

Abstract
Drug accumulation in the malignant tissue is a prerequisite for effective cancer treatment. However, most drug molecules and their formulated particles are blocked en route to the destiny tissue due to the existence of multiple biological and physical barriers including the tumor microvessel endothelium. Since the endothelial cells on the surface of the microvessel wall can be modulated by inflammatory cytokines and chemokines secreted by the tumor or stromal cells, an effective drug delivery approach is to enhance interaction between the drug particles and the unique spectrum of surface proteins on the tumor endothelium. In this study, we performed in vivo screening for thioaptamers that bind to the bone marrow endothelium with specificity in a murine model of lymphoma with bone marrow involvement (BMI). The R1 thioaptamer was isolated based on its high homing potency to bones with BMI, and 40–60% less efficiency in accumulation to healthy bones. In cell culture, R1 binds to human umbilical vein endothelial cells (HUVEC) with a high affinity (Kd ≈ 3 nM), and the binding affinity can be further enhanced when cells were treated with a mixture of lymphoma cell and bone marrow cell conditioned media. Cellular uptake of R1 is through clathrin-mediated endocytosis. Conjugating R1 on to the surface of liposomal doxorubicin nanoparticles resulted in 2–3 fold increase in drug accumulation in lymphoma BMI. Taking together, we have successfully identified a thioaptamer that preferentially binds to the endothelium of lymphoma BMI. It can serve as an affinity moiety for targeted delivery of drug particles to the disease organ.
INTRODUCTION
Lymphoma is a common malignancy in the lymphatic system, and bone marrow plays an important role in lymphoma development and progression. Clinical studies have revealed that bone marrow involvement (BMI) is common in patients with advanced stages of lymphoma [1–3]. BMI is a key indicator for negative prognosis, and is associated with significantly shorter survival in patients with intermediate- to high-grade non-Hodgkin’s lymphoma [2]. Numerous strategies have been tried to block proliferation and survival of lymphoma cells in the bone marrow, lymph nodes, and other lymphatic organs, such as chemotherapy, radiation therapy, and bone marrow transplantation [4,5]. One common practice in clinic is to overwhelm the body with a large quantity of drugs with the hope to send a sufficient amount of drug molecules to the bone; however, such an approach leads to severe systemic toxicity caused by drug accumulation in non-disease organs [6]. Another strategy is to package drug molecules into nanometer-size particles, such as liposomes and micelles [7–9]. It has been demonstrated that the microvessels inside the tumor tissue are leaky, which allows accumulation and retention of the drug particles [10,11]. However, there are many biological barriers for the drug molecules to overcome en route to the destiny organ/tissue/cell [12], nanoformulation only enable them to pass a number of them. Most drug nanoparticles will be blocked at the rest checkpoint steps. A recent analysis revealed that, on average, less than 1% of the total injected drug particles could reach the tumor tissues successfully [13], highlighting the challenge in development of effective cancer nanotherapeutic agents [14].
The unique structure of the bone marrow poses an additional barrier to target the lymphoma cells. Inside the bone marrow, the sinusoidal vessels are surrounded by a tight layer of perivascular reticular cells [15]. Although affinity moieties have been identified to target specific elements in bone marrow environment including osteoclasts [16], osteoblasts[7] and bone hydroxyapatite [17], the drug particles will need to pass the vascular barrier before reaching the malignant cells that reside in the endosteal and vascular niches [15]. Thus, effective approach for drug delivery to the lymphoma BMI is to target the unique features in the bone marrow vasculature.
Aptamers are single strand DNA or RNA sequences with unique 3-dimentional structures that are capable of recognizing and binding to their targets with high affinity. With a technique called systematic evolution of ligands by exponential enrichment (SELEX) [18,19], aptamers can be easily selected from a pooled oligonucleotide library towards defined targets. By screening aptamers based on protein or cells in vitro, a series of aptamers have been identified on lymphoma targeting and therapies [20–26]. However, the composition and spatial arrangement of abnormal microenvironment in bone marrow induced by lymphoma may provide an extra shield to hide the designated targets, and make them unreachable. Furthermore, aptamers cross-binding to blood components, which may cause the failure of the aptamer candidates, will be quickly eliminated during blood circulation.
Alternatively, in vivo SELEX in live animal, which a library was injected into a murine model of disease, and then organs or cells of interest were harvested for aptamer selection, was applied to select organ specific aptamers [26–28]. Although the actual protein or cell targets probably stays unknown, selected aptamers are more likely able to bind to the indicated organ and accumulate thereupon. Furthermore, the selected aptamer may serve as delivery guidance of drugs or diagnosis probes due to its natural homing ability.
In this study, live animal based SELEX technique was applied to isolate thioaptamers against Burkitt’s lymphoma. To identify thioaptamers with enhanced affinity to bone marrow involvement of Burkitt’s lymphoma, a DNA thioaptamer library, in which thiol modification ensures the oligo stability in vivo [29], was injected to mice with Raji lymphoma BMI in vivo, and the bone marrow was collected for thioaptamer screening. After 10 rounds of selection, several thioaptamer candidates were selected for validation of bone homing ability. Accumulation in healthy bones was also tested and compared. One of the candidates was found highly interacted with the endothelial cells in bone marrow, and the uptake depended on the presence of lymphoma, which suggested a drug recruitment in diseased tissues. After conjugation with liposome, the thioaptamer successfully promoted the accumulation of doxorubicin in femur, tibia and spine with lymphoma involvement.
MATERIALS and METHODS
Cell culture
Human Burkitt’s lymphoma cell lines Raji and Daudi, human T-anaplastic large cell lymphoma cell line KARPAS 299 (K299), human mantle cell lymphoma cell line Mino, acute monocytic leukemia THP-1 and non-small cell lung cancer cell line H358 were cultured in RPMI 1640 (Corning, USA) supplemented with 10% fetal bovine serum (FBS, GIBCO, USA) and 100 U/mL penicillin-streptomycin. Raji cells were transduced with a Plenti6/V5-DEST-luciferase lentivirus and selected in 3 ^g/mL blasticidin (InvivoGen, USA) to establish luciferase-expressing clones (Raji/Luc). Human MDA-MB-231 breast cancer cells were maintained in Dulbecco’s modified eagle medium (DMEM, Corning, USA) with 10% FBS and 100 U/mL penicillin-streptomycin. Human umbilical vein endothelial cells (HUVEC) were obtained from PromoCell (Germany) and maintained in Medium 200 with low serum growth supplement (GIBCO, USA). All cell cultures were maintained at 37°C with 5% CO2.
Murine models of human Burkitt’s lymphoma bone marrow involvement
All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Houston Methodist Research Institute in Houston, Texas. To generate murine models of Burkitt’s lymphoma, each of the 6–10 week old female SCID/beige mice (Charles River Laboratories) was inoculated with 1×106 Raji/Luc cells by tail vein injection. Tumor growth was monitored with a Xenogen IVIS-200 imaging system. All mice developed bone marrow involvement two weeks later, and lymph node enlargement was detected in half number of them. Mice with intensive luciferase signals from the femur, tibia and spine 3 weeks after inoculation were applied for in vivo SELEX. Mice with similar tumor growth status were grouped randomly for ex vivo and in vivo biodistribution studies.
