

Abstract
Our recent studies have revealed that microRNA-1291 (miR-1291) is downregulated in pancreatic cancer (PC) specimens and restoration of miR-1291 inhibits tumorigenesis of PC cells. This study is to assess the efficacy and underlying mechanism of our bioengineered miR-1291 prodrug monotherapy and combined treatment with chemotherapy. AT-rich interacting domain protein 3B (ARID3B) was verified as a new target for miR-1291, and miR-1291 prodrug was processed to mature miR-1291 in PC cells which surprisingly upregulated ARID3B mRNA and protein levels. Co-administration of miR-1291 with gemcitabine plus nab-paclitaxel (Gem-nP) largely increased the levels of apoptosis, DNA damage and mitotic arrest in PC cells, compared to mono-drug treatment. Consequently, miR-1291 prodrug improved cell sensitivity to Gem-nP. Furthermore, systemic administration of in vivo-jetPEI-formulated miR-1291 prodrug suppressed tumor growth in both PANC-1 xenograft and PC patients derived xenograft (PDX) mouse models to comparable degrees as Gem-nP alone, while combination treatment reduced tumor growth more ubiquitously and to the greatest degrees (70–90%), compared to monotherapy. All treatments were well tolerated in mice. In conclusion, biologic miR-1291 prodrug has therapeutic potential as a monotherapy for PC, and a sensitizing agent to chemotherapy.
Keywords: miR-1291, pancreatic cancer, PDX model, ARID3B, gemcitabine plus nab-paclitaxel
1. Introduction
Pancreatic cancer (PC) is one of the most lethal malignances with a 5-year survival of less than 8% [1, 2]. Less than 20% of PC patients are diagnosed with surgically resectable disease and are potentially curable. The remaining large majority of PC patients are diagnosed with advanced disease that is either unresectable or metastatic [3]. Gemcitabine has been a standard chemotherapeutic treatment for advanced PC since the late 1990s [4]. After more than a decade of active investigation, the first clinical improvement to gemcitabine-based treatment was seen with addition of paclitaxel albumin-stabilized nanoparticle (nab-paclitaxel) formulation (Gem-nP) which increased overall median survival (8.5 months) compared to gemcitabine monotherapy (6.7 months) [5]. Compared to most other solid tumors, advance in the treatment of PC has been slow, thus more effective treatment strategies with minimal toxicity are urgently needed for PC [6–9].
MicroRNAs (miRNAs) have been revealed as a family of noncoding RNAs (ncRNA) in the control of tumor initiation and progression [10, 11]. Furthermore, multiple miRNAs display tissue specific aberrant expression in cancer development, suggesting the potential of developing miRNA based anticancer therapies besides serving as diagnostic or prognostic markers [12, 13] including those for PC [14–16]. Recently, we have identified that miR-1291 is significantly downregulated in PC tissues compared with normal pancreatic tissues [17]. Our studies have also demonstrated that restoration of miR-1291 expression/function suppresses the proliferation, migration, invasion, and tumorigenesis of PC cells through the induction of apoptosis and cell cycle arrest, as well as alteration of PC cell metabolome [17, 18]. In addition, miR-1291 sensitizes PC cells to chemotherapy via direct targeting of multidrug resistance-associated protein 1 (MRP1) overexpressed in the cells [19]. Other investigators have also demonstrated that miR-1291 suppresses renal cell carcinoma and esophageal squamous cell carcinoma growth by downregulation of glucose transporter protein type 1 (GLUT1) and mucin 1 (MUC1) [20, 21]. The above reports suggest the potential of miR-1291 as a novel therapy for PC.
To explore miR-1291 based therapeutic strategies for the treatment of PC, we have successfully established a novel approach to producing large quantities of pre-miR-1291 agents in bacteria by using a sephadex aptamer tagged methionyl-tRNA (MSA) scaffold [22]. Further studies demonstrated that chimeric MSA/mir-1291 or “miR-1291 prodrug” was precisely processed into mature miR-1291 in human cells, and subsequently regulated target protein expression and suppressed the growth of PC cells [22]. It is noteworthy that bioengineered miRNA agents produced in living cells are distinguished from conventional miRNA agents made in test tubes by chemical synthesis or enzymatic reactions. Therefore, our biologic miRNA agents may better capture the natural characteristics of cellular RNA molecules [23].
The objective of current study was to investigate the utility of bioengineered miR-1291 prodrug for the control of PC and its underlying molecular mechanisms. Following the identification of a new target AT-rich interacting domain protein 3B (ARID3B) for miR-1291, which was rather surprisingly upregulated by miR-1291, we delineated the independent actions of miR-1291, gemcitabine and nab-paclitaxel on the apoptosis, DNA damage and mitosis arrest of PC cells, respectively, as well as optimal effects when the drugs were combined together. Therapy studies in PANC-1 xenograft and three different PC patient derived xenograft (PDX) tumor mouse models revealed that miR-1291 prodrug alone showed comparable levels of efficacy as Gem-nP in the suppression of PC tumor growth, and combination treatment with miR-1291 and Gem-nP inhibited PC tumor growth to the greatest degrees.
2. Material and Methods
2.1. Animals
All animal experiments were performed according to our protocol approved by the Institutional Animal Care and Use Committee at UC Davis. 5- to 6-week-old female athymic nude mice (NU/J) and NOD.CB17-Prkdcscid/J mice (The Jackson Laboratory, Bar Harbor, ME) were used to establish PANC-1 xenograft mouse models and PDX mouse models, respectively. The mice were maintained in sterile cages at constant temperature and humidity, with free access to food and water.
Materials and procedures of production of biologic miR-1291 prodrug (MSA/mir-1291) and control RNA MSA, cell culture and treatment, luciferase reporter gene assay, reverse transcription quantitative real-time PCR, immunoblot analysis, immunofluorescence, animal model establishment, therapy study, and statistical analysis were described in detail in Supplementary Materials.
3. Results
3.1. ARID3B is a direct target of miR-1291
Our recent studies have demonstrated that miR-1291 suppresses proliferation and tumorigenesis of PC cells [17]. To further delineate the molecular mechanisms through which miR-1291 controls PC cell growth, computational analysis was conducted to predict potential targets of miR-1291. Among a set of putative targets, ARID3B was a top candidate consisting of four miRNA response elements (MREs) for miR-1291 within its 3’UTR (Fig. 1A). An ARID3B 3’UTR luciferase reporter plasmid was thus constructed to evaluate the interactions between miR-1291 and ARID3B 3’UTR. Surprisingly, treatment with bioengineered miR-1291 significantly increased ARID3B-3’UTR-luciferase reporter activities in AsPC-1 (Fig. 1B) and HEK293 (data not shown) cells, as compared to controls. Introduction of miR-1291 into cells with miR-1291-expressing plasmid showed the same results (Supplementary Fig. S1A), whereas ARID3B-3’UTR-luciferase reporter activities were decreased in cells treated with miR-1291 antagomir (Supplementary Fig. S1B). These results suggest that miR-1291 targets ARID3B 3’UTR and may positively regulate the expression of ARID3B.
