

Abstract
A precise balance of DNA methylation and demethylation is required for epigenetic control of cell identity, development, and growth. DNA methylation marks are introduced by de novo DNA methyltransferases DNMT3a/b and are maintained throughout cell divisions by DNA methyltransferase 1 (DNMT1), which adds methyl groups to hemimethylated CpG dinucleotides generated during DNA replication. Ten Eleven Translocation (TET) dioxygenases oxidize 5-methylcytosine (mC) to 5-hydroxymethylcytosine (hmC), 5-formylcytosine (fC), and 5-carboxylcytosine (caC), a process known to induce DNA demethylation and gene reactivation. In the present study, we investigated the catalytic activity of human DNMT1 in the presence of oxidized forms of mC. A mass spectrometry-based assay was employed to study the kinetics of DNMT1-mediated cytosine methylation in CG dinucleotides containing C, mC, hmC, fC, or caC across from the target cytosine. Homology modeling, coupled with molecular dynamics simulations, was carried out to explore the structural consequences of mC oxidation in regard to the geometry of protein-DNA complexes. DNMT1 enzymatic activity was strongly affected by the oxidation status of mC, with catalytic efficiency decreasing in the order mC > hmC > fC > caC. Molecular dynamics simulations revealed that DNMT1 forms an unproductive complex with DNA duplexes containing oxidized forms of mC as a consequence of altered interactions of the target recognition domain of the protein with the C-5 substituent on cytosine. Our results provide a new structural and mechanistic insight into TET mediated DNA demethylation.
Graphical Abstract
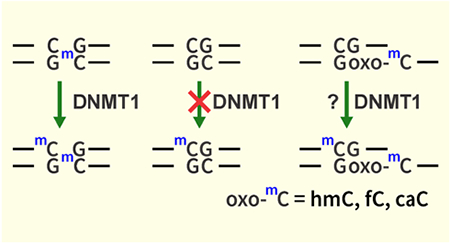
Introduction
5-Methylcytosine (mC) is a stable epigenetic mark most commonly found at CpG dinucleotides of DNA, where cytosine bases in both strands are methylated.1 Cytosine methylation typically has a repressive effect, leading to reduced levels of gene expression.2, 3 Methylated CpG sites within gene promoter regions interfere with transcription factor binding and instead are recognized by methyl-CpG binding proteins, promoting the recruitment of histone deacetylases and the formation of closed chromatin.2, 3
In human cells, DNA methylation marks are introduced by de novo methyltransferases DNMT3a/b and are subsequently preserved by maintenance DNA methyltransferase 1 (DNMT1).4 The activity of DNMT1 as a maintenance methyltransferase during replication is necessary to ensure accurate transmission of epigenetic methylation marks to progeny cells. DNMT1 is recruited to the replication fork by UHRF1 and acts at hemimethylated CpG sites generated during DNA replication through recognition of the methylated CG sites on the template strand of the DNA.5, 6 DNMT1 specifically recognizes and methylates hemi-methylated CG sites.3, 7 Formation of an enzymatically active complex requires mC binding to a concave hydrophobic pocket within the target recognition domain (TRD) of DNMT1.3, 9 This induces local melting of the DNA duplex, allowing unmethylated cytosine in the opposite strand of DNA to be actively “flipped” out of the DNA duplex stack to enter the protein active site.3, 5, 9, 10 The reversible addition of a thiolate from an active site cysteine residue of DNMT1 to the C-6 position of cytosine activates the C-5 position of the nucleobase, allowing it to accept a methyl group from the S-adenosylmethionine (SAM) cofactor.3, 11 The covalent DNA-protein complex is reversed via base catalyzed removal of the H-5 proton of the nucleobase and re-aromatization via elimination of the covalently attached cysteine (Scheme 1).3
Scheme 1:
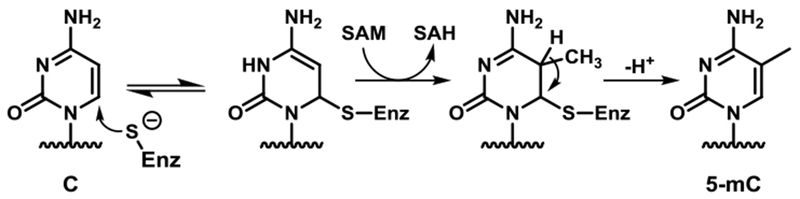
Enzymatic mechanism of DNMT1.10, 11 SAM= S-adenosylmethionine, SAH = S-adenosylhomocysteine
Crystal structures reveal two distinctive modes of DNMT1-DNA binding.3, 7 In productive DNMT1-DNA complex containing a hemi-methylated CG dinucleotide (PDB:4DA4), hydrophobic amino acid side chains of M1535, C1501, L1502 and L1515 form a target recognition domain (TRD) involved in interactions with hemimethylated mCpG.3 As a hemimethylated CpG site emerges from the replication complex, the TRD of DNMT1 specifically recognizes mC and facilitates maintenance methylation. A different binding mode is revealed in the crystal structure of DNMT1 interacting with unmethylated DNA (PDB:3PTA).7 In the unproductive DNMT1-DNA complex, the zinc finger-containing CXXC domain of the protein prevents de novo methylation by positioning the CXXC-BAH1 linker region of the protein between the DNA and the catalytic active site.7 Furthermore, the TRD of the protein assumes a retracted position that prevents it from direct interaction with the DNA.7 This autoinhibitory mechanism protects DNA from unintended de novo methylation by DNMT1.7
Ten Eleven Translocation (TET) dioxygenases sequentially oxidize the methyl group of mC in DNA to give 5-hydroxymethylcytosine (hmC), 5-formylcytosine (fC), and 5-carboxylcytosine (caC) (Scheme 2).15–17 Both fC and caC can be excised by thymine DNA glycosylase (TDG) and replaced with cytosine via TDG-mediated base excision repair pathway, leading to active demethylation.18 Additionally, oxidized forms of mC promote passive DNA demethylation by interfering with DNMT1 activity at hemimethylated CpG sites.19 The relative contributions of passive and active DNA demethylation are dependent on the cell type, developmental factors, and the stage of the cell cycle.20, 21 However, to our knowledge, the kinetics of DNMT1-mediated methyl transfer in the presence of oxidized forms of mC (oxo-mC) has not been elucidated, and the structural origins of reduced activity of DNMT1 protein in the presence of oxo-mC are not well understood.
Scheme 2:
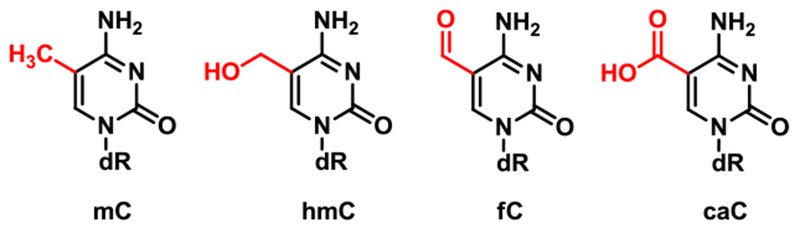
Epigenetic modifications of cytosine in DNA.
