

Abstract
Evofosfamide is a cytotoxic small‐molecule prodrug preferentially activated under hypoxic conditions. The cytotoxicity of evofosfamide impacted the generation of in vitro drug‐drug interaction (DDI) data, especially in vitro induction results. Therefore, a novel physiologically based pharmacokinetic (PBPK) approach was used, which involved available in vitro and clinical data of evofosfamide and combined it with induction data from the prototypical cytochrome P450 (CYP)3A inducer rifampicin. The area under the concentration‐time curve (AUC) ratios of midazolam were above 0.80, indicating that induction of CYP3A by evofosfamide administered weekly is unlikely to occur in humans. Moreover, static and PBPK modeling showed no clinically relevant inhibition via CYP2B6, CYP2D6, and CYP3A4. In conclusion, PBPK models were used to supplement in vitro information of a cytotoxic compound. This approach may set a precedent for future studies of cytotoxic drugs, potentially reducing the need for clinical DDI studies and providing more confidence in the clinical use of approved cytotoxic compounds for which DDI information is sparse.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Evofosfamide is a cytotoxic small‐molecule prodrug in development. PBPK and static modeling are established tools to estimate the DDI potential of new molecules based on in vitro data.
WHAT QUESTION DID THIS STUDY ADDRESS?
Cytotoxicity limited the generation of in vitro induction results. This study aimed to develop a PBPK approach by which the induction potential could still be addressed.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
A novel PBPK approach was used, which involved available in vitro and clinical data of evofosfamide and combined it with induction data from the prototypical strong CYP3A inducer rifampicin. The approach was successful in indicating that induction of CYP3A by evofosfamide administered weekly is unlikely to occur in humans. Potential DDI liabilities as inhibitor were also excluded.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
This approach may set a precedent for induction evaluations of other cytotoxic drugs.
Evofosfamide ([1‐methyl‐2‐nitro‐1H‐imidazole‐5‐yl] methyl N, N’‐bis [2‐bromoethyl] diamidophosphate, previously known as TH‐302) is a small‐molecule drug preferentially activated under hypoxic conditions. This leads to the formation of bromo‐isophosphoramide mustard, a potent cytotoxic DNA‐alkylating agent that causes DNA cross‐linking resulting in the inhibition of DNA replication and transcription, cell‐cycle arrest, and cell death in hypoxic tumor areas.1 Hypoxia is a prevalent feature of cancer and is associated with tumor invasiveness and poor prognosis, whereas it is rarely observed in normal tissues.2, 3 Although evofosfamide did not meet its primary efficacy end point in two recent phase III trials in soft tissue sarcoma4 and pancreatic adenocarcinoma,5 further development of the drug in more select treatment settings is ongoing.
In oncology, pharmacokinetic drug‐drug interactions (DDIs), which occur when one drug affects (inhibits or induces) the metabolism of one or more co‐administered drugs, are under‐researched.6 Polypharmacy leading to an increased potential for DDI is highly relevant for patients with cancer who are normally treated with multiple drugs, both against the malignant disease and in the management of concomitant conditions.7, 8, 9 Hence, knowledge about DDI is critical for reducing adverse drug reactions that may cause delays or even failure of therapeutics in drug development.10, 11, 12 As a result, the US Food and Drug Administration and the European Medicines Agency agencies have updated their guidelines,6, 13, 14, 15, 16 which require the DDI potential to be investigated early during the development of new molecular entities.
Cytochrome P450 (CYP) isozymes are implicated in many clinically relevant DDIs as they are involved in the metabolism of ~60% of marketed small molecule drugs. Reversible and irreversible inhibition17, 18, 19 and induction of CYPs20 can have serious clinical consequences, such as a reduction in the therapeutic margin of co‐administered drugs or respective loss of efficacy.
Cytotoxicity especially influences the in vitro assessment of the induction potential on CYPs. The prolonged exposure of human hepatocyte cultures to the cytotoxic drug, which is necessary to evaluate its impact on gene transcription and protein expression, limits the concentration range in which it can be used in these cell‐based systems. In contrast, assessment of the inhibition potential involves use of human liver microsomes or recombinant enzyme preparations, which are not dependent on cellular viability.
Numerous mathematical models have been developed to predict clinical DDI from in vitro data.21, 22, 23, 24 The simplest model is one in which a static score of degree of induction is calculated from the average plasma concentration of an inducer using in vitro half‐maximal effective concentration and maximum effect (Emax) estimates.25, 26, 27 The potential for induction‐based DDIs is then predicted based upon the extent of clearance associated with each induced enzyme. In contrast, dynamic models consider differences in the levels of enzyme activity and, thus, clearance of a drug as a result of increased enzyme synthesis in an inducer‐concentration dependent manner.28, 29, 30
For cytotoxic drugs, there are often substantial knowledge gaps regarding their DDI potential. This is unfortunate, because this information is particularly important for drugs with a narrow therapeutic range. DDI studies for cytotoxic anticancer drugs are typically conducted in patients with cancer because traditional chemotherapies are too toxic to be studied in healthy volunteers and, thus, face many challenges, such as restrictions on the study design or higher variability of the data.31, 32
This article describes the development of a physiologically based pharmacokinetic (PBPK) model33 using interaction parameters obtained from in vitro studies to assess the potential clinical effects on CYPs caused by evofosfamide as a perpetrator. The limitations using in vitro data due to cytotoxicity of evofosfamide are overcome by a novel approach that creates a worst‐case scenario for CYP induction, which is reported hereinafter. This approach combines the lower limit for half‐maximal induction (IndC50) with the maximum fold induction of rifampicin, a recognized very strong inducer of CYP3A.17, 34 To our knowledge, this is the first time such an approach has been used to evaluate the DDI of a cytotoxic drug.