Fig. 1.
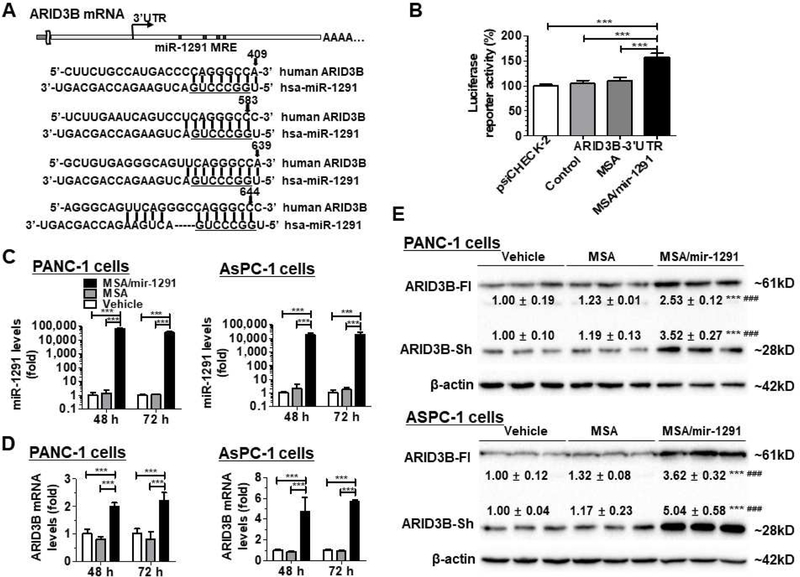
MiR-1291 targets ARID3B and upregulates its expression in human pancreatic cancer cells. (A) Computational analysis identified four putative MRE sites for miR-1291 within the 3’UTR of ARID3B mRNA. Underlined is the seed sequence of miR-1291. (B) Dual luciferase reporter assay indicated that ARID3B 3’UTR luciferase activities were increased about 50% in AsPC-1 cells treated with MSA/mir-1291, as compared to controls. (C) qPCR analyses revealed that MSA/mir-1291 was selectively processed to mature miR-1291 in PANC-1 and AsPC-1 cells, and subsequently upregulated ARID3B mRNA levels (D). Values are mean ± SD (N = 3). ***P < 0.001, compared to corresponding control (1- or 2-way ANOVA). (E) Immunoblot analyses showed that ARID3B protein levels were elevated in PANC-1 and AsPC-1 cells after transfection with bioengineered miR-1291. Both the full-length ARID3B (ARID3B-Fl, ~61 kD) and the short-form ARID3B (ARID3B-sh, ~28 kD) were upregulated in PANC-1 and AsPC-1 cells (72 h post-transfection), as compared to vehicle and MSA controls. β-actin was used as a loading control, and then the protein levels were normalized to vehicle group for comparison. Values are mean ± SD; ***P < 0.001, compared to vehicle; ### P < 0.001, compared to MSA (unpaired Student’s t-test).
3.2. MiR-1291 upregulates the mRNA and protein levels of ARID3B in PC cells
To define the effects of miR-1291 on the expression of ARID3B, we first verified the production of high levels of mature miR-1291 from bioengineered MSA/mir-1291 in PANC-1 and AsPC-1 cells (Fig. 1C). We then compared ARID3B mRNA levels in PC cells treated with MSA/mir-1291 and MSA or vehicle control. Compared to vehicle control, MSA did not alter ARID3B mRNA levels. Treatment with MSA/mir-1291 (20 nM) led to a 1.0- and 1.2-fold increase in ARID3B mRNA levels in PANC-1 cells, as compared to vehicle control, at 48 h and 72 h post-treatment, respectively (Fig. 1D). Similarly, MSA/mir-1291 (5 nM) caused a 3.7-fold and 4.6-fold upregulation of ARID3B mRNA levels in AsPC-1 cells (Fig. 1D), at 48 h and 72 h post-treatment, respectively.
We further conducted Western blots to examine the impact of miR-1291 on ARID3B protein levels in PC cells. As found in other types of cells by other investigators [24], we observed two different ARID3B bands in both PANC-1 and AsPC-1 cells which are designated as full-length ARID3B (ARID3B-Fl, ~61 kD) and short-form ARID3B (ARID3B-sh, ~28 kD), respectively (Fig. 1E). Our data showed that ARID3B-FL protein levels significantly increased in PANC-1 cells at 72 h post-transfection with 20 nM miR-1291 prodrug. A higher degree of increase of ARID3B-FL protein levels was found in AsPC-1 cells at both 48 h and 72 h post-treatment with 5 nM miR-1291. Interestingly, impact of miR-1291 on ARID3B-Sh protein levels appeared to follow the same pattern as ARID3B-Fl in both cell lines (Fig. 1E, Supplementary Fig. S2). These results indicate that miR-1291 upregulates ARID3B expression in PC cells.
3.3. Individual and combined actions of miR-1291 prodrug and Gem-nP on DNA damage, mitosis arrest, and apoptosis
Historically, single drug exerted very limited efficacy for the treatment of PC. Therefore, we aimed at examining combination effects (Fig. 2A) while assessing miR-1291 monotherapy and comparing it to Gem-nP, the first-line chemotherapy for PC. Individual and combined actions of miR-1291 prodrug and Gem-nP on their corresponding target or marker proteins were first investigated in PANC-1 and AsPC-1 cells by Western blots (Fig. 2B and C). Our data showed that co-administration of Gem-nP did not alter miR-1291-controlled upregulation of ARID3B in PANC-1 cells but enhanced the effects in AsPC-1 cells, again suggesting distinct sensitivities of the two cell lines. Immunoblot (Fig. 2B and C) and immunofluorescence studies were further conducted to determine single and combined drug effects on DNA damage (γH2A.X foci), apoptosis (cleaved caspase-3/7, c-caspase-3/7) and mitotic arrest (H3PS10) (Fig. 2D–F; Supplementary Fig. S3–4). The results showed that, in addition to the induction of apoptosis (c-caspase-3/7) in both PANC-1 and AsPC-1 cell lines, miR-1291 alone surprisingly elicited obvious DNA damage (formation of γH2A.X foci) in AsPC-1 cells. On the other hand, Gem-nP largely provoked DNA damage and mitotic arrest in both PC cell lines, as manifested by an upregulation of γH2A.X and H3PS10, respectively. Most importantly, combination treatment with miR-1291 prodrug and Gem-nP caused the greatest extents of DNA damage, mitosis and apoptosis in both cell lines, which were indicated by γH2A.X, H3PS10, and c-caspase-3/7, respectively. These results demonstrate that miR-1291 induces apoptosis and possibly DNA damage, and suggest that combination therapy with miR-1291 and Gem-nP may produce optimal outcomes.