In the present study, a mass spectrometry based quantitative assay developed in our laboratory was used to examine the kinetics of DNMT1-mediated maintenance methylation in the presence of mCG, hmCG, fCG, and caCG, while molecular dynamics simulations were conducted to examine the structural origins of the reduced DNMT1 activity in the presence of oxidized forms of 5-methylcytosine.22 Our results support a model in which mC oxidation to hmC, fC, and caC prevents the formation of a productive DNMT1- DNA complex by weakening hydrophobic interactions between the modified cytosine and the TRD of the DNMT1 protein, leading to reduced maintenance methylation rates and allowing for passive DNA demethylation.
Materials and Methods
Materials:
All nucleoside phosphoramidites including 5-methyl-dC, 5-hydroxymethyl-dC, 5-formyl-dC-III, 5-carboxy-dC, Ac-dC, dT, dA, dG, and dmf-dG, reagents, and controlled pore glass solid support for oligodeoxynucleotide synthesis were acquired from Glen Research Corporation (Sterling, VA). Human recombinant DNA methyltransferase 1 (DNMT1) and Δ580-DNMT18 (missing the PCNA,23 DNMT3A/B interaction domains23, 24) were purchased from New England BioLabs (Ipswich, MA). Phosphodiesterase I, phosphodiesterase II, and DNase I were acquired from Worthington Biochemical Corporation (Lakewood, NJ). Bovine intestinal alkaline phosphatase was procured from Sigma Aldrich Chemical Company (Milwaukee, WI). All remaining laboratory chemicals and solvents were purchased from ThermoFisher Scientific (Waltham, MA) and Sigma-Aldrich (Milwaukee, WI). The synthesis of 13C1015N2-5-Methyl-2′-deoxycytidine was described previously.22
Synthesis of mC, hmC, fC and caC Containing Oligodeoxynucleotides:
Synthetic DNA oligodeoxynucleotides (Table S1) were assembled by solid phase synthesis on an ABI 394 DNA synthesizer (Applied Biosystems, Grand Island, NY) according to the manufacturer’s instructions. mC and its oxidized forms (mC, hmC, fC, and caC) were added via manual coupling.25 mC containing strands were cleaved from solid support and deprotected using 30% ammonium hydroxide for 16 h at room temperature. hmC containing DNA was cleaved from support and deprotected using 30% ammonium hydroxide at 75 °C for 16 h. fC containing DNA was cleaved from solid support by incubation in 30% ammonium hydroxide for 16 h, followed by desalting using Illustra Nap-5 cartridges (GE Healthcare, Buckinghamshire, UK). The 5-(1,3-dioxane-2-yl) protecting group on fC was cleaved using glacial acetic acid for 6 h at room temperature. Synthetic DNA strands containing caC were cleaved and deprotected using 0.4 M methanolic sodium hydroxide (80% methanol:20% 2 M sodium hydroxide) overnight.
Synthetic DNA strands were purified by reverse phase HPLC using an Agilent 1100 HPLC system interfaced with a UV variable wavelength detector set at 260 nm. A Varian Pursuit C-18 HPLC column (5 μm, 250 × 10.0 mm) was eluted at flow rate of 3 mL/min with a gradient of 100 mM triethylammonium acetate pH 7.0 (A) and acetonitrile (B). In method A, solvent composition was linearly changed from 8.4% to 12% B over 30 min, increased to 17.5% over 10 min, further to 32.5% over 5 min. Solvent composition returned to initial conditions over 2 min, followed by re-equilibration for 15 min. In method B, solvent composition was changed from 9% to 16.3% B over 30 min, increased to 20% over 10 min, further to 32.5% over 5 min, and returned to initial conditions over 2 min, followed by equilibration over 15 min. Method A was used to purify C, mC, hmC, and caC containing strands while method B was used to purify fC containing DNA.
Following HPLC purification, synthetic DNA strands were desalted using Illustra Nap-5 cartridges (GE Healthcare) according to the manufacturer’s instructions. The presence of mC, hmC, fC, or caC in synthetic DNA strands was confirmed by HPLC-ESI−-MS analyses on an Agilent MSD Ion Trap MS interfaced with an Agilent 1100 HPLC system (Table S1). A Zorbax 300-SB C-18 column (5 μm, 150 × 0.5 mm) was eluted with a gradient of 15 mM ammonium acetate (A) and acetonitrile (B). Solvent composition was held at 2% B for 3 min, then linearly increased to 40% over 16 min, further to 55% over 1 min, followed by a return to initial conditions over 1 min and equilibration for 13 min. DNA concentrations were determined by quantitation of the 2′-deoxyguanosine in enzymatic digests.26–28
DNA Methyltransferase experiments:
Human DNMT1 (0.75U, New England BioLabs, Ipswich, MA) was incubated with synthetic DNA duplexes (250 – 1500 fmol) containing a single, centrally located CpG site with mC, hmC, fC, or caC opposite the target C (Table 1). Enzymatic reactions were conducted in commercial DNMT buffer in the presence of 0.1 mg/mL bovine serum albumin (BSA) and 160 μM S-adenosylmethionine (SAM) for 15 min at 37 °C. The reactions were quenched by placing samples on dry ice, followed by enzyme inactivation at 65 °C for 40 min. DNA was digested to 2′-deoxynucleotides in the presence of PDE I (55 mU), PDE II (63 mU), DNase I (28 U), and alkaline phosphatase (48 U) in a solution containing 10 mM Tris-HCl pH 7.0 and 15 mM magnesium chloride. Samples were spiked with 13C1015N2-5-methyl-2′-deoxycytidine (internal standard for mass spectrometry, 1.33 pmol) and purified by offline HPLC. A Waters T3 column (3 μm, 4.6 mm × 150 mm) was eluted with a gradient of 5 mM ammonium formate pH 4.0 (A) and methanol (B). Solvent composition was linearly changed from 3% B to 20% B in 15 min, further to 40% B over 5 min, increased to 80% over 5 min, held at 80% for 2 min, and returned to initial conditions over 2 min, followed by column re-equilibration for 8 minutes. HPLC fractions containing mC and its internal standard (8.9 – 10.2 min) were collected, concentrated under reduced pressure, and reconstituted in 12 μL of 15 mM ammonium acetate buffer prior to HPLC-ESI+-MS/MS analysis.