Methods
In vitro measurements of inhibition and induction parameters
Standard methods were used to determine the log of octanol/water partition coefficient (logP), the blood‐plasma concentration ratio, and the fraction unbound in plasma. The logP was determined by shaking 4 mL of octanol‐saturated water containing evofosfamide (~26 mg/mL) with 4 mL of water‐saturated octanol (23°C, 1 hour) and subsequent quantification of evofosfamide via high‐performance liquid chromatography in the two separate phases. The blood‐plasma‐ratio in human blood (fresh, pH‐stabilized) was investigated by centrifugation following incubation with test item (0.2 and 2 μM; 0.5 hours at 37°C) and subsequent quantification by liquid chromatography coupled with tandem mass spectrometry (LC‐MS/MS) in the separated plasma. Plasma‐protein‐binding in fresh human plasma was determined by ultrafiltration following incubation with test item (5, 10, and 75 μg/mL; 0.5 hours at 37°C) and subsequent LC‐MS/MS analysis in the ultrafiltrate and the originally spiked plasma.
Direct inhibition of several CYP enzymes by evofosfamide was characterized by determination of inhibition constants (Ki). The CYPs of interest and their probe substrates reactions included CYP2D6 (via dextromethorphan O‐demethylation), and CYP3A4/5 (via testosterone 6β‐hydroxylation, nifedipine oxidation, and midazolam 1′‐hydroxylation). Evofosfamide (50–5,000 μM) was incubated with probe substrates (at five different concentrations) and pooled human liver microsomes. The reaction was stopped after 5 minutes and probe substrate specific metabolites were determined using LC‐MS/MS to obtain formation rates. These formation rates at all inhibitor concentrations were fitted according to Michaelis‐Menten, to obtain the Ki value.
The time‐dependent inhibition of CYP2B6 and CYP3A4/5 was quantified by measuring inactivation rate constants (kinact) for CYP2B6 (efavirenz 8‐hydroxylation) and CYP3A4/5 (midazolam 1′‐hydroxylation). Evofosfamide was preincubated with pooled human liver microsomes and a nicotinamide adenine dinucleotide phosphate‐generating system over 45 and 30 minutes for CYP2B6 and CYP3A4/5, respectively. Following preincubated, an aliquot of the mixture was transferred to a second tube containing the marker substrate and a nicotinamide adenine dinucleotide phosphate‐generating system resulting in a 10‐fold dilution of the inhibitor, which minimized any direct inhibitory effect. The residual CYP2B6 and CYP3A4/5 enzymatic activities were then quantified. Experiments were repeated for different inhibitor concentrations. These data were evaluated in a two‐step method. First, the apparent slope of inactivation was determined for each inhibitor concentration and then kinact and Kapp (i.e., the maximum inactivation rate and the inhibition constant corresponding to the concentration causing 50% kinact), were determined from the dependency of the apparent slope from the inhibitor concentrations.
Primary cultures of cryopreserved human hepatocytes were used to measure the induction potential of evofosfamide. Cells from white donors (n = 3 independent evaluations) were treated once daily for 3 days with evofosfamide (1, 2.5, and 10 μM), vehicle control, and negative and positive controls (25 μM flumazenil and 20 μM rifampicin). Cells were lysed and total RNA harvested, followed by reverse transcription polymerase chain reaction analysis to quantify the effect of evofosfamide on CYP3A4 mRNA levels. Cell integrity was assessed by measuring release of lactate dehydrogenase into the culture medium, and by microscopic evaluation of cell morphology and confluency. The lactate dehydrogenase release was comparable to control incubations at 1 and 2.5 μM evofosfamide, but higher at 10 μM, indicating that cell integrity was maintained up to 2.5 μM. Microscopic evaluations showed no signs of toxicity in two of the three donor cells at 1 μM on day 3, whereas some signs of toxicity were observed in the third preparation. Toxicity was observed in all donor cells at 2.5 and 10 μM incubations. Therefore, the results at 2.5 and 10 μM were considered unreliable and, hence, 1 μM was defined as the highest evaluable concentration. Evofosfamide (1 μM) treatment of the hepatocytes for three consecutive days caused no increase in CYP3A4 mRNA in any of the donors measured.
PBPK modeling
Initially, a compound model for evofosfamide describing its pharmacokinetics (PKs) was developed and validated with clinical data from the phase I clinical trial of evofosfamide.35 The compound model was then developed further using an inhibitor model by inclusion of direct and/or mechanism‐based inhibition processes for CYP2D6, 2B6, or 3A4/5, as applicable (Table 1). A second evofosfamide model was developed by combining the evofosfamide PK model with the induction processes and the respective parameters, allowing separate DDI evaluations for both inhibition and induction.
Table 1.
Input parameter values used for evofosfamide
| Compound model parameters used for inhibition and induction | ||
|---|---|---|
| Parameter | Value | Method |
| Mol. wt. (g/mol) | 449 | |
| logP | 0.92 | In vitro |
| fu,p | 0.455 | In vitro |
| B:P ratio | 1.61 | In vitro |
| Vss (L/kg) | 0.43 (CV = 79%) | In vivo |
| CLIV (L/h) | 63.6 (CV = 54%) | In vivo |
| Hepatic intracellular accumulation (fold) | 1–4 | In vitro |
| Parameters used in the inhibition models only | ||
|---|---|---|
| Parameter | Value (± SE) | Method |
| For CYP3A4 and CYP3A5 (midazolam) | ||
| Ki (μM) (direct inh.) | 141 (± 18) | In vitro |
| kinact (h−1) (MBI) | 1.5 (± 0.18) | In vitro |
| Kapp (μM) (MBI) | 370 (± 90) | In vitro |
| For CYP2D6 | ||
| Ki (μM) (direct inh.) | 85.3 (± 8.5) | In vitro |
| For CYP2B6 | ||
| kinact (h−1) (MBI) | 1.92 (± 0.66) | In vitro |
| Kapp (μM) (MBI) | 3200 (± 2200) | In vitro |
| Parameters used in the induction models only | ||
|---|---|---|
| Parameter | Value (± SE) | Method |
| For CYP3A4 and CYP3A5 | ||
| IndC50 (μM) | 2.5 (CV = 30%) | In vitro, see METHODS |
| Indmax (fold) | 8.0 (CV = 30%) | Rifampicin in Simcyp Library |
B:P ratio, blood to plasma concentration ratio; CLIV, clearance after intravenous administration; CV, coefficient of variation; CYP, cytochrome P450 enzyme; fu,p, fraction unbound in plasma; IndC50, concentration that gives half maximal fold induction; Indmax, maximal fold induction; Kapp, concentration at 50% kinact; kdeg, degradation rate constant; Ki, direct inhibition constant; kinact, maximal inactivation rate; logP, octanol‐water partition coefficient; MBI, mechanism‐based inhibition; mol. wt., molecular weight; Vss, volume of distribution at steady state.