Preparation of duplex thioaptamer library
A library of random single strand DNA (ssDNA) oligonucleotides without backbone modifications was obtained from The Midland Certified Reagent Company (Midland, TX). This library consisted of a 30 base pair random region flanked by a 21 base 5’-primer (5’-CGCTCGATAGATCGAGCTTCG-3’) and a 23 base 3’-primer (5’-GTCGATCACGCTCTAGAGCACTG-3’). To synthesize a duplex thioaptamer library, the ssDNA random library was annealed with a reverse primer and incubated with Taq DNA polymerase and a mixture of dATP, dTTP, dCTP, and dGTP (800 μM each) for 5 hours at 37 °C to synthesize the reverse strand. The library (500 nM) was then amplified by PCR using forward primer (400 nM), 5’-biotinylated reverse primer (400 nM), MgCl2 (2.5 mM), Taq DNA polymerase (5 units) and a mixture of αS-dATP, dTTP, dCTP, and dGTP (300 μM each). PCR was run for 20 cycles (94 °C for 1 min, 59 °C for 1 min, and 72 °C for 1 min). This resulted in a library containing monothiophosphate substitutions on the 5’ side of every dA residue except for those in the forward and reverse PCR primers. PCR amplification was repeated using the selected ssDNA oligonucleotides following each selection cycle. PCR products were filtered through a Millipore YM-30 filter to remove excess reagents and isolate dsDNA product. The dsDNA product was combined with streptavidin coated magnetic beads in a buffer containing 10 mM Tris-HCl, 2M NaCl, and 1 mM EDTA (pH 7.5) and was incubated for 15 min. Biotin on the reverse strand of the dsDNA binds to the streptavidin beads. The beads were washed 3 times with buffer to remove any unbound DNA. Melting solution (0.1 M NaOH) was then added to separate the two DNA strands. Biotinylated reverse strands remained bound to the beads while the nonbiotinylated forward strands were collected in the supernatant. The forward strands were filtered through Millipore YM-10 filter and collected for selection rounds.
In vivo SELEX for lymphoma-homing thioaptamers
To initiate in vivo SELEX, a library of 10 μg thioaptamers was injected via tail vein into SCID/beige mice with Raji lymphoma. Mice were sacrificed 4 hours later, and femur and tibia were flushed with PBS to collect bone marrow and lymphoma cells. Cell-bound thioaptamers were isolated and applied to generate a mini-library for the next round in vivo SELEX screening. After 10 rounds of in vivo SELEX enrichment, cell-bound thioaptamers were PCR amplified using normal dNTPs with extended primers that contained one of the three short additional sequences (bar codes) on one of the primers, and the resulting sequence mixture was sent to SeqWright Genomic Services for next generation sequencing. The fastq file was analyzed using the Aptaligner software and a library design file [30]. Bottom-up clustering of the top 250 aptamers was automatically performed with the Aptaligner software. Structure predictions for the aptamers were determined with Mfold [31].
Synthesis and purification of individual thioaptamers
Individual thioaptamers-of-interest were synthesized on an Expedite 8909 Oligo Synthesizer using standard phosphoramidite chemistry and manufacturer-recommended monomer coupling times, as previously described [29]. Deprotection of dye-labeled thioaptamers was conducted for 24 hours at room temperature with ammonium hydroxide to preserve the dye. All thioaptamers were purified by reverse phase chromatography on a semi-prep scale Hamilton PRP-1 column running on an AKTA-10 purifier system (GE) at a flow rate of 2 ml/min. They were loaded in 100% Buffer A (100 mM triethylamine acetate, pH 8.4, TEAA), stepped up to 8 to 12% Buffer B (acetonitrile), and eluted with increasing Buffer B gradient at the rate of 1% per 3–4 minutes up to ~25–30%. The fractions corresponding to well-resolved thioaptamer peaks were analyzed with 15% polyacrylamide gel electrophoresis to determine their purity and integrity prior to pooling and concentration by freezing drying or centrifugation in Amicon 3kD WMCO filters. The final concentrations of thioaptamer solutions and dye labeling efficiencies were determined spectrophotometrically by measuring OD at 260nm (DNA) and 650 nm (Cy5), and using calculated extinction coefficients (OligoCalc at Northwestern University) for the thioaptamers and values for the dyes suggested by the Glen Research website [32].
Binding specificity of thioaptamer to bone marrow cells
Bone marrow cells were prepared to determine binding specificity of selected thioaptamers to different cell types. Briefly, femur and tibia were collected from mice with Raji lymphoma and sheared into small pieces. The tissue samples were digested with 250 unit/mL collagenase type III (Worthington Biochem, USA) at 37°C for 2 hours to prepare single cell suspension. Cells were incubated with 25 nM Cy5-thioaptamer at 37 C for 20 minutes, and then stained with cell specific antibodies from Tonbo Biosciences (USA) or Biolegend (USA) on ice for 30 minutes (T cell: CD45+/CD3+, B cell: CD45+/B220+, Raji cell: HLA-ABC+, endothelial cell: CD45−/CD31+, macrophage: CD11b+/F4/80+/Ly6G−, granulocyte: CD11b+/Ly6G+). All antibodies were 1:100 diluted in PBS containing 2% FBS. Thioaptamer binding potency was measured using a BD LSR Fortessa analyzer and analyzed with the Flowjo 7.6.1 software (Tree Star, USA).
Uptake of thioaptamer by vascular endothelial cells
To analyze thioaptamer-binding affinity to endothelial cells, 1×104 HUVECs were seeded in a 48-well plate and cultured overnight. On the next day, thioaptamers were added into cell culture and cells were kept incubated for another 20 minutes before they were detached from plate with 20 mM EDTA and analyzed with flow cytometry. To identify route of cell entry by the thioaptamer, HUVEC cells were co-incubated with the following inhibitors before co-culture with thioaptamer: cytochalasin D (phagocytosis inhibitor, 100 μM, MP Biomedicals), amiloride (macropinocytosis inhibitor, 500 μM, Sigma-Aldrich), chlorpromazine (inhibitor of clathrin mediated endocytosis, 10 μM, 20 μM, 30 μM and 60 μM, MP Biomedicals), genistein (inhibitor of caveolae mediated endocytosis, 100 μM, Sigma- Aldrich) and dynasore (inhibitor of dynamin-dependent endocytosis, 100 μM, 300 μM and 500 μM, ApexBio Technology). After a 30-minute treatment with the inhibitors, cells were mixed with 12.5 nM of the R1 thioaptamer (R1) and incubated for 20 minutes before they were harvested for flow cytometry analysis. All samples for flow cytometry analysis were prepared in triplicate.
The above procedure was also applied to HUVECs that were activated with conditioned media (CM) for 6 hours. Due to the high binding affinity after stimulation, HUVECs were incubated with a reduced concentration (1 nM) of the R1 thioaptamer. For the competition assay, a 50-fold excess of non-labeled R1 was mixed with Cy5-R1 before incubation with HUVECs. To visualize intracellular trafficking of thioaptamer, cells were seeded in an 8-well chamber slide cell culture system, and then incubated with the thioaptamer (100 nM for naive HUVECs, or 5 nM for activated HUVECs). They were then fixed with 10% phosphate buffered formalin and permeabilized with 0.1% triton X-100. Anti- EEA1 (early endosome marker), anti-Rab7 (late endosome marker) and anti-LAMP1 (lysosome marker) antibodies (Abcam, UK) were 1:300 diluted and incubated with HUVECs overnight at 4°C, followed by incubation with an AlexaFluor-488 conjugated secondary antibody (1:400 dilution, Life Technologies). Images were captured with a Nikon Eclipse 80i fluorescent microscope.
Impact of cell conditioned medium on thioaptamer uptake by vascular endothelial cells
Conditioned media from bone marrow cells isolated from tumor-free SCID/beige mice (4 million cells per mL) and tumor cells (0.5 million cells per mL) were applied to supplement HUVEC growth before cells were used to test thioaptamer binding. Briefly, HUVECs were grown in fresh RPMI-1640 medium either without CM supplement or supplemented with tumor CM (CMT), bone marrow CM (CMBM) or a mixture of CMT and CMBM at a 1:1 (v/v) ratio for 6 hours or otherwise indicated. Cells were washed with PBS and then incubated for 20 minutes with 12.5 nM R1 aptamer in a binding buffer containing 1 mg/mL bovine serum albumin (BSA), 4.5 mg/mL glucose, 0.1 mg/mL yeast t-RNA, and 5 mM MgCl2 in PBS. R1 uptake by HUVECs was assessed with a BD Accuri C6 flow cytometer. All samples were run in triplicate.
