

Abstract
Background –
Hypertrophic cardiomyopathy (HCM) is a major cause of sudden death in young athletes and one of the most common inherited cardiovascular diseases affecting 1 in 500 individuals. Often viewed as a disease of the cardiac sarcomere, mutations in genes encoding myofilament proteins are associated with disease pathogenesis. Despite a clinically available genetic test, a significant proportion of HCM patients remain genetically unexplained. We sought to determine the spectrum of mutations in PLN-encoded phospholamban in a large cohort of HCM cases as a potential cause of mutation-negative HCM.
Methods –
Comprehensive genetic interrogation of the promoter and coding region of PLN was conducted utilizing polymerase chain reaction, denaturing high performance liquid chromatography, and direct DNA sequencing.
Results –
One L39X nonsense mutation was identified in 1 out of 1064 HCM proband cases with a family history of HCM, previously found to be negative for the current HCM genetic test panel. This mutation cosegregated with incidence of HCM in a multi-generational family. Compared to similar studies, we identified an overall yield of PLN-HCM mutations of 0.65%, similar to three genes which are part of current HCM genetic test panels. We did not observe any PLN coding sequence genetic variation in 600 reference alleles.
Conclusions –
Overall, mutations in PLN are rare in frequency, yet the small size of the genetic locus may make it amenable to inclusion on HCM gene test panels especially since the frequency of background genetic variation among otherwise healthy subjects appears negligible. The exact role of mutations in PLN, and other calcium-handling proteins, in the development of HCM warrants further investigation.
Keywords: Hypertrophic cardiomyopathy, phospholamban, PLN, genetics, mutation, calcium
INTRODUCTION
Hypertrophic cardiomyopathy (HCM), defined as unexplained cardiac hypertrophy, affects approximately 1 in 500 persons and is one of the most common genetic cardiovascular diseases1. HCM is one of the most common causes of sudden cardiac arrest in young athletes and a significant cause of sudden death in the young in general2,3. Often viewed as a disease of the cardiac sarcomere, mutations in the genes encoding sarcomeric proteins of the heart, such as MYH7-encoded β-myosin heavy chain and MYBPC3-encoded cardiac myosin binding protein C, cause pathological cardiac hypertrophy. This group of myofilament-encoding genes is the centerpiece of clinically available genetic tests for HCM with MYH7- and MYBPC3-HCM being by far the two most common HCM genotypes. However, a large proportion of patients with HCM remain genetically and mechanistically elusive4–6.
Several independent studies have identified rare genetic mutations in genes encoding calcium (Ca2+)-handling or Ca2+-regulatory proteins in individuals with myofilament negative-HCM including junctophilin 2 (JPH2)7, calreticulin (CALR3)8, and troponin C (TNNC1)9. All told, the prevalence of mutations in these genes constitute a small minority of patients with mutation-negative HCM, yet may expand understanding of the role of Ca2+ in the pathogenesis of HCM.
Coordinated, highly regulated Ca2+ flux and homeostasis within cardiocytes is critical for efficient excitation-contraction coupling and proper functioning of the beating heart. In cardiocytes, voltage-gated L-type Ca2+ channels at the sarcolemma allow for an influx of extracellular Ca2+, which triggers a relatively large Ca2+ release from the sarco/endoplasmic reticulum via intracellular Ca2+ release channels known as ryanodine receptors. This process, known as Ca2+-induced Ca2+-release, is the molecular initiator of myofilament-based myocyte contraction. Systolic contraction is terminated, and diastolic relaxation is initiated, by uptake of cytosolic Ca2+ back into the SR through the action of the energy-expending SR Ca2+ ATPase (SERCA2). PLN-encoded phospholamban (PLN, also known as PLB) is a small, 52 amino acid protein which negatively-regulates the Ca2+ uptake action of SERCA2. Upon protein kinase A or calmodulin-dependent protein kinase 2 phosphorylation at serine 16 and threonine 17, respectively, this inhibition is relieved, leading to increased Ca2+ removal from the cytosol into the SR increasing diastolic relaxation10,11.