Table 1:
DNA duplexes used to investigate the effects of TET oxidation and local sequence on the rate of cytosine methylation mediated by DNMT1.
| Duplex Name | Duplex sequence | Melting temperature Tm ± std. dev. (°C) |
|---|---|---|
| (−)C | 5′-TCAGATTCGCGCCGGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCGGCCGACGCTATTCGA-5′ |
77.4 ± 0.2 |
| (−)mC | 5′-TCAGATTCGCGCC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCGGmCCGACGCTATTCGA-5′ |
77.0 ± 0.6 |
| (−)hmC | 5′-TCAGATTCGCGCC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCGGhmCCGACGCTATTCGA-5′ |
76.6 ± 0.9 |
| (−)fC | 5′-TCAGATTCGCGCC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCGGfCCGACGCTATTCGA-5′ |
77.5 ± 0.6 |
| (−)caC | 5′-TCAGATTCGCGCC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCGGcaCCGACGCTATTCGA-5′ |
78.0 ± 0.4 |
| mCGT | 5′-TCAGATTCGCGAC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCTGmCCGACGCTATTCGA-5′ |
75.7 ± 0.3 |
| mCGA | 5′-TCAGATTCGCGTC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCAGmCCGACGCTATTCGA-5′ |
75.5 ± 0.7 |
| mCGC | 5′-TCAGATTCGCGGC GGCTGCGATAAGCT-3′ 3′-AGTCTAAGCGCCGmCCGACGCTATTCGA-5′ |
77.3 ± 0.4 |
HPLC-ESI+-MS/MS:
Quantification of mC was carried out by isotope dilution HPLC-ESI+-MS/MS using 13C1015N2-mC as an internal standard. A Thermo Dionex Ultimate3000 HPLC system was coupled to a Thermo TSQ Vantage mass spectrometer (Thermo Fisher Scientific, Waltham, MA). A Thermo Hypercarb column (3 μm, 100 × 0.5 mm) was maintained at 60 °C and eluted with a gradient of 15 mM ammonium acetate (A) and acetonitrile (B). Solvent composition was changed from 15 to 60% B over 10 min, further to 95% over 2 min, held at 95% B for 3 min, and returned to initial conditions over 2 min, followed by re-equilibration for 7 min. HPLC eluent was directed into the mass spectrometer during 2-12 min of the chromatographic run. Typical MS parameters were as follows: spray voltage, 3200 V; sheath gas pressure, 20 psi; capillary temperature 350 °C; collision energy, 8; declustering voltage, 22 V; collision gas pressure, 1.5 mTorr; tuned S-Lens, 93; Q1 (full width at half maximum), 0.4; Q3 (full width at half maximum), 0.7; scan width, 0.4; scan time, 0.1 s. The mass spectrometer parameters were optimized upon direct infusion of authentic standards. The instrument was operated in the selected reaction monitoring mode by following the transitions m/z 242.1 [M+H]+ →126.1 [M + H - dR]+ for mC and m/z 254.1[M+H]+ →133.1 [M + H -dR]+ for 13C1015N2-mC. mC amounts in each sample were determined from HPLC-ESI+-MS/MS peak areas corresponding to the analyte and its internal standard using calibration curves constructed with authentic standards.
Methylation velocity (VmC, M/min) was calculated from the HPLC-ESI+-MS/MS areas corresponding to mC and internal standard according to Equation 1:
where AAN and AIS are the areas under the HPLC-ESI+-MS/MS peaks corresponding to mC (analyte, AN) and its 13C, 15N-labeled internal standard (IS), respectively, CIS is the amount of internal standard used in pmol, V is the volume in microliters, and t is the reaction time in min. Steady-state kinetic parameters (Km and Vmax) for methyl transfer reaction were determined by plotting the calculated velocities vs. substrate concentration.
Kinetic Analyses:
The mC amounts determined by HPLC-ESI+-MS/MS were plotted against DNA concentrations using Prism 6 software from Graphpad Software, Inc. (La Jolla, CA). The kinetic curves were fitted to the Michaelis-Menton equation using non-linear regression to give the values of Km and Vmax.
Electrophoretic Mobility Shift Assay (EMSA) to study DNMT1-DNA binding:
DNA strands (Table S1) (50 pmol) were radiolabeled by incubation with T4 polynucleotide kinase (20 U, New England BioLabs, Beverly, MA) and γ32P-ATP (5 μCi, PerkinElmer Life Sciences, Boston, MA) in 1X polynucleotide kinase reaction buffer for 1 hour at 37 °C. The enzyme was inactivated by heating at 65 °C for 10 min, and free γ32P-ATP was removed by Illustra MicroSpin G-25 Column (GE Healthcare, Pittsburgh, PA). Following radiolabeling, complementary DNA strands were annealed by heating to 90 °C for 5 min, followed by slow cooling to room temperature to obtain double stranded substrates (Table 1).
32P-end-labeled DNA duplexes (+ strand, 5′-AGTCTAAGCGCGGXCGACGCTATTCGA-3′, where X= mC, hmC, fC, or caC) (2 nM) were incubated with purified human Δ580-Dmnt1 (0-128 nM, New England BioLabs, Beverly, MA), and 1X gel shift assay buffer (10 mM HEPES, 50 mM KCl, 0.1 mM EDTA, 1 mM DTT 2.5 mM MgCl2, 0.2% Triton X-100, and 10% glycerol) at 37 °C for 30 min. The mixture was loaded onto 4 % polyacrylamide gel (37.5:1 acrylamide:bisacrylamide ratio, prepared with 0.5X TBE) while running at 300 V for 10 minutes. The gels were electrophoresed at 140 V for an additional 40 minutes at 4 °C. Gels were imaged with a Typhoon FLA 7000 instrument (GE Healthcare)..
Homology Modeling:
All molecular modeling was performed using the Schrödinger modeling suite package (Schrödinger, LLC, NY).29 Homology modeling was carried out using Schrodinger’s Prime as described previously.22 In brief, homology modeling of the human DNMT1 (hDNMT1) utilized the crystal structure of mouse DNMT1 (mDNMT1) in complex with hemi-methylated DNA (PDB 4DA4) was carried out based on the human reference sequence (NP_001124295.1).3 mDNMT1 shares an 85% sequence similarity with the hDNMT1 reference sequence (Figure S3). The DNA sequence was then modified accordingly to match the sequence used experimentally in determining DNA methylation rates. Each oxidized form of 5-methylcytosine (hmC, fC, caC) was subsequently modeled within the DNA template.
Molecular Dynamics:
Desmond30 was used to simulate each oxidized forms of 5-methylcytosine (hmC, fC, caC) within the modeled DNA template. As reported previously, each of the modeled hDNMT1 – DNA complexes was subjected to standard protein preparation protocols.22 DNMT1-DNA complexes containing mC or caC at a central CpG site were solvated with a 15 Å buffer region from its outer edge inside a rectangular box of TIP3P explicit solvent model.31 150 mM Na+ and Cl− counter ions were added to electroneutralize the final system. Each MD simulation was carried out using Desmond with default protocol for initialization, followed by 100 ns of unrestrained production simulation run under isothermal isobaric (NPT) conditions at 310 K and 1 atm with the OPLS3 force field. The long-range electrostatic interactions were evaluated by the Particle-Mesh Ewald method under periodic boundary conditions with a dielectric constant of 1. The stability of the protein-DNA complex was assessed by evaluating the protein CαRMSD and DNA backbone phosphorus RMSD (P_RMSD)32 with respect to the minimized starting structure (Figures 1A and 1B).
Figure 1.