Fig. 2.
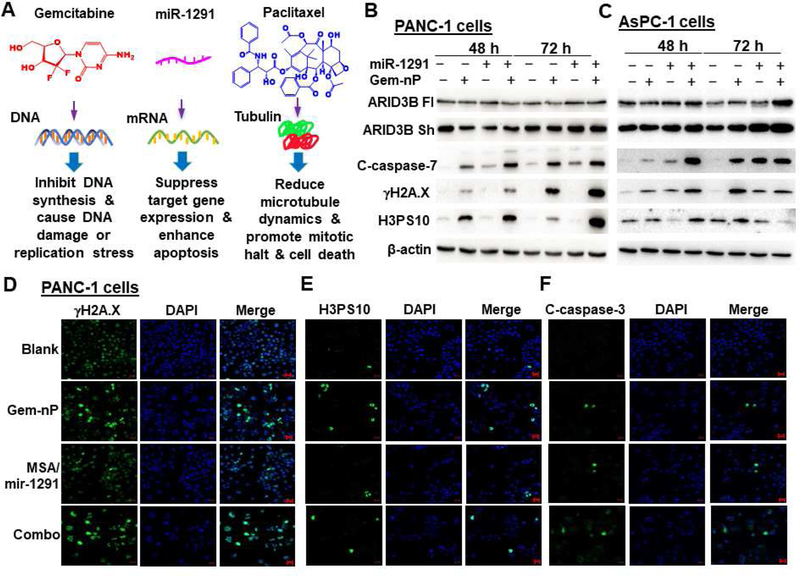
Independent and combined actions of miR-1291 and gemcitabine plus nab-paclitaxel (Gem-nP) in human pancreatic cancer cells. (A) Gemcitabine, miR-1291 and paclitaxel may act on specific targets and thus interfere with particular cellular processes. Immunoblot (B and C) and immunofluorescence (D-F; scale bar, 20 µm) studies showed that combination (combo) treatment with miR-1291 prodrug (10 nM in PANC-1 cells, 3 nM in AsPC-1 cells) and Gem-nP exhibited the greatest degrees of DNA damage, mitosis and apoptosis in PANC-1 and AsPC-1 cells, which were indicated by γH2A.X, H3PS10, and cleaved caspase-3/7 (c-caspase-3/7), respectively. β-actin was used as a loading control. C-caspase-7 images in PANC-1 cells are provided in Supplementary Fig. S3, and individual biomarkers in AsPC-1 cells are shown in Supplementary Fig. S4.
3.4. Bioengineered miR-1291 prodrug enhances the sensitivity of PC cells to chemotherapeutic drugs
To assess whether miR-1291 prodrug could increase the sensitivity of pancreatic cancer cells to Gem-nP, the anti-proliferative activity of Gem-nP in the presence of miR-1291 prodrug or control MSA was evaluated in PANC-1 and AsPC-1 cells by CellTiter-Glo assay. The results showed that miR-1291 treated PANC-1 and AsPC-1 cells were much more sensitive to Gem-nP, as compared to MSA treated cells (Fig. 3). The enhanced sensitivity was also manifested by the lower EC50 value in miR-1291 treated PANC-1 cells (52.3 ± 20.3 nM) than that in MSA treated cells (155 ± 33 nM, *P < 0.05). In addition, miR-1291 transfected AsPC-1 cells also showed a significantly lower EC50 value (14.6 ± 5.5 nM) than MSA treated cells (40.4 ± 1.8 nM, **P < 0.01) (Fig. 3E). These results show that co-administration of miR-1291 is able to sensitize PC cells to chemotherapies.
Fig. 3.
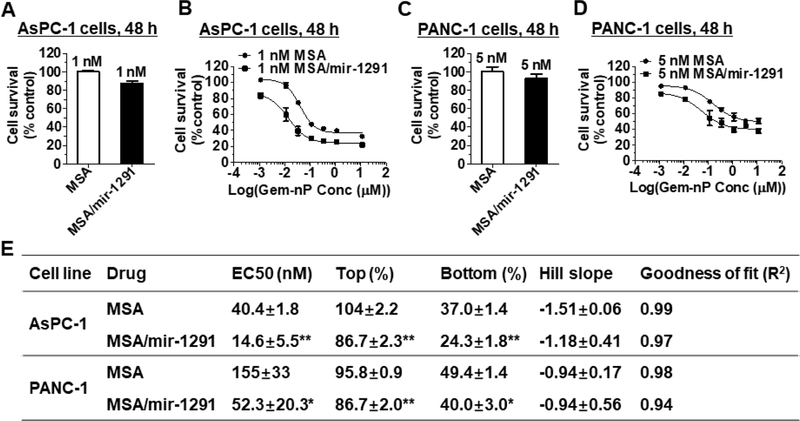
Bioengineered miR-1291 prodrug sensitizes human pancreatic cancer cells to Gem-nP. Compared to the MSA control, a low dose of MSA/miR-1291 had minimal effects on AsPC-1 (A) and PANC-1 (C) cell proliferation, whereas it significantly (*P < 0.001; 2-way ANOVA with Bonferroni posttests) improved the sensitivity of AsPC-1 (B) and PANC-1 (D) cells to Gem-nP, which was also indicated by the estimated pharmacodynamics parameters (E). AsPC-1 and PANC-1 cells were treated with MSA/mir-1291 or MSA control alone (A, C) or in combination with various concentrations of Gem-nP (B, D; shown are total concentrations of Gem-nP at a fixed ratio of 8: 1) for 48 h, and cell viability was determined by CellTiter-Glo assay. Values are mean ± SD (N = 3). *P < 0.05; **P < 0.01, compared to corresponding MSA control treatment (1-way ANOVA).