Recently, familial mutations and common polymorphisms in the coding sequence and promoter regions of PLN have been implicated in human disease including dilated cardiomyopathy (DCM) and HCM8,12. Based on these studies, and the recent identification of a potential Ca2+-mishandling HCM genetic subtype, we sought to determine the prevalence of PLN mutations in a large cohort of HCM patients and to summarize the literature to date.
METHODS
Study populations
Between April 1997 and April 2007, 1064 unrelated index cases were evaluated in the Hypertrophic Cardiomyopathy Clinic at Mayo Clinic, Rochester, Minnesota, and consented for HCM genetic testing. Following receipt of written consent for this Mayo Foundation Institutional Review Board-approved protocol, DNA was extracted from peripheral blood lymphocytes using the Purgene DNA extraction kit (Gentra, Inc, Minneapolis, Minnesota).
Clinical data was collected on all HCM patients including physical examination, pertinent personal and family history, 12-lead electrocardiogram (ECG) analysis, and echocardiographic testing to determine mean left ventricular wall thickness (MLVWT) and maximum left ventricular outflow tract gradient before myectomy. Each of the subjects met the clinical diagnostic criteria for HCM (i.e. MLVWT greater than 13 mm in the absence of other confounding diagnoses). A panel of 300 control samples, including 200 Caucasian subjects and 100 African American subjects, were obtained from the Coriell Institute for Medical Research (Camden, New Jersey) to serve as ostensibly healthy control individuals.
Mutational Analysis
Comprehensive genetic analysis of the promoter and coding region of PLN was conducted using polymerase chain reaction amplification followed by denaturing high performance liquid chromatography (DHPLC, Transgenomic, Omaha, Nebraska) heteroduplex analysis. Promoter sequence was based on the NCBI accession number AF177763. Primer sequences, PCR and DHPLC conditions are available upon request. Abnormal DHPLC elution profiles were directly sequenced (ABI Prism 377, Applied Biosystem, Foster City, California) to characterize the difference between the wild type and variant alleles. PLN-positive subjects were analyzed for mutations in nine myofilament-HCM associated genes (MYBPC, MYH7, TNNT2, TNNI3, TNNC1, TPM1, ACTC, MYL2, and MYL3) as well as the HCM phenocopy-associated genes PRKAG2, GLA, and LAMP2.
Pedigree Analysis
Pedigree expansion of PLN mutation-positive probands was done in accordance with the Mayo Foundation Institutional Review Board. Clinical history and genomic DNA extracted from peripheral blood lymphocytes was obtained from all participating family members and mutation status for each individual was determined.
Statistics
Cohort demographics, where appropriate, were expressed as mean ± standard deviation.
Funding Sources
The analyses were performed with support from the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program, a Leducq Fondation program grant “Alliance for Calmodulin Kinase II Signaling in Heart Disease,” and the National Institutes of Health 1PO1HL094291. The authors are solely responsible for the design and content of this study, all study analyses, the drafting and editing of the paper, and its final contents.
RESULTS
Genetic Analysis
The demographics of our proband-based HCM cohort are shown in Table 1. Briefly, our cohort of 1064 cases (~60% male) were diagnosed with HCM at 44.4 ± 18.6 years with a mean ventricular septal thickness of 20.9 ± 5.9 mm, and a mean resting left ventricular outflow tract gradient of 43.7 ± 43.5 mmHg. Genetic analysis of this cohort revealed a single open reading frame nucleotide alteration in a single index case. As depicted in Figure 1, this T to G transition resulted in a leucine (TTA) to termination codon (TGA) nonsense mutation at position 39 (L39X) in a heterozygous manner. This Caucasian patient was negative for mutations in all nine myofilament genes that have been associated with HCM and are currently part of commercially available genetic tests. This mutation was absent in 600 reference alleles (200 African American, 400 Caucasian American). Further, no amino acid altering genetic variants were observed in the PLN coding sequence in any of these 600 reference alleles.