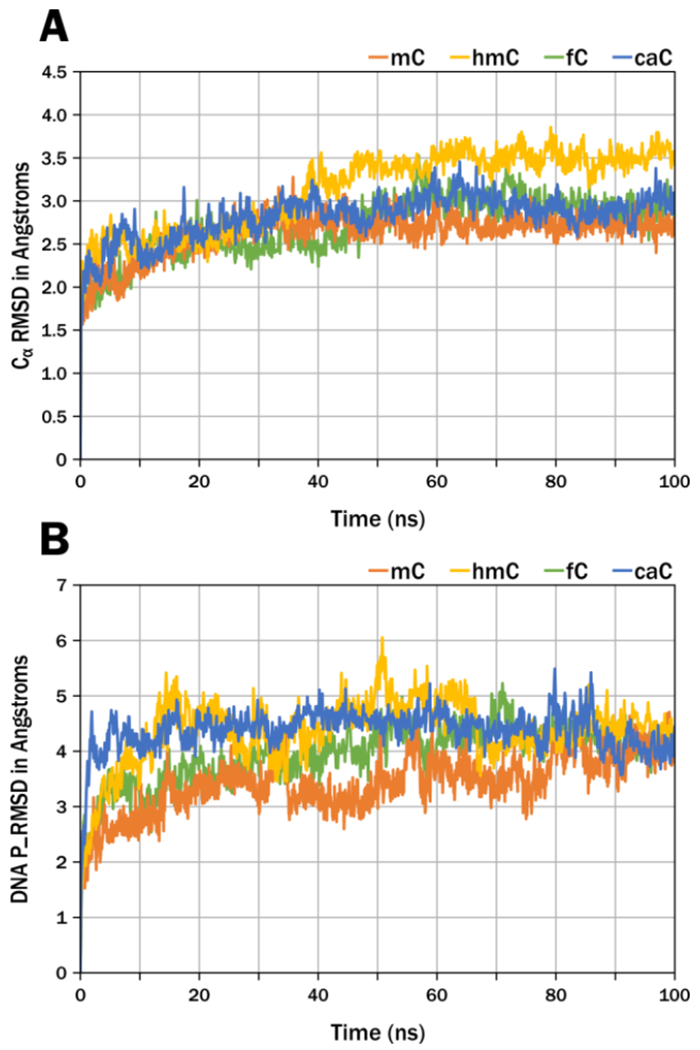
Stability of the DNA-Protein complex. A. Cα RMSD demonstrates stability of the protein backbone during the MD simulation. B. The P_RMSD of the DNA phosphorus backbone shows the stability of the DNA duplex with respect to the starting minimized structure.
Results
Kinetics of DNMT1 Mediated Cytosine Methylation in the Presence of mC and its Oxidized Forms
To examine the kinetics of DNMT1 mediated cytosine methylation in the presence of mC and its oxidized variants, synthetic DNA duplexes (5′-AGCTTATCGCAGC XG GCGCGAATCTGA-3′) containing a single mC, hmC, fC, or caC residue (X) at the central CpG site were prepared (Table 1). In the resulting double stranded DNA substrates (Table 1), a single centrally located CpG site contains C (negative control), mC (positive control), hmC, fC, or caC opposite unsubstituted cytosine. The ability of these 27-mer duplexes to serve as DNMT1 substrates was established by electrophoretic gel mobility shift (EMSA) assays, in which radiolabeled DNA duplexes were incubated with increasing amounts of human Δ580-DNMT1 protein (0-128 nM), followed by separation on 4% non-denaturing polyacrylamide gel to detect DNA-protein complexes8 (Figure S2). An electrophoretic mobility shift characteristic of the formation of DNA-protein complexes was observed, consistent with DNMT1 binding to synthetic DNA duplexes.
To establish the kinetics of DNMT1-mediated methyl transfer in the presence of mC and its oxidized forms, a mass spectrometry based assay developed in our laboratory was employed (Scheme S1).22 Following in vitro methylation reactions in the presence of human recombinant DNMT1 and S-adenosylmethionine cofactor, the amounts of newly formed mC were determined by capillary HPLC-ESI+-MS/MS using 13C1015N2-mC internal standard.22 In brief, DNA was spiked with known amounts of 13C1015N2-mC and enzymatically digested to 2′-deoxynucleosides, which were analyzed by HPLC-ESI+-MS/MS as reported previously.22 Steady-state kinetic parameters (Km and Vmax) for methyl transfer reaction were determined by plotting the methylation velocity at a particular substrate concentration and using non-linear regression to fit the data to the Michaelis Menten equation.22 The methylation velocity was calculated using Equation 1 as shown in the supplementary Scheme S2.
We found that DNMT1 methylation kinetics at CpG sites was strongly affected by the oxidation status of mC in the opposite strand. The highest value of Vmax (190 × 10−11 M/min) was observed for DNA duplexes containing mCG dinucleotide (Figure 2A), which is similar to previous reports (27.2 – 163 × 10−11 M/min) that also observed a large effect of sequence context on methyl transfer rates.33 Much lower rates of methyl transfer were observed for CpG sites containing hmC (41 × 10−11 M/min), fC (11.0 × 10−11 M/min), and caC (0.77 × 10−11 M/min) (Table 2). In general, DNMT1-mediated methylation rates decreased upon oxidation of mC in the opposite strand (Figure 2A, Table 2). Indeed, the Vmax values for methylation of cytosines placed opposite hmC, fC, and caC were 4-, 17-, and 240-fold lower than for mC containing DNA, respectively (Table 2). The Km values for mC, hmC, fC, and caC were 28, 20, 13, and 1.1 nM respectively (Table 2). Catalytic efficiencies (Vmax/Km) for methyl transfer to cytosine residues opposite C, mC, hmC, fC, and caC in CpG dinucleotides were calculated as 0.56, 6.7, 2.1, 0.85, and 0.71 (×10−2 min−1M−1) respectively (Table 2). Overall, our results indicate that the ability of mC to direct DNMT1-mediated maintenance methylation of CpG sites is reduced upon its oxidation to hmC, fC, and caC. This reduced maintenance methylation activity is likely to lead to passive DNA demethylation.34 In contrast, DNMT1 efficiency was only weakly affected by local sequence context, with Vmax/Km values of 8.1, 12, 6.2, and 8.4 (×10−2 min−1M−1) for mCGG, mCGC, mCGA, and mCGT, respectively (Figure 2B, Table S1).
