

Abstract
Activation of EGFR (epidermal growth factor receptor) and Eph receptor often exerts opposing effects on cell functions. In this issue of Science Signaling, Stallaert et al. reveal how cells maintain sustained response to EGF stimulation by replenishing EGFR at the plasma membrane and how conflicting signals from the EphA-ephrin system and EGFR are integrated to coordinate cellular responses, including cell migration and proliferation.
One-sentence summary:
EphA activation upon cell-cell contact balances EGFR degradation and recycling to ensure sustained responsiveness to EGF (Stallaert et al., in 31 July 2018 issue).
Cells in living organisms must cope with the continuous bombardment of many fluctuating environmental signals that are often conflicting in nature. Shedding light on how cells decipher these signals and orchestrate appropriate responses to maintain homeostasis and contain deleterious outputs, Stallaert et al. reveal cross-talk between EphA receptor tyrosine kinases (RTKs) and epidermal growth factor receptor (EGFR) that may constitute one such coping mechanism (1). EphA-mediated inhibition of Akt upon cell-cell contact balances EGFR degradation with constitutive recycling to ensure sustained responsiveness to EGF (Fig. 1A). Because Eph receptors and EGFR mediate opposing effects, this study provides insight into how signals are integrated to control cell migration and proliferation.
Fig. 1. Constitutive recycling and induced endocytosis of EGFR and their inhibition by EphA-ephrinA.
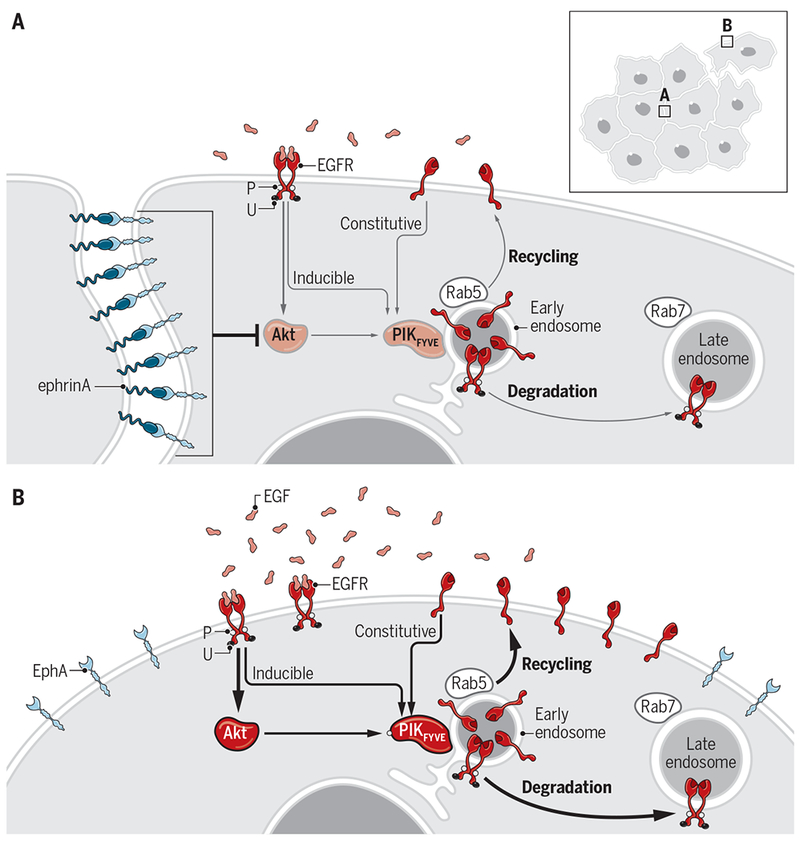
(A) In addition to endocytosis and degradation of EGF-stimulated EGFR that is tyrosine phosphorylated (P) and ubiquitinated (U), Stallaert et al. report a constitutive recycling mechanism of the ligand-free EGFR. The recycling maintains EGFR abundance at the PM. Both pathways depend on Akt. EphA-ephrinA interactions upon cell-cell contact inhibit Akt and suppress EGFR recycling. (B) In the absence of cell-cell contact, such as at the leading edge of migrating cells, the lack of EphA-ephrinA engagement unleashes Akt activation and promotes both recycling and endocytosis of EGFR, which can be further enhanced by increased EGF levels during wound healing and metastasis.
In the ephrin signaling system, the GPI-anchored ephrinA and transmembrane ephrinB ligands generally bind to EphA and EphB receptors, respectively (2). Because both receptors (Ephs) and ligands (ephrins) are membrane proteins, Eph-ephrin interactions occur upon cell-cell contact and mediate bidirectional signaling to both receptor- and ligand-bearing cells. The Eph-ephrin system guides growth cones and cell migration through mostly repulsive and sometimes adhesive responses.
In addition to migration, Eph receptors control other cellular processes, including cell proliferation, differentiation, and survival. EphA2 can function as either a tumor suppressor or an oncogenic protein, which is dictated by the cellular context, including ligand occupancy status and environmental cues. Ligand-stimulated EphA2 inhibits Akt in PTEN-deficient prostate cancer cells, glioma cells (3), and other cell types (4, 5). Moreover, liganded EphA2 can counteract Akt activation by various growth factors, including EGF, which correlates with inhibition of growth factor-induced chemotactic cell migration (3) through poorly understood mechanisms. Filling this gap, Stallaert et al. report that liganded EphA2 and possibly other EphA receptors suppress the constitutive and EGF-induced recycling of EGFR to the plasma membrane (PM), leading to EGFR accumulation in early endosomes and suppression of cell migration.