Model development and simulations were performed with the Simcyp population‐based simulator version 13 release 2 (Simcyp, Certara) using the North European white population. The age range of 18–80 years was set with an equal men/women split to resemble the targeted patient population.
PBPK compound models
The compound model development focused initially on the description of the concentration‐time profiles for evofosfamide. Minimal models rather than detailed mechanistic descriptions (i.e., simpler models), were applied to describe the distribution and elimination, as these models are sufficient for evaluations of the perpetrator potential. Model input parameters included compound specific in vitro parameters (Table 1). The distribution and elimination of evofosfamide were described based on clinical data from the initial phase I monotherapy dose‐escalation study in patients with solid tumors (NCT00495144) using the minimal PBPK model (Table 1). Due to its short elimination half‐life of 0.81 hours,35 no accumulation of evofosfamide was observed upon repeat once weekly administration. Therefore, multiple‐dose and single‐dose concentration‐time profiles were combined to construct the PBPK model (Figure 1). Overlay plots demonstrate that the compound model predicts the PK observed in this study as expected. The compound model for evofosfamide was further qualified based on a second independent study that included evofosfamide at the target dose level of 340 mg/m2 in combination with gemcitabine, docetaxel, or pemetrexed in patients with advanced solid tumors (NCT00743379). Note S1 provides the overlay plots relating to the two studies, as well as additional details on the qualification of the Simcyp simulator for the characterization of drug interactions.
Figure 1.
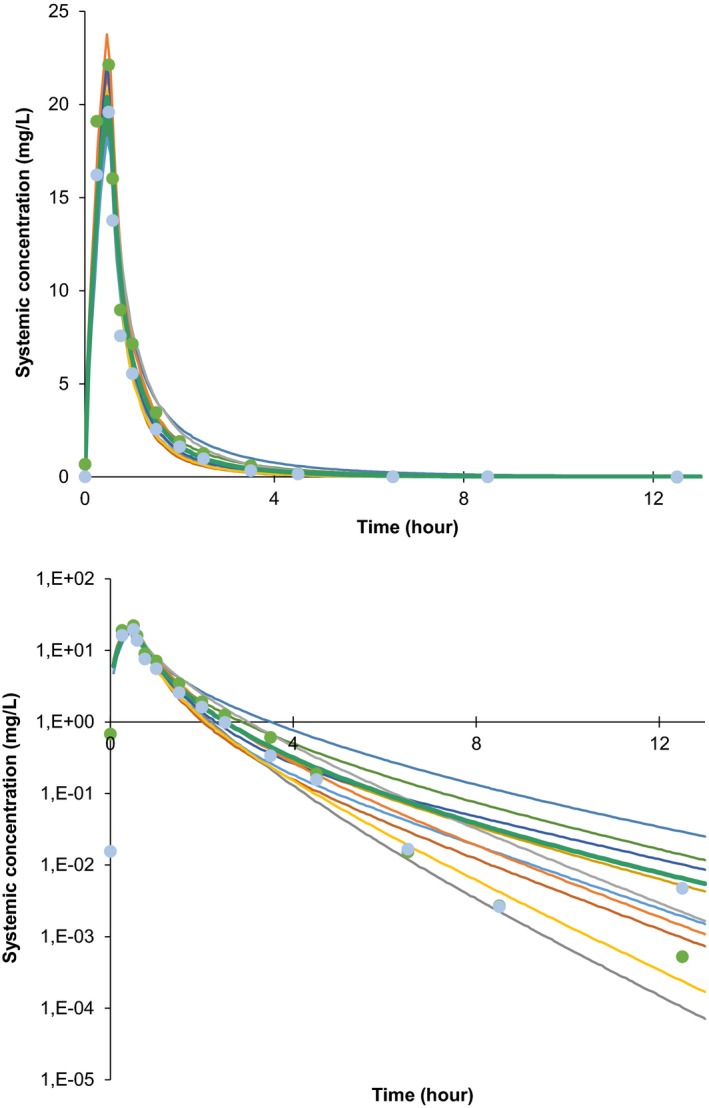
Final compound model simulations: Simulated (lines) and observed (dots) mean plasma concentration‐time profiles of evofosfamide given as intravenous infusion of 480 mg/m2 over 30 minutes. Ten trials of 22 subjects each were simulated; the lines reflect the means for each trial. The two sets of observed clinical data plotted reflect the measured values after the first (green dots) and third infusion (light blue dots), respectively. The graphs show the data on linear (top) and semi‐logarithmic scale (bottom). For comparison, 2.5 μM (used as half‐maximal induction of cytochrome P450 (CYP)3A induction) corresponds to 1.12 mg/L.
Given evofosfamide's cytotoxicity, a “hybrid” model was constructed using a lower limit for the drug's IndC50 and rifampicin's maximum fold induction (Indmax). The in vitro viability of the hepatocytes was monitored and found to be maintained up to evofosfamide concentrations of 1 μM, but not at higher concentrations. No induction was observed at this concentration. Based on these data, an IndC50 of 2.5 μM was conservatively assumed for evofosfamide. The compound model for induction combined the IndC50 with the Indmax of the prototypical strong CYP3A inducer, rifampicin, which was taken from the Simcyp compound library. Thus, a “hybrid” model for CYP3A induction was developed for evofosfamide using the Indmax value of rifampicin. The hybrid model underestimates the CYP3A IndC50, and likely overestimates the Indmax value for evofosfamide. Therefore, this approach is considered to yield a worst‐case scenario when simulating the potential of evofosfamide to induce CYP3A.
PBPK interaction models and simulations
The PKs and oral doses of dextromethorphan, bupropion, and midazolam, as sensitive probe substrates for CYP2D6, 2B6, and 3A, respectively, were described using the Simcyp compound library files “SV‐Dextromethorphan,” “SV‐Bupropion,” and “Sim‐Midazolam,” respectively. The target posology of evofosfamide was 300 mg/m2 of drug on days 1 and 8 of a 21‐day cycle in soft tissue sarcoma, and 340 mg/m2 evofosfamide on days 1, 8, and 15 of a 28‐day cycle for pancreatic cancer. Simulations were performed at the higher dose of 340 mg/m2 evofosfamide, and weekly dosing up to day 15. Evofosfamide infusion time of 30 minutes and concomitant administration of the oral probe substrates at the start of infusion were used in the simulations.