Figure 2:
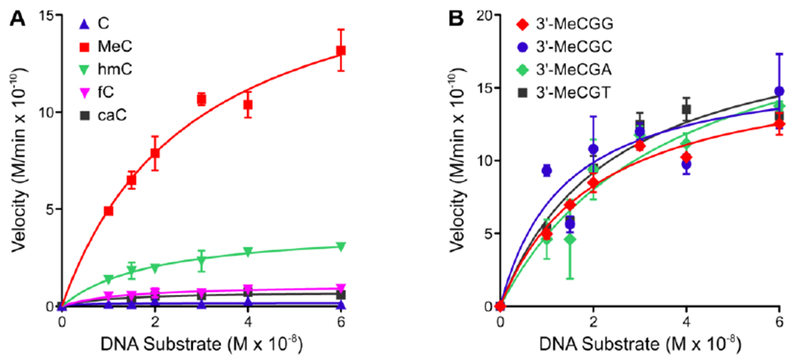
Michaelis-Menton plots for full-length DNMT1 mediated methylation for DNA duplexes containing central CG, mCG, hmCG, fCG, and caCG dinucleotides DNA duplexes were incubated with hDNMT1 and SAM for 15 min at 37 °C. After quenching, DNA was digested to nucleosides, spiked with 13C1015N2-mC and analyzed for mC by LC-MS/MS. The methylation velocity was plotted against DNA concentrations. (A) Influence of oxidation state of the methyl group on the rates of maintenance methylation in DNA duplexes 5′-AGCTTATCGCAGC XG GCGCGAATCTGA-3′ (X =C, mC, hmC, fC, or caC). (B) Influence of 3′-neighboring nucleobase on methyl transfer rates for DNA duplexes of the sequence (5′-AGCTTATCGCAGC mCGX CGCGAATCTGA-3′ where X = C, G, T, or A).
Table 2:
Kinetic parameters of the DNMT1 mediated methylation of 27-mer duplexes containing modified cytosines.a
| DNA Duplex | Vmax (×10−11min−1) | Km (nM) | Vmax/Km (×10−2 min−1M−1) |
|---|---|---|---|
| (−)C | 1.8 ± 0.61 | 3.2 ± 8.6 | 0.56 ± 1.5 |
| (−)mC | 190 ± 18 | 28 ± 5.7 | 6.7 ± 1.5 |
| (−)hmC | 41 ± 5.1 | 20 ± 6.2 | 2.1 ± 0.69 |
| (−)fC | 11 ± 1.1 | 13 ± 4.0 | 0.85 ± 0.33 |
| (−)caC | 0.77 ± 0.19 | 1.1 ± 1.0 | 0.71 ± 0.67 |
The Vmax and Km values were determined via nonlinear regression using data from three or more individual points. The ranges in Vmax and Km are the standard error for regression analysis. Error was propagated for Vmax/Km using the equation: ; where a, b, and c are Vmax, Km, and Vmax/Km respectively.
Molecular modeling of DNMT1-DNA complexes containing oxidized forms of mC
To establish the structural origins of reduced DNMT1 activity in the presence of oxidized forms of mC, molecular models of DNMT1-DNA complexes were considered. For this purpose, homology models of the productive hDNMT1 complex with DNA duplexes containing mC, hmC, fC, and caC were created (Figure 3). Molecular modeling of hDNMT1 protein was carried out using the published crystal structure of mDNMT1 in complex with hemi-methylated DNA.3 The associated DNA duplex was modeled to reflect the sequence employed in our experimental studies (Table 1). The homology model was based upon the sequence alignment in Figure S3. Alignment of the hDNMT1 reference sequence with the sequence of mDNMT1 reveals an 85% identity. Importantly, the residues making up the hydrophobic binding pocket of the TRD (C1501, L1502, W1512, and M1535) are conserved between mDNMT1 and hDNMT1 (Figure S4).
Figure 3:
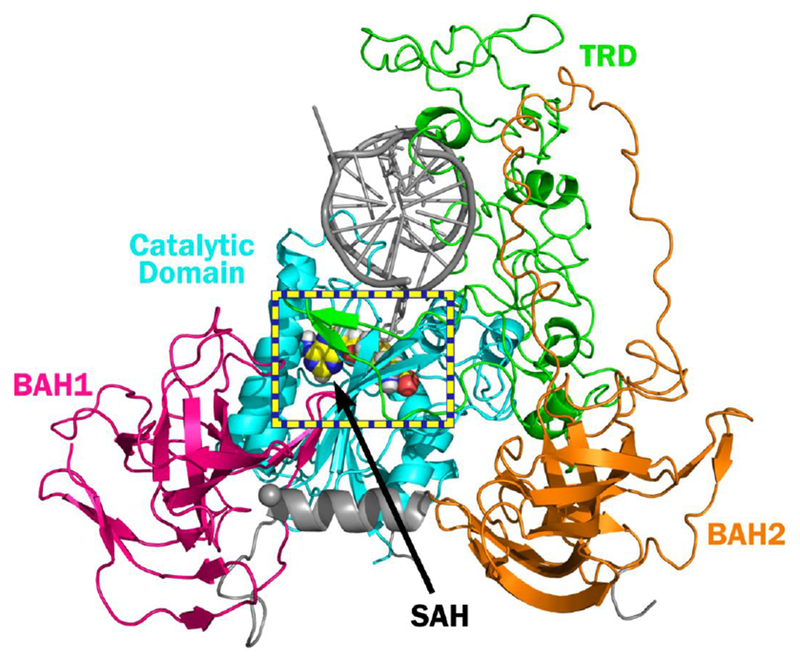
Homology model of the productive human DNMT1 – DNA complex. Homology modeling was performed using the Schrödinger modeling suite package and the crystal structure of mouse DNMT1 in complex with hemi-methylated DNA (PDB: 4DA4)3 coupled with the reference sequence of hDNMT1.
In order to examine the structural effects of TET-mediated oxidation of mC on DNA-protein binding, mC was sequentially replaced with hmC, fC, and caC. Molecular dynamics (MD) simulations of hDNMT1 in complex with DNA containing either mC, hmC, fC, or caC were performed to determine how oxidized forms of mC influence the recognition of hemi-methylated CpG sites in DNA by the TRD of DNMT1. The TRD of human DNMT1 contains a hydrophobic binding pocket consisting of M1535, C1501, L1502 and W1512.3 This pocket harbors the 5-methyl group of mC and is involved in the recognition of the hemi-methylated mCpG in productive hDNMT1-DNA complexes.3
MD simulations were carried out using the previously developed homology model of hDNMT1 in complex with hemi-methylated DNA.22 The root-mean-square deviation (RMSD) was used to determine the stability of the protein and DNA structures over the MD simulation. The RMSD is a similarity measure widely used in the analysis of macromolecular structures and dynamics as it measures the total structural deviation from the starting position. The stability of the enzyme-DNA complex for each modification is demonstrated by the RMSD of the protein backbone (Cα RMSD) and the RMSD of the DNA phosphorous backbone (Figure 1A and 1B). The rise of the RMSD represents the equilibration from the coordinates of the initial model then the RMSD of mC and its oxidized forms remains stable over the 100 ns of simulation. Monitoring the interactions of mC and its oxidized forms with amino acid residues within the hydrophobic pocket of the TRD, we observed an iterative increase in distance between the oxidized forms of mC and DNMT1 residues including Cys1501, Leu1502, and Met1535 (Table 3, Figure 4, Figure 5). This suggests that oxidation to caC may lead to a conformational change to the unproductive mode of binding. Our results demonstrate that as mC is oxidized to hmC, fC, and caC, the increased size and hydrophilicity of the C-5 substituent induces a spatial displacement of the oxidized mC from the TRD binding pocket, disrupting the hydrophobic interactions between DNMT1 and DNA and leading to a loss of enzymatic activity. Our model supports the experimentally observed trend for hDNMT1 enzymatic activity that caC leads to the largest perturbation of the TRD, causing a significant loss in activity. (Figure 2A, Table 1).