Preparation and characterization of thioaptamer-conjugated liposomal doxorubicin (Dox)
Thioaptamer conjugation: R1 or a control thioaptamer was chemically conjugated to the distal end of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000 (DSPE- PEG-Mal, Avanti Polar Lipids, USA) via reaction of thiol with maleimide group (R1 -lipid or Ctrl Apt- lipid, respective). Briefly, thioaptamers with a 5’ thiol group (Integrated DNA Technologies, USA) were treated with tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Thermo Fisher Scientific, USA) to convert any disulfide aptamer dimers back into thiol monomers, and then mixed with DSPE-PEG-Mal micelle in PBS (pH7.0, 50 mM) at room temperature overnight with gentle shaking. Liposomal Dox preparation: Dox-loaded long circulating liposomes were prepared by following a previously described ammonium sulfate gradient method [33]. Briefly, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000 (DSPE-PEG2000) (Corden Pharma International GmbH, Germany) and the above thioaptamer-micelle (in aqueous solution) were mixed at the molar ratio of 63:32:4.5:0.5. The mixture was lyophilized to remove solvent. Lipid powder was then hydrated with 150 mM ammonium sulfate and sonicated until clear. Unencapsulated ammonium sulfate was removed by dialysis against 1 L 20 mM HEPES buffered saline (pH 7.4) for 4 hours in a dialysis tubing with 10,000 Da molecular weight cut off (Thermo Fisher Scientific, USA). Next, Dox was added into empty liposomes at a drug-to-lipid ratio of 1:10 (w/w) and incubated at 60 °C for 1 hour with gentle shaking. Unencapsulated Dox was removed by dialysis as described above. Liposomal Dox characterization: To calculate drug-loading efficiency, a 2-fold (Vol.) 1% triton X-100 in methanol was added to the liposomes, and Dox content was measured based on absorbance at 480 nm. Dynamic light scattering (DLS) measurement with a Zetasizer Nano ZS (Malvern, UK) was applied to monitor size distribution of the liposomes.
Biodistribution of thioaptamers and bone marrow-targeted liposomal doxorubicin
To determine biodistribution and bone marrow-homing efficiency of selected thioaptamers, each mouse with Raji lymphoma was treated i.v. with 0.5 nmol Cy5-labeled thioaptamer (n = 3–4 mice/group). Mice were euthanized 4 hours post injection, and major organs including the bone were collected. Thioaptamer content in each organ was quantified based on fluorescent intensity from Cy5 with an IVIS-200 imaging system. Thioaptamer distribution in the femur and tibia was further evaluated based on immunohistochemical staining. Briefly, tissue samples were fixed in 10% formalin overnight, decalcified with phosphate buffered 500 mM ethylenediaminetetraacetic acid (EDTA, pH 7.4) and processed for tissue staining with an anti-mouse E-selectin antibody (Abcam, USA). Unlike endothelial cells in other organs, one of the important features of bone marrow endothelial cells is the constitutive expression of E-selectin [34,35].
To determine biodistribution of bone marrow-targeted liposomal doxorubicin, SCID/beige mice with Raji (n = 4 mice/group) were treated i.v. with 6 mg/kg dox-equivalent thioaptamer-conjugated liposomal Dox. Mice were sacrificed 28 hours post drug treatment, and Dox was extracted from blood and major organs. Dox content was analyzed by high performance liquid chromatography (HPLC) with a Zorbax 300SB-C18 reverse phase column (Agilent, USA) and a fluorescence detector (Hitachi LaChrom Elite, Japan), and daunorubicin was used as an internal standard [36].
Statistical analysis
Statistical analysis was performed with the GraphPad Prism 5 program (GraphPad Software, Inc, California, USA). Comparison between two values was evaluated with a two-tailed, unpaired Student’s t-test. Pairs of values in multiple groups were analyzed by one-way ANOVA with Tukey’s correction method. When comparing multiple values to a single value, such as negative control or R1, one-way ANOVA was used with Dunnett’s correction for multiplicity adjustment. P <0.05 (*) and P<0.01 (**) indicated statistically significant and very significant, respectively. Data are expressed as means ± SD. Sample size was pre-determined based on preliminary results and the number of qualified mice.
RESULTS
In vivo SELEX identifies lymphoma bone marrow involvement-enriched thioaptamers
Lymphoma mice with BMI were systematically characterized before they were applied for in vivo SELEX. Intensive Raji/Luc tumor growth could be detected in the lymph nodes, spine, femur and tibia based on bioluminescence (Fig. 1A), and confirmed by histological analysis (Fig. 1B). Thioaptamers were enriched in the lymphoma-bearing organs after 10 rounds of in vivo SELEX (Fig. 1C), and four oligos (R1, R2, R3 and R4) with the highest level of bone marrow accumulation based on deep sequencing were listed (Fig. 1D). The thioaptamers were resistant to plasma deoxyribonuclease (DNase) digestion in an in vitro setting, and intact fragments could still be detected by gel electrophoresis after more than 7 hours of incubation in peripheral blood (Supplementary Fig. 1). Inside the body, high plasma R1 concentration was maintained within the first hour (Supplementary Fig. 2).
Figure 1:
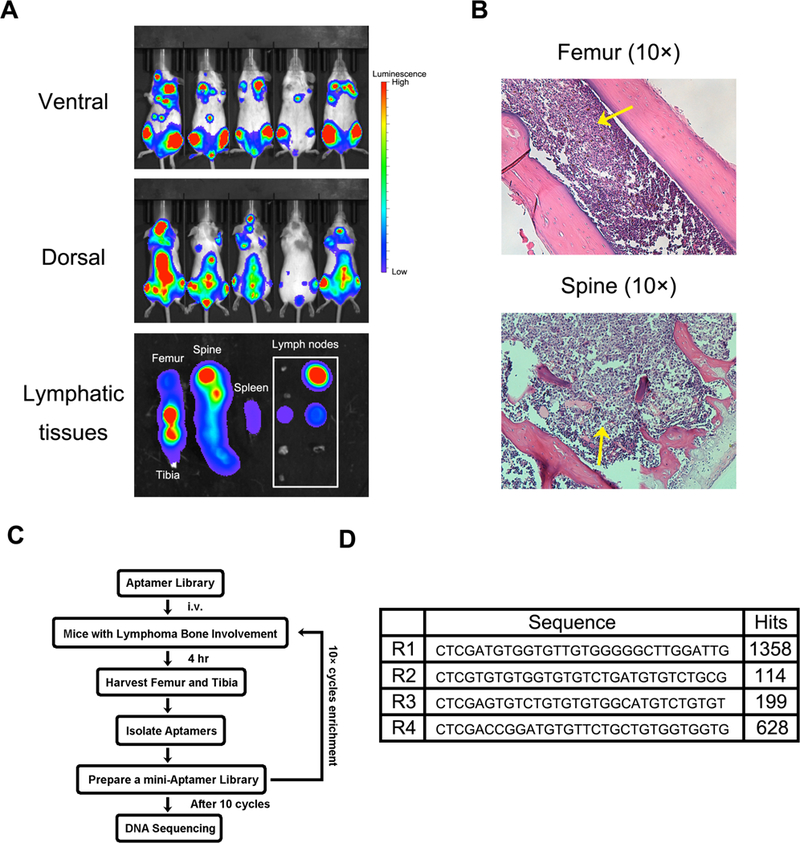
Murine model of human Burkitt’s lymphoma with bone marrow involvement for in vivo SELEX of aptamer. (A). Growth of Raji cells with luciferase gene in SCID/Beige mice was monitored by an Xenogen IVIS-200 system. Pictures were taken on both ventral and dorsal sides. The ex vivo images showed lymphoma in lymphatic tissues (femur, tibia, spine, spleen and lymph nodes). Lymphoma cells widely invaded into bones and lymph nodes, but less were observed in spleen. (B). Histological analysis of Raji lymphoma in medullary cavities of spine and femur. Raji cells invaded into bone marrow and effaced normal tissues. Yellow arrows indicate the tumor nodules. (C). Schematic diagram of in vivo SELEX process.
Biodistribution analysis was performed to confirm enrichment of the selected thioaptamers in lymphoma with BMI. Overall, all 4 thioaptamers accumulated more in the relevant organs (spine, femur, tibia) comparing to the negative control (Fig. 2A). In addition, high thioaptamer concentration was detected in kidney, indicating a rapid renal clearance due to their relatively small size (24 kD molecule weight) and high solubility (Fig. 2A-B). Quantitative analysis revealed high enrichment in the spine (2.6 folds and 2.3 folds, respectively) and femur and tibia (3.4 folds and 2.6 folds, respectively) by R1 and R4 over the control thioaptamer (Fig. 2C). R1 also displayed a preference to the disease over the healthy organs (Fig. 2A), and its level was elevated by 2.3 folds in the spine and 1.8 folds in the femur and tibia in lymphoma mice over lymphoma-free mice (Fig. 2D). Although R2 and R3 also accumulated in lymphoma with BMI, the degree of enrichment was not as significant as R1 and R4, a result confirmed by histological analysis (Fig. 2E).