The perpetrator properties of evofosfamide regarding CYP3A were applied to both CYP3A4 and CYP3A5. Interactions solely based on direct inhibition were investigated by simulating trials lasting for 24 hours, those including induction or time‐dependent inhibition were described using simulations extended to 360 hours. The area under the concentration‐time curve up to 24 hours (AUC0–24) and maximum concentration (Cmax) of the probe substrates after administration on trial day 1 or 15 were used for evaluations, respectively. DDI ratios across the simulated populations of 100 subjects (10 trials with 10 subjects each) were summarized using the geometric mean and its 95% confidence interval (CI). Maximum impact of induction was observed with midazolam dosing 3 hours after the start of evofosfamide infusion and, therefore, this setting was used in respective sensitivity analyses. Details of the base model are summarized in Note S2 .
PBPK sensitivity analyses
Sensitivity analyses were performed for the in vitro data characterizing the inhibition and induction processes covering at least a twofold change in parameters. A twofold change was selected to represent the typical variability we have experienced with these in vitro systems. A more conservative approach was used for any model parameter for which the SD exceeded 50% of the mean (Kapp for mechanism‐based inactivation of CYP2B6). For these model parameters, the mean minus one SD was used as the lower end instead of 50% of the mean (i.e., the value indicating stronger inhibition was included in the analysis). Hepatocyte uptake experiments found intracellular total concentrations of evofosfamide to be onefold to fourfold higher than the extracellular concentrations (data on file). Therefore, sensitivity analyses ranged from onefold to fivefold increase in the intracellular concentrations of evofosfamide.
Static modeling
Standard static modeling was performed according to Eqs. (1) and (2) for direct and time‐dependent inhibition, respectively. Fractions escaping gut metabolism in the presence and absence of inhibitors were set to unity for CYP2B6 and 2D6, because expression of these enzymes in the gastrointestinal tract is negligible,36 and to unity and 0.57 for midazolam in line with the prominent gut expression of CYP3A.37 The static equation for a single CYP isozyme simplifies to:
| (1) |
where AUCʹ and AUC are the area under the concentration time curve in the presence and absence of the inhibitor, and f G are the fractions escaping gut metabolism in the presence and absence of inhibitor, [I] is the estimated unbound inhibitor concentration as discussed below, Ki is the reversible inhibition constant, and fmCYPi represents the fraction of the probe drug metabolized by CYPi. Similarly, the equation for irreversible inhibition is:
| (2) |
with kinact as the maximal inactivation rate constant, Kapp the inhibitor concentration at 50% of kinact, and kdeg the endogenous degradation rate constant of the CYP enzyme. Parameters were adopted from Simcyp for consistency. Parameters specific to the CYP enzymes and probe substrates are presented in Table 2. The inhibition constants for evofosfamide were also used in the PBPK modeling approach given in Table 1. The maximum unbound systemic concentration (Imax) was 13.6 μM and the average unbound inhibitor concentration (Iav) was 0.0639 μM, based on estimations from the model simulation after administration of a 340 mg/m2 dose of evofosfamide. Standard approaches, as outlined in guidance documents, were designed as a worst‐case approach, putting the patient safety first by using the maximum concentration.
Table 2.
Parameters specific to the CYP enzymes and probe substrates
| CYP | kdeg | Substrate | fmCYP |
|---|---|---|---|
| CYP2D6 | Not applicable | Dextromethorphan | 0.72 |
| CYP2B6 | 0.000362 min−1 = 0.0217 h−1 | Bupropion | 0.90 |
| CYP3A4 | 0.000322 min−1 = 0.0193 h−1 | Midazolam | 0.96 |
CYP, cytochrome P450 enzyme; fmCYP, fraction of substrate metabolized by CYP; kdeg, first order degradation rate constant.
Results
PBPK modeling
A PBPK model describing the PK of evofosfamide was successfully developed and validated (Figure 1) and then used for inhibitor or inducer interaction in trial simulations with sensitive probe substrates. The AUC changes observed for the probes were evaluated as a measure of evofosfamide's drug interaction potentials.
Evofosfamide as a CYP inhibitor
In vitro data indicated a potential for direct, but not time‐dependent inhibition of CYP2D6. The static calculation indicated that the interaction potential is negligible, with AUC ratios for the sensitive substrate dextromethorphan of 1.11 and 1.0005 based on Imax and Iav, respectively. These values are well below 1.25, the threshold for inhibition according to regulatory guidelines. In conjunction to the static modeling, PBPK modeling was constructed and validated the results obtained from the static model as shown in Tables 3 and 4.
Table 3.
Interaction simulations – results for simulations of evofosfamide as inhibitor or inducer, respectively, using PBPK modeling
| Probe | Probe Cmax ratio | Probe AUC ratio | ||||
|---|---|---|---|---|---|---|
| GeoMean | 95% CI | GeoMean | 95% CI | |||
| Evofosfamide as inhibitor | ||||||
|
Dextromethorphan CYP2D6 (DI) | ||||||
| Day 1 | 1.04 | 1.03 | 1.04 | 1.04 | 1.03 | 1.04 |
|
Bupropion CYP2B6 (MBI) | ||||||
| Day 1 | 1.001 | 1.001 | 1.001 | 1.001 | 1.001 | 1.001 |
| Day 15 | 1.001 | 1.001 | 1.001 | 1.001 | 1.001 | 1.002 |
|
Midazolam CYP3A4 and CYP3A5 (DI + MBI) | ||||||
| Day 1 | 1.02 | 1.01 | 1.02 | 1.03 | 1.03 | 1.04 |
| Day 15 | 1.02 | 1.02 | 1.02 | 1.03 | 1.03 | 1.04 |
| Evofosfamide as inducer | ||||||
|
Midazolam CYP3A4 and CYP3A5 (Ind) | ||||||
| Day 1 | 0.95 | 0.93 | 0.96 | 0.91 | 0.89 | 0.92 |
| Day 15 | 0.94 | 0.93 | 0.95 | 0.90 | 0.88 | 0.92 |
AUC, area under the concentration‐time curve; CI, confidence interval; Cmax, peak plasma concentration; CYP, cytochrome P450 enzyme; DI, direct inhibition; GeoMean, geometric mean; Ind, induction; MBI, mechanism‐based inhibition; PBPK, physiologically based pharmacokinetic.