Table 3:
Average distance (Å) between the C5 methyl carbon of mC, hmC, fC or caC and the closest heavy atom of amino acid residues in the TRD (Cys1501: Sulfur atom; Leu1502: nearest terminal carbon atom; Met1535: Sulfur atom ) of DNMT1 in DNMT1-DNA complexes as calculated by the molecular dynamics simulation. Distances are averaged from 50-100 ns of MD simulations.
| Residue | mC distance (Å) | hmC distance (Å) | fC distance (Å) | caC distance (Å) |
|---|---|---|---|---|
| Cys1501 | 4.44 ± 0.49 | 6.47 ± 0.47 | 5.42 ± 0.46 | 10.12 ± 0.39 |
| Leu1502 | 4.01 ± 0.36 | 5.47 ± 0.51 | 4.39 ± 0.35 | 8.85 ± 0.47 |
| Met1535 | 4.39 ± 0.34 | 4.39 ± 0.30 | 5.88 ± 0.49 | 9.34 ± 0.75 |
Figure 4.
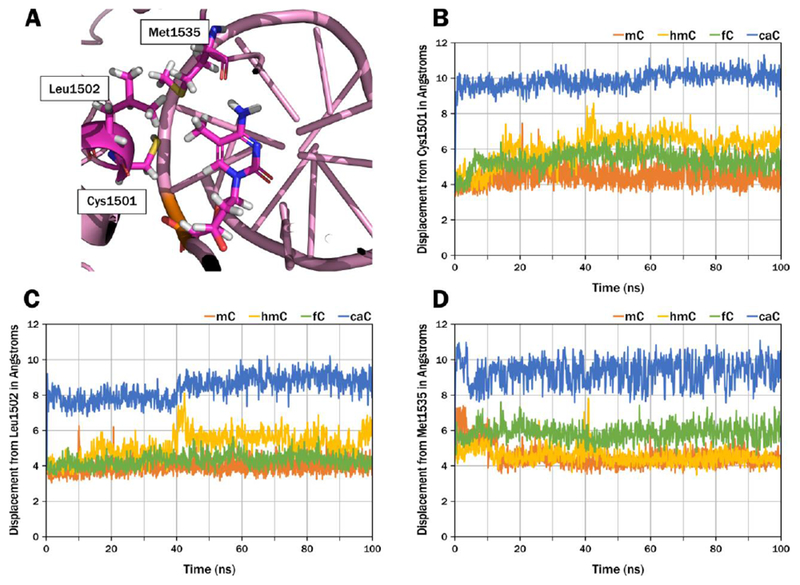
Molecular dynamics (MD) simulations demonstrate an incremental spatial displacement of oxo-mC from the TRD hydrophobic binding pocket. A. Residues Cys1501, Leu1502, and Met1535 make up the target recognition domain (TRD) and harbor the methyl group of mC, providing the specificity of DNMT1 for hemi-methylated DNA. The MD simulations quantify the displacement of the oxidized forms of mC from these residues in the TRD: B. Cys1501 C. Leu1502 D. Met1535
Figure 5.
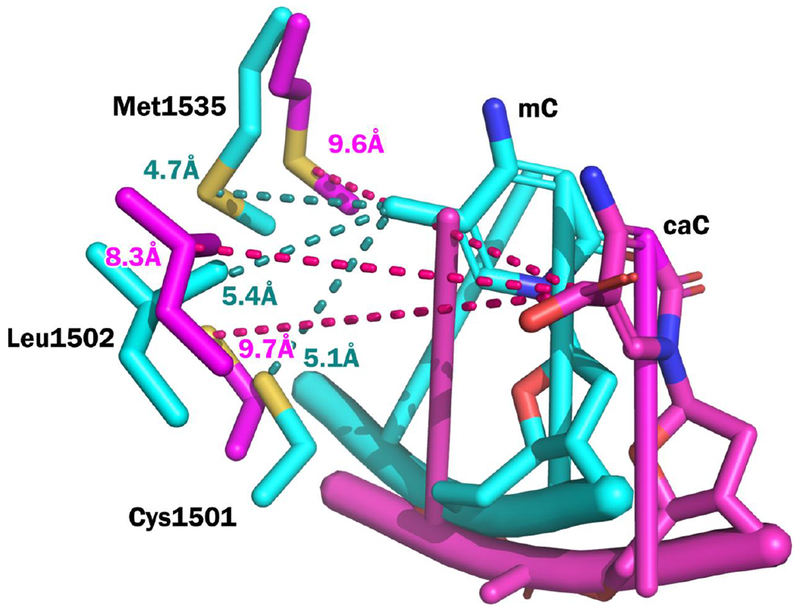
Overlay of mC and caC structures after 100 ns MD simulation. The mC structure is shown in cyan and the caC structure is shown in magenta. Distances between key residues which make up the TRD (Cys1501, Leu1502, Met1535) and the C5 position of each base are shown in dashed lines (mC: cyan, caC: magenta). The increased distances between caC and the residues in the TRD suggest a structural change in the DNMT1-DNA mode of binding.
Discussion
Epigenetic DNA methylation marks (mC) must be removed as part of normal development,35 neuronal plasticity,36 and memory formation, resulting in chromatin remodeling and gene reactivation.36 DNA demethylation can be accomplished via passive or active DNA demethylation processes.14 Active demethylation is mediated by the iterative oxidation of mC to hmC, fC, and caC by Ten Eleven Translocation (TET) proteins, followed by excision of fC and caC by TDG and their replacement with C via base excision repair mechanism.15–18 Passive demethylation occurs when DNMT1 fails to methylate hemi-methylated DNA sequences generated during DNA replication.37 Recent studies have shown that oxidized forms of mC may participate in passive DNA demethylation by reducing the activity of maintenance methyltransferase (DNMT1).19, 38 However, the structural origins and the mechanistic details for reduced DNMT1 activity in the presence of oxidized mC variant remained unknown.
Two distinct modes of DNMT1-DNA binding are known. The protein adopts an unproductive mode of binding in complex with unmethylated DNA (PDB: 3PTA) and a productive mode of binding when bound to hemi-methylated (PDB:4DA4) DNA (Figure 6).3, 7, 22 In the unproductive DNMT1-DNA complex, the double stranded DNA retains its base pairing, and the auto inhibitory mechanism described earlier prevents DNMT1 from performing de novo DNA methylation and directs its activity to hemimethylated sites.7 In the productive DNMT1-DNA complex formed with CpG sites containing a single mC, both the enzyme and its DNA substrate undergo a large conformational change, which is initiated by mC binding in a hydrophobic segment within the TRD (Figure 6).3 This recognition of mC by the TRD results in the insertion of amino acid sidechains from the catalytic and recognition domains of DNMT1 into both grooves of the DNA.3 This productive mode of binding undergoes a local melting of the DNA duplex, rotating the target cytosine out of the DNA helix into the catalytic pocket and allowing for methyl transfer to take place.3 The side chain of Met1235 inserts into the DNA from the minor groove and occupies the space vacated by the target cytosine.3
Figure 6.