EGFR regulates many processes, including cell proliferation, migration, and apoptosis. EGF stimulation induces EGFR dimerization and catalytic activation characterized by transphosphorylation on tyrosine residues (Fig. 1B), leading to the activation of PI3K/Akt and Ras/ERK/p90RSK pathways. Ubiquitination of active EGFR recruits the endocytic machinery to initiate clathrin-mediated internalization, endocytosis, and degradation (Fig. 1B). Er et al. have shown that the latter processes are facilitated by Akt, which activates PIKfyve in early endosomes to drive EGFR trafficking from early to late endosomes and ultimately lysosomal degradation (5). The study by Stallaert et al. showed that this signaling cascade also promoted recycling of unliganded EGFR from early endosomes to the PM. EphA2 stimulation potently inhibited Akt, reducing PIKfyve activation in early endosomes that is required for EGFR recycling (Fig. 1A) (1).
The coupling of EGFR activation to vesicular recycling generated a positive feedback loop to maintain free EGFR at the PM and drive Akt-PIKfyve activation, leading to increased EGFR degradation and enhanced recycling. Replenishing EGFR ensured sustained responses to EGF. This feedback mechanism would be expected to operate during chemotactic cell migration, in which continuous recycling and activation of EGFR ensure sustained directional cell motility in response to EGF. Indeed, EGF induced migration in mouse embryonic fibroblasts and MDA-MB-231 breast cancer cells in a manner dependent on Akt activation and EGFR recycling. Consistent with the mediation of contact inhibition of locomotion by liganded EphA receptors, EphA activation by either cell contact or soluble ephrinA suppressed cell migration.
Interestingly, Stallaert et al. noted differential effects of liganded EphA2 on EGFR-mediated activation of Akt and ERK. Whereas Akt was inactivated at the PM by liganded EphA, EGF-bound EGFR continued to signal from endosomes to activate ERK and support cell proliferation. However, previous studies have shown transient inhibition of the Ras-ERK pathway by EphA2 (6) and EphB2 (7) upon ligand exposure. In primary keratinocytes and hepatoma cells, ephrinA1 exposure suppresses ERK in wild-type but not EphA2 knockout cells (6). These discrepancies may be due to cell type differences, such as the presence of other EphA receptors with opposing functions on ERK.
Ligand-activated EphA2 itself undergoes rapid internalization and degradation through endocytic trafficking (8). Similar to EGFR, EphA2 is endocytosed upon ligand binding through clathrin- or caveole-dependent pathways and continues to signal in early endosomes. A fraction of the endocytosed EphA2 is also recycled to the PM. It remains to be explored whether the liganded EGFR and EphA receptors share endocytosis machineries and whether the two sets of receptors interact at the endosomes.
Finally, the cross-talk between EGFR and EphA2 is likely reciprocal. Not only does ligand activation of EphA2 inhibit EGFR signaling to Akt, EGFR activation also indirectly targets free EphA2 by stimulating Akt and p90RSK. Phosphorylation of Ser897 by these kinases (3, 9) effectively converts EphA2 from a tumor suppressor into an oncogenic protein (3). Since this phosphorylation event drives cell migration and invasion, it may synergistically signal with EGFR recycling driven by Akt-PIFfyve to promote cell locomotion, a scenario that may take place in processes such as wound healing and tumor metastasis. Future studies are needed to test this possibility.
The results of Stallaert et al. highlight a mechanism by which regulation of vesicular dynamics by one RTK (EphA2) alters the spatial distribution and functional outcome of another RTK (EGFR) without requiring the physical interactions of these two receptor types (Fig. 1A) (1). A key question remains as to whether the cross-talk occurs in vivo. Nonetheless, both Eph receptors and EGFR are attractive therapeutic targets for the disease processes in which they are involved. Characterization of the crosstalk mechanism could lead to new strategies to curb tumor cell development and dissemination.
Acknowledgments:
We thank members of the Wang laboratory for helpful comments on the manuscript.
Funding: B.W. is supported by NIH grants R01 NS096956 and R01 CA155676 and by NCI Cancer Center support grant P30 CA043703.
REFERENCES AND NOTES
- 1.Stallaert W, Brüggemann Y, Sabet O, Baak L, Gattiglio M, Bastiaens PIH, Contact inhibitory Eph signaling suppresses EGF-promoted cell migration by decoupling EGFR activity from vesicular recycling. Sci. Signal 11, eaat0114 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Eph Nomenclature Committee, Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 90, 403–404 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Miao H, Li D-Q, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B, EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 16, 9–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang N-Y, Fernandez C, Richter M, Xiao Z, Valencia F, Tice DA, Pasquale EB, Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells. Cell. Signal 23, 201–212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin B, Yin T, Wu YI, Inoue T, Levchenko A, Interplay between chemotaxis and contact inhibition of locomotion determines exploratory cell migration. Nat. Commun 6, 6619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, Wang B, Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 66, 7050–7058 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Elowe S, Holland SJ, Kulkarni S, Pawson T, Downregulation of the Ras-mitogen-activated protein kinase pathway by the ephb2 receptor tyrosine kinase is required for ephrin-induced neurite retraction. Mol. Cell. Biol 21,7429–7441 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boissier P, Chen J, Huynh-Do U, EphA2 signaling following endocytosis: Role of Tiam1. Traffic 14, 1255–1271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I, Sakurai H, Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat. Commun 6, 7679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]