Table 4.
AUC ratios for the respective probe substrates, calculated based on Imax and Iav, respectively, using static modeling
| CYP | AUC ratio (Imax; direct inhibition) | AUC ratio (Imax; TDI) | AUC ratio (Iav; direct inhibition) | AUC ratio (Iav; TDI) |
|---|---|---|---|---|
| Dextromethorphan | 1.11 | N/A | 1.0005 | N/A |
| Bupropion | N/A | 1.32 | N/A | 1.002 |
| Midazolam | 1.91 | 5.93 | 1.76 | 1.78 |
AUC, area under the concentration‐time curve; CYP, cytochrome P450 enzyme; Iav, average inhibitor concentration; Imax, maximum inhibitor concentration; N/A, not applicable; TDI, time dependent inhibition.
The geometric mean ratios (95% CI) for AUC and Cmax with and without evofosfamide were 1.04 (1.03; 1.04), and 1.04 (1.03; 1.04), respectively. Sensitivity analyses showed that the AUC ratio (95% CI) both at half the measured Ki value of 1.07 (1.06; 1.08) and at a 5‐fold increased intracellular concentration of evofosfamide 1.16 (1.13; 1.18) were well below the threshold of 1.25.
The time‐dependent inhibition of CYP2B6 was predicted using static modeling. The results predicted evofosfamide as a borderline weak inhibitor based on the maximum inhibitor concentration with a ratio of 1.32, whereas the calculation based on the average concentration yielded an AUC ratio of 1.002, making a clinically relevant DDI again very unlikely.
PBPK modeling was used to refine the analyses. The results shown in Table 3 indicated a < 1% change in both AUC and Cmax values following evofosfamide administration. The AUC ratios stayed below 1.02 for all simulated scenarios, including sensitivity analyses. The AUC ratios also stayed below 1.02 for repeat dosing of evofosfamide, in which CYP2B6 was only minimally inhibited. More than 99.5% of isozyme remained active, leading to bupropion AUC ratios of 1.00 (1.00; 1.00), indicating a lack of potential inhibition of CYP2B6 by evofosfamide in the clinic.
The most complex case was the inhibition of CYP3A4/5, which consists of simultaneous direct and time‐dependent inhibition (TDI). Static modeling predicted AUC ratios above the threshold of 1.25; the values are presented in Table 4. PBPK modeling was used to refine the results. No inhibition by evofosfamide was predicted for CYP3A4/5 with AUC ratios close to unity (Table 3). Sensitivity analyses for Ki (direct inhibition), and Kapp and kinact (mechanism‐based inhibition (MBI)) showed AUC ratios below 1.10, which is well below the threshold of 1.25 for evofosfamide to be considered as a weak inhibitor. Assuming a 5‐fold accumulation of evofosfamide in hepatocytes, the AUC ratio was 1.14 (1.12; 1.17), again below the threshold of 1.25. Because the in vitro studies indicated a onefold to fourfold possible accumulation of evofosfamide in hepatocytes, these simulations imply a lack of clinically relevant CYP3A4/5 inhibition by evofosfamide.
Again, no clinically relevant inhibition was anticipated following repeat once‐weekly dosing of evofosfamide, in which only marginal declines in CYP3A4 and 3A5 activities were predicted after the first and third doses (Figure 2). Interaction ratios were similar between days 1 and 15 (Table 3). In addition, accumulations of any inhibitory effects were negligible, given the once weekly dosing of evofosfamide (Figure 2), and, therefore, repeat dosing did not lead to a further decrease in enzyme activity. This was also evident between treatment cycles given that the third dose was followed by a 1‐week treatment break, thus the subsequent cycle began as the previous one with no meaningful enzyme inhibition. Therefore, the duration of the simulated trials of 360 hours was found to be sufficient to characterize the inhibition potential after multiple dosing of evofosfamide.
Figure 2.
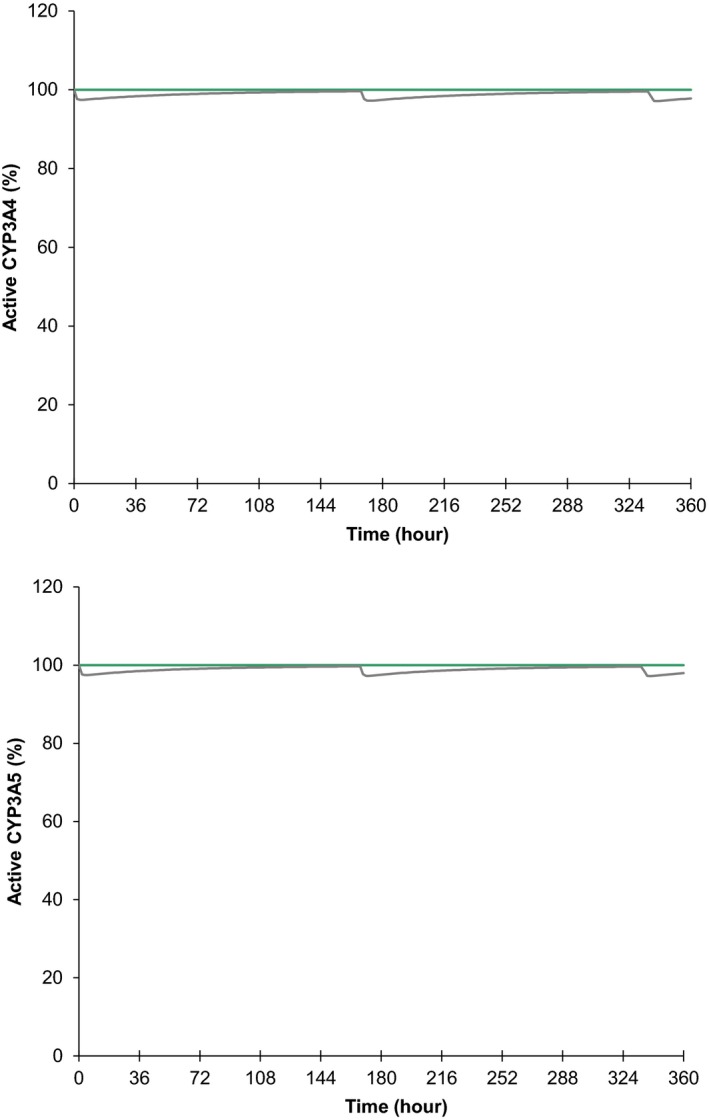
Relative amount of active cytochrome P450 (CYP)3A4 (top) and CYP3A5 (bottom) in the liver simulated for inhibition effect only over 360 hours in the absence (straight lines at 100%) and presence (curved lines) of evofosfamide administrations. The 340 mg/m2 evofosfamide was infused over 30 minutes starting at 0, 168, and 336 hours. The lines are the means for the population (n = 100).