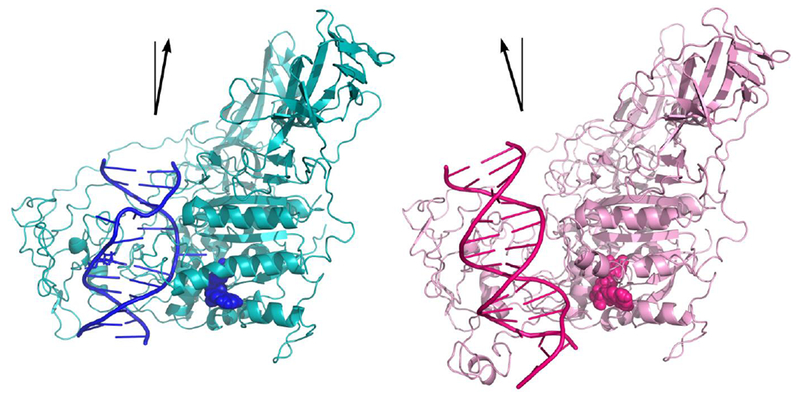
Crystal structures of DNMT1 bound to methylated DNA (teal, PDB: 4DA4)3 or unmethylated DNA (pink, PDB: 3PTA)7 reveal a productive and unproductive mode of binding, respectively. Shift in the angle of the DNA binding mode is highlighted by the black arrow. Cofactor, SAH, is shown as spheres.
The enzymatic mechanism of DNMT1 enzyme is well understood and involves nucleophilic addition of a cysteine in the active site to the C6 position of cytosine, followed by methyl transfer from the SAM cofactor to the C5 position.3 Following methyl transfer, an excess proton from the C5 position is abstracted, and the covalent bond between the enzyme and cytosine base is cleaved to liberate the methylated DNA (Scheme 1).10, 11 Nucleophilic addition of the cysteine residue of DNMT1 to DNA is fast and reversible, while the following methyl transfer is the rate-limiting step during the formation of mC.39
The purpose of the present work was to establish kinetic parameters for DNMT1 mediated methylation in the presence of mC, hmC, fC, and caC and to elucidate the structural mechanisms of their effects on methyl transfer kinetics. A novel mass spectrometry based assay developed in our laboratory was used to follow the kinetics of methyl transfer.22 We found that the rates of methyl transfer drastically decreased as the oxidation state of the methyl group on mC increased from mC to hmC, fC, and caC (Vmax 190, 41, 11, and 0.77 (×10−11min−1), for mC, hmC, fC, and caC respectively – see Figure 2, Table 2). Taken together with previous reports by Ji et al.38 and Valinluck et al.,19 our results show that unlike mC, its oxidized forms fail to effectively direct DNMT1 enzyme to methylate the cytosine in the opposite strand. The Vmax/Km values also decreased according to the oxidation status hmC > fC > caC, indicating that the overall efficiency of enzymatic methylation was reduced. The catalytic efficiency for methyl transfer in the presence of for hmC, fC, and caC decreased 3.3-, 7.9-, and 9.5-fold relative to mC (Table 2). In addition, mC oxidation to hmC, fC, and caC leads to lower Km values for DNMT1-DNA binding (Table 2), indicative of the formation of tightly bound unproductive complexes. Indeed, previous reports by Pradhan et al. demonstrated that DNMT1 binds unmethylated DNA with higher affinity than hemi-methylated DNA.8
In contrast, local nucleotide sequence context had a minimal effect on methylation transfer kinetics (Figure 2B, Table S2) When the 3′ neighboring base was altered (mCGX), this did not change the kinetic parameters for methyl transfer. The efficiency of methyl transfer was slightly higher in mCGC context (Figure 2B, Table S2). This of interest because mC is commonly found in promoter CpG islands of inactive genes.40, 41
In our earlier study, extending the aliphatic side chain on C-5 of cytosine beyond methyl (5-ethyl-dC, 5-propyl-dC) resulted in a loss of DNMT1 maintenance methylation.22 In that study, Vmax and Km values for mC-containing DNA were determined as 9.6 × 10−2 nM/min and 21.8 nM using the sequence 5′-CGCGGA[mC]GCGGGTGCCGGG-3′.22 As the length of the C5-alkyl chain increased, DNMT1-DNA binding via the TRD was disrupted, causing the enzyme to adopt an unproductive mode of binding to DNA.22 Specifically, when the C5 alkyl chain on cytosine was extended from methyl to ethyl, we observed a 4-fold loss in Vmax.22 Further increase of the C-5 substituent size to propyl completely abolished methylation.22 Since 5-ethyl-dC and hmC are of comparable size, but DNMT1 activity is 2-3 fold less efficient for hmC-containing duplexes, this suggests that in addition to steric effects, oxidation of the methyl group of mC leads to a loss of hydrophobic interactions with the TRD of the protein.
To examine the structural basis for reduced DNMT1 methylation activity in the presence of oxo-mC, a computational model of hDNMT1-DNA complex was developed. We found that as mC was oxidized to hmC, fC, and caC, hydrophobic interactions responsible for DNMT1 recognition of hemimethylated CpG sites in DNA were disrupted. The increased distances between caC and the residues in the TRD (Figure 4) suggest a structural change in the DNMT1-DNA mode of binding. In the presence of oxo-mC, the loss of key hydrophobic interactions prevents the formation of a productive DNA-protein complex and instead DNMT1 forms tightly bound unproductive DNMT1-DNA complexes characterized by lower Km value (Figure 5, Figure 6, Table 2).3, 8, 22 Overall, our results confirm that oxidized forms of mC participate in passive DNA demethylation and provide further kinetic and structural details for this important epigenetic process.
Supplementary Material
Experimental scheme for the HPLC-ESI+-MS/MS analysis of mC (Scheme S1)
example calculation for methylation velocity from LC-MS/MS peak areas (Scheme S2)
nucleotide sequences and their mass spectrometry characterization (Table S1)
kinetic data for nucleotide sequence context (Table S2)
example chromatogram of mC and internal standard (Figure S1)
DNA gel shifts for DNA binding to Δ580-DNMT1 (Figure S2)
sequence alignment of hDNMT1 aomology model (Figure S3)
sequence alignment of target recognition domain (Figure S4)
Acknowledgments
Funding information
This work was supported by the U.S. National Cancer Institute [2R01 CA-095039].
