

Abstract
Lentiviral vectors (LVs) pseudotyped with the measles virus hemagglutinin (H) and fusion (F) glycoproteins have been reported to more efficiently transduce hematopoietic stem and progenitor cells (HSPCs) compared with vesicular stomatitis virus glycoprotein (VSV-G) pseudotyped LVs. However, a limit to H/F LV use is the low titer of produced vector. Here we show that measles receptor (CD46) expression on H/F transfected HEK293T vector-producing cells caused adjacent cell membrane fusion, resulting in multinucleate syncytia formation and death prior to peak vector production, leading to contaminating cell membranes that co-purified with LV. H/F LVs produced in CD46 null HEK293T cells, generated by CRISPR/Cas9-mediated knockout of CD46, produced 2-fold higher titer vector compared with LVs produced in CD46+ HEK293T cells. This resulted in approximately 2- to 3-fold higher transduction of HSPCs while significantly reducing target cell cytotoxicity caused by producer cell contaminates. Improved H/F LV entry into HSPCs and distinct entry mechanisms compared with VSV-G LV were also observed by confocal microscopy. Given that vector production is a major source of cost and variability in clinical trials of gene therapy, we propose that the use of CD46 null packaging cells may help to address these challenges.
Keywords: lentiviral vector, enhanced gene delivery, stem cells, measles lentiviral vector, syncytia, CD46, CD46 knockout, measles glycoprotein, pseudotyping, vector production
Graphical Abstract
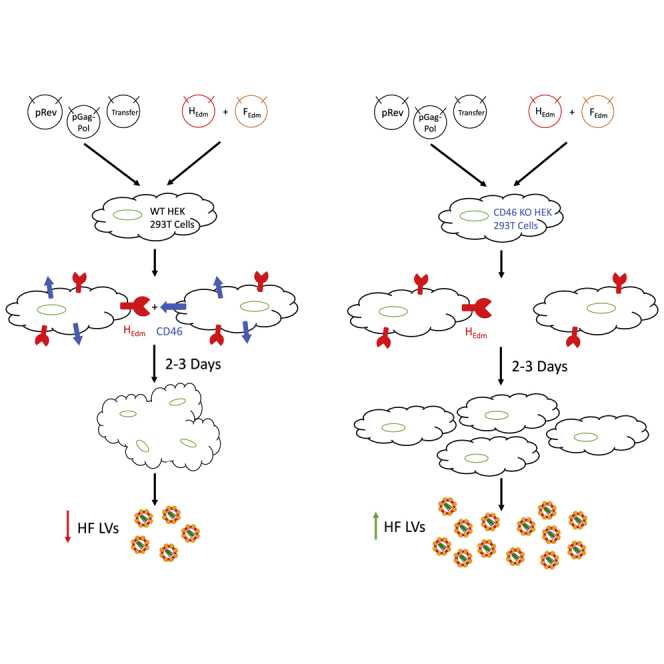
Introduction
Gene delivery to hematopoietic stem and progenitor cells (HSPCs) holds promise to correct a large range of monogenic, oncologic, and infectious diseases.1 Recent clinical trials targeting this cell population have made meaningful impacts in patient outcomes for several diseases including sickle cell anemia,2 beta-thalassemia,3 metachromatic leukodystrophy,4 Wiskott-Aldrich syndrome,5 and X-linked adrenoleukodystrophy.6
HIV-1-based self-inactivating lentiviral vectors (LVs) have proven to be an optimal platform for modification of HSPCs, given their improved integration safety profile compared with gamma-retroviral vectors and their ability to mediate long-lasting disease correction in frequently dividing cells.7 Replacement of the native HIV-1 envelope glycoprotein with other viral glycoproteins, in a process known as pseudotyping, greatly expands the potential targetable cell types modifiable by these vectors, as well as increases their stability, allowing for ultracentrifuge-based concentration and therefore improved vector titers.8
The standard viral envelope for pseudotyping remains the Indiana strain of the vesicular stomatitis virus glycoprotein (VSV-G), which recognizes the low-density lipoprotein receptor (LDL-R) for cellular attachment and entry.9 VSV-G has been shown to require trafficking through the cellular endosomal network followed by a low-pH-mediated fusion event to eject viral capsid contents into the cytoplasm, prior to reverse transcription, viral DNA migration into the nucleus, and integration.10, 11 Variation in the level of LDL-R expression has been shown to reduce the efficiency with which VSV-G pseudotyped LVs (VSV-G LVs) are able to mediate gene delivery to several therapeutically important cells, including T and B cells and HSPCs.12
In an effort to improve gene delivery to difficult-to-transduce cell types, several groups have investigated pseudotyping with alternate viral glycoproteins. Baboon endogenous virus, feline endogenous retrovirus, Nipah virus, and other rhabdoviruses have all shown improvements over VSV-G in gene delivery to HSPCs.13, 14, 15, 16 However, each of these vectors has demonstrated variability in the ability to produce high-titer vector. Prior reports have shown the promise of pseudotyping with the measles hemagglutinin and fusion glycoproteins (H/F), demonstrating high levels of gene delivery to therapeutically relevant and typically difficult-to-transduce cells, including resting T and B lymphocytes and monocyte-derived dendritic cells.17, 18, 19, 20 Modifications of the measles H protein with single-chain antibodies and designed ankyrin repeat proteins (DARPins) have further widened the potential targetable cell types by these vectors.21, 22
Wild-type (WT) measles virus H protein recognizes either the nectin-4 or signaling lymphocytic activation molecule (SLAM, CD150) proteins as cellular receptors, while the cell culture-adapted Edmonston vaccine strain predominantly binds the ubiquitously expressed CD46 complement regulatory protein.23, 24, 25 Multinucleate syncytia formation after measles virus infection is observed in both cell lines and primary human tissues in vitro, and is thought to be important for virus spread.25, 26 WT measles pseudotyped LVs can achieve comparable titers to VSV-G LVs but, given the limited range of cells that express CD150 or nectin-4, are therapeutically less useful.27 Conversely, Edmonston strain pseudotyped LVs (H/F LVs) have consistently demonstrated approximately 2-log fold lower titers, making approaches to improve production of these vectors necessary for the field.28
Herein we demonstrate that LV pseudotyping with the Edmonston strain H/F envelopes in HEK293T cells promotes syncytia formation, producer cell death, reduction of LV titer, and the presence of CD46-positive cell membrane-H/F LV complexes in purified LV preparations. Vector titer, transduction efficacy, and target cell cytotoxicity from producer cell contaminates were all improved when H/F LV was produced in CD46 gene-disrupted (CD46 null) HEK293T cells. Confocal analysis and small-molecule studies also showed differing viral trafficking patterns in H/F LVs compared with VSV-G LVs that help to explain enhanced transduction efficiency. The use of CD46 null packaging cells may lower the cost and improve the efficiency of generating homogeneous preparations of H/F LVs for both research and clinical applications.
Results
Measles H and F Glycoprotein Pseudotyped LV Production in HEK293T Cells Results in Syncytia Formation and Producer Cell Death
To directly compare H/F and VSV-G LVs, we generated GFP-expressing LVs (FG-12 UbiC-EGFP), while keeping all packaging components consistent between vector types, varying only the H/F and VSV-G envelope plasmids. After co-transfection of plasmids in HEK293T cells, transgene-derived GFP expression increased, peaking approximately 3 days later (Figure 1A). HEK293T cells producing VSV-G LVs remain as isolated cells and continued to grow and proliferate until limited by cell density (Figure 1A, middle). By contrast, H/F LV-producing HEK293T cells begin to show large multi-nucleate syncytia formation approximately 12 hr after transfection, with cell death occurring within 3 days post-transfection (Figure 1A, right). We reasoned that syncytia formation, known to occur with measles infections,29 and premature cell death may be a major reason for low titers in H/F LV production.