3.5. Bioengineered miR-1291 prodrug monotherapy and combination therapy with Gem-nP are effective to control tumor growth in PANC-1 xenograft mouse models, while they are well tolerated in mice
To determine the anti-tumor efficacy of miR-1291 prodrug monotherapy and combination therapy with Gem-nP in vivo, we first established PANC-1 xenograft mouse models (Fig. 4A). Systematic administration of a single dose of in vivo-jetPEI formulated miR-1291 prodrug was distributable to PANC-1 xenograft tumor tissues, as indicated by high levels of tumoral miR-1291 at 24 h after drug administration (Supplementary Fig. S5A). Compared to buffer or MSA treatment, miR-1291 prodrug alone significantly suppressed PANC-1 tumor growth to a similar degree as Gem-nP, while combination treatment with miR-1291 and Gem-nP inhibited tumor growth to the greatest extent (Fig. 4B). Visual inspection and weights of the dissected tumors (Fig. 4C–D) further demonstrated the remarkably optimal tumor suppressive effects for combination therapy. These results demonstrated the effectiveness of miR-1291 prodrug monotherapy in the control of PANC-1 xenograft tumor progression as well as an optimal outcome for combination therapy with miR-1291 and Gem-nP.
Fig. 4.
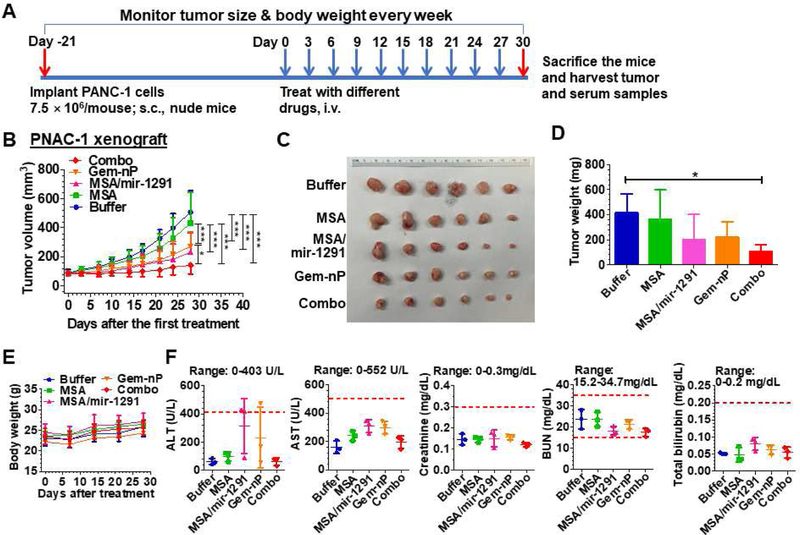
Combination therapy with miR-1291 prodrug and Gem-nP is the most effective in suppressing PANC-1 xenograft tumor growth in mice and all therapies are well tolerated. (A) Timeline of the establishment of PANC-1 xenograft mouse model and drug treatment. (B) PANC-1 xenograft tumor growth was reduced to the greatest degree by combination treatment with miR-1291 prodrug and Gem-nP. *P < 0.05, ***P < 0.001 (2-way ANOVA with Bonferroni posttests). (C) Visual comparison of dissected tumors from mice with different treatments. (D) Weights of the dissected xenograft tumors. *P < 0.05 (1-way ANOVA). (E) Body weights were not altered by drug treatment. (F) Blood biochemistry profiles including alanine transaminase (ALT), aspartate transaminase (AST), creatinine, blood urea nitrogen (BUN), and total bilirubin showed no significant difference by different treatments. Values are mean ± SD (N = 6 per group, except N = 3 for blood chemistry profiles). The ranges of individual markers (derived from BALB/c mice; Comparative Pathology Laboratory at UC-Davis) were marked as references.
All treatments were well tolerated in mice as animal body weights showed no significant differences among different groups (Fig. 4E). To further examine the safety of drug treatments, blood biochemistry profiles were determined (Fig. 4F). All markers of liver and kidney functions including alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin, blood urea nitrogen (BUN) and creatinine were within the normal ranges, except ALT levels in two mice (one from miR-1291 monotherapy and one from Gem-nP monotreatment) slightly exceeded the normal range. However, there was no significant difference in each blood biomarker between any treatment groups, suggesting that therapies did not cause any hepatic or renal toxicity. Together, the results indicate that systemic administration of therapeutic doses of miR-1291 prodrug or Gem-nP alone, or in combination are well tolerated in PANC-1 xenograft mouse models.
3.6. Efficacy of miR-1291 prodrug treatment alone and in combination with Gem-nP chemotherapy in three different PDX mouse models
Compared to xenograft models derived from cancer cell lines, PDX tumor models may better preserve the heterogeneity and histological characteristics of the original tumors [25–27]. Therefore, we established three PDX models from clinical PC samples and employed them to further assess miR-1291 prodrug therapies. The first PDX model (PA-0387, Fig. 5) was subjected to the same dose regimens as those used in PANC-1 xenograft mouse models. Similarly, RT-qPCR analyses confirmed high levels of miR-1291 in PDX tissues at 24 h after systemic administration of a single dose of miR-1291 prodrug (Supplementary Fig. S5B). Therapy data showed that treatment with miR-1291 prodrug or Gem-nP alone significantly reduced PDX PA-0387 tumor growth, as compared to buffer or MSA control; and combination treatment showed the highest degree of inhibition (Fig. 5A). Likewise, visual inspection of dissected tumors (Fig. 5B) and examination of final tumor weights (Fig. 5C) supported the effectiveness of miR-1291 prodrug alone, Gem-nP alone, and their combination in the control of PDX PA-0387, while there was no statistical difference between mono- and combination therapy. H&E staining demonstrated that PDXs indeed showed the histologic phenotypic characteristics close to clinical pancreatic adenocarcinomas (Supplementary Fig. S6). Furthermore, immunohistochemistry studies showed that there was no difference in cell proliferation (Ki-67 staining) between different treatment groups, while tumors from combination group showed the highest levels of apoptosis (c-caspase-3) (Fig. 5D), supporting the induction of apoptosis as a major mechanism behind their anti-tumor activities. In addition, none of animals showed any signs of stress, and there was no significant difference in body weights (Fig. 5E) and blood biochemistry profiles (Fig. 5F) among different treatments, suggesting that all drug treatments were safe to PDX-bearing mice.
Fig. 5.