Evofosfamide as a CYP inducer
The CYP induction results were based on the hybrid model combining a lower limit for IndC50 determined experimentally with the maximum fold‐induction of rifampicin. Simulations of evofosfamide as an inducer provided a geometric mean AUC ratio (95% CI) in this simulation of 0.91 (0.89; 0.92) for CYP3A. The AUC and Cmax ratios (Table 3) of midazolam are near unity and well above the threshold of 0.80 for consideration of evofosfamide as a weak inducer of CYP3A.
Interaction ratios were found to be similar between days 1 and 15 (Table 3). Enzyme levels had essentially returned to baseline within the dosing interval, with < 101% of CYP3A being active prior to the next dose. Therefore, repeat once weekly dosing of evofosfamide did not result in an increase in enzyme activity. The interaction potential was covered by simulations for up to week 3, accordingly.
The sensitivity analyses yielded AUC ratios (95% CI) of 0.82 (0.80; 0.85) assuming 2‐fold higher Indmax and 0.88 (0.86; 0.90) assuming 2‐fold lower IndC50, and are, therefore, above the regulatory threshold of 0.80 in the sensitivity analyses regarding the induction parameters.
Discussion
Mathematical models have been developed to predict clinical DDI early from in vitro data.22, 23, 24, 25, 26 These range from simple static models to more sophisticated mechanistic PBPK models. Although static modeling requires fewer resources, it is limited by the use of constant concentration data. Considering the once weekly dosing regimen of evofosfamide and its short elimination half‐life of 0.81 hours,35 the use of constant maximum inhibitor concentrations in static models likely overestimates evofosfamide's DDI potential. Hence, a dynamic model was applied because it can simultaneously model plasma concentration‐time profiles of both the perpetrator drug and the model substrate. Furthermore, PBPK modeling was used to overcome the problem of cytotoxicity that frequently limits the use of sufficiently high compound concentrations required to derive reliable in vitro estimates for IndC50 and Indmax. Therefore, a “hybrid” model was constructed using a lower limit of evofosfamide's IndC50 derived from experimental data combined with Indmax from rifampicin, which is considered one of the strongest CYP3A inducers.34 The values chosen for the modeling underestimated the actual CYP3A IndC50 for evofosfamide and likely overestimated the Indmax value for evofosfamide. This approach is considered to yield a worst‐case scenario when simulating the potential of evofosfamide to induce CYP3A. In addition, sensitivity analyses showed that the respective regulatory thresholds are not reached neither when considering uncertainty in enzyme kinetic parameters nor when considering possible accumulation of evofosfamide in hepatocytes. Wagner et al.38 reported that PBPK predictions of induction by rifampicin can be improved by increasing the maximum induction effect from 8‐fold to 11.5‐fold. The present modeling approach for evofosfamide included a sensitivity analysis of a maximum induction effect up to 16‐fold showing that no induction is present across the whole range.
Although the number of conservative assumptions included in our modeling approach provides confidence in the reliability of the current prediction, the lack of available clinical DDI data of evofosfamide limited further refinement and potential improvement of the model's predictivity, a recommended part of model qualification.39, 40 Furthermore, the present research does not revisit the qualifications of the Simcyp platform nor the verifications of the substrate models. This would include the qualification of the computational framework, the description of the virtual population, as well as the parameters used to describe the human physiology. We only present the development and verification of the evofosfamide compound file for use as a perpetrator model. We made several conservative assumptions and were still able to demonstrate absence of clinically relevant interaction potential, due to the properties of evofosfamide. However, this might not be possible for other compounds, in which more precise descriptions might be needed, thus limiting the approach to certain circumstances. In other situations, more thorough model development might be needed, for instance to more precisely capture the concentration time profile. There is also a limitation of the approach related to the combination of Indmax and IndC50 from two different drugs. Accordingly, the quantification of the induction potential can only be a surrogate value, which provides information about a boundary for the induction liability of the drug in question. The case presented in this study refers to the complete exclusion of induction liability and may only be applicable for drugs with short half‐lives or intermittent dosing, such as evofosfamide. Nevertheless, the approach could in principle also be used for other situations. For example, it could be of value for an exclusion of strong induction as opposed to moderate or weak induction.
This approach can derive information about the DDI potential of cytotoxic drugs, which otherwise could hardly be generated.41 PBPK simulations of DDI mediated by drug metabolizing enzymes is the area in which substantial scientific and regulatory experience has been gathered so far.40, 41, 42, 43 In addition, predictive confidence has been demonstrated for interactions via CYPs, so that these approaches can be used for modeling and simulation with high regulatory impact,44, 45, 46, 47 and generate essential information also for the safe use of cancer drugs in light of the limited studies in humans.32, 48
The current approach is novel combining the lower limit of IndC50 of evofosfamide with the maximum fold induction of rifampicin as the basis for PBPK modeling. Thereby, it was possible to predict that evofosfamide is unlikely to cause a clinically relevant DDI due to induction of CYP3A, despite the absence of reliable in vitro induction data above clinical peak concentrations. PBPK modeling should be considered when one key assumption of static models does not hold, namely maintaining inhibitory concentrations over an extended period of time). This approach is expected to work particularly well for compounds with short elimination half‐life and/or intermittent dosing regimen. This novel PBPK modeling approach may set a precedent for future studies of cytotoxic drugs and allows filling the knowledge gaps regarding their DDI potential.