Figure 1.

H/F LV Production Causes Syncytia Formation in Packaging Cells and Cell Debris Remains in Vector Preparations
(A) 10× bright-field (left) and 488-nm fluorescent (middle and right) microscopy images of transiently transfected HEK293T cells generating either VSV-G LV (middle) or H/F LV (right) 3 days after co-transfection with respective packaging plasmids. (B) 64× confocal microscopy images of umbilical cord blood (UCB)-derived CD34+ cells transduced with either H/F LV (left) or VSV-G LV (right). Cytokine pre-stimulated cells were treated with either LV for 15 min, then fixed before staining for EEA1 (green), vector Gag p24 protein (red), and nuclei with Hoechst (blue).
Producer cell cytotoxicity can result in the release of contaminating cellular components in LV preparations, possibly impacting target cells during transduction. To determine whether cell debris remains in concentrated LV preparations, harvested vector supernatant was pelleted and filtered through a 0.45-μm filter, before concentration by 20% sucrose cushion ultracentrifugation. Umbilical cord blood (UCB)-derived CD34+ HSPCs were then treated with either H/F or VSV-G LVs and analyzed by confocal microscopy. H/F LVs or VSV-G LVs were used at MOI of 5 and 15, respectively, to result in consistent final transduction levels. Cells were fixed 15 min and 1 hr after LV addition and then stained for nuclei (Hoechst), vector capsid p24 protein, and early endosome antigen 1 (EEA1) to evaluate vector endosome trafficking. Upon imaging, large p24 antigen-positive staining, non-nucleated HEK293T membrane fragments were observed on slides, often in close association with nucleated CD34+ HSPCs in H/F LV-treated conditions (Figure 1B, left, arrows), which was not observed in VSV-G LV-treated cells (Figure 1B, right). Thus, despite purification, these findings indicate that current commonly used vector production schemes are insufficient to remove contaminating HEK293T cellular components.
CD46 Knockout in HEK293T Cells Prevents Syncytia Formation and Improves H/F LV Titer
Other reports evaluating measles pseudotyped LVs have demonstrated that H-receptor engagement is thought to be sufficient to induce F glycoprotein conformational change and subsequent viral-host membrane fusion.21 Consequently, we reasoned that disrupting cell-surface expression of CD46 may be sufficient to avoid syncytia formation and LV interaction with packaging cells during LV production. To generate CD46 null cells, a CRISRP/Cas9-mediated knockout of CD46 in HEK293T cells was undertaken. Three appropriate PAM sequences specific to exon 1 of the human CD46 gene were predicted using the web-based software tool described in Hsu et al.30 to evaluate guide specificity (Figure S1A). HEK293T cells were then transfected with pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid containing three distinct CD46 gRNAs, selected with puromycin and evaluated for CD46 expression. Post-selection, cells transfected with the gRNA1-containing plasmid showed the greatest percentage of CD46 null (∼17% of cells CD46 null from gRNA1; Figure S1B) (4% null gRNA2, 9% gRNA3). Puromycin selected cells were then flow sorted based on the absence of CD46 expression (Figure S1C). The resulting cell population was approximately 98% CD46-negative, and this lack of CD46 expression was maintained after 6 months of culture (Figure S1D, red). By contrast, WT HEK293T cells were found to maintain >99% CD46 expression (Figure S1D, blue). Importantly, knocking out CD46 in HEK293T cells did not alter cell replication kinetics, viability, or cell morphology (Figure S1E).
To evaluate whether CD46 expression mediates H/F LV transduction, we treated WT and CD46 null 293T cells with VSV-G or H/F LVs. WT and CD46 null 293T cells transduced with VSV-G LV (MOI 10) showed identical transduction efficiency (Figure S1F, top). Conversely, only WT 293T cells were transducible by H/F LV (MOI 1) (Figure S1F, bottom). Of the cells sorted for the absence of CD46 expression, only the small remnant population of CD46-expressing cells that were not completely removed demonstrated any EGFP expression 3 days later. The transduction capacity of this limited, remnant population supports previous evidence from other studies that CD46 is a necessary receptor for Edmonston strain H/F LV transduction of HEK293T cells.17
To determine whether CD46 null packaging cells improve vector production, we generated H/F LV in CD46 WT and null cells. Multinucleate syncytia formation was observed in WT cells 24 hr after transfection, with major cytotoxicity after 3 days (Figure 2A). By contrast, CD46 null cells showed no syncytia formation during the same culture period, with limited cytotoxicity and continued cellular proliferation (Figure 2B). Equivalent vector production, as shown by similar p24 protein amounts, was produced 2 days after packaging plasmid transfection of WT and CD46 null cells. However, LV Gag p24 protein production dramatically decreased in WT cells, concomitant with the decrease in cell viability 3 days post-transfection (Figure 3A). By contrast, CD46 null cells produce approximately 2-fold higher p24 protein, indicating increased vector production in these cells (Figure 3A). Propidium iodide (PI) staining and flow cytometry analysis of HEK293T cells transfected to produce H/F LV confirmed that vector production was somewhat cytotoxic (∼22% ± 5.8% PI staining cells, n = 3) to CD46 null producer cells, whereas almost all WT HEK293T cells were dead 3 days after transfection (Figure 3B).
Figure 2.

H/F LV Production in CD46 Null HEK293T Cells Is Non-cytotoxic and Does Not Form Syncytia Observed in Wild-Type HEK293T Cells
(A) 10× bright-field (left) and 488-nm (right) fluorescence microscopy of HEK293T cells transiently co-transfected with packaging plasmids on day 0 to generate H/F LV. Limited syncytia formation was observed in wild-type HEK293T cells after 1 day, with major syncytia apparent by 2 days and cytotoxicity observed after 3 days when supernatant was collected. (B) CD46 null HEK293T cells transfected for LV generation and imaged as above. Cells form no observable syncytia throughout the 3-day culture period and continue to grow and proliferate without observable cell cytotoxicity. Representative images from n = 7 LV productions.
Figure 3.

H/F LV Purity and Titer Increased When Generated in CD46 Null HEK293T Cells, Compared with Wild-Type Producer Cells
(A) Wild-type or CD46 null HEK293T cells were co-transfected with H/F LV plasmids, and cell viability (dotted lines) and total p24 protein production per day (solid lines) in supernatant were determined each day for 1 week after transfection. Data are presented as line graphs (mean ± SD p24 ng/mL or % viable cells). (B) Wild-type (blue) or CD46 null HEK293T cells (red) were transfected on day 0 to generate H/F LV, and producer cell viability was evaluated by propidium iodide (PI) exclusion using flow cytometry 3 days post-transfection. Data are presented as a FACS plots from a representative experiment (n = 3). (C) Average functional (transducing units [TU]/mL) and physical (μg/mL) titer of vector preps (n = 7), measured by serial dilution flow cytometric assay of wild-type HEK293T cell transduction or by anti-p24 ELISA. (D) Comparison of average functional titer (TU/mL) of H/F LV produced in wild-type HEK293T cells, CD46 null HEK293T cells, and VSV-G LV, by serial transduction on indicated cell lines (n = 3). (E) Representative SDS-PAGE gel of H/F LV containing supernatant from wild-type and CD46 knockout HEK293T cells collected 24 (D1), 48 (D2), and 72 hr (D3) post-transfection. Collected supernatant was also concentrated by ultracentrifugation over 20% sucrose, before matching for total protein and visualization by Coomassie stain.