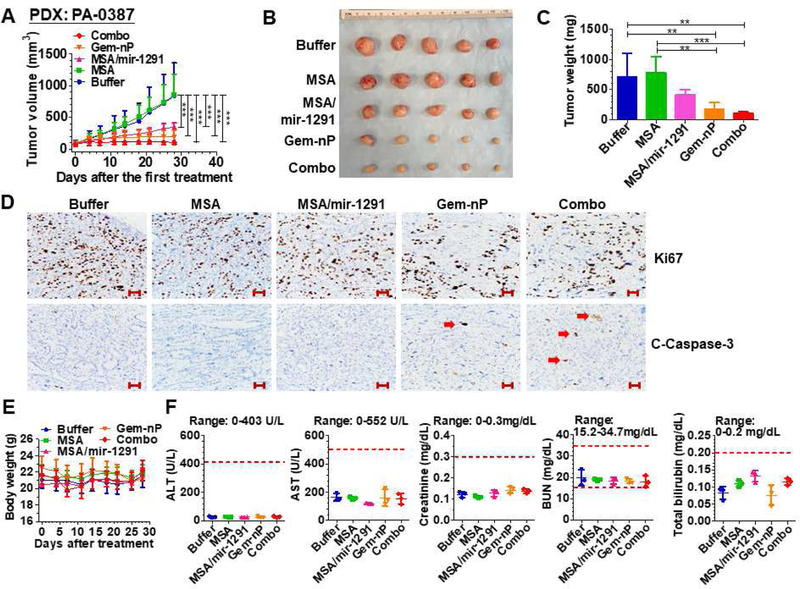
Bioengineered miR-1291 prodrug monotherapy and combination therapy with Gem-nP in PDX mouse model derived from clinical PDAC tissues (PA-0387). (A) PDX tumor growth was significantly suppressed by miR-1291 monotherapy or combination therapy, as compared to MSA or buffer control. *P < 0.05, **P < 0.01, and ***P < 0.001 (2-way ANOVA with Bonferroni post-tests). (B) Comparison of dissected tumor from mice with different treatments. (C) Weights of the dissected xenograft tumors. **P < 0.01, ***P < 0.001 (1-way ANOVA). (D) Representative IHC of PDX tumors stained with Ki-67 or cleaved caspase-3 antibodies. Combination treatment induced the highest degree of apoptosis (Red arrow: caspase-3 staining) while cell proliferation did not differ much among different treatment groups. Scale bar indicates 100µm. (E) Body weights were not different from each treatment. (F) Blood biochemistry profiles were not altered by any drug treatment. Values are mean ± SD (N = 5 per group, except N = 3 for blood chemistry profiles). The ranges of individual markers were marked as references.
Another PDX model, PA-0375, was utilized to critically assess the efficacy of miR-1291 prodrug monotherapy and combination therapy with Gem-nP, by following the same dosing regimens for PANC-1 xenograft models (Fig. 4A). Our data showed that treatment with either miR-1291 prodrug or Gem-nP, alone or in combination, was able to significantly suppress PA-0375 PDX tumor growth in mice (Fig. 6A–C). While it was not statistically significant different between mono- and combination therapy, combination therapy obviously produced the greatest extent of inhibition.
Fig. 6.
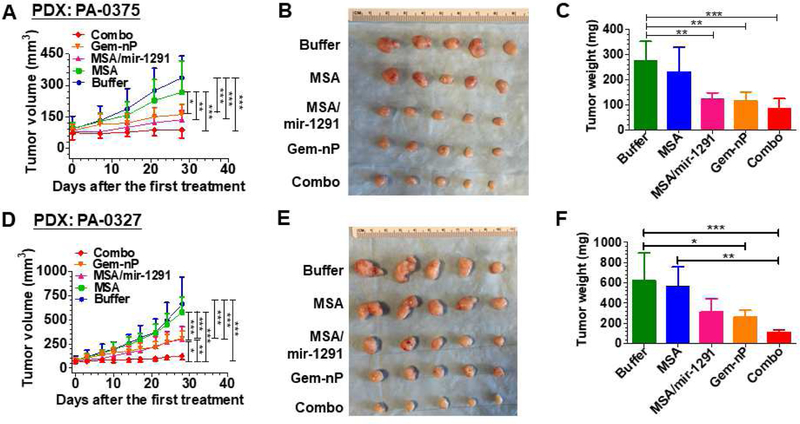
Combination therapy with miR-1291 prodrug and Gem-nP is proven the most effective in controlling tumor growth in two other PDX mouse models established with patients’ PDAC tissues (PA-0375: A, B, and C; PA-0327: D, E, and F). (A) and (D) PDX tumor growth was significantly suppressed by miR-1291 monotherapy or combination therapy, as compared to MSA or buffer control. *P<0.05, **P < 0.01, and ***P < 0.001 (2-way ANOVA with Bonferroni post-tests). (B)and (E)Comparison of dissected tumor from mice with different treatments. (C) and (F) Weights of the dissected xenograft tumors. *P < 0.05, **P < 0.01, ***P < 0.001 (1-way ANOVA). Values are mean ± SD (N = 5 per group). Because PA-0327 was more aggressive, dose of miR-1291 prodrug was increased to 20 µg/mouse for both monotherapy and combination therapy in this PDX model which, surprisingly, produced optimal outcomes.
Because the third PDX model, PA-0327, was more aggressive than the other two PDX models, we refined the dosing regimens by increasing miR-1291 prodrug dose to 20 µg/mouse for both mono- and combination therapy, while using the same dose of Gem-nP. Optimal outcomes were surprisingly observed (Fig. 6D–F). Compared to buffer and MSA treatment, monotherapy with miR-1291 prodrug or Gem-nP significantly reduced PA-0327 PDX to a similar level (~50%), which was indicated by tumor growth over time (Fig. 6D), visual inspection of dissected tumors (Fig. 6E) and quantitative measurement of tumor weights (Fig. 6F) at the end of the study. Most importantly, co-administration of miR-1291 prodrug and Gem-nP chemotherapeutics could suppress PDX progression to the greatest degree (> 80%) that was also significantly different from monotherapy (Fig. 6D). The strongest anti-tumor effects of combination therapy were also demonstrated by visual inspection (Fig. 6E) and weighting (Fig. 6F) of the dissected tumors. In addition, body weight of PDX-bearing animals did not show any significant difference among different treatment groups (Supplementary Fig. S7), indicating that all treatments were well tolerated in mice.