Footnotes
Publisher's Disclaimer: This document is the unedited Author’s version of a Submitted Work that was subsequently accepted for publication in Biochemistry,
References
- (1).Jin B, Li Y, and Robertson KD (2011) DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Panning B, and Jaenisch R (1998) RNA and the epigenetic regulation of X chromosome inactivation. Cell 93, 305–308. [DOI] [PubMed] [Google Scholar]
- (3).Song J, Teplova M, Ishibe-Murakami S, and Patel DJ (2012) Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335, 709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chedin F, Lieber MR, and Hsieh CL (2002) The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by DNMT3a. Proc Natl Acad Sci USA 99, 16916–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Jones PA, and Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070. [DOI] [PubMed] [Google Scholar]
- (6).Sharif J, and Koseki H (2011) Recruitment of DNMT1 roles of the SRA protein Np95 (UHRF1) and other factors. Prog Mol Biol Transl Sci 101, 289–310. [DOI] [PubMed] [Google Scholar]
- (7).Song J, Rechkoblit O, Bestor TH, and Patel DJ (2011) Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pradhan M, Esteve PO, Chin HG, Samaranayke M, Kim GD, and Pradhan S (2008) CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 47, 10000–10009. [DOI] [PubMed] [Google Scholar]
- (9).Matje DM, Zhou H, Smith DA, Neely RK, Dryden DT, Jones AC, Dahlquist FW, and Reich NO (2013) Enzyme-promoted base flipping controls DNA methylation fidelity. Biochemistry 52, 1677–1685. [DOI] [PubMed] [Google Scholar]
- (10).Matje DM, Krivacic CT, Dahlquist FW, and Reich NO (2013) Distal structural elements coordinate a conserved base flipping network. Biochemistry 52, 1669–1676. [DOI] [PubMed] [Google Scholar]
- (11).Svedruzic ZM, and Reich NO (2004) The mechanism of target base attack in DNA cytosine carbon 5 methylation. Biochemistry 43, 11460–11473. [DOI] [PubMed] [Google Scholar]
- (12).Das PM, and Singal R (2004) DNA methylation and cancer. J Clin Oncol 22, 4632–4642. [DOI] [PubMed] [Google Scholar]
- (13).Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB, and Herman JG (1999) Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res 59, 67–70. [PubMed] [Google Scholar]
- (14).Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, and Ecker JR (2013) Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, and Xu GL (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Maiti A, and Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Valinluck V, and Sowers LC (2007) Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 67, 946–950. [DOI] [PubMed] [Google Scholar]
- (20).Seisenberger S, Peat JR, and Reik W (2013) Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr Opin Cell Biol 25, 281–288. [DOI] [PubMed] [Google Scholar]
- (21).Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W, Li W, Zhou Q, Aluru N, Tang F, He C, Huang X, and Liu J (2014) Programming and inheritance of parental DNA methylomes in mammals. Cell 157, 979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kotandeniya D, Seiler CL, Fernandez J, Pujari SS, Curwick L, Murphy K, Wickramaratne S, Yan S, Murphy D, Sham YY, and Tretyakova NY (2018) Can 5-methylcytosine analogues with extended alkyl side chains guide DNA methylation? Chem Commun (Camb) 54, 1061–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, and Li BF (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277, 1996–2000. [DOI] [PubMed] [Google Scholar]
- (24).Kim GD, Ni J, Kelesoglu N, Roberts RJ, and Pradhan S (2002) Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J 21, 4183–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Guza R, Kotandeniya D, Murphy K, Dissanayake T, Lin C, Giambasu GM, Lad RR, Wojciechowski F, Amin S, Sturla SJ, Hudson RH, York DM, Jankowiak R, Jones R, and Tretyakova NY (2011) Influence of C-5 substituted cytosine and related nucleoside analogs on the formation of benzo[a]pyrene diol epoxide-dG adducts at CG base pairs of DNA. Nucleic Acids Res 39, 3988–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Guza R, Rajesh M, Fang Q, Pegg AE, and Tretyakova N (2006) Kinetics of O6-Me-dG repair by O6-alkylguanine DNA-alkyltransferase within K-ras gene derived DNA sequences. Chem. Res. Toxicol 19, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Guza R, Ma L, Fang Q, Pegg AE, and Tretyakova NY (2009) Cytosine methylation effects on the repair of O6-methylguanines within CG dinucleotides. J. Biol. Chem 284, 22601–22610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rajesh M, Wang G, Jones R, and Tretyakova N (2005) Stable isotope labeling-mass spectrometry analysis of methyl- and pyridyloxobutyl-guanine adducts of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in p53-derived DNA sequences. Biochemistry 44, 2197–2207. [DOI] [PubMed] [Google Scholar]
- (29).(2015) Schrodinger Modeling Suite Package, Schrodinger LLC, New York, NY. [Google Scholar]
- (30).D. E. Shaw Research, N. Y., NY. (2018) Desmond Molecular Dynamics System.
- (31).Jorgensen WL, Chandrasekhar J, Madura JD (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926. [Google Scholar]
- (32).Zhou YB, Wu YY, Pokholenko O, Grimsrud M, Sham Y, Papper V, Marks R, and Steele T (2018) Aptamer adaptive binding assessed by stilbene photoisomerization towards regenerating aptasensors. Sensor Actuat B-Chem 257, 245–255. [Google Scholar]
- (33).Pradhan S, Bacolla A, Wells RD, and Roberts RJ (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem 274, 33002–33010. [DOI] [PubMed] [Google Scholar]
- (34).Kohli RM, and Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mayer W, Niveleau A, Walter J, Fundele R, and Haaf T (2000) Demethylation of the zygotic paternal genome. Nature 403, 501–502. [DOI] [PubMed] [Google Scholar]
- (36).Lister R, and Mukamel EA (2015) Turning over DNA methylation in the mind. Front Neurosci 9, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wu X, and Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 18, 517–534. [DOI] [PubMed] [Google Scholar]
- (38).Ji D, Lin K, Song J, and Wang Y (2014) Effects of Tet-induced oxidation products of 5-methylcytosine on DNMT1- and DNMT3a-mediated cytosine methylation. Mol Biosyst 10, 1749–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Aranda J, Zinovjev K, Swiderek K, Roca M, and Tunon I (2016) Unraveling the reaction mechanism of enzymatic C5-cytosine methylation of DNA. A combined molecular dynamics and QM/MM study of wild type and Gln119 variant. Acs Catal 6, 3262–3276. [Google Scholar]
- (40).Deaton AM, and Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jeziorska DM, Murray RJS, De Gobbi M, Gaentzsch R, Garrick D, Ayyub H, Chen T, Li E, Telenius J, Lynch M, Graham B, Smith AJH, Lund JN, Hughes JR, Higgs DR, and Tufarelli C (2017) DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc Natl Acad Sci USA 114, E7526–E7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental scheme for the HPLC-ESI+-MS/MS analysis of mC (Scheme S1)
example calculation for methylation velocity from LC-MS/MS peak areas (Scheme S2)
nucleotide sequences and their mass spectrometry characterization (Table S1)
kinetic data for nucleotide sequence context (Table S2)
example chromatogram of mC and internal standard (Figure S1)
DNA gel shifts for DNA binding to Δ580-DNMT1 (Figure S2)
sequence alignment of hDNMT1 aomology model (Figure S3)
sequence alignment of target recognition domain (Figure S4)