H/F LV produced in CD46 null cells resulted in approximately 2-fold higher titer on average compared with CD46 WT-produced LV (Figure 3C). This 2-fold higher titer was consistent when LVs were evaluated on monocyte, promyeloblast, and B cell lines (THP-1, KG-1a, and Raji, respectively) (Figure 3D). H/F LV titers were 2- to 3-fold lower than VSV-G LVs for KG-1a and THP-1, and higher for Raji cells, consistent with reported enhanced transduction efficiency in B cells.18
H/F LV Production in CD46 Null Cells Improves Vector Purity
To determine the purity of produced vector, H/F LVs generated in CD46 WT and null cells were evaluated by SDS-PAGE. Unconcentrated supernatant from vector-producing cells was collected 24, 48, and 72 hr post-transfection, as well as after ultracentrifugation and 20% sucrose-cushion purification. Matching for total loaded protein, supernatant and concentrated vector were separated on a 10% Bis-Tris polyacrylamide gel. Coomassie staining of concentrated vector indicated a relative abundance of high-molecular-weight proteins present in the concentrated vector produced in WT HEK293T cells, which are absent in the concentrated vector produced in CD46 null cells (Figure 3E). Anti-Gag western blotting confirmed that both uncleaved Gag species and fully cleaved p24 protein were present in both the WT and CD46 null cell vector preparations (Figure S2A). Three vector preps each of WT or CD46 null HEK293T-generated H/F LV were subsequently evaluated by total protein stain and anti-Gag western blot (Figure S2B), and mature p24 capsid signal was quantified as a percent of total protein and total Gag (Figure S2C). Mature p24 capsid represented less of the overall Gag signal in WT versus CD46 null-generated H/F LV preps (WT H/F LV preps versus CD46 null H/F LV preps, mean signal 64% versus 86%). Mature p24 capsid also represented less of the total protein signal in WT versus CD46 null-generated H/F LV preps (mean signal 3.12% versus 6.4%) (Figure S2C). Together these findings support the production of H/F LV in CD46 null cells as a method for improving vector purity.
H/F LV Produced in CD46 Null Cells Has Improved Transduction Ability Compared with VSV-G LVs and WT-Produced H/F LVs
We next compared VSV-G LVs with H/F LVs produced in both WT and CD46 null cell lines, for the ability to transduce UCB-derived CD34+ cells, as well as granulocyte colony-stimulating factor (G-CSF) mobilized peripheral blood (mPB)-derived CD34+ cells. mPB CD34+ cells are a convenient source for autologous stem cell therapy during myeloablative chemotherapy, as well as for allogeneic transplantation, and so are an attractive target for genetic modification.31 WT cell-produced H/F LV demonstrated 2- to 3-fold improved transduction efficiency over VSV-G LV, for both UCB and mPB-derived CD34+ transduction at low MOI (Figure 4A; Figure S3A). Interestingly, H/F LV produced in CD46 null cells demonstrated 6- to 8-fold improved transduction efficiency compared with VSV-G LV and a 2-fold improvement compared with WT-produced H/F LV at equivalent MOI. We observed a larger increase in transduced cells with increasing vector in UBC-derived CD34+ cells than with mPB-derived CD34+ cells with all vectors, consistent with other findings that mPB-derived CD34+ cells transduce less efficiently.32
Figure 4.

H/F LV Produced in CD46 Null Cells Has Increased Transduction Efficiency and Less Cytotoxicity Than LV Produced in Wild-Type Cells on CD34+ HSPCs
(A) UCB-derived CD34+ cells were transduced with either H/F LV derived from WT (blue) or CD46 null HEK293T cells (red) or VSV-G LV (gray) at indicated MOI, and evaluated by flow cytometry for EGFP expression after 7 days. (B) Cell viability of mock or LV-treated UCB-derived CD34+ cells matched for MOI of VSV-G LV, or H/F LV derived from either WT or CD46 null HEK293T cells. CD34+ cell viability was calculated as percent of trypan blue excluding cells. (C) Vector copy number quantification of UCB-derived CD34+ HSPCS treated with specified vector at indicated MOI. (D) Comparison of EGFP expression 7 days after transduction in UCB-derived CD34+ HSPCs after treatment with VSV-G LV or CD46 null cell-produced H/F LV with vector matched for physical titer (p24 protein). All data presented in bar and line graphs show mean ± SD (n = 2, 2 donors, 2 vector lots). Statistical analysis using two-way ANOVA with Tukey’s multiple comparisons test. *p < 0.0332; **p < 0.0021; ***p < 0.0002; ****p < 0.0001. ns, not significant.
We next evaluated whether improved transduction efficiency may be related to differences in cytotoxicity of the vector preparations. UCB-derived CD34+ HSPCs were treated with VSV-G-, H/F null-, and WT H/F-produced LVs matched for MOI, and cell viability was assessed by trypan blue exclusion (Figure 4B). H/F null LV and VSV-G LV both showed a small reduction in viability 1 day after treatment that was comparable with that observed in mock-treated cells, with recovery 3 days post-vector addition. By contrast, H/F LV produced in WT HEK293T cells dramatically reduced CD34+ HSPC viability after 1 day, with viability not returning to mock-treated levels until 1 week after treatment. Due to the difficulty of distinguishing non-trypan excluding dead target cells from producer cell debris by visual morphology alone, UCB-derived CD34+ cells were also analyzed 1 day after LV treatment by flow cytometry (Figure S3B). Live/Dead cell analysis of cells treated with MOI 1 of VSV-G-, H/F null-, and WT H/F-produced LVs again demonstrated significantly higher cytotoxicity among cells treated with H/F WT vector (Figure S3C). Thus, the improved transduction of CD46 null H/F LV preparations compared with WT H/F LV may be partially the result of a reduced cytotoxicity of target cells.
Given the variation observed in transduction efficiency across primary cells and several cell lines, we additionally sought to compare integrated vector copy number (VCN) by qPCR in UCB-derived CD34+ HSPCs. Transduction efficiency as measured by flow cytometry was consistent with VCN for low MOI vector inputs, with 2- to 3-fold improved transduction comparing VSV-G with H/F LV produced in WT cells, and 6- to 8-fold improvements compared with CD46 null-produced H/F LV (Figure 4C). Additionally, H/F LV CD46 null cell-produced vector matched for input p24 protein showed 2-fold improvement in transduction compared with VSV-G LV (Figure 4D). When matched for a final transduction level of ∼25%, UCB-derived CD34+ cells plated for colony-forming unit (CFU) assay showed no difference in plating efficiency (Figure S3D) or colony type distribution, when comparing vector treatment (Figure S3E), indicating no effect on HSPC differentiation capacity.
H/F LV Shows Distinct Entry Mechanisms and Improved Loading in UCB CD34+ Cells Compared with VSV-G LV
Measles virus has been reported to enter cells through a non-pH-mediated macropinocytosis-like mechanism at the cell surface or through an endocytosis-like pathway, depending on cell type and receptor engagement.29, 33 However, less is known concerning measles entry via CD46 into hematopoietic cells. We first compared the subcellular trafficking of H/F LV produced in WT HEK293T cells with VSV-G LV in activated UCB-derived CD34+ HSPCs through staining for EEA1 and by co-localization of LV p24 protein by confocal microscopy (Figure 5A). VSV-G was shown to co-localize with EEA1+ endosomes 15 min after LV addition and decrease after 1 hr, consistent with VSV-G LV’s reported dependence on endosomal maturation for core escape (Figure 5B).34 By contrast, WT H/F LV p24 showed minimal co-localization with EEA1+ signal at both time points, supporting a non-endosome-mediated entry mechanism.
Figure 5.