Figure 2.
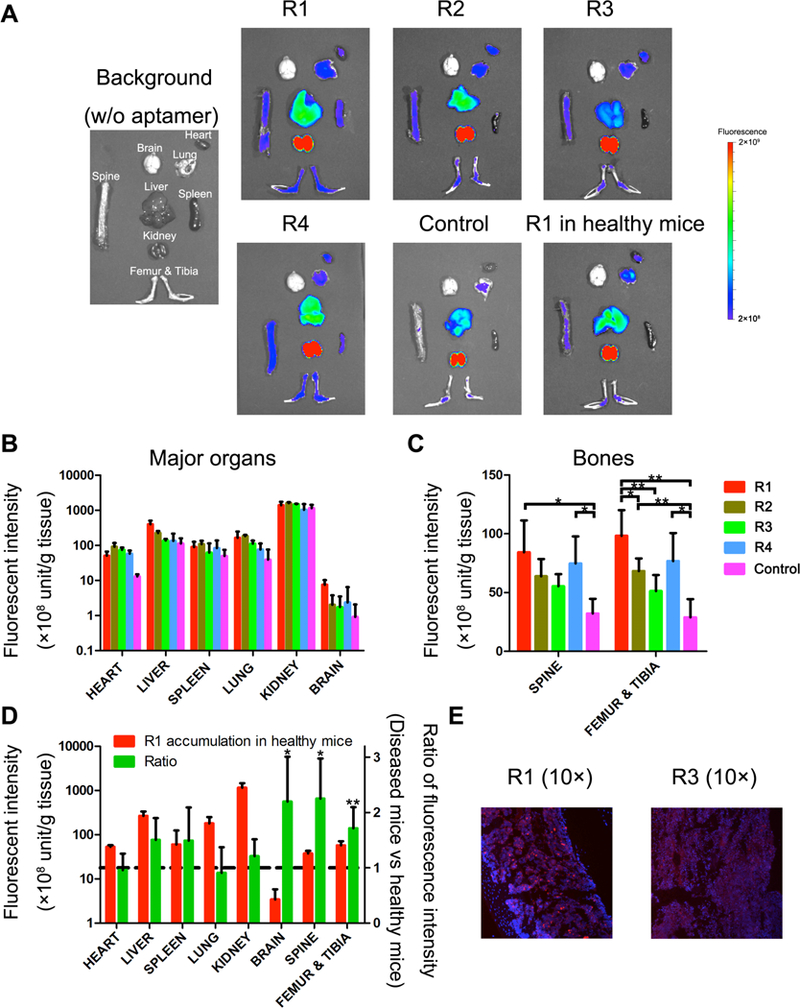
Accumulation of Cy5-aptamers in major organs. Aptamers selected from in vivo SELEX and control aptamers were intravenously injected into mice with Raji lymphoma BMI (0.5 nmol per mouse, n=3–4). Mice were sacrificed 4 hours post injection, and tissues of interest were collected for fluorescence analysis of aptamer content. R1 was also dosed to healthy mice, and mice without aptamer administration were measured for adjustment of figure setting and subtraction of autofluorescence background. (A). Representative figures of fluorescence imaging. Fluorescence intensity of soft organs (B) and bones (C) ex vivo were quantified and plotted. Data were normalized by weight of the organs. (D). Fluorescent signals on healthy mice and diseased mice were compared. Left Y-axis: Fluorescence intensity, right Y-axis: ratio of aptamer in diseased mice versus healthy mice. (E). Histological analysis of Cy5-R1 and Cy5-R3 aptamers in the bone marrow of femur samples. Red: Cy5 labeled aptamer, Blue: DAPI stained nuclei.
Tihioaptamers bind to bone marrow endothelium with high affinity and specificity
There exist multiple cell types inside the bone marrow in a lymphoma-bearing mouse including adipocytes, endothelial cells, fibroblasts, leukocytes, osteoblasts and osteoclasts. In order to identify cells that preferentially interacted with the thioaptamers, we treated lymphoma mice with Cy5-labled thioaptamers, and analyzed cell populations from in spine, femur and tibia. While Raji cells and the hematopietic cells did not show a significant binding to the thioaptamers, a large number of endothelial cells were associated with the thioaptamers, especially with R1 (Fig. 3A), indicating that the bone marrow endothelium might be the target for the thioaptamers.
Figure 3.
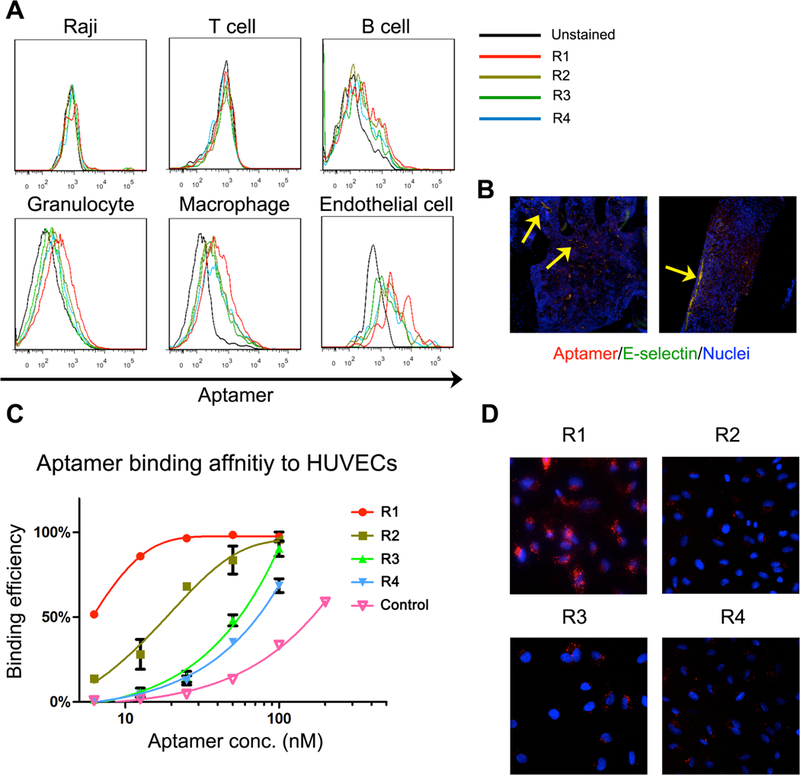
Identification of targeted cells of selected aptamers. (A). Representative figures of flow cytometry analysis. Bone marrow cells were stained with Cy5-aptamers, and different cell populations were isolated by flow cytometry for aptamer analysis. R1 aptamer was found bound to endothelial cells with higher affinity. (B) Distribution of R1 aptamer in bone marrow. Blood vessels were costained with E-selectin antibody. Yellow arrows indicated the binding of R1 on bone marrow endothelium (yellow spots). (C). Binding affinity of aptamers to HUVECs was evaluated by flow cytometry. R1 was able to bind to HUVECs with high affinity. (D). Fluorescence microscope images of HUVECs with Cy5-aptamers. Red: Cy5 labeled aptamer, Blue: DAPI stained nuclei.
We measured thioaptamer-binding affinity to HUVEC, an endothelial cell lines that has been widely used in endothelium-related studies [37]. R1 had the highest binding affinity to HUVECs with an equilibrium dissociation constant (Kd) of 3 nM, while R2 had a moderate binding affinity (Kd =13.8 nM), and other two aptamers showed weaker binding to the cells with bind affinities above 50 nM (Fig. 3B). Consistent with the binding assay results, fluorescent imaging revealed that R1 had the strongest interaction with HUVECs among all 4 thioaptamers (Fig. 3C, 3D). In addition, R1 binding did not trigger HUVEC cell death (Supplementary Fig. 3). Thus, R1 was chosen for all further studies.