Table 1:
Summary of the Demographics and Clinical Characteristics of the 1064-proband HCM cohort
| Clinical Characteristics | HCM Cohort |
|---|---|
| No. of individuals | 1064 |
| Sex, male/female | 635/429 |
| Age at diagnosis (years) | 44.4 ± 18.6 |
| Mean LV wall thickness (mm) | 20.9 ± 5.9 |
| Resting LVOTO (mmHg) | 43.7 ± 43.5 |
| Positive fam hx for HCM | 29% |
| Positive fam hx for SCD | 12% |
| Surgical myectomy | 39% |
| Atrial fibrillation | 21% |
| Ablation | 9% |
| ICD | 18% |
| Pacemaker | 19% |
LV, left ventricular; LVOTO, left ventricular outflow tract obstruction; fam hx, family history in a first degree relative; HCM, hypertrophic cardiomyopathy; SCD, sudden cardiac death/arrest; ICD, implantable cardioverter-defibrillator
Figure 1 – L39X Nonsense Mutation Identification.
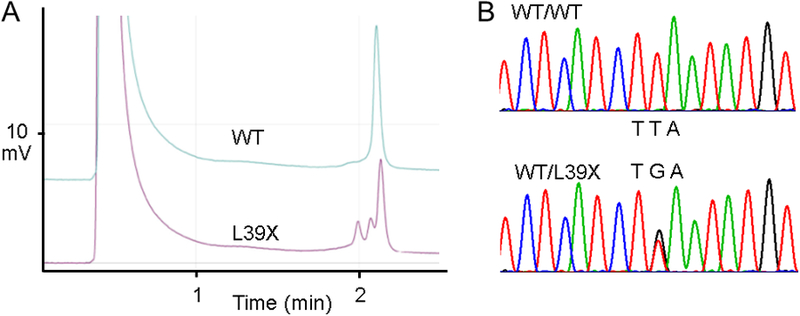
A) Denaturing high performance liquid chromatography elution profile of the PCR product for the L39X mutation compared to wild-type. B) Direct DNA sequencing chromatogram of the heterozygous L39X mutation demonstrating a TTA to TGA mutation compared to wild-type.
In addition, promoter variants were identified in 5 out of 1064 probands which were not identified in 600 reference alleles. Two heterozygous common polymorphisms were also identified in the promoter (A>C −36: 85/1025, 8.3% of HCM probands; 2/100, 2% of African Americans; and 15/200, 7.5% of Caucasian Americans) and in the 5’ untranslated region (G>A; 8/1025, 0.78% of HCM probands; and 9/100, 9% of African Americans).
PLN-L39X Proband
The PLN-L39X-nonsense mutation was identified in a 58-year-old, Caucasian male with septal and apical HCM. He was diagnosed at 51 years of age presenting with palpitations and was maintained on atenolol and amiodarone for approximately two years when he developed recurrent palpitations and chest discomfort with normal angiography. At this time, he was diagnosed with Wolff-Parkinson-White (WPW) syndrome due to abnormalities on resting ECG. He was also diagnosed with thyrotoxicosis secondary to amiodarone and was placed on propylthiouracil after discontinuing the amiodarone. He was placed on candesartan cilexetil and sotalol.
Over the next month he continued to develop episodic palpitations of increasing frequency, presyncope, dyspnea, and left-sided chest discomfort radiating to the left shoulder and arm, particularly while physically active and occasionally while lying supine. At this time, ECG (Figure 2A) and echocardiographic (Figure 2B) analysis revealed sinus bradycardia with paroxysmal atrial fibrillation/flutter, a MLVWT of 24 mm and left atrial enlargement with mild mitral valve regurgitation without resting or valsalva-induced left ventricular outflow tract obstruction. He demonstrated a normal ejection fraction of 68% at rest increasing to 90% at peak physical exertion. He was found to have conduction block as well as ventricular ectopy with symptomatic non-sustained ventricular tachycardia for which he received an automatic internal cardioverter defibrillator. He reported a positive family history of HCM involving his mother, one sister, and one grandchild who have been diagnosed with HCM echocardiographically. He reported no family history of sudden cardiac death, DCM, or heart failure.