4. Discussion
Despite several decades of investigation into biology and treatment of PC, there is still a lack of deep understanding of the causes and pathogenesis of PC and more effective therapeutics, making PC one of the most lethal malignancies. Recent findings on the association of dysregulation of miRNAs with pathogenesis and progression of PC offer clue to developing miRNA-based therapies [12–16, 28]. After revealing a significant downregulation of miR-1291 in human PC tissues and a tumor suppressive action of miR-1291 [17], we demonstrated in the present study that miR-1291 prodrug monotherapy (10–20 µg/mouse or 0.5–1 mg/kg, i.v.) was as effective as Gem-nP (300–40 µg/mouse; 7.5/1 ratio; i.v.) for the control of PC growth in PANC-1 xenograft and PDX mouse models, while combination therapy offered the greatest degrees of suppression. The optimal outcome of combination treatment with miR-1291 and Gem-nP was associated with an increased level of apoptosis.
Research and development of new miRNA therapeutics are limited to the use of miRNA mimics made in test tubes by chemical synthesis, as well as the access to large quantities of miRNA agents required for animal and human studies [23]. Distinguished from the conventional synthetic miRNA agents, the present study investigated the efficacy of a bioengineered miR-1291 prodrug that was produced in living cells, and purified by a fast protein liquid chromatograph (FPLC) method to high degree of homogeneity on large scale [22, 29]. Biologic miR-1291 prodrug was selectively processed to mature miR-1291 in human PC cells and xenograft tumor tissues, which consequently modulated target gene expression and improved the efficacy of Gem-nP.
PANC-1 cell line is well known for its resistance to chemotherapy which is at least partly due to the overexpression of efflux transporter ABCC1/MRP1, and our previous studies have demonstrated the suppression of ABCC1 by miR-1291 in PANC-1 cells [19]. Most importantly, PANC-1 is a cell line directly derived from human pancreas/ducts with epithelioid carcinoma [30], whereas the AsPC-1 cell line is derived from mouse xenografts established with ascites cells of a patient with pancreatic cancer [31]. Therefore, the PANC-1 cell line should be closer and more relevant to human PC and were chosen establish xenograft mouse models for therapy studies. While both miR-1291 and Gem-nP monotherapy showed an overall effectiveness in controlling PANC-1 xenograft tumor growth, intra-individual variation was obvious. Even with a small sample size of six mice per group, three subjects were sensitive to miR-1291 and Gem-nP monotherapy, whereas the other three showed relatively poor responses. In contrast, combination therapy with miR-1291 and Gem-nP, while well tolerated in mice, was able to ubiquitously suppress tumor growth and to a greater extent than either Gem-nP or miR-1291 alone, demonstrating the advantage of combination treatment than monotherapy.
To better recapitulate the properties of original patient tumors and reflect the efficiency of new therapies in patients, an increasing number of PDX models have been used for studying cancer biology and assessing new drugs [25, 32–35]. In current study, PDX models from three different PC patients were established and utilized to evaluate miR-1291 monotherapy and combination treatment with Gem-nP. Consistent with findings from PANC-1 xenograft mouse models, miR-1291 prodrug was effective to reduce PDX tumor growth and improve the efficacy of Gem-nP, while histopathology analysis indicated that PDX tumor indeed better preserved the histological features of clinical PC than PANC-1 xenograft tumors (data no shown). As manifested by the increased c-caspase-3 levels, reduction of PDX progression by combination therapy was attributable to the induction of apoptosis, which is also in accordance with in vitro data. Moreover, different PDX tumor models unsurprisingly showed variable sensitivities to miR-1291 and Gem-nP treatment alone. The third PDX, PA-0327, seemed to be the most invasive, showed lower sensitivity to both Gem-nP and miR-1291 monotherapy. Generally, combination treatment with miR-1291 and Gem-nP reduced the final tumor sizes of PA-0327 by > 80%, supporting combination treatment including dose tailoring as an optimal strategy to combat PC. Nevertheless, subcutaneous PDX mouse models used in current study might not be as comprehensive and persuasive as orthotopic tumor models when considering pharmacokinetics properties of a drug, although subcutaneous PDX animal models are able to recapitulate the histologic and genotypic characteristics of human tumor and indicative of tumor responses to therapeutic drugs. To better demonstrate the pharmacokinetics and pharmacodynamics as well as therapeutic potential of miR-1291 mono- and combination treatment, further studies are highly warranted by using orthotopic PDX animal models before clinical investigation.
Optimal outcomes of miR-1291 plus Gem-nP combination treatment are inevitably due to multi-targeting in PC cells. Consistent with our previous findings [17], current study showed that miR-1291 alone enhanced apoptosis, as manifested by higher c-caspase-3/7 levels, which increased to even greater degrees when Gem-nP was co-administered. Gemcitabine is a nucleoside analogue that is converted to gemcitabine triphosphate, and subsequently inhibits DNA synthesis by incorporating into DNA, leading to G1/S cell cycle arrest and apoptosis [36, 37]. Paclitaxel reduces cell mitosis through stabilization of microtubes [38]. As indicated by the increase in γH2A.X and H3PS10 levels, actions of Gem-nP on DNA damage and mitotic arrest were obvious in PC cell lines. Likewise, co-administration of miR-1291 enhanced the levels of DNA damage and mitotic arrest, providing a good explanation of the sensitization of PC cells to Gem-nP by miR-1291. It is also notable that AsPC-1 cells were more sensitive to miR-1291 and chemotherapies than PANC-1 cells, in agreement with the finding on more striking increases in the expression of marker proteins at earlier time points (48 h post-treatment) in AsPC-1 cells.
The present study also validated a new target for miR-1291, ARID3B, an addition to those reported previously [17, 19–22]. While a miRNA generally reduces target gene expression, ARID3B was rather upregulated in PC cells by miR-1291. Although the precise mechanisms are unknown, there is growing evidence that miRNAs are also able to stimulate the expression of target genes through direct or indirect actions [39]. In addition, the role of ARID3B in cancer remains controversial although there are only a limited number of reports. Some studies showed that ARID3B promoted cancer cell proliferation or tumorigenesis [40–43], whereas other studies demonstrated that ARID3B played an important role in the induction of apoptosis [24, 44]. These studies differ much in the types of cancer cells investigated and reagents used, as well as study designs. The upregulation of ARID3B by miR-1291 not only agrees with the function of ARID3B in the induction of apoptosis showed by others [24, 44] but also the role of miR-1291 in the enhancement of apoptosis in PC cells reported by us very recently [17]. Therefore, the upregulation of pro-apoptotic ARID3B is likely one of many possible mechanisms behind the antitumor function of miR-1291, and further studies are warranted to improve our understanding of ARID3B functions in PC.