H/F LVs Demonstrate Distinct Cellular Trafficking Patterns and Improved Transduction Ability in Cytokine-Stimulated CD34+ HSPCs
(A) Experimental workflow of transduction of UCB-derived CD34+ HSPCs by VSV-G or H/F LV. CD34+ cells were isolated and transduced as described in the Materials and Methods. (B) Co-localization analysis of confocal images for EEA1+ and p24+ vesicles, at 15 min and 1 hr after vector addition. Data are presented as dot plots (mean ± SD). Pearson’s correlation coefficient was compared by Kruskal-Wallis test with Dunn’s multiple comparison correction; ***p < 0.0002; ****p < 0.0001. (C) UCB-derived CD34+ HSPCs were pre-treated for 4 hr with the indicated concentration of prostaglandin E2 (PGE-2), EGA, or DMSO vehicle control, before 24-hr co-treatment with H/F (blue) or VSV-G LV (gray). Data are presented as bar graphs (mean ± SD). *p = 0.0332; ****p < 0.0001 by two-tailed Student’s t test. (D) Representative plots of UCB-derived CD34+ HSPCs (n = 3) freshly isolated (blue) or treated with stem cell transduction-promoting cytokines for 24 hr (red), then analyzed for CD150 or CD46 expression by flow cytometry. (E) Freshly isolated UCB-derived CD34+ HSPCs were cultured with stem-cell-promoting cytokines for 24 hr before transduction with VSV-G or H/F LV at the indicated MOI (n = 3 donors). 24 hr after LV addition, vector was removed and cells were cultured in cytokine-containing media, before analysis by flow cytometry 7 days later. (F) UCB-derived CD34+ HSPCs were freshly isolated, then immediately transduced with either VSV-G or H/F pseudotyped LV as above, without pre-stimulation with cytokines. 24 hr after LV addition, LV was removed and cells were cultured and evaluated as above (n = 2 donors). Data are presented as bar graphs (mean ± SD). Statistical analysis using unpaired Student’s t test; *p < 0.0332; **p < 0.0021.
To further evaluate this difference in the cellular entry mechanism, we evaluated studies using co-incubation of both pseudotyped LVs with transduction modifying small molecules. We and others have recently identified prostaglandin E2 (PGE-2) supplementation as an enhancer of LV gene delivery, acting in a step distinct from endosomal fusion.35 While UCB CD34+ HSPCs treated with PGE-2 and VSV-G LVs demonstrated modest transduction enhancement, WT H/F LVs showed non-significant changes in transduction efficiency compared with vehicle controls (Figure 5C). A recently reported small-molecule screen for anthrax lethal toxin inhibitors identified 4-bromobenzaldehyde N-(2,6-dimethylphenyl)semicarbazone (EGA) as a potent inhibitor of endosome maturation that does not alter endosome pH.36 Co-incubation of UCB-derived CD34+ HSPCs with EGA and VSV-G demonstrated nearly complete abolishment of transduction, while co-incubation with H/F LVs showed no effect (Figure 5B). These findings further indicate that unlike VSV-G, H/F LVs enter and traffic through a non-endosome maturation-dependent cellular pathway.
It has been reported that WT H/F LVs transduce quiescent or resting cells, including CD34+ HPSCs, T and B lymphocytes, and monocyte-derived dendritic cells, without requiring cell activation.17, 18, 19 To examine the role that receptor expression influenced transduction efficiency, we evaluated freshly isolated and cytokine-stimulated UCB CD34+ cells for measles receptor expression. Both freshly isolated and cytokine-stimulated UCB CD34+ cells showed little to no expression of CD150 (SLAM) (Figure 5D, left), as we have reported previously,37 which is in accordance with this protein’s reported expression primarily by a subset of B cells, activated and memory T cells, and immature thymocytes.38 By contrast, >90% of freshly isolated UCB CD34+ cells expressed CD46, with mean fluorescence intensity (MFI) increasing 2- to 3-fold upon 24 hr of cytokine stimulation. These findings are consistent with the notion that increased receptor density may be a factor in the improved transduction efficiency observed with H/F LVs upon cell stimulation (Figure 5D, right), as has also been observed with VSV-G LV and LDL-R expression.12
To evaluate whether CD46 expression upregulation with cytokine stimulation impacts transduction efficiency, UCB CD34+ cells were either pre-stimulated with stem cell supporting cytokines or in basal media for 24 hr, before transduction with either H/F or VSV-G LVs. Vector was removed after 24 hr by cell washes, and transduction was evaluated 7 days later by flow cytometry, after expansion in serum-containing medium. Both H/F and VSV-G LVs showed enhanced transduction capacity in cytokine-stimulated cells (Figure 5E) compared with non-cytokine pre-stimulated cells (Figure 5F), although H/F LVs showed a distinct advantage in transduction efficiency, as has been recently reported.39 Thus, the distinct entry mechanism used by H/F LV may be useful for modifying non-cytokine pre-stimulated CD34+ HPSCs.
We next evaluated the trafficking of H/F LV produced in HEK293T WT and CD46 null cells in UCB CD34+ HSPCs by confocal microscopy. Unlike WT-produced H/F LV (Figure 1B), very limited cell debris was observed in images of cells treated with CD46 null-produced H/F LV (Figure S3F). Analysis of subcellular trafficking showed different patterns for each vector. EEA1+ endosome number per cell decreased from 15 min to 1 hr after LV addition, for all vector conditions, indicating that LV treatment may induce endosome cycling in UCB-derived CD34+ HSPCs (Figure 6A). Interestingly, WT-generated H/F LV treatment induced a large increase in EEA1+ endosome number compared with VSV-G LV and null-produced H/F LV, a result presumably related to the presence of HEK293T cell debris. We also quantified the number, volume, and staining intensity of p24+ intracellular compartments for each LV type (potentially macropinosomes, termed vesicles below). The number of p24+ staining vesicles within cells was significantly greater in both types of H/F LVs compared with VSV-G LV, indicating that the observed improvement in transduction efficiency may be related to improved vector entry (Figure 6B). Finally, both p24+ vesicle volume and staining intensity were significantly greater in CD46 null cell-line-produced H/F LV, compared with WT-produced H/F LV and VSV-G LV (Figures 6C and 6D). This large increase in signal intensity and vesicle volume is indicative of more H/F LV loaded per vesicle, which may be responsible for the improved transduction efficiency observed.
Figure 6.

H/F LV Generated in Wild-Type or CD46 Null HEK293T Shows Distinct Effects on the Early Endosome Compartment and Enters Cells with Improved Efficiency Compared with VSV-G LV
(A) UCB-derived CD34+ HSPCs were treated for 15 min or 1 hr with VSV-G LV (MOI 15) or H/F LV generated in either WT or CD46 null HEK293T cells (MOI 5). 50 cells for each condition were imaged by confocal microscopy, quantifying (A) EEA1+ endosome number and (B) Gag p24+ intracellular vesicle number, (C) mean vesicle volume, and (D) mean vesicle staining intensity. Data are presented as dot plots (mean ± SD). Statistical analysis using Kruskal-Wallis test with Dunn’s multiple comparisons test; *p < 0.0332; **p < 0.0021; ***p < 0.0002; ****p < 0.0001. EEA1, early endosome antigen-1.
Discussion
Gene therapy of hematopoietic stem cells has demonstrated success as an approach for the correction of monogenic diseases of the blood system, and is an increasingly appealing method for the treatment of multifactorial diseases like autoimmune or oncologic disorders.40, 41 However, a large barrier to the widespread use of these techniques remains the ability to efficiently target repopulating stem cells without diminishing their engraftment potential.
The findings described here follow from an early report demonstrating that infectious measles virus is able to enter and replicate in HSPCs using the CD46 receptor.37 Recent work by Lévy et al.39 has demonstrated that pseudotyping LV with the Edmonston strain measles virus H and F glycoproteins allows for highly efficient transduction of both cytokine-stimulated and quiescent HSPCs. Our findings here confirm this observation. Moreover, serial HSPC transfer studies utilizing NSG mice cells by Lévy et al.39 demonstrated maintenance of high levels of marking after successive rounds of engraftment, consistent with LV hematopoietic stem cell transduction. Enhanced transduction efficiency by H/F LV may be related to the observed lack of colocalization of H/F LVs with the early endosome compartment, unlike VSV-G LV, which requires an acidification step for core escape into the cytoplasm. Measles virus has been proposed to fuse at the surface of the cell, thereby potentially allowing for avoidance of restriction factors or bottlenecks that limit VSV-G LVs.42 However, investigations into host factors that inhibit VSV-G and H/F LV entry and transport of the reverse-transcribed DNA to the nucleus will be required to maximize transduction of HSPCs.