Figure 2 – L39X Proband Clinical Characteristics.
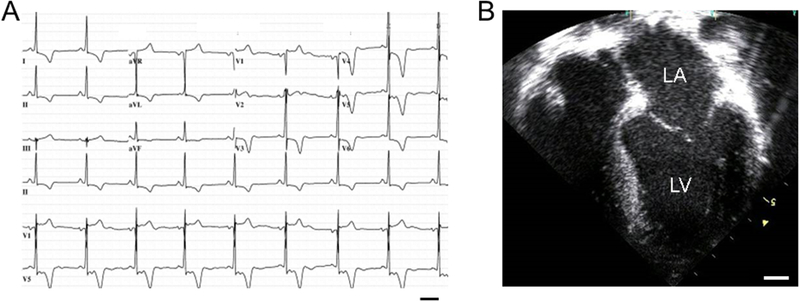
A) 12-lead electrocardiogram demonstrating criteria for left ventricular hypertrophy and T-wave abnormalities. Bar, 0.4s. B) Still image of end diastole from an echocardiogram demonstrating asymmetric apical and septal hypertrophy with left atrial dilation. LA, left atrium; LV, left ventricle; Bar, 10mm.
Given the proband’s previous diagnosis of WPW, and the association between ventricular preexcitation and specific hypertrophic cardiomyopathy disease phenocopies, we next explored the possibility that a compound mutation in PRKAG2-encoded γ2 regulatory subunit of AMP-activated protein kinase, GLA-encoded α -galactosidase A, or LAMP2-encoded lysosome-associated membrane protein 2 might account for this disease phenotype. While, mutations in PRKAG2 have been associated with development of WPW, the proband was found to be PRKAG2 mutation negative13,14. In addition, the proband did not host a mutation in Fabry’s disease-associated GLA or Danon’s syndrome-associated LAMP215–17.
PLN-L39X Pedigree
In an attempt to investigate whether this nonsense mutation might co-segregate with the proband’s family history of HCM, we obtained genomic DNA from as many first and second degree relatives as possible (Figure 3). The proband has two daughters, ages 39 and 38 years. The older daughter is HCM phenotype-negative with a phenotype-negative son (14 years old) and daughter (12 years old) who are all PLN-L39X mutation negative. Conversely, the younger daughter, who does not demonstrate features of HCM currently, has a 3 year-old, HCM phenotype-positive daughter. Both host the PLN-L39X mutation. Unfortunately, no autopsy tissue was available on the proband’s deceased HCM-positive mother to permit a PLN-L39X confirmatory postmortem genetic test.
Figure 3 – L39X Proband Pedigree.