In summary, the present study demonstrated that a first-of-a-kind biologic miR-1291 prodrug was effective as Gem-nP in the control of PC tumor growth in PANC-1 xenograft and different PDX mouse models, while combination therapy with miR-1291 and Gem-nP suppressed xenograft tumor growth to the greatest degrees. Furthermore, all treatments were well tolerated in mice without any signs of hepatic and renal toxicity. Optimal efficacy of combination treatment was attributable to the enhanced induction of apoptosis, DNA damage and mitotic arrest. In addition, the induction of apoptosis by miR-1291 was associated with upregulation of ARID3B. These results suggest that biologic miR-1291 prodrug may be developed as a new anti-tumor agent for the treatment of PC, and co-administration of miR-1291 may augment the efficacy of current standard chemotherapy Gem-nP.
Supplementary Material
Highlights.
Bioengineered miR-1291 prodrug sensitizes pancreatic cancer cells to chemotherapy.
Induction of apoptosis by miR-1291 is associated with the upregulation of ARID3B.
MiR-1291 alone suppresses tumor growth in PANC-1 xenograft and PDX mouse models.
MiR-1291 augments the efficacy of chemotherapy for pancreatic cancer in vivo.
Acknowledgements
This study was supported by the grant (U01CA175315 to A.-M. Yu) from the National Cancer Institute and in part by the grant (R01GM113888 to A.-M. Yu) from the National Institute of General Medical Sciences, National Institutes of Health, as well as the 111 project (Grant: B16047) and the National Key Research and Development Program (Grant: 2017YFE0109900 to H. Bi).
Abbreviations:
- miR-1291
microRNA-1291
- PC
pancreatic cancer
- ARID3B
AT-rich interacting domain protein 3B
- Gem-nP
gemcitabine plus nab-paclitaxel
- PDX
patients derived xenograft
- miRNAs
microRNAs
- ncRNA
noncoding RNAs
- MRP1
multidrug resistance-associated protein 1
- GLUT1
glucose transporter protein type 1
- MUC1
mucin 1
- MSA
sephadex aptamer tagged methionyl-tRNA
- MREs
miRNA response elements
- c-caspase-3/7
cleaved caspase-3/7
- FPLC
fast protein liquid chromatograph
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2018, CA. Cancer J. Clin 68 (2018) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Tempero MA, Malafa MP, Al-Hawary M, Asbun H, Bain A, Behrman SW, Benson AB 3rd, Binder E, Cardin DB, Cha C, Chiorean EG, Chung V, Czito B, Dillhoff M, Dotan E, Ferrone CR, Hardacre J, Hawkins WG, Herman J, Ko AH, Komanduri S, Koong A, LoConte N, Lowy AM, Moravek C, Nakakura EK, O’Reilly EM, Obando J, Reddy S, Scaife C, Thayer S, Weekes CD, Wolff RA, Wolpin BM, Burns J, Darlow S, Pancreatic Adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology, J Natl Compr Canc Netw 15 (2017) 1028–1061. [DOI] [PubMed] [Google Scholar]
- [3].Hidalgo M, Pancreatic cancer, N. Engl. J. Med 362 (2010) 1605–1617. [DOI] [PubMed] [Google Scholar]
- [4].Burris H 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial, J. Clin. Oncol 15 (1997) 2403–2413. [DOI] [PubMed] [Google Scholar]
- [5].Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine, N. Engl. J. Med 369 (2013) 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mohammed A, Janakiram NB, Madka V, Li M, Asch AS, Rao CV, Current Challenges and Opportunities for Chemoprevention of Pancreatic Cancer, Curr. Med. Chem 25 (2018) 2535–2544. [DOI] [PubMed] [Google Scholar]
- [7].Tsai S, Evans DB, Therapeutic Advances in Localized Pancreatic Cancer, JAMA Surg 151 (2016) 862–868. [DOI] [PubMed] [Google Scholar]
- [8].Narayanan V, Weekes CD, Molecular therapeutics in pancreas cancer, World J Gastrointest Oncol 8 (2016) 366–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH, Therapeutic developments in pancreatic cancer: current and future perspectives, Nat Rev Gastroenterol Hepatol 15 (2018) 333–348. [DOI] [PubMed] [Google Scholar]
- [10].Ambros V, The functions of animal microRNAs, Nature 431 (2004) 350–355. [DOI] [PubMed] [Google Scholar]
- [11].Bartel DP, MicroRNAs: genomics, biogenesis, mechanism, and function, Cell 116 (2004) 281–297. [DOI] [PubMed] [Google Scholar]
- [12].Rupaimoole R, Slack FJ, MicroRNA therapeutics: towards a new era for the management of cancer and other diseases, Nat Rev Drug Discov 16 (2017) 203–222. [DOI] [PubMed] [Google Scholar]
- [13].Yu AM, Tian Y, Tu MJ, Ho PY, Jilek JL, MicroRNA Pharmacoepigenetics: Posttranscriptional Regulation Mechanisms behind Variable Drug Disposition and Strategy to Develop More Effective Therapy, Drug Metab. Dispos 44 (2016) 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yonemori K, Kurahara H, Maemura K, Natsugoe S, MicroRNA in pancreatic cancer, J. Hum. Genet 62 (2017) 33–40. [DOI] [PubMed] [Google Scholar]
- [15].Sicard F, Gayral M, Lulka H, Buscail L, Cordelier P, Targeting miR-21 for the therapy of pancreatic cancer, Mol. Ther 21 (2013) 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li J, Wu H, Li W, Yin L, Guo S, Xu X, Ouyang Y, Zhao Z, Liu S, Tian Y, Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-κB signaling, Oncogene 35 (2016) 5501–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tu M-J, Pan Y-Z, Qiu J-X, Kim EJ, Yu A-M, MicroRNA-1291 targets the FOXA2-AGR2 pathway to suppress pancreatic cancer cell proliferation and tumorigenesis, Oncotarget 7 (2016) 45547–45561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bi H-C, Pan Y-Z, Qiu J-X, Krausz KW, Li F, Johnson CH, Jiang C-T, Gonzalez FJ, Yu A-M, N-methylnicotinamide and nicotinamide N-methyltransferase are associated with microRNA-1291-altered pancreatic carcinoma cell metabolome and suppressed tumorigenesis, Carcinogenesis, 35 (2014) 2264–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pan Y-Z, Zhou A, Hu Z, Yu A-M, Small nucleolar RNA-derived microRNA hsa-miR-1291 modulates cellular drug disposition through direct targeting of ABC transporter ABCC1, Drug Metab. Dispos 41 (2013) 1744–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yamasaki T, Seki N, Yoshino H, Itesako T, Yamada Y, Tatarano S, Hidaka H, Yonezawa T, Nakagawa M, Enokida H, Tumor‐suppressive microRNA‐1291 directly regulates glucose transporter 1 in renal cell carcinoma, Cancer Sci 104 (2013) 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Luo H, Guo W, Wang F, You Y, Wang J, Chen X, Wang J, Wang Y, Du Y, Chen X, miR-1291 targets mucin 1 inhibiting cell proliferation and invasion to promote cell apoptosis in esophageal squamous cell carcinoma, Oncol. Rep 34 (2015) 2665–2673. [DOI] [PubMed] [Google Scholar]