This study demonstrates that modification of LV packaging cell lines proves to be a viable approach for improving vector titers and LV transduction efficacy. CD46 expression on HEK293T packaging cells was necessary for H/F-mediated cell death during the critical vector production period after transient transfection of H/F plasmids. Although we were able to demonstrate that syncytia formation and cell death significantly limit vector production, the titers of Edmonston strain H/F LV generated in CD46 null cells was still lower than VSV-G LVs and those reported for WT H/F LV.27 Differences in transduction efficiency, and therefore functional titer between Edmonston strain H/F LVs and WT measles H/F LVs, may be related to the lower binding affinity to CD46 compared with CD150 by the Edmonston strain measles H protein.43, 44 The 2-log fold higher titers we observed of Edmonston H/F LVs on Raji cells, which express CD150, support this possibility.
However, given that producer cell cytotoxicity is dramatically reduced with CD46 knockout, it might be expected that H/F LV titers could increase more than the 2-fold increase we observed. Our findings demonstrating continued H/F LV production (Figure 3A) in transfected CD46 null cells suggest that it is transfected plasmid concentration and vector protein expression rather than cytotoxicity that limit H/F LV production. Given that other cell lines have been developed for the stable expression of LVs, our results suggest that constitutive expression of measles H and F glycoproteins, along with other vector packaging plasmids by CD46 null cells, may be a viable system for further increasing production of H/F LV.45 Marino et al.28 have reported on a method for concentrating and purifying H/F LVs without the need for ultracentrifugation. The production of H/F LV in CD46 null cell lines is compatible with this approach and should allow integration into commercial-scale vector production systems.
Finally, this study highlights the need for additional research into factors that limit efficient gene delivery to HSPCs at both the packaging cell and HSPC level. The improvement in transduction efficiency to HSPCs observed with H/F LV produced in CD46 null cells compared with vector produced in WT HEK293T cells indicates that vector production techniques can have a meaningful effect on final LV transduction efficiency. The close association of HSPCs to HEK293T cell debris in confocal analysis may indicate that debris prevents receptor access by free LV, which may be a limiting factor in HSPC transduction. The removal of cell debris from the vector preparation allowed for an increase in the number and staining intensity of p24+ vesicles within cells. The observed increase in early endosome number with impure H/F LV addition in HSPCs may also indicate that non-specific uptake of cell debris components is a transduction-limiting process that is reduced with improved vector purity. Additionally, H/F LV produced in CD46 null cells was observed to be markedly less cytotoxic to CD34+ HSPCs than WT HEK293T-produced LV. Of note, this difference was observed during liquid culture of HSPCs, but not in plating frequency of CFU assays, a result potentially explained by the normalization of input cell number after transduction while setting up the CFU assay. Given that the number of hematopoietic progenitor cells engrafted in patients after myeloablative transplantation is directly related to the time to recovery of immunocompetence,46 the reduction of cytotoxicity with CD46 null H/F LV further supports clinical evaluation of this vector.
These studies demonstrate an effective and underexplored method for the improvement of gene delivery to HSPCs by LVs. The modification of packaging cell lines may prove to be a more feasible approach than vector modification, given the limited number of proteins and genetic elements available in vector systems. H/F LVs have shown an enhanced ability to transduce therapeutically relevant cells, and further studies are under way to improve LV production.
Materials and Methods
Cells
HEK293T (GTX) cells were provided as a kind gift from Kenneth Cornetta (Indiana University School of Medicine, Indianapolis, IN, USA). Cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, CA, USA), 1% penicillin and streptomycin, and 1× L-glutamine. Human cord blood was provided by the Cleveland Cord Blood Center (Cleveland, OH, USA), under approved institutional protocols and in accordance with the Declaration of Helsinki. Primary human CD34+ cells were isolated using the EasySep Human Cord Blood CD34 Positive Selection Kit according to manufacturer’s instructions (STEMCELL Technologies, Vancouver, BC, Canada). Sorted cell purity was checked by flow cytometry, and only cells with >95% CD34+ were used in further experiments. Following purification and purity check, cells were frozen in FBS/10% DMSO by liquid nitrogen until later use. THP-1 and KG-1a cells were obtained from the American Type Culture Collection. Raji cells were a kind gift from David Rawlings (Seattle Children’s Research Institute, Seattle, WA, USA). Frozen G-CSF mPB CD34+ cells were purchased from the Co-Operative Center for Excellence in Hematology at the Fred Hutchinson Cancer Research Center (Seattle, WA) under approved institutional protocols.
Plasmids
Measles Edmonston strain H (GenBank: AB583749.1) and F (GenBank: U03657.1) glycoprotein plasmids were a kind gift from Els Verhoeyen. Both glycoproteins were inserted into pCG plasmid driven by the cytomegalovirus early promoter (#51476; Addgene). The cytoplasmic tail of the H and F glycoproteins was deleted by truncation of 24 and 30 amino acids (aa), respectively, and has been previously described.17 For use in CD46 null studies, pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Feng Zhang (Addgene plasmid 62988).47
Vector Preparation
Vectors were produced by polyethylenimine (PEI)-mediated transient co-transfection of WT or CD46 null HEK293T cells. In brief, 5 × 106 cells were plated on poly-L-lysine-coated 10-cm2 tissue culture-treated dishes in 10% FBS containing DMEM. 24 hr later, media were exchanged for 3% FBS containing media before addition of plasmids. 3 μg/plate of the ubiquitin-C-driven EGFP-expressing transfer vector FG12 (#14884; Addgene), 1.95 μg/dish of the packaging constructs pMDLg/RRE (#12251; Addgene), 0.75 μg/dish of the Rev-containing construct pRSV-Rev (#12253; Addgene), and 0.9 μg/dish each of the measles H and F glycoprotein constructs or 1.8 μg/dish VSV-G envelope construct pMD2.G (#12259; Addgene) were mixed with 22.5 μL of 1 mg/mL branched PEI in 1 mL of OptiMEM (GIBCO; Life Technologies, Waltham, MA, USA). DNA:PEI mix was then vortexed and incubated for 15 min, before being added to 10-cm2 dishes of cells, dropwise. Supernatant from transfections was harvested 24 and 48 hr post-transfection and concentrated by ultracentrifugation at 50,000 × g for 2 hr 20 min on a 20% sucrose cushion. Vector was then resuspended in IMDM at 200-fold concentration and stored long term at −80°C.
Vector Functional Titer
Vector functional titer was determined by serial dilution on HEK293T cells in duplicate, with evaluation for GFP expression 72 hr post-transduction, as described previously.48 In brief, WT HEK293T cells were plated in 24-well plates at 4 × 104 cells per well and incubated overnight at 37°C. Serial dilutions of concentrated vector preps were applied in duplicate to cells and inoculated for 24 hr. Vector inoculum was then removed and replaced with fresh medium. Cells were incubated for an additional 48 hr before dissociated from plates and evaluated for GFP expression. MOI was calculated from the vector dilution that yielded %GFP+ levels between 5% and 25%.