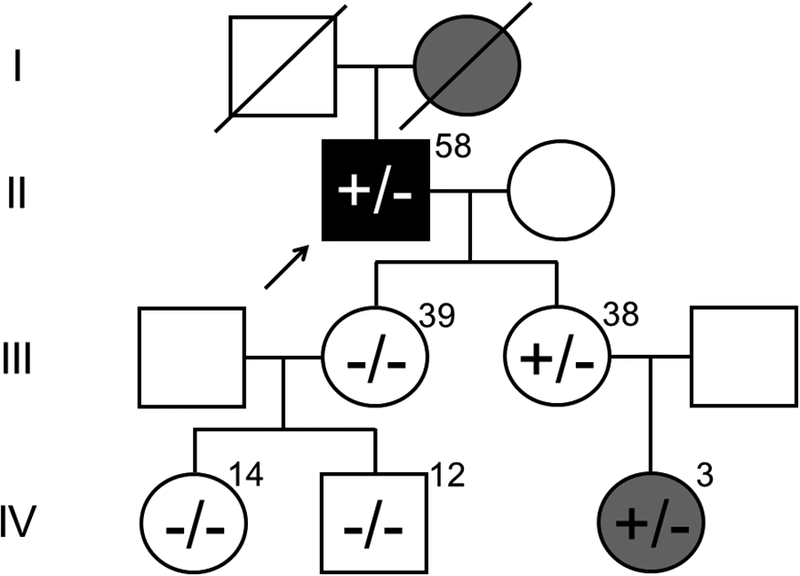
The PLN-L39X proband (arrow) with a history of HCM and atrial fibrillation has an echocardiography-proven HCM-affected mother (deceased, diagonal line), sister, and granddaughter. One daughter of the proband with no evidence of HCM does not host the L39X mutation nor do her two phenotype-negative daughters. The second daughter of the proband, who has yet to develop signs of HCM, hosts the PLN-L39X mutation as does her daughter who is HCM phenotype-positive. Numbers indicate age of family members at time of study. White, no cardiovascular phenotype; gray, HCM; black, HCM and atrial fibrillation; +, PLN-L39X positive; −, wild-type PLN.
DISCUSSION
PLN Mutations in HCM and DCM
Over the past few years, promoter and coding region variants of PLN have been associated with DCM/heart failure and HCM. For example, a C to G conversion at position −42 (C>G −42) promoter variant has been described in one HCM case in a study that included 186 HCM and DCM patients21. This variant, found in a female, diagnosed with HCM at 67 years of age with atrial fibrillation, had reduced penetrance in a small familial pedigree. A second promoter variant, an A>G −77 mutation, was identified in one out of 87 HCM patients22. As with the C>G −42 variant, the proband hosting this variant was female, diagnosed with HCM at 56 years of age, and demonstrated paroxysmal atrial fibrillation. In addition to unique variants in the promoter, a common A>C −36 variant has been controversially found in higher frequency among patients with DCM23,24. However, in this study, we did not observe over-representation of this variant in HCM probands (8.3%) compared to ostensibly healthy, ethnically-matched individuals (2% of African Americans, and 7.5% of Caucasian Americans).
In addition to promoter variants, mutations in the coding region of PLN have been associated with cardiovascular disease, mainly DCM or heart failure. The PLN-R9C mutation, identified in a large family of DCM patients25, and a deletion of arginine 14 (PLN-R14del), found in a large cohort of 1,203 DCM cases26, have been described. Notably, Haghighi and colleagues described the PLN-L39X mutation in a large family of hereditary heart failure and demonstrated that genetic “dosage” correlated with progression of individuals towards heart failure12. Individuals within the family homozygous for PLN-L39X either demonstrated, or quickly progressed to, heart failure, while individuals hosting one PLN-L39X mutation were either unaffected or demonstrated HCM. Chiu et al. recently demonstrated this same nonsense mutation in one out of 252 HCM cases8. In close similarity with previously described HCM-associated PLN promoter variants, the L39X proband hosting this heterozygous mutation was a female and diagnosed late in life at 61 years of age with HCM upon development of palpitations, syncope and dyspnea, and demonstrated atrial fibrillation.
In our HCM cohort, heterozygous PLN-L39X was identified in a male, diagnosed at 58 years of age, demonstrating paroxysmal atrial fibrillation. The proband was genotype-negative for the nine canonical HCM-associated myofilament genes as well as the three HCM phenocopy-associated genes. Furthermore, while HCM phenocopy diseases can present with isolated left ventricular hypertrophy, the 58-year-old proband did not demonstrate additional clinical findings which might suggest a non-HCM diagnosis such as skeletal muscle myopathy, mental retardation, or ophthalmic abnormalities commonly associated with Danon’s disease18, and acroparesthesias, angiokeratoma, corneal and lenticular opacities, and anhidrosis variably associated with Fabry’s disease19,20.