R1 enters endothelial cells through clathrin-mediated endocytosis
Cy5-labeled R1 (Cy5-R1) was incubated with HUVECs, and cell uptake of the thioaptamer was monitored. The red fluorescent Cy5-R1 could be co-localized with the EEA1-positive early endosomes (in green) 30 minutes after incubation (Fig. 4A), indicating that the thioaptamer entered endothelial cells through the vesicular transport system. Between the 1h and 3 h time points, we observed increasing separation of the red Cy5-R1 from the green early endosomes, and most red fluorescent Cy5-R1 dots did not co-localize with the Rab7-positive late endosomes or LAMP1-positive lysosomes (Fig. 4A), suggesting that the thioaptamers had gradually left vesicles and entered the cytosol. To identify routes of R1 internalization, we pre-treated HUVECs with inhibitors of different cell entry pathways followed by aptamer co-incubation. R1 uptake by HUVECs was significantly blocked by the clathrin-mediated endocytosis inhibitors dynasore and chlorpromazine, but not affected by inhibitors of phagocytosis (cytochalasin D), macropinocytosis (amiloride) or caveolar-mediated endocytosis (genistein) (Fig. 4B).
Figure 4.
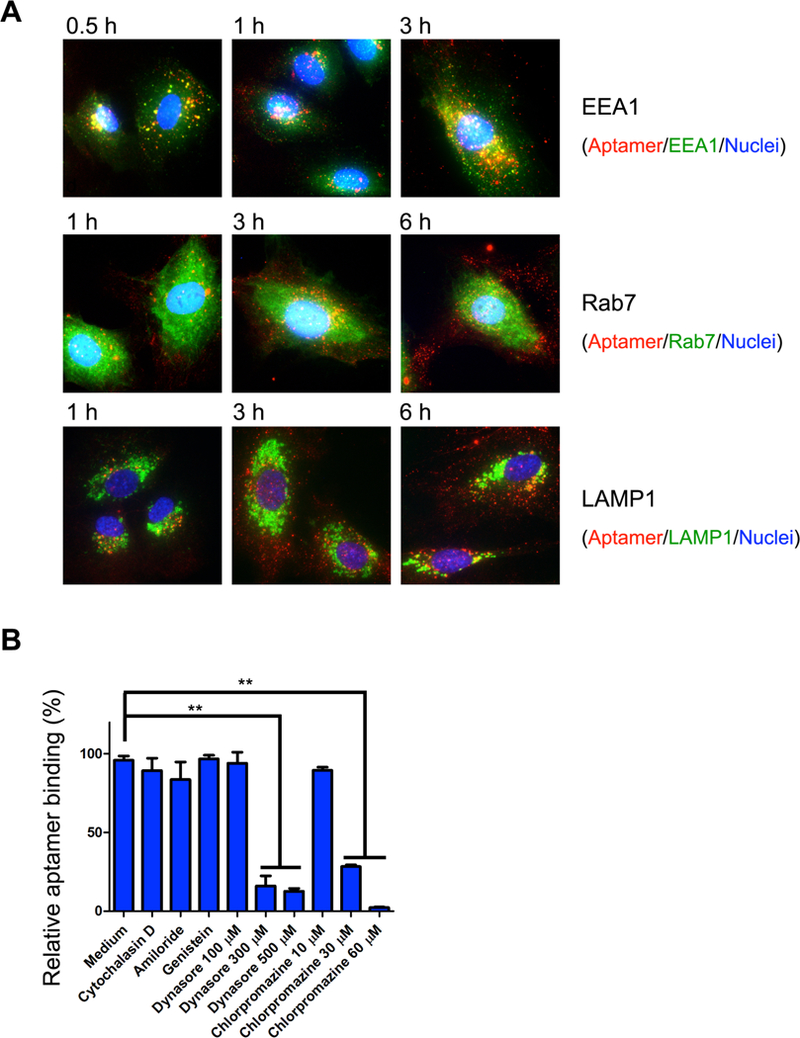
Internalization of R1 aptamer in HUVECs. (A) HUVECs were incubated with Cy5-R1 aptamer, and then fixed and co-stained with EEA1 (early endosome marker), Rab7 (late endosome marker) or LAMP1 (lysosome marker) antibodies, in the top, middle, and bottom rows, respectively. Red: Cy5-R1 aptamer, Green: organelle markers (EEA1, Rab7 or LAMP1), Blue: DAPI stained nuclei. (B) Identification of internalization pathway. HUVECs were pre-treated with endocytosis inhibitors, and then incubated with Cy5-R1 aptamer. Dose dependent attenuation of R1 uptake was found after chlorpromazine or dynasore pre-treatments, which indicated R1 uptake is mediated by the clathrin dependent pathway.
Tumor and stromal cell-secreted factors promote R1 binding by endothelial cells.
We have previously shown that, inside the tumor-growing bone marrow, tumor cell-secreted inflammatory cytokines promote expression of endothelial cell surface proteins, such as E-selectin, which serve as docking sites for targeted delivery of drug particles to the bone [38]. In order to determine whether R1 binding to and uptake by endothelial cells could also be modulated in a lymphoma BMI microenvironment, we incubated HUVECs with bone marrow cell conditioned medium (BM CM), Raji cell conditioned medium (Raji CM), or a mix of BM CM and Raji CM, and measured R1-HUVEC binding affinity. BM CM or Raji CM alone only had a low to moderate effect on R1 binding. Surprisingly, R1 binding was dramatically enhanced by HUVECs pretreated with a mixture of BM CM and Raji CM (Fig. 5A). In addition, the ratio between the two CM had an impact on R1 binding, with an equal ratio providing the highest capacity (Fig. 5B).
Figure 5.
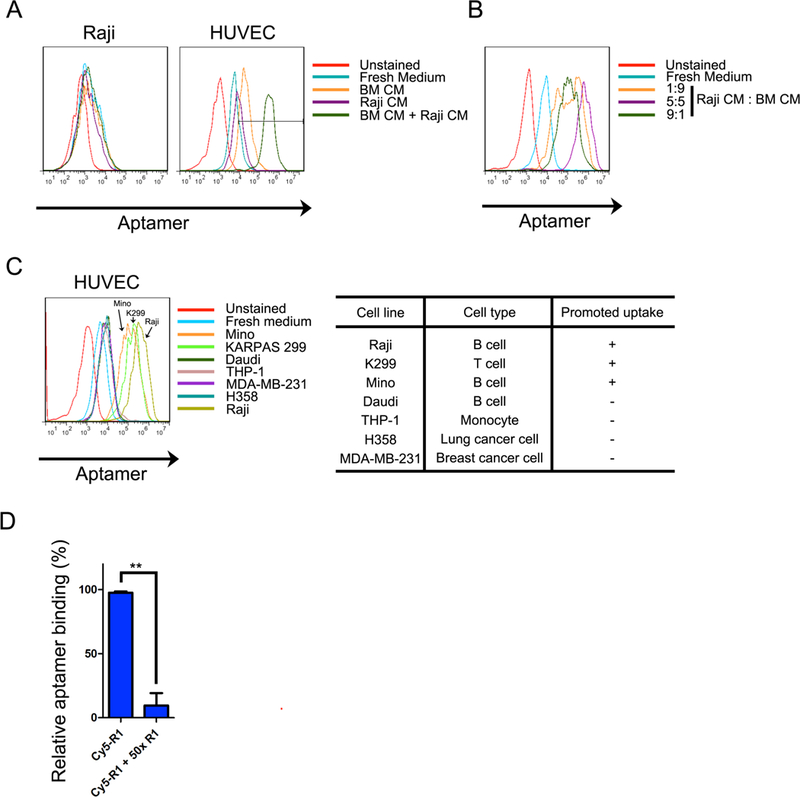
Accelerated uptake of R1 aptamer on HUVECs treated with mixed Raji/BM CMs. (A). Raji or HUVECs were treated with Raji CM, BM CM or their mixture thereof prior to R1 incubation. R1 uptake was assessed with flow cytometry, and representative figures are shown. R1 uptake of HUVECs was enhanced with co-incubation of mixed Raji/BM CMs. Such phenomenon cannot be found in Raji cells. (B). Impact of CM mixture ratio. (C). Enhanced R1 uptake on HUVECs activated with BM CM and CMs from selected cells. Most of lymphoma CMs, but not myeloid leukemia or solid tumor cell lines, can accelerate R1 uptake. (D) Internalization of R1 on CM activated HUVECs. HUVECs were stimulated by mixed Raji CM and BM CM, followed by R1 incubation for different periods. Cells were then co-stained with anti-EEA1 antibody to visualize early endosomes. (E) Identification of internalization pathways. Activated HUVECs were treated with different endocytosis inhibitors, and incubated with 1 nM Cy5-R1 afterwards. (F) Competitive inhibition of cellular uptake by flow cytometry. Binding of Cy5-R1 was measured with or without co-incubation of 50-fold non-labeled R1.