Vector Physical Titer
Vector Gag p24 protein was quantified using an in-house-developed p24 ELISA modified from Aalto Bio HIV-1-gp120 DIY ELISA protocols. In brief, anti-HIV p24 polyclonal antibody (D7320; Aalto Bio) diluted in 100 mM NaHCO3 (pH 8.5) to 10 μg/mL was bound to high-bind ELISA plates (Corning, Costar) overnight at room temperature. After binding, plates were washed three times with Tris-buffered saline (TBS) wash buffer containing 0.05% Empigen BB detergent (30326; Sigma), then blocked with 20 mg/mL BSA diluted in 1× TBS for 30 min. Vector supernatant was lysed in 1% Empigen BB detergent at 56°C for 30 min, diluted in TBS/E/S (10% lamb serum, 1× TBS, 1% Empigen BB) and added to a 96-well plate. After 2 hr at 37°C, the wells were washed six times with wash buffer. Biotinylated anti-HIV antibody (BC 1071-BIOT; Aalto Bio) was diluted to 0.5 μg/mL in TMT/SS (1× Tween 20 and TBS [T-TBS], 2% BSA, 20% lamb serum) and applied to the washed plate, followed by 2-hr incubation at room temperature. Plates were then washed six times with wash buffer and incubated for 1 hr at room temperature with streptavidin-tagged horseradish-peroxidase-bound antibody (STAR5B; Serotec) diluted to 0.1 μg/mL in TMT/SS. After a final six washes with 0.05% Empigen BB detergent wash buffer, the plate was developed in 1-step Ultra TMB-ELISA substrate (34028; Pierce) for 10 min and quenched with 0.5M H2SO4. Absorbance at 450 nM was read on a BioTek μQuant Universal Microplate Spectrophotometer and compared with a four-parameter calculated standard curve generated by serial dilution of recombinant HIV p24 standard (Ag 6054; Aalto Bio), diluted in TBS/E/S.
Western Blotting
LV prep lysates were measured for protein concentration with Pierce BCA Protein Assay Kit (catalog [Cat] #23227) and normalized for loading. Samples were processed with LDS Sample Buffer (Cat #B0007) and Sample Reducing Agent (Cat #B0004). Lysates were run on NuPAGE 10% Bis-Tris Bolt Precast Gel from Invitrogen (Cat #NP0301BOX) and transferred to an activated polyvinylidene difluoride fluorescence (PVDF FL) membrane. Total protein in gel was visualized by InstantBlue Coomassie stain (Cat #ISB1L; Sigma-Aldrich). Total protein on membrane was visualized by Revert Total Protein Stain (Cat #926-11010; Li-Cor). Membranes were incubated with anti-p24 antibody (MAB880-A, clone 7A8.1; Millipore). Membranes were then incubated with a horseradish peroxidase (HRP) secondary or goat anti-mouse IR800 (AC2135; Azure Biosystems) and imaged using the Azure c600 imaging system (Azure Biosystems). Western blot signal quantification was conducted using AzureSpot analysis software (Azure Biosystems).
Human HSPC Transduction
Frozen cord blood-derived CD34+ cells were thawed and then cultured in IMDM supplemented with 20% BIT 9500 (STEMCELL Technologies). Cells were pre-stimulated for 24 hr with 50 ng/mL each of human interleukin-6 (IL-6) and thrombopoietin, 150 ng/mL human stem cell factor (hSCF), and 100 ng/mL FLT-3 ligand (PeproTech, Rocky Hills, NJ, USA). After pre-stimulation, 2 × 104 cells per well were transduced at indicated MOIs in the presence of 8 μg/mL polybrene. In studies involving small-molecule transduction enhancers or inhibitors, drugs were reconstituted in DMSO first, then diluted in respective media. If used, cells were pre-treated with small molecules for 4 hr before addition of vectors, with the equivalent concentration of DMSO as the respective vehicle control. All small molecules and vectors were washed out of culture 24 hr later. After removal of vector inoculum, cells were cultured at growth phase for 7 days in IMDM supplemented with 10% FBS (Omega Scientific, Tarzana, CA, USA), 50 ng/mL interleukin-3 (IL-3) and IL-6, and 100 ng/mL hSCF. Expression levels of EGFP were evaluated by flow cytometry 7 days after vector addition.
Frozen G-CSF mPB CD34+ cells were thawed and then cultured in GMP-Stem Cell Growth Medium (SCGM; CellGenix, Freiburg, Germany). Cells were pre-stimulated for 48 hr with 100 ng/mL each of human thrombopoietin (TPO), hSCF, and FLT-3 ligand (PeproTech, Rocky Hills, NJ, USA). After pre-stimulation, cells were transduced as above for UCB CD34+ HSPCs. After removal of vector inoculum, cells were cultured at growth phase for 7 days in SCGM supplemented with 100 ng/mL each of TPO, hSCF, FLT-3, IL-3, and IL-6. Expression levels of EGFP were evaluated by flow cytometry 7 days after vector addition.
Cell Viability and Growth, and CFU Assessment
Human cord blood CD34+ cells were transduced with FG12-UbiC-EGFP vectors, pseudotyped with H/F or VSV-G envelopes. Cells were then washed and seeded, in duplicate, in serum-containing expansion medium for up to 14 days. Cells were resuspended in FACS buffer containing trypan blue, and viability was determined by hemocytometer as the percentage of dye excluding cells. For growth assessment, live cells from each sample, in duplicate, were counted on a hemocytometer, and cell numbers were normalized to mock-transduced sample. CFU assessment of CD34+ cells and EGFP assessment was previously described.48 MethoCult H4034 media for CFU assessment was purchased from STEMCELL Technologies.
Flow Cytometry
Flow cytometry analysis was performed on either BD Biosciences LSR II or ACEA Biosciences Novocyte flow cytometers equipped with UV, violet, blue, and red lasers. Doublets and dead cells were excluded by forward scatter (FSC) and side scatter (SSC) and propidium iodide stain, and non-specific binding was avoided by preincubation with normal mouse serum (Thermo Scientific) and human TruStain FcX (BioLegend) Fc block. Antibodies used for flow cytometric and cell sorting analysis were CD46-allophycocyanin (APC; E4.3; BD Biosciences), CD34-fluorescein isothiocyanate (FITC; 581; BD Biosciences), CD38-APC (HIT2; BD Biosciences), and CD46-APC (HI30; BD Biosciences), and live/dead cell stain for transduction studies was LIVE/DEAD Fixable Aqua Dead Cell Stain Kit, for 405 nm excitation (Cat #L34957; Invitrogen).
Confocal Microscopy
Cord blood-derived CD34+ HSPCs were isolated and pre-stimulated with cytokines as per transduction protocols above. 1 × 105 CD34+ cells were transduced per vector condition. Vector was incubated with cells for either 15 min or 1 hr in 48-well plates, before cells were washed three times in 2% FBS + PBS buffer to remove surface-bound virus. After washing, cells were spun via cytospin onto positively charged confocal grade microscopy slides at 450 × g for 5 min. After slide plating, cells were fixed in 3.7% confocal grade paraformaldehyde (50-980-487; Fisher Scientific) for 8 min and washed three times with confocal wash buffer (PBS + 0.1% saponin and 1% BSA). After fixing, cells were stained overnight at 4°C with Mouse Anti-EEA1 (14/EEA1; BD Biosciences), diluted 1:200 in confocal wash buffer. After incubation, slides were again washed three times with wash buffer and incubated for 1 hr with Alexa Fluor 488 Donkey Anti-Mouse antibody (H+L, A-21202; Thermo Fisher Scientific) at room temperature. Finally, cells were incubated with Mouse anti-HIV-1 core antigen-PE (KC57-RD1, 6604667; Beckman Coulter) antibody at 1:50 dilution overnight at 4°C. After antibody staining, slides were stained for 30 min at room temperature with Hoechst stain at 1:2,000, before mounting with ProLong Gold Antifade Reagent (P36930; Thermo Fisher).