In agreement with previous findings, our PLN-L39X proband demonstrated cardiac hypertrophy with a family history suggestive of an autosomal dominant mode of inheritance as both his mother, sister, and granddaughter have HCM. The PLN-L39X mutation cosegregates with incidence of HCM in this pedigree, and absence of an HCM phenotype in the proband’s genotype-positive daughter at the present time indicates incomplete penetrance of the disease. While all HCM-positive members of this pedigree host the PLN-L39X mutation, the relatively small size of the reported family, and the number of individuals available for genotyping, prevents formal quantification of the strength of this cosegregation with a logarithm of the odds score. The absence of a family history of heart failure or DCM in this pedigree, as well as a normal ejection fraction by the proband, supports the conclusion that heterozygous PLN-L39X is an HCM-predisposing mutation specifically. Importantly, future studies identifying the PLN-L39X variant in a larger family with HCM are required to validate this possibility. In addition, given that the frequency of atrial fibrillation in our cohort is 21%, the identification of this arrhythmia in all four PLN-HCM mutation cases described to date is notable. Again, further studies are required to elucidate any potential association between PLN function and atrial fibrillation.
Our data further suggests that genetic variation in PLN is rare and might even contain a relative ‘hot-spot’-termination mutation at position 39. In an effort to determine the prevalence of PLN mutations in HCM patients across multiple cohorts, we identified several studies in the literature which genetically interrogated the coding region of PLN across multiple index-case cohorts from variable ethnic regions. These results are summarized in Table 2. Four studies which genotyped small HCM cohorts of Japanese22, Spanish21, Northern Greek27, and European Individuals (Swiss and Germany)28 did not identify coding region mutations in PLN. Three putative mutations, not found in healthy individuals, were identified in the promoter region of PLN including the previously mentioned 1/87 Japanese case (A>G-77)22, 1/101 Spanish case (C>G −42)21, and 1/252 Australian case (L39X)8.
Table 2:
Summary of PLN coding region mutations identified across multiple proband-based cohorts
| HCM Cohort | Promoter | Coding Region | ||||
|---|---|---|---|---|---|---|
| N | Ethnicity | Mutations | Prevalence (% Frequency) | Mutations | Prevalence (% Frequency) | Reference |
| 87 | Japanese | A>G −77 | 1/87 (1.15) | - | 0/87 | 22 |
| 101 | Spanish | C>G −42 | 1/101 (0.99) | - | 0/101 | 21 |
| 53 | Greek | - | 0/53 (0) | - | 0/53 | 27 |
| 38 | European | - | 0/38 (0) | - | 0/38 | 28 |
| 252 | Australian | Not reported | Not reported | L39X | 1/252 (0.40) | 8 |
| 1064 | North American | C>T −235, A>C −198, A>G −120, T>C −114, G>T −47 | 5/1064 (0.47) | L39X | 1/1064 (0.094) | |
| Overall | 7/1343 (0.52) | 2/1595 (0.13) | ||||
N, number of probands genotyped for PLN
PLN Genotyping in HCM
Across the six independent cohorts we identified in the literature, including our own, we identified 2 probands hosting the L39X premature truncation among 1605 cases genotyped – a yield of 0.13%. Incorporation of promoter variants identified which were not found in healthy control populations (7/1343, 0.52%) brings the overall yield of PLN genetic interrogation to 0.65% in HCM. We have shown previously that the prevalence of mutations in some canonical sarcomeric-HCM genes including TNNC1 (0.4%), TPM1-encoded alpha-tropomyosin (0.5%), and ACTC-encoded actin (0.3%) are similar.