Funding
The study was funded by Merck KGaA, Darmstadt, Germany, and Threshold Pharmaceuticals, South San Francisco, CA, USA, respectively.
Conflict of Interest
C.L., S.E.B., H.D., S.H., A.J. are employees of Merck KGaA, Darmstadt, Germany. M.D. is an employee of EMD Serono, Billerica, USA. O.v.R, D.G., and D.J. were employees of Merck KGaA, Darmstadt, Germany, and Threshold Pharmaceuticals, South San Francisco, CA, USA, respectively.
Author Contributions
C.L., O.v.R., S.E.B., H.D., and A.J. wrote the manuscript. C.L., O.v.R., S.H., and A.J. designed the research. C.L., O.v.R., D.G., and D.J. performed the research. C.L., M.D., O.v.R., H.D., D.G., S.H., and A.J. analyzed the data.
Supporting information
Note S1 Model development and qualification.
Note S2 Evofosfamide compound file and trial simulation settings.
Acknowledgments
The authors gratefully acknowledge Sheila Peters for scientific discussions and thank Jack McQueen Allen for assistance with manuscript preparation, funded by Merck KGaA.
References
- 1. Meng, F. et al Molecular and cellular pharmacology of the hypoxia‐activated prodrug TH‐302. Mol. Cancer Ther. 11, 740–751 (2012). [DOI] [PubMed] [Google Scholar]
- 2. Wilson, W.R. & Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410 (2011). [DOI] [PubMed] [Google Scholar]
- 3. Sun, J.D. et al Selective tumor hypoxia targeting by hypoxia‐activated prodrug TH‐302 inhibits tumor growth in preclinical models of cancer. Clin. Cancer Res. 18, 758–770 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Tap, W.D. et al Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft‐tissue sarcoma (TH CR‐406/SARC021): an international, multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 18, 1089–1103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Cutsem, E. et al Evofosfamide (TH‐302) in combination with gemcitabine in previously untreated patients with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma: primary analysis of the randomized, double‐blind phase III MAESTRO study. J. Clin. Oncol. 34, suppl, abstract 4007 (2016). [Google Scholar]
- 6. Zhang, L. , Zhang, Y.D. , Zhao, P. & Huang, S.M. Predicting drug‐drug interactions: an FDA perspective. AAPS J. 11, 300–306 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al‐Lazikani, B. , Banerji, U. & Workman, P. Combinatorial drug therapy for cancer in the post‐genomic era. Nature Biotechnol. 30, 679–692 (2012). [DOI] [PubMed] [Google Scholar]
- 8. Li, F. , Zhao, C. & Wang, L. Molecular‐targeted agents combination therapy for cancer: developments and potentials. Int. J. Cancer 134, 1257–1269 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Melero, I. , Berman, D.M. , Aznar, M.A. , Korman, A.J. , Perez Gracia, J.L. & Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 15, 457–472 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Edwards, I.R. & Aronson, J.K. Adverse drug reactions: definitions, diagnosis, and management. Lancet 356, 1255–1259 (2000). [DOI] [PubMed] [Google Scholar]
- 11. Dechanont, S. , Maphanta, S. , Butthum, B. & Kongkaew, C. Hospital admissions/visits associated with drug‐drug interactions: a systematic review and meta‐analysis. Pharmacoepidemiol. Drug Saf. 23, 489–497 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Butlen‐Ducuing, F. et al Regulatory watch: challenges in drug development for central nervous system disorders: a European Medicines Agency perspective. Nat. Rev. Drug Discov. 15, 813–814 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Zhang, J.G. , Ho, T. , Callendrello, A.L. , Crespi, C.L. & Stresser, D.M. A multi‐endpoint evaluation of cytochrome P450 1A2, 2B6 and 3A4 induction response in human hepatocyte cultures after treatment with beta‐naphthoflavone, phenobarbital and rifampicin. Drug Metab. Lett. 4, 185–194 (2010). [DOI] [PubMed] [Google Scholar]
- 14. U.S. Food and Drug Administration , Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) . Guidance for industry: in vitro metabolism‐ and transporter ‐ mediated drug‐drug interaction studies. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf> (2017).
- 15. U.S. Food and Drug Administration , Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) . Guidance for industry: clinical drug interaction studies ‐ study design, data analysis, and clinical implications. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf> (2017).
- 16. European Medicines Agency . Committee for Human Medicinal Products (CHMP), Guideline on the investigation of drug interactions CPMP/EWP/560/95/Rev. 1 Corr. 2** <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf> (2012).