Confocal microscopy analysis of vector and endosomal markers was performed using a Zeiss 710 laser-scanning confocal microscope at 64× oil-immersion magnification at a 2-fold digital zoom with a 1,024 × 1,024 field size. For establishing voltage and aperture for confocal microscopy, snap images were captured for each channel using control slides stained only with secondary antibody or no vector added. z stack depth was set by nuclei imaging, with images collected in 16 slice intervals, 0.2 μm per image, collected at ∼1 image per minute.
z stacks were analyzed using Imaris Software (Bitplane) using the ImarisCell analysis module, quantifying number, volume, and intensity of EEA1 and p24+ vesicles. Cellular boundaries were selected and Pearson’s correlation coefficients were calculated across EEA1 and p24 channels using the Imaris Colocalization module, with thresholding set by unstained or secondary antibody-stained control images.
CRISPR/Cas9 Knockout of CD46 and Cell Sorting
PAM sequences specific to exon 1 of the human CD46 gene were predicted using the web-based software tool described in Hsu et al.30 to evaluate guide specificity and predict potential off-target modification. The top three sequences were 5′-CACCGCAATTGTGTCGCTGCCATCG-3′ (guide RNA [gRNA]1), 5′- CACCGCGATTTCCAGTAGTCGAAAA-3′ (gRNA2), and 5′-CACCGTTTAAAGGATCCCGTATATA-3′ (gRNA3). Oligonucleotides were designed containing the recognition sequence for BbsI. Oligonucleotides were annealed, cut with BbsI, and then ligated into BbsI cut pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid.
CD46 expression on WT HEK293T GTX cells was evaluated before transfection by flow cytometry and found to be >99% CD46+. gRNA-containing PX459 plasmids were transfected by PEI transfection with 7.5 μg DNA/dish with 22.5 μL of 1 mg/mL PEI as above. One day after transfection, cells were selected in 1.5 μg/mL puromycin for 3 days before cell sorting for CD46 expression.
Cell Sorting
Puromycin selected CRISPR/Cas9-transfected cells were stained for CD46 expression (CD46-APC, E4.3; BD Biosciences), then sorted for CD46-negative cells using a FACSAria IIU (BD Bioscience). Cells were analyzed after 20 passages for continued lack of CD46 expression, with stable null phenotype observed for >6 months.
VCN Quantification
Total DNA was extracted from flash-frozen pelleted cells using QIAGEN DNeasy Blood and Tissue Kit (Cat #69506) and treated with DpnI (New England Biolabs) for 2 hr to eliminate residual packaging plasmid DNA if present. qPCR was carried out according to a previously published protocol with probes specific to integrated U5 to Ψ regions of the HIV genome (primers MH531, MH532, LTR-P).49 Reactions were run on a Roche LightCycler 480, using LightCycler 480 Probes Master (Roche) as previously described.50
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). Numbers described in the Results section and figures with bar graphs and dot plots indicate mean ± SD. All data presented in bar graphs in Figures 4 and S3 were analyzed using 2-way ANOVA with Tukey’s multiple comparisons test. Dot plots in Figures 5 and 6 were analyzed using Kruskal-Wallis test with Dunn’s multiple comparison correction. Data presented as bar graphs in Figure 5 were analyzed by unpaired two-tailed Student’s t test.
Author Contributions
B.E.T., E.V., and S.O. conceived the project. B.E.T., S.O., P.M., and N.D.T. directed the studies. S.O., C.X.C., E.S., and O.G. performed experiments and analyzed molecular data. P.M. and N.D.T. provided assistance and advice on gene knockout studies. E.V. gave important feedback regarding measles vector production. S.O. and B.E.T. wrote the manuscript. All authors edited and approved the manuscript.
Conflicts of Interest
B.E.T. was a consultant for Rocket Pharma. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Dr. Mary Laughlin and the Cleveland Cord Blood Center (Cleveland, OH, USA) for providing human cord blood. These studies were supported by the NIH (grants 1R01HL116221, 2U54GM103368, and 2P30AI036214 to B.E.T.). N.D.T. was supported by NIH/NIAID grant 5T32AI007354. S.O. was supported by NIH/NHLBI grant 1F30HL137563. This is publication number 29790 from The Scripps Research Institute.
Footnotes
Supplemental Information includes three figures and can be found with this article online at https://doi.org/10.1016/j.omtm.2018.11.006.
Supplemental Information
References
- 1.Holt N., Wang J., Kim K., Friedman G., Wang X., Taupin V., Crooks G.M., Kohn D.B., Gregory P.D., Holmes M.C., Cannon P.M. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 3.Negre O., Eggimann A.-V., Beuzard Y., Ribeil J.-A., Bourget P., Borwornpinyo S., Hongeng S., Hacein-Bey S., Cavazzana M., Leboulch P., Payen E. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-globin gene. Hum. Gene Ther. 2016;27:148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 5.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 7.Naldini L., Trono D., Verma I.M. Lentiviral vectors, two decades later. Science. 2016;353:1101–1102. doi: 10.1126/science.aah6192. [DOI] [PubMed] [Google Scholar]
- 8.Naldini L., Blömer U., Gallay P., Ory D., Mulligan R., Gage F.H., Verma I.M., Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 9.Finkelshtein D., Werman A., Novick D., Barak S., Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Blanc I., Luyet P.-P., Pons V., Ferguson C., Emans N., Petiot A., Mayran N., Demaurex N., Fauré J., Sadoul R. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 2005;7:653–664. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim I.S., Jenni S., Stanifer M.L., Roth E., Whelan S.P.J., van Oijen A.M., Harrison S.C. Mechanism of membrane fusion induced by vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA. 2017;114:E28–E36. doi: 10.1073/pnas.1618883114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amirache F., Lévy C., Costa C., Mangeot P.E., Torbett B.E., Wang C.X., Nègre D., Cosset F.L., Verhoeyen E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood. 2014;123:1422–1424. doi: 10.1182/blood-2013-11-540641. [DOI] [PubMed] [Google Scholar]
- 13.Girard-Gagnepain A., Amirache F., Costa C., Lévy C., Frecha C., Fusil F., Nègre D., Lavillette D., Cosset F.L., Verhoeyen E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood. 2014;124:1221–1231. doi: 10.1182/blood-2014-02-558163. [DOI] [PubMed] [Google Scholar]
- 14.Sandrin V., Boson B., Salmon P., Gay W., Nègre D., Le Grand R., Trono D., Cosset F.L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- 15.Witting S.R., Vallanda P., Gamble A.L. Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther. 2013;20:997–1005. doi: 10.1038/gt.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu S., Mohan Kumar D., Sax C., Schuler C., Akkina R. Pseudotyping of lentiviral vector with novel vesiculovirus envelope glycoproteins derived from Chandipura and Piry viruses. Virology. 2016;488:162–168. doi: 10.1016/j.virol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frecha C., Costa C., Nègre D., Gauthier E., Russell S.J., Cosset F.-L., Verhoeyen E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood. 2008;112:4843–4852. doi: 10.1182/blood-2008-05-155945. [DOI] [PubMed] [Google Scholar]