Importantly, across the 600 reference alleles which were genotyped for PLN genetic variants, we did not identify any amino acid altering variation, nor did we identify any rare promoter variants which might be considered “false positive” results for PLN genetic testing. Indeed, identification of two well-documented common polymorphisms in the promoter and 5’ UTR constituted all “healthy” genetic variability in PLN. Further, while the yield of genetic interrogation of PLN in HCM probands is low, comprehensive genotyping can be accomplished in just two amplicons, which may argue favorably for inclusion on the genetic test for HCM especially since the interpretative signal-to-noise ratio for a particular PLN mutation would be favorable29.
CONCLUSIONS
Mutations in PLN, such as the L39X, are rare among patients with HCM. However, despite the low yield of PLN-associated HCM genetic testing, the small size of PLN, and the paucity of genetic variation in PLN among healthy subjects, warrant consideration for its inclusion in clinically available HCM gene test panels. Whether or not perturbations in phospholamban are directly responsible for the observed phenotype of HCM with atrial fibrillation in the PLN-positive subjects requires further investigation.
Footnotes
DISCLOSURES
MJA is a consultant for Medtronic, St. Jude Medical, Inc., Boston Scientific, and PGxHealth and serves on PGxHealth’s Scientific Advisory Board.
REFERENCES
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults : Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Circulation. 1995; 92:785–789. [DOI] [PubMed] [Google Scholar]
- 2.Maron B, Roberts W, McAllister H, Rosing D, Epstein S. Sudden death in young athletes. Circulation. 1980; 62:218–229. [DOI] [PubMed] [Google Scholar]
- 3.Maron B, Epstein S, Roberts W. Causes of sudden death in competitive athletes. J Am Coll Cardiol. 1986; 7:204–214. [DOI] [PubMed] [Google Scholar]
- 4.Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004; 44:602–610. [DOI] [PubMed] [Google Scholar]
- 5.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003; 108:445–451. [DOI] [PubMed] [Google Scholar]
- 6.Van Driest SL, Vasile VC, Ommen SR, Will ML, Gersh BJ, Nishimura RA, Tajik AJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004; 44:1903–1910. [DOI] [PubMed] [Google Scholar]
- 7.Landstrom AP, Weisleder N, Batalden KB, Martijn Bos J, Tester DJ, Ommen SR, Wehrens XHT, Claycomb WC, Ko J-K, Hwang M, Pan Z, Ma J, Ackerman MJ. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007; 42:1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiu C, Tebo M, Ingles J, Yeates L, Arthur JW, Lind JM, Semsarian C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2007; 43:337–343. [DOI] [PubMed] [Google Scholar]
- 9.Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, Bos JM, Tester DJ, Ommen SR, Potter JD, Ackerman MJ. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J Mol Cell Cardiol. 2008; 45:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. Journal of Biological Chemistry. 1986; 261:13333–13341. [PubMed] [Google Scholar]
- 11.Inui M, Chamberlain BK, Saito A, Fleischer S. The nature of the modulation of Ca2+ transport as studied by reconstitution of cardiac sarcoplasmic reticulum. J Biol Chem. 1986; 261:1794–1800. [PubMed] [Google Scholar]
- 12.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan G-C, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW II, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Invest 2003; 111:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gollob MH, Green MS, Tang ASL, Gollob T, Karibe A, Hassan A-S, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Tapscott T, Gonzales O, Begley D, Mohiddin S, Roberts R. Identification of a Gene Responsible for Familial Wolff-Parkinson-White Syndrome. N. Engl. J. Med 2001; 344:1823–1831. [DOI] [PubMed] [Google Scholar]