To determine whether enhanced aptamer binding capacity was Raji cell-specific, we collected conditioned media from a group of malignant cells including lymphoma, leukemia and solid tumor cells, and mixed them individually with BM CM to treat HUVECs. We observed dramatically enhanced HUVEC binding of the R1 aptamer in media from 3 of the 4 lymphoma cell lines (Raji, K299, and Mino); in comparison, media from the 2 epithelial tumor cell lines (MDA-MB-231 and H358) or one leukemia cell line (THP-1) did not have a comparable effect (Fig. 5C). Thus, enhanced R1 binding by endothelial cells is promoted by lymphoma cell-derived factors only. In addition, binding of the Cy5- labeled R1 aptamers (Cy5-R1) to HUVECs could be blocked by an excessive amount of unlabeled R1 aptamers (Fig. 5D).
R1 serves as an affinity-targeting moiety for lymphoma BMI-specific drug delivery.
Lymphomas are traditionally treated with cytotoxic chemotherapy drugs such as the R-CHOP containing rituximab, cyclophosphamide, doxorubicin (Dox), vincristine and prednisone [39]. However, heart accumulation of Dox induces severe cardiac toxicity, which limits the overall life time dosage a patient can receive for the drug. The unique binding specificity from R1 to the tumor endothelium indicates that the thioaptamer may serve as an affinity moiety for targeted delivery of therapeutic agents to lymphoma BMI, which not only changes the overall biodistribution pattern of the therapeutic agent but also reduces dosage needed for cell killing activity.
A lymphoma BMI-targeted liposomal Dox (R1-lipo Dox) and a control liposomal Dox formulation (Ctrl-lipo Dox) were prepared by incorporating the R1 or 0tamer-conjugated lipid in the formulation. The average size of liposomes from both formulations was similar (R1 Apt-Dox liposome: 116.7 nm; Ctrl Apt-Dox liposome: 115.2 nm), and both had a narrow polydispersity index (PDI) (Table 1). With an active remote loading technique [40], over 90% of Dox was encapsulated in the liposomes. Comparing to Ctrl-lipo Dox, R1-lipo Dox bound to activated HUVECs with a much higher affinity (Fig. 6A). Drug formulation did not compromise nuclear accumulation of the active drug ingredient. In cell culture, the red Dox molecules co-localized with the green fluorescenr R1 aptamer within the first 30 minutes (pointed with a yellow arrow). Nuclear accumulation of Dox could be visualized at the 1 hour time point, and red fluorescence from Dox in the nuclei was even stronger 3 hours after incubation (Fig. 6B).
Table 1.
Characterization of aptamer conjugated Dox liposome
| Size (nm) | PDI | LE | T1/2α(h) | T1/2β(h) | T1/2Kel (h) | AUC (μg h/mL) | |
| R1 | 116.7 ± 1.1 | 0.250 ± 0.010 | 92.10% | 0.181 | 7.25 | 3.99 | 284.6 |
| Ctrl | 115.2 ± 2.1 | 0.251 ± 0.029 | 96.70% | 0.224 | 8.11 | 6.05 | 487.7 |
Figure 6.
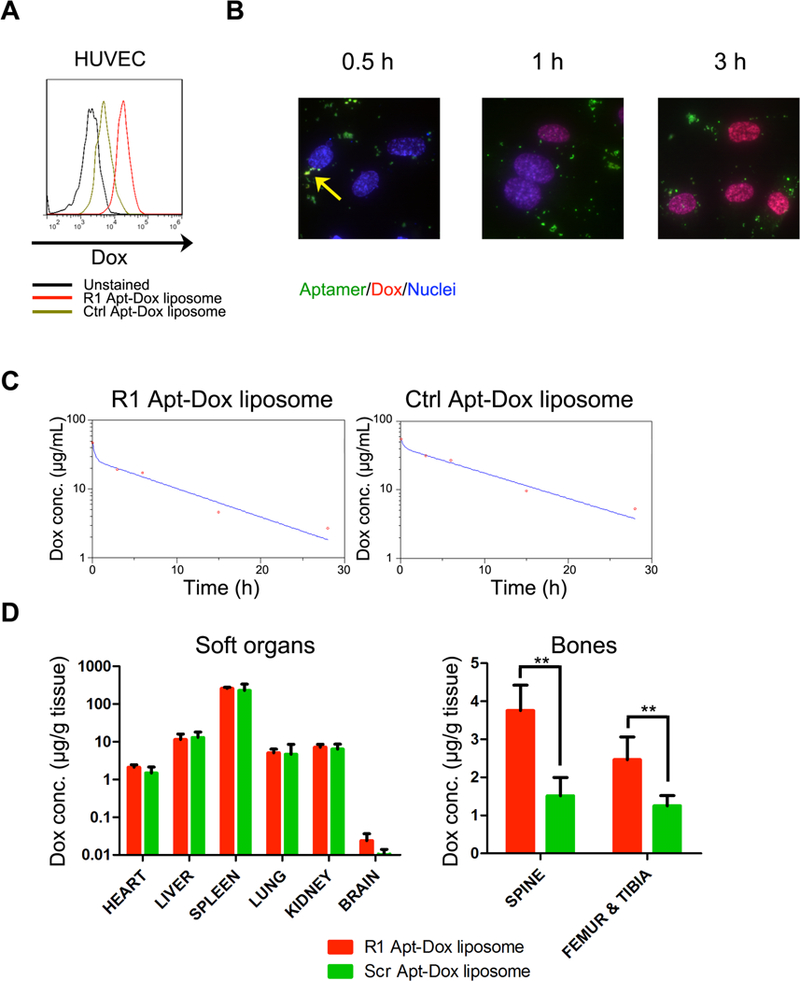
Pharmacokinetics and biodistribution studies of R1 aptamer conjugated liposomal doxorubicin. Mice with lymphoma BMI were dosed with 6 mg/kg Ctrl Apt-Dox liposome or R1 Apt-Dox liposome (n=4). (A). Conjugation of R1 on liposomal Dox promotes uptake by HUVECs in vitro. (B) Intracellular trafficking and drug release of R1 Apt-Dox liposome. Time dependent Dox concentration in peripheral blood (C) and biodistribution of Dox content in soft organs and bones at 28 h post treatment (D) were analyzed.
Pharmacokinetics and biodistribution of the R1 -lipo Dox and Ctrl-lipo Dox were compared in SCID/beige mice with Raji lymphoma. Comparing to Ctrl-lipo Dox, the R1 -lipo Dox showed a faster distribution phase and a similar elimination phase (Table 1, Fig. 6C). Biodistribution in the major organs was similar between the two formulations; however, the bones with lymphoma involvement received a significantly higher level of drug in mice treated with R1-lipo Dox over those with Ctrl-lipo Dox. There was a 2 to 3 fold enhancement in spine, femur and tibia in mice treated with R1 -lipo Dox (Fig. 6D).