- 18.Frecha C., Lévy C., Costa C., Nègre D., Amirache F., Buckland R., Russell S.J., Cosset F.L., Verhoeyen E. Measles virus glycoprotein-pseudotyped lentiviral vector-mediated gene transfer into quiescent lymphocytes requires binding to both SLAM and CD46 entry receptors. J. Virol. 2011;85:5975–5985. doi: 10.1128/JVI.00324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humbert J.M., Frecha C., Amirache Bouafia F., N’Guyen T.H., Boni S., Cosset F.L., Verhoeyen E., Halary F. Measles virus glycoprotein-pseudotyped lentiviral vectors are highly superior to vesicular stomatitis virus G pseudotypes for genetic modification of monocyte-derived dendritic cells. J. Virol. 2012;86:5192–5203. doi: 10.1128/JVI.06283-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lévy C., Amirache F., Costa C., Frecha C., Muller C.P., Kweder H., Buckland R., Cosset F.L., Verhoeyen E. Lentiviral vectors displaying modified measles virus gp overcome pre-existing immunity in in vivo-like transduction of human T and B cells. Mol. Ther. 2012;20:1699–1712. doi: 10.1038/mt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Funke S., Maisner A., Mühlebach M.D., Koehl U., Grez M., Cattaneo R., Cichutek K., Buchholz C.J. Targeted cell entry of lentiviral vectors. Mol. Ther. 2008;16:1427–1436. doi: 10.1038/mt.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Münch R.C., Mühlebach M.D., Schaser T., Kneissl S., Jost C., Plückthun A., Cichutek K., Buchholz C.J. DARPins: an efficient targeting domain for lentiviral vectors. Mol. Ther. 2011;19:686–693. doi: 10.1038/mt.2010.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mühlebach M.D., Mateo M., Sinn P.L., Prüfer S., Uhlig K.M., Leonard V.H.J., Navaratnarajah C.K., Frenzke M., Wong X.X., Sawatsky B. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011;480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatsuo H., Ono N., Tanaka K., Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 25.Dörig R.E., Marcil A., Chopra A., Richardson C.D. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 26.Singh B.K., Hornick A.L., Krishnamurthy S., Locke A.C., Mendoza C.A., Mateo M., Miller-Hunt C.L., Cattaneo R., Sinn P.L. The Nectin-4/Afadin protein complex and intercellular membrane pores contribute to rapid spread of measles virus in primary human airway epithelia. J. Virol. 2015;89:7089–7096. doi: 10.1128/JVI.00821-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funke S., Schneider I.C., Glaser S., Mühlebach M.D., Moritz T., Cattaneo R., Cichutek K., Buchholz C.J. Pseudotyping lentiviral vectors with the wild-type measles virus glycoproteins improves titer and selectivity. Gene Ther. 2009;16:700–705. doi: 10.1038/gt.2009.11. [DOI] [PubMed] [Google Scholar]
- 28.Marino M.P., Panigaj M., Ou W., Manirarora J., Wei C.H., Reiser J. A scalable method to concentrate lentiviral vectors pseudotyped with measles virus glycoproteins. Gene Ther. 2015;22:280–285. doi: 10.1038/gt.2014.125. [DOI] [PubMed] [Google Scholar]
- 29.Takeuchi K., Miyajima N., Nagata N., Takeda M., Tashiro M. Wild-type measles virus induces large syncytium formation in primary human small airway epithelial cells by a SLAM(CD150)-independent mechanism. Virus Res. 2003;94:11–16. doi: 10.1016/s0168-1702(03)00117-5. [DOI] [PubMed] [Google Scholar]
- 30.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croop J.M., Cooper R., Seshadri R., Fernandez C., Graves V., Kreissman S., Smith F.O., Cornetta K., Williams D.A., Abonour R. Large-scale mobilization and isolation of CD34+ cells from normal donors. Bone Marrow Transplant. 2000;26:1271–1279. doi: 10.1038/sj.bmt.1702720. [DOI] [PubMed] [Google Scholar]
- 32.Pollok K.E., van Der Loo J.C., Cooper R.J., Hartwell J.R., Miles K.R., Breese R., Williams E.P., Montel A., Seshadri R., Hanenberg H., Williams D.A. Differential transduction efficiency of SCID-repopulating cells derived from umbilical cord blood and granulocyte colony-stimulating factor-mobilized peripheral blood. Hum. Gene Ther. 2001;12:2095–2108. doi: 10.1089/10430340152677430. [DOI] [PubMed] [Google Scholar]
- 33.Gonçalves-Carneiro D., McKeating J.A., Bailey D. The measles virus receptor SLAMF1 can mediate particle endocytosis. J. Virol. 2017;91:e02255-16. doi: 10.1128/JVI.02255-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun X., Roth S.L., Bialecki M.A., Whittaker G.R. Internalization and fusion mechanism of vesicular stomatitis virus and related rhabdoviruses. Future Virol. 2010;5:85–96. doi: 10.2217/FVL.09.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heffner G.C., Bonner M., Christiansen L., Pierciey F.J., Campbell D., Smurnyy Y., Zhang W., Hamel A., Shaw S., Lewis G. Prostaglandin E2 increases lentiviral vector transduction efficiency of adult human hematopoietic stem and progenitor cells. Mol. Ther. 2018;26:320–328. doi: 10.1016/j.ymthe.2017.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gillespie E.J., Ho C.L., Balaji K., Clemens D.L., Deng G., Wang Y.E., Elsaesser H.J., Tamilselvam B., Gargi A., Dixon S.D. Selective inhibitor of endosomal trafficking pathways exploited by multiple toxins and viruses. Proc. Natl. Acad. Sci. USA. 2013;110:E4904–E4912. doi: 10.1073/pnas.1302334110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manchester M., Smith K.A., Eto D.S., Perkin H.B., Torbett B.E. Targeting and hematopoietic suppression of human CD34+ cells by measles virus. J. Virol. 2002;76:6636–6642. doi: 10.1128/JVI.76.13.6636-6642.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sintes J., Romero X., Marin P., Terhorst C., Engel P. Differential expression of CD150 (SLAM) family receptors by human hematopoietic stem and progenitor cells. Exp. Hematol. 2008;36:1199–1204. doi: 10.1016/j.exphem.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lévy C., Amirache F., Girard-Gagnepain A., Frecha C., Roman-Rodríguez F.J., Bernadin O., Costa C., Nègre D., Gutierrez-Guerrero A., Vranckx L.S. Measles virus envelope pseudotyped lentiviral vectors transduce quiescent human HSCs at an efficiency without precedent. Blood Adv. 2017;1:2088–2104. doi: 10.1182/bloodadvances.2017007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg S.A., Restifo N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 42.Rasbach A., Abel T., Münch R.C., Boller K., Schneider-Schaulies J., Buchholz C.J. The receptor attachment function of measles virus hemagglutinin can be replaced with an autonomous protein that binds Her2/neu while maintaining its fusion-helper function. J. Virol. 2013;87:6246–6256. doi: 10.1128/JVI.03298-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massé N., Barrett T., Muller C.P., Wild T.F., Buckland R. Identification of a second major site for CD46 binding in the hemagglutinin protein from a laboratory strain of measles virus (MV): potential consequences for wild-type MV infection. J. Virol. 2002;76:13034–13038. doi: 10.1128/JVI.76.24.13034-13038.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santiago C., Björling E., Stehle T., Casasnovas J.M. Distinct kinetics for binding of the CD46 and SLAM receptors to overlapping sites in the measles virus hemagglutinin protein. J. Biol. Chem. 2002;277:32294–32301. doi: 10.1074/jbc.M202973200. [DOI] [PubMed] [Google Scholar]
- 45.Schweizer M., Merten O.W. Large-scale production means for the manufacturing of lentiviral vectors. Curr. Gene Ther. 2010;10:474–486. doi: 10.2174/156652310793797748. [DOI] [PubMed] [Google Scholar]
- 46.Le Blanc K., Barrett A.J., Schaffer M., Hägglund H., Ljungman P., Ringdén O., Remberger M. Lymphocyte recovery is a major determinant of outcome after matched unrelated myeloablative transplantation for myelogenous malignancies. Biol. Blood Marrow Transplant. 2009;15:1108–1115. doi: 10.1016/j.bbmt.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyoshi H., Smith K.A., Mosier D.E., Verma I.M., Torbett B.E. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 49.Mbisa J.L., Delviks-Frankenberry K.A., Thomas J.A., Gorelick R.J., Pathak V.K. Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol. Biol. 2009;485:55–72. doi: 10.1007/978-1-59745-170-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C.X., Sather B.D., Wang X., Adair J., Khan I., Singh S., Lang S., Adams A., Curinga G., Kiem H.P. Rapamycin relieves lentiviral vector transduction resistance in human and mouse hematopoietic stem cells. Blood. 2014;124:913–923. doi: 10.1182/blood-2013-12-546218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.