- 14.Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R. Novel PRKAG2 Mutation Responsible for the Genetic Syndrome of Ventricular Preexcitation and Conduction System Disease With Childhood Onset and Absence of Cardiac Hypertrophy. Circulation. 2001; 104:3030–3033. [DOI] [PubMed] [Google Scholar]
- 15.Sakuraba H, Oshima A, Fukuhara Y, Shimmoto M, Nagao Y, Bishop DF, Desnick RJ, Suzuki Y. Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am J Hum Genet. 1990; 47:784–789. [PMC free article] [PubMed] [Google Scholar]
- 16.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000; 406:906–910. [DOI] [PubMed] [Google Scholar]
- 17.Yang Z, McMahon CJ, Smith LR, Bersola J, Adesina AM, Breinholt JP, Kearney DL, Dreyer WJ, Denfield SW, Price JF, Grenier M, Kertesz NJ, Clunie SK, Fernbach SD, Southern JF, Berger S, Towbin JA, Bowles KR, Bowles NE. Danon Disease as an Underrecognized Cause of Hypertrophic Cardiomyopathy in Children. Circulation. 2005; 112:1612–1617. [DOI] [PubMed] [Google Scholar]
- 18.Charron P, Villard E, Sebillon P, Laforet P, Maisonobe T, Duboscq-Bidot L, Romero N, Drouin-Garraud V, Frebourg T, Richard P, Eymard B, Komajda M. Danon’s disease as a cause of hypertrophic cardiomyopathy: a systematic survey. Heart. 2004; 90:842–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoigné P, Attenhofer Jost CH, Duru F, Oechslin EN, Seifert B, Widmer U, Frischknecht B, Jenni R. Simple criteria for differentiation of Fabry disease from amyloid heart disease and other causes of left ventricular hypertrophy. Int. J. Cardiol 2006; 111:413–422. [DOI] [PubMed] [Google Scholar]
- 20.Ackerman MJ, Landstrom AP. Detection of Subclinical Fabry Disease in Patients Presenting With Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2007; 50:2404–2405. [DOI] [PubMed] [Google Scholar]
- 21.Medin M, Hermida-Prieto M, Monserrat L, Laredo R, Rodriguez-Rey JC, Fernandez X, Castro-Beiras A. Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN - 42 C>G mutation. Eur J Heart Fail. 2007; 9:37–43. [DOI] [PubMed] [Google Scholar]
- 22.Minamisawa S, Sato Y, Tatsuguchi Y, Fujino T, Imamura S, Uetsuka Y, Nakazawa M, Matsuoka R. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun 2003; 304:1–4. [DOI] [PubMed] [Google Scholar]
- 23.Santos D, Medeiros A, Brum P, Mill J, Mansur A, Krieger J, Pereira A. No evidence for an association between the −36A>C phospholamban gene polymorphism and a worse prognosis in heart failure. BMC Cardiovasc Disord. 2009; 9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haghighi K, Chen G, Sato Y, Fan G-C, He S, Kolokathis F, Pater L, Paraskevaidis I, Jones WK, Dorn II GW, Kremastinos DT, Kranias EG. A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids. Hum Mutat. 2008; 29:640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003; 299:1410–1413. [DOI] [PubMed] [Google Scholar]
- 26.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan G-C, Tsiapras D, Parekh RR, Dorn GW II, MacLennan DH, Kremastinos DT, Kranias EG. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA. 2006; 103:1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalemi T, Efthimiadis G, Zioutas D, Lambropoulos A, Mitakidou A, Giannakoulas G, Vassilikos V, Karvounis H, Kotsis A, Parharidis G, Louridas G. Phospholamban Gene Mutations Are Not Associated with Hypertrophic Cardiomyopathy in a Northern Greek Population. Biochem. Genet 2005; 43:637–642. [DOI] [PubMed] [Google Scholar]
- 28.Fokstuen S, Lyle R, Munoz A, Gehrig C, Lerch R, Perrot A, Osterziel KJ, Geier C, Beghetti M, Mach F, Sztajzel J, Sigwart U, Antonarakis SE, Blouin J-L. A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum Mutat. 2008; 29:879–885. [DOI] [PubMed] [Google Scholar]
- 29.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AAM, Ackerman MJ. Genetic Testing for Long-QT Syndrome: Distinguishing Pathogenic Mutations From Benign Variants. Circulation. 2009; 120:1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]