DISCUSSION
Aptamers are short oligonucleotides with low immunogenicity and toxicity, and can be manipulated in large scale with simplicity. In addition, the high diversity of sequences and related structures provide a very large pool to screen for ligands that bind targets with high affinity and specificity. Thus, aptamers are emerging diagnostic and therapeutic agents for clinic applications [26,41,42]. Pegaptanib (Macugen™ from Pfizer) is the first aptamer-based drug approved by the Food and Drug Administrations in the United States. It is a single stranded aptamer that binds to the vascular endothelial growth factor with specificity, and is used to treat age-related macular degeneration [43]. The thioaptamers used in the current study contain monothiophosphate substitutions on the 5’ side of every dA residue in the sequence, which provides more resistance to the plasma and tissue DNases comparing to the conventional aptamers [44] [45].
Lymphoma is a group of heterogeneous diseases consisting of more than 60 different subtypes, and the interaction and regulation of the components in the tumor microenvironment are diverse and complicated [46,47]. Depending on their unique characteristics, lymphoma cells may either hide behind normal tissues or efface normal tissues due to abnormally fast growth and autonomous survival [48], which is the case in the murine model of Raji Burkitt’s lymphoma. Infiltrating leukocytes, endothelial cells, fibroblasts, adipocytes and extracellular matrix are all actively participating in the cross-regulation process with lymphoma cells. On one hand, it raises the requirement of therapeutic strategy that not only kills the cancer cells, but also conquers the dysfunction of stromal cells. On the other hand, it provides a unique environment that differs from healthy tissues for targeted drug delivery. In this study, we have identified a thioaptamer that preferentially binds the vascular endothelium in bone marrow with lymphoma over the healthy medullary cavity. We have also demonstrated the feasibility of applying R1 as an affinity moiety to achieve lymphoma BMI-targeted delivery of doxorubicin, a chemotherapy drug that is commonly applied in lymphoma treatment but may cause severe toxic side effects [49]. Targeted delivery of the drug may provide the benefit of enhanced therapeutic efficacy with significantly reduced toxicity, as we have recently shown in solid epithelial tumors [36].
Our study has indicated that R1 binding to the inflammatory endothelium is subjected to regulation by soluble factors secreted by both endothelial and lymphoma cells. Although the identity of such factors is not known, it is highly likely that a group of cytokines and chemokines are involved in the process. To support this notion, we have revealed that E-selectin is overexpressed by the vascular endothelial cells in a tumor microenvironment [38]. A group of tumor and stromal cell-secreted cytokines can promote E-selectin expression, such as TNF-α and interleukin-1 β [50]. Since neither conditioned medium from the bone marrow cells and lymphoma cells could provide the strongest R1 binding to HUVEC (Fig. 5), expression of the cell surface docking protein must be regulated by a combination of factors from both media.
In conclusion, we have successfully isolated a novel thioaptamer that can be applied for drug enrichment to lymphoma BMI. R1 preferentially binds to endothelial cells, and R1-endothelium interaction is enhanced when the vascular endothelial cells are stimulated by a combination of factors secreted from the lymphoma and stromal cells. In a proof-of-principal study, we have applied R1 as an affinity moiety to develop a lymphoma BMI-targeted drug formulation, and successfully demonstrated drug enrichment in the malignant tissues. This thioaptamer should provide a powerful tool in the development of effective treatments for lymphoma BMI.
Supplementary Material
Acknowledgments
Financial support: This work was supported by National Cancer Institute grants 1R01CA193880–01A1 (HS), U54CA151668 (MF and DG), U54CA210181 (MF), and Department of Defense grant W81XWH-12–1-0414 (MF). MF is an Ernest Cockrell Jr. distinguished endowed chair.
Footnotes
Conflict of interest disclosure statement: The authors declare no competing financial interests.
REFERENCES
- [1].Cabezas-Quintario MA, Gomez P, Yuste-Del Pozo V, Valencia-Mesa AL, Sosa G, Ricard P, Hijas-Gomez AI, Pinedo F, Arguelles M, Bone marrow trephine biopsy involvement by lymphoma: Pattern of involvement and concordance with flow cytometry, in 10 years from a single institution, Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico (2015). [DOI] [PubMed] [Google Scholar]
- [2].Conlan MG, Bast M, Armitage JO, Weisenburger DD, Bone marrow involvement by non-hodgkin’s lymphoma: The clinical significance of morphologic discordance between the lymph node and bone marrow. Nebraska lymphoma study group, J Clin Oncol 8(7) (1990) 1163–1172. [DOI] [PubMed] [Google Scholar]
- [3].Shi YF, Li XH, Song YQ, Song WW, Lai YM, Involvement of bone marrow in lymphoma: Pathological investigation in a single-center from northern china, International journal of clinical and experimental pathology 8(6) (2015) 7102–7111. [PMC free article] [PubMed] [Google Scholar]
- [4].Wilson WH, Treatment strategies for aggressive lymphomas: What works?, Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2013((2013) 584–590. [DOI] [PubMed] [Google Scholar]
- [5].Zelenetz AD, Gordon LI, Wierda WG, Abramson JS, Advani RH, Andreadis CB, Bartlett N, Byrd JC, Czuczman MS, Fayad LE, Fisher RI, Glenn MJ, Harris NL, Hoppe RT, Horwitz SM, Kelsey CR, Kim YH, Krivacic S, LaCasce AS, Nademanee A, Porcu P, Press O, Rabinovitch R, Reddy N, Reid E, Saad AA, Sokol L, Swinnen LJ, Tsien C, Vose JM, Yahalom J, Zafar N, Dwyer M, Sundar H, n. National comprehensive cancer, Non-hodgkin’s lymphomas, version 4.2014, Journal of the National Comprehensive Cancer Network : JNCCN 12(9) (2014) 1282–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sou K, Goins B, Oyajobi BO, Travi BL, Phillips WT, Bone marrow-targeted liposomal carriers, Expert Opin Drug Deliv 8(3) (2011) 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liang C, Guo B, Wu H, Shao N, Li D, Liu J, Dang L, Wang C, Li H, Li S, Lau WK, Cao Y, Yang Z, Lu C, He X, Au DW, Pan X, Zhang BT, Lu C, Zhang H, Yue K, Qian A, Shang P, Xu J, Xiao L, Bian Z, Tan W, Liang Z, He F, Zhang L, Lu A, Zhang G, Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel rna interference-based bone anabolic strategy, Nature medicine 21(3) (2015) 288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mu CF, Xiong Y, Bai X, Sheng YJ, Cui J, Codelivery of ponatinib and sar302503 by active bone-targeted polymeric micelles for the treatment of therapy-resistant chronic myeloid leukemia, Molecular pharmaceutics 14(1) (2017) 274–283. [DOI] [PubMed] [Google Scholar]
- [9].Sou K, Goins B, Leland MM, Tsuchida E, Phillips WT, Bone marrow-targeted liposomal carriers: A feasibility study in nonhuman primates, Nanomedicine 5(1) (2010) 41–49. [DOI] [PubMed] [Google Scholar]
- [10].Maeda H, Wu J, Sawa T, Matsumura Y, Hori K, Tumor vascular permeability and the epr effect in macromolecular therapeutics: A review, Journal of controlled release : official journal of the Controlled Release Society 65(1–2) (2000) 271–284. [DOI] [PubMed] [Google Scholar]
- [11].Maeda H, Toward a full understanding of the epr effect in primary and metastatic tumors as well as issues related to its heterogeneity, Advanced drug delivery reviews 91((2015) 3–6. [DOI] [PubMed] [Google Scholar]
- [12].Blanco E, Shen H, Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nature biotechnology 33(9) (2015) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Analysis of nanoparticle delivery to tumours, Nature Reviews Materials 1((2016) 16014. [Google Scholar]
- [14].Park K, Questions on the role of the epr effect in tumor targeting, Journal of controlled release : official journal of the Controlled Release Society 172(1) (2013) 391. [DOI] [PubMed] [Google Scholar]
- [15].Doan PL, Chute JP, The vascular niche: Home for normal and malignant hematopoietic stem cells, Leukemia 26(1) (2012) 54–62. [DOI] [PubMed] [Google Scholar]
- [16].Chi B, Park SJ, Park MH, Lee SY, Jeong B, Oligopeptide delivery carrier for osteoclast precursors, Bioconjugate chemistry 21(8) (2010) 1473–1478. [DOI] [PubMed] [Google Scholar]