- [22].Li MM, Addepalli B, Tu MJ, Chen QX, Wang WP, Limbach PA, LaSalle JM, Zeng S, Huang M, Yu AM, Chimeric MicroRNA-1291 Biosynthesized Efficiently in Escherichia coli Is Effective to Reduce Target Gene Expression in Human Carcinoma Cells and Improve Chemosensitivity, Drug Metab. Dispos 43 (2015) 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ho PY, Yu AM, Bioengineering of noncoding RNAs for research agents and therapeutics, Wiley Interdiscip Rev RNA 7 (2016) 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Joseph S, Deneke VE, Dahl KDC, ARID3B induces tumor necrosis factor alpha mediated apoptosis while a novel ARID3B splice form does not induce cell death, PLoS ONE, 7 (2012) e42159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG, Patient-derived tumour xenografts as models for oncology drug development, Nat Rev Clin Oncol 9 (2012) 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Siolas D, Hannon GJ, Patient-derived tumor xenografts: transforming clinical samples into mouse models, Cancer Res 73 (2013) 5315–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Voskoglou-Nomikos T, Pater JL, Seymour L, Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models, Clin. Cancer Res 9 (2003) 4227–4239. [PubMed] [Google Scholar]
- [28].Rachagani S, Macha MA, Heimann N, Seshacharyulu P, Haridas D, Chugh S, Batra SK, Clinical implications of miRNAs in the pathogenesis, diagnosis and therapy of pancreatic cancer, Adv. Drug Deliv. Rev 81 (2015) 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen QX, Wang WP, Zeng S, Urayama S, Yu AM, A general approach to high-yield biosynthesis of chimeric RNAs bearing various types of functional small RNAs for broad applications, Nucleic Acids Res 43 (2015) 3857–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lieber M, Mazzetta J, Nelson‐Rees W, Kaplan M, Todaro G, Establishment of a continuous tumor‐cell line (PANC‐1) from a human carcinoma of the exocrine pancreas, Int. J. Cancer 15 (1975) 741–747. [DOI] [PubMed] [Google Scholar]
- [31].Chen W, Horoszewicz J, Leong S, Shimano T, Penetrante R, Sanders W, Berjian R, Douglass H, Martin EW, Chu T, Human pancreatic adenocarcinoma: in vitro and in vivo morphology of a new tumor line established from ascites, In Vitro 18 (1982) 24–34. [DOI] [PubMed] [Google Scholar]
- [32].Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, An in vivo platform for translational drug development in pancreatic cancer, Clin. Cancer Res 12 (2006) 4652–4661. [DOI] [PubMed] [Google Scholar]
- [33].Knudsen ES, Balaji U, Mannakee B, Vail P, Eslinger C, Moxom C, Mansour J, Witkiewicz AK, Pancreatic cancer cell lines as patient-derived avatars: genetic characterisation and functional utility, Gut 67 (2018) 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang L, Wang P, Qin Y, Cong Q, Shao C, Du Z, Ni X, Li P, Ding K, RN1, a novel galectin-3 inhibitor, inhibits pancreatic cancer cell growth in vitro and in vivo via blocking galectin-3 associated signaling pathways, Oncogene 36 (2017) 1297. [DOI] [PubMed] [Google Scholar]
- [35].Delitto D, Pham K, Vlada AC, Sarosi GA, Thomas RM, Behrns KE, Liu C, Hughes SJ, Wallet SM, Trevino JG, Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements, Am. J. Pathol 185 (2015) 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yip-Schneider MT, Sweeney CJ, Jung S-H, Crowell PL, Marshall MS, Cell cycle effects of nonsteroidal anti-inflammatory drugs and enhanced growth inhibition in combination with gemcitabine in pancreatic carcinoma cells, J. Pharmacol. Exp. Ther . 298 (2001) 976–985. [PubMed] [Google Scholar]
- [37].Huang P, Plunkett W, Induction of apoptosis by gemcitabine, Semin. Oncol (22) 1995, pp. 19–25. [PubMed] [Google Scholar]
- [38].Horwitz SB, Cohen D, Rao S, Ringel I, Shen H-J, Yang C, Taxol: mechanisms of action and resistance, J. Natl. Cancer Inst. Monographs (1993) 55–61. [PubMed]
- [39].Vasudevan S, Posttranscriptional upregulation by microRNAs, Wiley Interdiscip Rev RNA 3 (2012) 311–330. [DOI] [PubMed] [Google Scholar]
- [40].Kobayashi K, Era T, Takebe A, Jakt LM, Nishikawa S, ARID3B induces malignant transformation of mouse embryonic fibroblasts and is strongly associated with malignant neuroblastoma, Cancer Res 66 (2006) 8331–8336. [DOI] [PubMed] [Google Scholar]
- [41].Cowden Dahl KD, Dahl R, Kruichak JN, Hudson LG, The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells, Neoplasia 11 (2009) 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bobbs A, Gellerman K, Hallas WM, Joseph S, Yang C, Kurkewich J, Cowden Dahl KD, ARID3B Directly Regulates Ovarian Cancer Promoting Genes, PLoS ONE 10 (2015) e0131961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liao TT, Hsu WH, Ho CH, Hwang WL, Lan HY, Lo T, Chang CC, Tai SK, Yang MH, let-7 Modulates Chromatin Configuration and Target Gene Repression through Regulation of the ARID3B Complex, Cell Rep 14 (2016) 520–533. [DOI] [PubMed] [Google Scholar]
- [44].Pratama E, Tian X, Lestari W, Iseki S, Ichwan SJ, Ikeda M-A, Critical role of ARID3B in the expression of pro-apoptotic p53-target genes and apoptosis, Biochem. Biophys. Res. Commun 468 (2015) 248–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.