ferdinandusse S, et al. Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J Inherit Metab Dis 2016;39:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gan-schreier H, Haas D, Langhans C, et al. Bile acid precursors compete with very long chain fatty acids for translocation into peroxisomes. J Gastroenterol Hepatol Res 2013;2:362–8. [Google Scholar]
- [17].Hillebrand M, Gersting SW, Lotz-Havla AS, et al. Identification of a new fatty acid synthesis-transport machinery at the peroxisomal membrane. J Biol Chem 2012;287:210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Braiterman LT, Zheng S, Watkins PA, et al. Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum Mol Genet 1998;7:239–47. [DOI] [PubMed] [Google Scholar]
- [19].Nury T, Zarrouk A, Ragot K, et al. 7-Ketocholesterol is increased in the plasma of X-ALD patients and induces peroxisomal modifications in microglial cells: potential roles of 7-ketocholesterol in the pathophysiology of X-ALD. J Steroid Biochem Mol Biol 2017;169:123–36. [DOI] [PubMed] [Google Scholar]
- [20].Jang J, Park S, Jin Hur H, et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun 2016;7:13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Faust PL, Kovacs WJ. Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 2014;98:75–85. [DOI] [PubMed] [Google Scholar]
- [22].Chu BB, Liao YC, Qi W, et al. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 2015;161:291–306. [DOI] [PubMed] [Google Scholar]
- [23].Nagase T, Ishikawa K, Suyama M, et al. Prediction of the coding sequences of unidentified human genes. XII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 1998;5:355–64. [DOI] [PubMed] [Google Scholar]
- [24].Spellmann I, Rujescu D, Musil R, et al. Pleckstrin homology domain containing 6 protein (PLEKHA6) polymorphisms are associated with psychopathology and response to treatment in schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry 2014;51:190–5. [DOI] [PubMed] [Google Scholar]
- [25].Dowler S, Currie RA, Campbell DG, et al. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J 2000;351(pt 1):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.