- [17].Ye WL, Zhao YP, Li HQ, Na R, Li F, Mei QB, Zhao MG, Zhou SY, Doxorubicin-poly (ethylene glycol)-alendronate self-assembled micelles for targeted therapy of bone metastatic cancer, Scientific reports 5((2015) 14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ellington AD, Szostak JW, Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures, Nature 355(6363) (1992) 850–852. [DOI] [PubMed] [Google Scholar]
- [19].Tuerk C, Gold L, Systematic evolution of ligands by exponential enrichment: Rna ligands to bacteriophage t4 DNA polymerase, Science 249(4968) (1990) 505–510. [DOI] [PubMed] [Google Scholar]
- [20].Parekh P, Kamble S, Zhao N, Zeng Z, Portier BP, Zu Y, Immunotherapy of cd30-expressing lymphoma using a highly stable ssdna aptamer, Biomaterials 34(35) (2013) 8909–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tang Z, Shangguan D, Wang K, Shi H, Sefah K, Mallikratchy P, Chen HW, Li Y, Tan W, Selection of aptamers for molecular recognition and characterization of cancer cells, Analytical chemistry 79(13) (2007) 4900–4907. [DOI] [PubMed] [Google Scholar]
- [22].Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W, Aptamers evolved from live cells as effective molecular probes for cancer study, Proceedings of the National Academy of Sciences of the United States of America 103(32) (2006) 11838–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shum KT, Zhou J, Rossi JJ, Nucleic acid aptamers as potential therapeutic and diagnostic agents for lymphoma, Journal of cancer therapy 4(4) (2013) 872–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mallikaratchy PR, Ruggiero A, Gardner JR, Kuryavyi V, Maguire WF, Heaney ML, McDevitt MR, Patel DJ, Scheinberg DA, A multivalent DNA aptamer specific for the b-cell receptor on human lymphoma and leukemia, Nucleic acids research 39(6) (2011) 2458–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou J, Rossi JJ, Cell-type-specific, aptamer-functionalized agents for targeted disease therapy, Molecular therapy Nucleic acids 3((2014) e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou J, Rossi J, Aptamers as targeted therapeutics: Current potential and challenges, Nat Rev Drug Discov (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mi J, Liu Y, Rabbani ZN, Yang Z, Urban JH, Sullenger BA, Clary BM, In vivo selection of tumor-targeting rna motifs, Nature chemical biology 6(1) (2010) 22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng C, Chen YH, Lennox KA, Behlke MA, Davidson BL, In vivo selex for identification of brain-penetrating aptamers, Molecular therapy Nucleic acids 2((2013) e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Volk DE, Yang X, Fennewald SM, King DJ, Bassett SE, Venkitachalam S, Herzog N, Luxon BA, Gorenstein DG, Solution structure and design of dithiophosphate backbone aptamers targeting transcription factor nf-kappab, Bioorganic chemistry 30(6) (2002) 396–419. [DOI] [PubMed] [Google Scholar]
- [30].Lu E, Elizondo-Riojas MA, Chang JT, Volk DE, Aptaligner: Automated software for aligning pseudorandom DNA x-aptamers from next-generation sequencing data, Biochemistry 53(22) (2014) 3523–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zuker M, Mfold web server for nucleic acid folding and hybridization prediction, Nucleic acids research 31(13) (2003) 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kibbe WA, Oligocalc: An online oligonucleotide properties calculator, Nucleic acids research 35(Web Server issue) (2007) W43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bolotin EM, Cohen R, Bar LK, Emanuel N, Ninio S, Lasic DD, and Barenholz Y, Ammonium sulfate gradients for efficient and stable remote loading of amphipathic weak bases into liposomes and ligandsomes, Journal of Liposome Research 4(1) (1994) 455–479. [Google Scholar]
- [34].Schweitzer KM, Drager AM, van der Valk P, Thijsen SF, Zevenbergen A, Theijsmeijer AP, van der Schoot CE, Langenhuijsen MM, Constitutive expression of e-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues, The American journal of pathology 148(1) (1996) 165–175. [PMC free article] [PubMed] [Google Scholar]
- [35].Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, Magnani JL, Levesque JP, Vascular niche e-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance, Nat Med 18(11) (2012) 1651–1657. [DOI] [PubMed] [Google Scholar]
- [36].Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S, Shen J, Liu H, Hu Z, Chen L, Huang Y, Koay E, Huang Y, Liu J, Ensor JE, Blanco E, Liu X, Ferrari M, Shen H, An injectable nanoparticle generator enhances delivery of cancer therapeutics, Nature biotechnology 34(4) (2016) 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gilles ME, Maione F, Cossutta M, Carpentier G, Caruana L, Di Maria S, Houppe C, Destouches D, Shchors K, Prochasson C, Mongelard F, Lamba S, Bardelli A, Bouvet P, Couvelard A, Courty J, Giraudo E, Cascone I, Nucleolin targeting impairs the progression of pancreatic cancer and promotes the normalization of tumor vasculature, Cancer research 76(24) (2016) 7181–7193. [DOI] [PubMed] [Google Scholar]
- [38].Mai J, Huang Y, Mu C, Zhang G, Xu R, Guo X, Xia X, Volk DE, Lokesh GL, Thiviyanathan V, Gorenstein DG, Liu X, Ferrari M, Shen H, Bone marrow endothelium-targeted therapeutics for metastatic breast cancer, Journal of controlled release : official journal of the Controlled Release Society 187((2014) 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mahadevan D, Fisher RI, Novel therapeutics for aggressive non-hodgkin’s lymphoma, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29(14) (2011) 1876–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lasic DD, Frederik PM, Stuart MC, Barenholz Y, McIntosh TJ, Gelation of liposome interior. A novel method for drug encapsulation, FEBS letters 312(2–3) (1992) 255–258. [DOI] [PubMed] [Google Scholar]
- [41].Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y, Oligonucleotide aptamers: New tools for targeted cancer therapy, Molecular therapy Nucleic acids 3((2014) e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhou J, Rossi JJ, The therapeutic potential of cell-internalizing aptamers, Current topics in medicinal chemistry 9(12) (2009) 1144–1157. [DOI] [PubMed] [Google Scholar]
- [43].Ng EW, Shima DT, Calias P, Cunningham ET Jr., Guyer DR, Adamis AP, Pegaptanib, a targeted anti-vegf aptamer for ocular vascular disease, Nature reviews Drug discovery 5(2) (2006) 123–132. [DOI] [PubMed] [Google Scholar]
- [44].Mann AP, Somasunderam A, Nieves-Alicea R, Li X, Hu A, Sood AK, Ferrari M, Gorenstein DG, Tanaka T, Identification of thioaptamer ligand against e-selectin: Potential application for inflamed vasculature targeting, PloS one 5(9) (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jhaveri S, Olwin B, Ellington AD, In vitro selection of phosphorothiolated aptamers, Bioorganic & medicinal chemistry letters 8(17) (1998) 2285–2290. [DOI] [PubMed] [Google Scholar]
- [46].Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES, The 2008 who classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications, Blood 117(19) (2011) 5019–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Scott DW, Gascoyne RD, The tumour microenvironment in b cell lymphomas, Nature reviews Cancer 14(8) (2014) 517–534. [DOI] [PubMed] [Google Scholar]
- [48].Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, Liu X, Powell J, Yang Y, Xu W, Zhao H, Kohlhammer H, Rosenwald A, Kluin P, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Ogwang MD, Reynolds SJ, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pittaluga S, Wilson W, Waldmann TA, Rowe M, Mbulaiteye SM, Rickinson AB, Staudt LM, Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics, Nature 490(7418) (2012) 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Limat S, Demesmay K, Voillat L, Bernard Y, Deconinck E, Brion A, Sabbah A, Woronoff-Lemsi MC, Cahn JY, Early cardiotoxicity of the chop regimen in aggressive non-hodgkin’s lymphoma, Annals of oncology : official journal of the European Society for Medical Oncology 14(2) (2003) 277–281. [DOI] [PubMed] [Google Scholar]
- [50].Laferriere J, Houle F, Huot J, Regulation of the metastatic process by e-selectin and stress-activated protein kinase-2/p38, Annals of the New York Academy of Sciences 973((2002) 562–572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.