- 17. Grub, S. , Bryson, H. , Goggin, T. , Ludin, E. & Jorga, K. The interaction of saquinavir (soft gelatin capsule) with ketoconazole, erythromycin and rifampicin: comparison of the effect in healthy volunteers and in HIV‐infected patients. Eur. J. Clin. Pharmacol. 57, 115–121 (2001). [DOI] [PubMed] [Google Scholar]
- 18. Josephson, F. Drug‐drug interactions in the treatment of HIV infection: focus on pharmacokinetic enhancement through CYP3A inhibition. J. Intern. Med. 268, 530–539 (2010). [DOI] [PubMed] [Google Scholar]
- 19. Lee, J.Y. , Lee, S.Y. , Oh, S.J. , Lee, K.H. , Jung, Y.S. & Kim, S.K. Assessment of drug‐drug interactions caused by metabolism‐dependent cytochrome P450 inhibition. Chem. Biol. Interact. 198, 49–56 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Foisy, M.M. , Yakiwchuk, E.M. & Hughes, C.A. Induction effects of ritonavir: implications for drug interactions. Ann. Pharmacother. 42, 1048–1059 (2008). [DOI] [PubMed] [Google Scholar]
- 21. Ito, K. , Iwatsubo, T. , Kanamitsu, S. , Ueda, K. , Suzuki, H. & Sugiyama, Y. Prediction of pharmacokinetic alterations caused by drug‐drug interactions: metabolic interaction in the liver. Pharmacol. Rev. 50, 387–412 (1998). [PubMed] [Google Scholar]
- 22. Kanamitsu, S. , Ito, K. & Sugiyama, Y. Quantitative prediction of in vivo drug‐drug interactions from in vitro data based on physiological pharmacokinetics: use of maximum unbound concentration of inhibitor at the inlet to the liver. Pharm. Res. 17, 336–343 (2000). [DOI] [PubMed] [Google Scholar]
- 23. Galetin, A. , Burt, H. , Gibbons, L. & Houston, J.B. Prediction of time‐dependent CYP3A4 drug‐drug interactions: impact of enzyme degradation, parallel elimination pathways, and intestinal inhibition. Drug Metab. Dispos. 34, 166–175 (2006). [DOI] [PubMed] [Google Scholar]
- 24. Kato, M. et al The quantitative prediction of CYP‐mediated drug interaction by physiologically based pharmacokinetic modeling. Pharm. Res. 25, 1891–1901 (2008). [DOI] [PubMed] [Google Scholar]
- 25. Kato, M. , Chiba, K. , Horikawa, M. & Sugiyama, Y. The quantitative prediction of in vivo enzyme‐induction caused by drug exposure from in vitro information on human hepatocytes. Drug Metab. Pharmacokinet. 20, 236–243 (2005). [DOI] [PubMed] [Google Scholar]
- 26. Peters, S.A. , Schroeder, P.E. , Giri, N. & Dolgos, H. Evaluation of the use of static and dynamic models to predict drug‐drug interaction and its associated variability: impact on drug discovery and early development. Drug Metab. Dispos. 40, 1495–1507 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Shou, M. et al Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab. Dispos. 36, 2355–2370 (2008). [DOI] [PubMed] [Google Scholar]
- 28. Almond, L.M. , Yang, J. , Jamei, M. , Tucker, G.T. & Rostami‐Hodjegan, A. Towards a quantitative framework for the prediction of DDIs arising from cytochrome P450 induction. Curr. Drug Metab. 10, 420–432 (2009). [DOI] [PubMed] [Google Scholar]
- 29. Grime, K. , Ferguson, D.D. & Riley, R.J. The use of HepaRG and human hepatocyte data in predicting CYP induction drug‐drug interactions via static equation and dynamic mechanistic modelling approaches. Curr. Drug Metab. 11, 870–885 (2010). [DOI] [PubMed] [Google Scholar]
- 30. Yang, J. et al Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr. Drug Metab. 9, 384–394 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Beijnen, J.H. & Schellens, J.H. Drug interactions in oncology. Lancet Oncol. 5, 489–496 (2004). [DOI] [PubMed] [Google Scholar]
- 32. Mani, S. , Ghalib, M. , Chaudhary, I. & Goel, S. Alterations of chemotherapeutic pharmacokinetic profiles by drug‐drug interactions. Expert Opin. Drug Metab. Toxicol. 5, 109–130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rowland, M. , Peck, C. & Tucker, G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 51, 45–73 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Yu, J. , Ritchie, T.K. , Mulgaonkar, A. & Ragueneau‐Majlessi, I. Drug disposition and drug‐drug interaction data in 2013 FDA new drug applications: a systematic review. Drug Metab. Dispos. 42, 1991–2001 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Weiss, G.J. et al Phase 1 study of the safety, tolerability, and pharmacokinetics of TH‐302, a hypoxia‐activated prodrug, in patients with advanced solid malignancies. Clin. Cancer Res. 17, 2997–3004 (2011). [DOI] [PubMed] [Google Scholar]
- 36. Madani, S. , Paine, M.F. , Lewis, L. , Thummel, K.E. & Shen, D.D. Comparison of CYP2D6 content and metoprolol oxidation between microsomes isolated from human livers and small intestines. Pharm. Res. 16, 1199–1205 (1999). [DOI] [PubMed] [Google Scholar]
- 37. Peters, S.A. , Jones, C.R. , Ungell, A.L. & Hatley, O.J.D. Predicting drug extraction in the human gut wall: assessing contributions from drug metabolizing enzymes and transporter proteins using preclinical models. Clin. Pharmacokinet. 55, 673–696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wagner, C. , Pan, Y. , Hsu, V. , Sinha, V. & Zhao, P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin. Pharmacokinet. 55, 475–483 (2016). [DOI] [PubMed] [Google Scholar]
- 39. Shebley, M. et al Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin. Pharmacol. Ther. 104, 88–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. European Medicines Agency . Committee for Human Medicinal Products (CHMP), Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. EMA/CHMP/458101/2016. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf> (2016).
- 41. Sinha, V. , Zhao, P. , Huang, S.M. & Zineh, I. Physiologically based pharmacokinetic modeling: from regulatory science to regulatory policy. Clin. Pharmacol. Ther. 95, 478–480 (2014). [DOI] [PubMed] [Google Scholar]
- 42. U.S. Food and Drug Administration , Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) . Guidance for industry: physiologically based pharmacokinetic analyses ‐ format and content. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM531207.pdf> (2016).
- 43. Sato, M. et al Quantitative modeling and simulation in PMDA: a Japanese regulatory perspective. CPT Pharmacometrics Syst. Pharmacol. 6, 413–415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wagner, C. et al Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 54, 117–127 (2015). [DOI] [PubMed] [Google Scholar]
- 45. Rowland, M. , Lesko, L.J. & Rostami‐Hodjegan, A. Physiologically based pharmacokinetics is impacting drug development and regulatory decision making. CPT Pharmacometrics Syst. Pharmacol. 4, 313–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shepard, T. , Scott, G. , Cole, S. , Nordmark, A. & Bouzom, F. Physiologically based models in regulatory submissions: output from the ABPI/MHRA forum on physiologically based modeling and simulation. CPT Pharmacometrics Syst. Pharmacol. 4, 221–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang, S.M. , Abernethy, D.R. , Wang, Y. , Zhao, P. & Zineh, I. The utility of modeling and simulation in drug development and regulatory review. J. Pharm. Sci. 102, 2912–2923 (2013). [DOI] [PubMed] [Google Scholar]
- 48. Saeheng, T. , Na‐Bangchang, K. & Karbwang, J. Utility of physiologically based pharmacokinetic (PBPK) modeling in oncology drug development and its accuracy: a systematic review. Eur. J. Clin. Pharmacol. 74, 1365–1376 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note S1 Model development and qualification.
Note S2 Evofosfamide compound file and trial simulation settings.